

Abstract
Congenital muscular dystrophies with hypoglycosylation of α-dystroglycan (α-DG) are a heterogeneous group of disorders often associated with brain and eye defects in addition to muscular dystrophy. Causative variants in 14 genes thought to be involved in the glycosylation of α-DG have been identified thus far. Allelic mutations in these genes might also cause milder limb-girdle muscular dystrophy phenotypes. Using a combination of exome and Sanger sequencing in eight unrelated individuals, we present evidence that mutations in guanosine diphosphate mannose (GDP-mannose) pyrophosphorylase B (GMPPB) can result in muscular dystrophy variants with hypoglycosylated α-DG. GMPPB catalyzes the formation of GDP-mannose from GTP and mannose-1-phosphate. GDP-mannose is required for O-mannosylation of proteins, including α-DG, and it is the substrate of cytosolic mannosyltransferases. We found reduced α-DG glycosylation in the muscle biopsies of affected individuals and in available fibroblasts. Overexpression of wild-type GMPPB in fibroblasts from an affected individual partially restored glycosylation of α-DG. Whereas wild-type GMPPB localized to the cytoplasm, five of the identified missense mutations caused formation of aggregates in the cytoplasm or near membrane protrusions. Additionally, knockdown of the GMPPB ortholog in zebrafish caused structural muscle defects with decreased motility, eye abnormalities, and reduced glycosylation of α-DG. Together, these data indicate that GMPPB mutations are responsible for congenital and limb-girdle muscular dystrophies with hypoglycosylation of α-DG.
Introduction
Congenital muscular dystrophy (CMD) represents a clinically and genetically heterogeneous group of neuromuscular disorders characterized by the onset of muscle weakness, often associated with limb contractures, at birth or within the first few months of life. A major subgroup of CMD is associated with mutations in genes involved in the glycosylation of α-dystroglycan (α-DG) and is commonly referred to as a secondary dystroglycanopathy. α-DG is a peripheral-membrane protein that is part of the dystrophin-associated glycoprotein complex, which provides a link between proteins located in the extracellular matrix and proteins located in the cytoplasm immediately beneath the plasma membrane. Dystroglycan is translated from a single mRNA DAG1 (MIM 128239) and cleaved into α and β subunits.1,2 Extracellular α-DG noncovalently binds to the transmembrane protein β-dystroglycan (β-DG), which in turn associates with intracellular proteins involved in force and signal transduction. In contrast, α-DG binds a number of extracellular ligands, including laminins, perlecan, agrin, neurexin, pikachurin, and slit.2–5 Extensive and tissue-specific glycosylation is essential for the binding of α-DG to these extracellular-matrix ligands.6–10 Recessive mutations in DAG1 have been identified in a single case of mild muscular dystrophy, suggesting the existence of a primary subset of dystroglycanopathies.11
Mutations in 14 genes are currently known to cause secondary dystroglycanopathies. POMT1 (MIM 607423), POMT2 (MIM 607439), and POMGNT1 (MIM 606822) encode proteins with confirmed involvement in O-mannosylation.3,4,12,13 DPM2 (MIM 603564), DPM3 (MIM 605951), and DOLK (MIM 610746) encode proteins involved in the synthesis of dolichol-phosphate mannose (Dol-P-Man), which is the essential mannose donor required for mannosylation.14–16 LARGE (MIM 603590) encodes a bifunctional enzyme with xylosyl and glucuronyltransferase activities.17,18 B3GNT1 (MIM 605581) and B3GALNT2 (MIM 610194) encode glycosyltransferases whose exact role in O-mannosylation remains unclear.19,20 On the basis of protein sequence homology, FKTN (MIM 607440), FKRP (MIM 606596), and GTDC2 (MIM 614828) are thought to encode glycosyltransferases, although their enzymatic function has not been conclusively demonstrated.21–23 Finally, mutations in the largely uncharacterized genes ISPD (MIM 614631) and TMEM5 (MIM 605862) can also cause secondary dystroglycanopathies.24–28
Mutations in the aforementioned genes were originally identified almost invariably in individuals affected by severe CMD variants—with associated structural brain and eye abnormalities—such as Walker-Warburg syndrome (WWS [MIM 236670]), Fukuyama CMD (FCMD [MIM 253800]), and muscle-eye-brain disease (MEB [MIM 253280]) or forms with exclusive skeletal-muscle involvement, such as CMD type 1C (MIM 606612). Subsequently, milder allelic mutations in several of these genes were identified in later-onset forms of limb-girdle muscular dystrophy (LGMD), such as in the common LGMD2I variant (MIM 607155). Muscle pathology in all of these conditions shares dystrophic changes and defective glycosylation of α-DG, which can be assessed on immunohistochemistry or immunoblot with IIH6 or VIA4-1 antibodies, which recognize disease-relevant glycoepitopes on α-DG.9
The dystroglycanopathies represent one of the most common forms of muscular dystrophy, and mutations in POMT1, POMT2, POMGNT1, FKRP, or ISPD are the most frequent cause.24,27 Mutations in the known genes cannot explain all cases.27,29–31 In this study, we identified mutations in guanosine diphosphate mannose (GDP-mannose) pyrophosphorylase B (GMPPB) in eight dystroglycanopathy cases affected by either CMD or LGMD phenotypes. Functional analyses using cellular and zebrafish models implicated GMPPB (also known as mannose-1-phosphate guanyltransferase beta [Enzyme Commission number 2.7.7.13]) in the glycosylation of α-DG.
Subjects and Methods
Clinical Presentation of Subjects
The main clinical features of the eight unrelated cases (P1–P8) reported in this manuscript are presented in Table 1. The spectrum of severity observed in these cases ranges from a child with a classical CMD presentation characterized by muscle weakness at birth and motor and cognitive developmental delays including ataxia, absent speech development, and inability to walk unsupported (P1) to children presenting in the first few months of life with hypotonia, muscle weakness, delayed acquisition of independent mobility, and mild intellectual disability but normal brain structure (P3 and P4) to children presenting in the first few years of life with mild limb-girdle weakness and mild intellectual disability (P2 and P8) to a child with normal cognitive function following a LGMD disease course (P7). Commonly associated features included epilepsy (P2, P5, P6, and P8), microcephaly (P1, P2, P3, and P8), cataracts (P3, P4, and P8), strabismus (P3, P4, and P5) (Figure S1, available online), and nystagmus (P4 and P8). Brain MRI showed a range of abnormalities, including structural defects such as cerebellar and pontine hypoplasia (Figure 1), but it was normal in P2, P3, P4, and P8 (it was not performed in P7), who were all affected by a milder variant. Progression of muscle weakness was clearly observed, and P4 lost ambulation during childhood. Evidence of combined cardiorespiratory compromise was evident by the age of 10 years in P8, and features of cardiac involvement, including a long QT interval and left ventricular dilation, were documented in P3 and P4, respectively. P5 and P6, affected by CMD with cerebellar involvement, have previously been described in detail (affected individuals 1 and 2, respectively, in Messina et al.32).
Table 1.
Clinical Features of Individuals with GMPPB Mutations
|
Case |
||||||||
|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5a | P6a | P7 | P8 | |
| Gender | male | female | male | female | female | female | male | male |
| Current age | 8 years | 12 years | 16 years | 13 years | died at 14 years | 10 years | 6 years | 18 years |
| Ethnicity | Pakistani | Indian | Mexican | Mexican | Italian | Italian | English | Egyptian |
| Prenatal findings | oligohydramnios | none | none | decreased fetal movement | decreased fetal movement | decreased fetal movement | none | none |
| Age at presentation | birth | birth | 2 weeks | birth | 4 months | 4 months | 4 years | 2.5 years |
| Presenting symptoms | increased tone; microcephaly; cleft palate; feeding difficulties | hypotonia; microcephaly | increased tone, then hypotonia; microcephaly; cataracts; torticollis; ileal atresia | hypotonia; feeding difficulties | poor head control | poor head control | mild exercise intolerance | difficulty in climbing stairs; microcephaly |
| Maximal motor ability | walking with support (at 3 years) | running | walking (at 3.5 years) | walking (at 3 years) | unable to sit | sitting (at 2 years) | running | running |
| Main neurological features | severe intellectual delay; sensorineural hearing loss; ataxia | mild intellectual delay; epilepsy | mild intellectual delay | mild intellectual delay | severe intellectual delay; drug-resistant epilepsy; motor delay | severe intellectual delay; drug-resistant epilepsy; motor delay | normal cognitive function | mild intellectual delay; epilepsy |
| Ophthalmologic findings | retinal dysfunction (on electroretinogram) | none | cataracts; strabismus; ptosis | cataracts; strabismus; intermittent nystagmus; ptosis | strabismus | none | none | cataracts; nystagmus |
| Cardiorespiratory findings | none | none | long QT syndrome | left ventricular dilatation | none | none | none | wandering atrial pacemaker; cardiomyopathy; respiratory insufficiency |
| Maximum CK (U/l) | 2,974 | 4,504 | 7,323 | 8,450 | 630 | 720 | 3,000 | 5,200 |
| Brain MRI findings | pontine and cerebellar hypoplasia | no structural abnormality | no structural abnormality | no structural abnormality | cerebellar hypoplasia | cerebellar hypoplasia | not performed | no structural abnormality |
| Diagnosisb | MEB- and/or FCMD-like | LGMD-MR | CMD-MR | CMD-MR | CMD-CRB | CMD-CRB | LGMD | LGMD-MR |
Abbreviations are as follows: CK, creatine kinase; CMD-CRB, congenital muscular dystrophy with cerebellar involvement; CMD-MR, congenital muscular dystrophy with mental retardation; FCMD, Fukuyama congenital muscular dystrophy; LGMD-MR, limb-girdle muscular dystrophy with mental retardation; MEB, muscle-eye-brain disease; and WWS, Walker-Warburg syndrome.
The phenotypes of P5 and P6 have previously been described in detail.32
Diagnostic categories have previously been described.31
Figure 1.
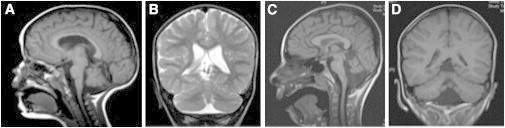
MRI Reveals Structural Brain Abnormalities in P1 and P6
On sagittal views of T1-weighted MRI, there is evidence of cerebellar hypoplasia in P6 (A) at 2 years of age and pontine and cerebellar hypoplasia in P1 (C) at 6 years of age. Coronal views reveal evidence of cerebellar inferior vermian hypoplasia on T2-weighted MRI in P6 (B) and on T1-weighted MRI in P1 (D).
The institutional review board (IRB) of the University College London Institute of Child Health and Great Ormond Street Hospital in the United Kingdom approved the study of P1, P2, and P7 and all cellular work. Informed consent and local-review-board approval were in accordance with the UK10K project ethical framework. We recruited and analyzed P3 under Children’s National Medical Center (CNMC) IRB protocol 2405, which was reviewed and approved by the Office for the Protection of Human Subjects at the CNMC, Washington, DC, USA. P3 provided informed consent for publication of clinical photographs. The IRB of the University of Iowa approved the study of P4 (IRB ID 200510769). The study of P5 and P6 was performed under ethical approval number 489/2012 and protocol number GUP1100A1. The study of P8 was approved by the Sydney Children’s Hospitals Network Human Research Ethics Committee.
Genetic Analysis
We sequenced the exome of P1 within the UK10K project and called and filtered variants as previously described.26
P3 underwent exome sequencing at the CNMC according to the following protocol: 1 μg of DNA was sheared to 200 bp with a Covaris S220 (Covaris) and was prepared with TruSeq DNA Sample Preparation and TruSeq Exome Enrichment Kits (Illumina) according to the manufacturer’s instructions. We sequenced it by using a HiScanSQ (Illumina) with 200 cycles and paired-end multiplexed sequencing with read lengths of 100 bp in each direction. The mean coverage was 50×. We used CASAVA 1.8.1 for initial demultiplexing and file conversion, and we aligned sequences to the human reference genome (UCSC Genome Browser hg19) with the use of NextGENe (SoftGenetics) and annotated calls by using the dbNSFP database.33
P5 and P6 were exome sequenced as previously described, and genetic variants were further prioritized on the basis of a filter consisting of glycosylation genes.24,34
For P8, exome capture was performed on genomic DNA with the Agilent Whole Exome SureSelect v.2 kit according to the manufacturer’s instructions. Captured exome DNA was then subjected to Illumina sequencing. Reads were processed by Picard and aligned to the human reference genome (hg19) with the Burrows-Wheeler Aligner,35,36 and then calling of single-nucleotide variants (SNVs) and small indels was performed with the GATK toolkit.37 Variants were annotated with a modified version of the Ensembl Variant Effect Predictor38 and filtered for inheritance patterns and predicted functional severity with the xBrowse web server.
We used standard Sanger-sequencing protocols to verify all the mutations identified by exome sequencing and to discover mutations in P2, P4, and P7 (data not shown). Primer sequences are available upon request.
Muscle Pathology and Immunoblots
Histological, histochemical, and immunohistochemical studies on skeletal-muscle biopsies were performed as described previously,20,39 except that in addition, an antibody against the 300 kDa fragment of laminin-α2 (4H8-2)40 was used.
Immunoblotting of P1 muscle protein lysate and P1, P2, P4, P5, and P6 fibroblast protein lysate was performed as previously described.20 For P4 muscle-biopsy samples, we enriched glycoproteins with wheat germ agglutinin (WGA) and immunoblotted these on polyvinylidene difluoride (PVDF) membranes as described.9 Membranes were developed with infrared-dye-conjugated secondary antibodies (Pierce Antibodies) and scanned with an Odyssey infrared imaging system (LI-COR Bioscience). Laminin-overlay assays were performed as previously described.9 The monoclonal antibodies to the fully glycosylated form of α-DG (IIH6), core α-DG (G6317), and β-DG (AP83) have been characterized previously.25,41,42
Expression Studies of GMPPB
In order to study the transcription profile of GMPPB, we reverse transcribed an adult tissue RNA panel (Life Technologies) and fetal tissue RNA panel (Agilent Technologies) by using a one-step RT-PCR kit (QIAGEN) according to the manufacturer’s instructions. Primers are listed in Table S1.
In order to assess the localization of wild-type GMPPB and GMPPB carrying the amino acid changes observed in affected individuals, we amplified its coding sequence (RefSeq accession number NM_021971.1) from a full-length cDNA IMAGE clone of human GMPPB (IRAUp969D1013D, Source Bioscience). The product was cloned into the pcDNA3.1/V5-His TOPO expression vector (Life Technologies) according to the manufacturer’s instructions. We introduced mutations identified in the individuals with CMD into GMPPB pcDNA 3.1/V5-His TOPO by using QuickChange II site-directed mutagenesis (Agilent Technologies). Primers are listed in Table S2. We assessed the localization of the V5-FITC-tagged wild-type and altered GMPPB constructs in cultured C2C12 myoblasts as previously described20 by using the V5-FITC antibody (Life Technologies).
Effect of GMPPB Mutations on α-DG Glycosylation Assessed by Flow Cytometry
In order to quantify the amount of α-DG glycosylation on fibroblasts of available affected individuals and healthy control fibroblasts, we used IIH6 (Merck Millipore) to perform flow cytometry as previously described.20 To calculate the mean fluorescence intensity (MFI) of each population of fibroblasts, we first subtracted the background-intensity values obtained when cells were incubated only with IgM (biotinylated) and streptavidin-PE. The addition of the primary antibody allowed us to obtain the intensity values (calculated with FlowJo software [Tree Star]) for the IIH6-positive fibroblasts. Statistical analysis was performed with the use of unpaired two-tailed t tests.
Zebrafish Knockdown
We extracted zebrafish RNA at different developmental stages and carried out reverse transcription and PCR as previously described.20 Primer sequences are in Table S3. To knock down zebrafish gmppb, we obtained a splice-blocking morpholino oligonucleotide (MO) (sequence 5′-GGACCAGCTGAAAACAGAAACAGAT-3′) from Gene Tools. This was injected into 1- to 4-cell stage Tuebingen Long Fin zebrafish embryos. Unless otherwise stated, it was coinjected with p53 MO. The sequences of the p53 and dag1 MOs have previously been described.43,44
To assess muscle pathology in zebrafish, we used immunofluorescent imaging as previously described44 to study the following proteins: filamentous actin with the use of Alexa-Fluor-594-conjugated phalloidin (Life Technologies) and β-DG (monoclonal, NCL-b-DG from Leica Microsystems). We also performed an Evans blue dye (EBD) assay and immunoblotting of zebrafish microsome-pellet proteins as previously described and quantified the immunoblot results by using ImageJ software.20,45 To assess the significance of the results of the EBD assay, we used unpaired two-tailed t tests.
Results
Identification and Characterization of GMPPB Variants in Individuals Affected by CMD
We performed exome sequencing in P1, who is affected by a severe form of CMD with brain involvement, as part of the UK10K rare cohort, and variants were filtered as outlined in Table S4. We identified 22 high-quality loss-of-function heterozygous variants in P1. Only two of these candidate genes showed possible compound heterozygosity and are therefore consistent with the expected recessive mode of inheritance with unrelated parents: alpha 1, 2-mannosidase (MAN1B1 [MIM 604346]) and GMPPB.
Mutations in MAN1B1 can cause nonsyndromic and syndromic intellectual disability without microcephaly.46 Because neither muscular dystrophy nor brain malformation is a feature of MAN1B1 mutations, it is unlikely that these variants explain the phenotype of P1. GMPPB catalyzes the formation of GDP-mannose from mannose-1-phosphate and GTP.47 GDP-mannose is required in four glycosylation pathways (Figure 2), including O-mannosylation of membrane and secretory glycoproteins, such as α-DG. Mutations in other members of this pathway are known to cause dystroglycanopathy.14,15 We therefore considered variants in GMPPB as the likely cause of disease in P1.
Figure 2.
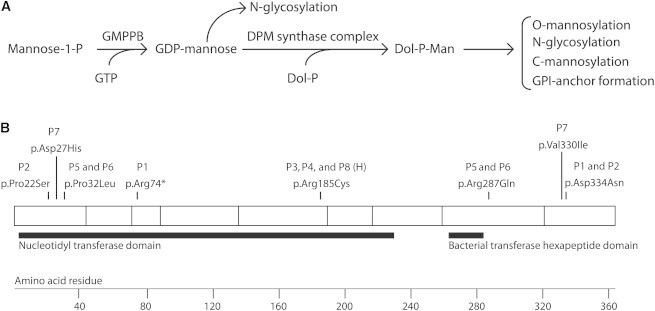
GMPPB Function, Structure, and Identified Substitutions
(A) The function of GMPPB in glycosylation pathways.
(B) GMPPB has 360 amino acids and two predicted Pfam functional domains: a nucleotidyl transferase domain and a bacterial transferase hexapeptide domain. In this schematic diagram, the blocks represent regions encoded by exons and the substitutions identified in individuals with dystroglycanopathy are shown. The following abbreviation is used: H, homozygous.
RT-PCR of human fetal and adult RNA from various tissues showed that GMPPB is transcribed as two isoforms in humans (RefSeq NM_013334.2 and NM_021971.1). In contrast to NM_013334.2, NM_021971.1 was strongly expressed in all tested tissues, including brain and skeletal muscle (Figure S2). All listed mutations correspond to NM_021971.1.
The GMPPB mutations in P1 are c.220C>T (p.Arg74∗) and c.1000G>A (p.Asp334Asn). Through an international collaborative effort, we identified recessive GMPPB mutations in seven further dystroglycanopathy cases (P2–P8) with phenotypes ranging from severe CMD with the inability to walk to severe intellectual disability and epilepsy to later-onset phenotypes resembling LGMD with only mild intellectual disability or no evidence of brain involvement. P2 has the compound heterozygous GMPPB mutations c.64C>T (p.Pro22Ser) and c.1000G>A (p.Asp334Asn). P3, P4, and P8 carry the homozygous c.553C>T (p.Arg185Cys) mutation. P5 and P6 carry the c.95C>T (p.Pro32Leu) and c.860G>A (p.Arg287Gln) mutations, and P7 has the heterozygous mutations c.79G>C (p.Asp27His) and c.988G>A (p.Val330Ile). No MAN1B1 variants were found in the other individuals who were exome sequenced (P3, P5, P6, and P8). None of these individuals are related, and although the recurrent mutations in individuals of related ethnic origin could be due to founder effects (i.e., P1 and P2 share one variant, and P3 and P4 are homozygous for the same change), we also found the latter change in P8, who is of a completely different origin. The asymptomatic parents of P1, P2, P5, P6, P7, and P8 and the mother of P4 were found to be heterozygous carriers of the variants described.
Of the eight variants described, five have not been previously reported. The three that have (c.860G>A, c.79G>C, and c.988G>A) are very rare and have a minor allele frequency of less than or equal to 0.001 according to data from the UK10K twins cohort or the ClinSeq study (Table S5). This is compatible with CMD’s frequency, which has been estimated to be around 1 in 50,000–100,000.48
We performed alignments by using the human GMPPB sequence against the genomes of five diverse eukaryotic species and found that each has a GMPPB ortholog that is at least 63.8% identical to the human sequence (Table S6 and Figure S3). We found that of the eight variants described, five (c.64C>T, c.95C>T, c.1000G>A, c.553C>T, and c.988G>A) affect amino acids that are conserved throughout all species tested, suggesting functional importance (Figure S3). Although the amino acid affected by the c.220C>T mutation in P1 is not highly conserved, it is located in the nucleotidyl transferase domain (Pfam ID PF00483) (Figure 2), and the mutation is predicated to cause a severely truncated protein in addition to nonsense-mediated mRNA decay. Of the eight variants described, five are within the nucleotidyl transferase domain. Interestingly, all affected individuals have at least one alteration in this domain (Figure 2).
Individuals with GMPPB Mutations Have Hypoglycosylated α-DG in Muscle Biopsy
To assess the muscle pathology, we examined muscle biopsies of all affected individuals except P5 and P8. In all cases, the muscle biopsies showed features of a muscular dystrophy with abnormal variation in fiber size, necrosis, regeneration, excess endomysial connective tissue, and an increase in internal nuclei and inflammatory infiltrates (Figure 3 and Figures S4 and S5). Immunolabeling of laminin-α2 with antibodies to both the 80 and 300 kDa fragments showed reduced labeling of some fibers, but not in all cases (data not shown). Labeling of β-DG was invariably normal, whereas labeling with the IIH6 antibody was variably reduced. In P3 and P4, the reduction of the glycosylated epitope on α-DG was also confirmed with the VIA4-1 antibody, whereas the staining obtained with α-DG GT20ADG, a goat polyclonal antibody, which recognizes the core protein, was normal (Figure S4).
Figure 3.
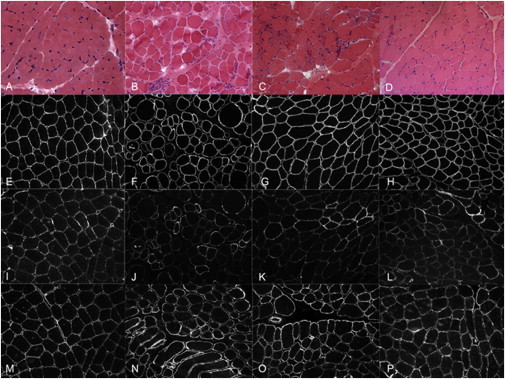
α-DG Glycosylation Is Reduced in Muscle Biopsies of Affected Individuals with GMPPB Mutations
An examination of skeletal-muscle cryosections from the control (A, E, I, and M), P1 (B, F, J, and N), P2 (C, G, K, and O), and P7 (D, H, L, and P) revealed that glycosylated α-DG immunolabeling (assessed with IIH6) was moderately reduced in individuals (P1, P2, and P7) with GMPPB mutations and that β-DG and core α-DG were well preserved. Stains are as follows: hematoxylin and eosin (A–D), β-DG (E–H), α-DG IIH6 (I–L), and α-DG core (M–P).
Immunoblots from skeletal-muscle biopsies confirmed the reduction in glycosylated α-DG in P1, P4, and P8 compared to the control (Figure 4 and Figure S6); in P4, the lower-molecular-weight band identified by the core α-DG antibody and the reduction with the laminin overlay also suggested hypoglycosylated α-DG (Figure 4). Immunoblotting of fibroblast lysate with the IIH6 antibody showed that there was substantially less α-DG glycosylation in P1, P2, P4, and P5 than in controls and only a lesser reduction in P6 (Figure 4). Flow cytometry showed that there was significantly less glycosylated α-DG in fibroblasts from P1, P2, P4, P5, P6, and P7 (p < 0.05) than in controls, as shown by the MFI of IIH6, as well as the percentage of fibroblasts that were positive for the IIH6 epitope (Figure 4).
Figure 4.
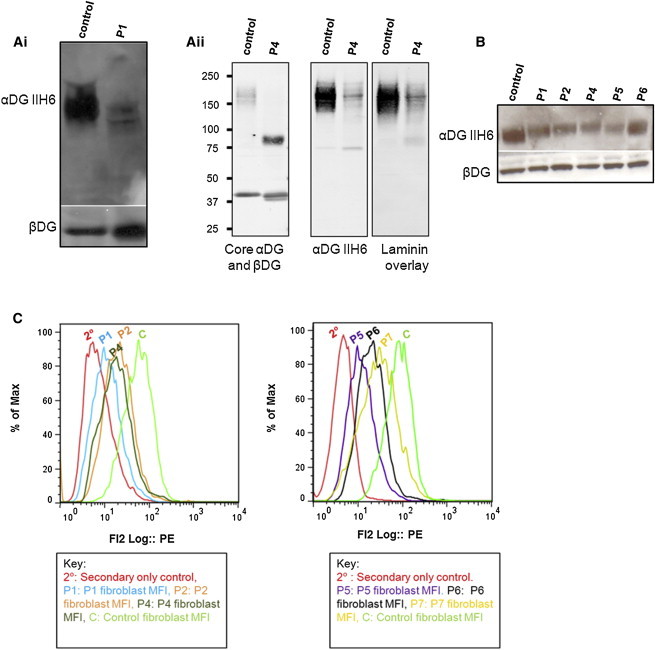
Immunoblots and Flow Cytometry Confirm that α-DG Glycosylation Is Reduced in Muscle and Fibroblasts of Affected Individuals with GMPPB Mutations
(A) Immunoblot analysis of skeletal-muscle protein lysates from P1 and P4. (Ai) The membrane was incubated with IIH6 and β-DG antibodies. P1 shows a reduction in α-DG glycosylation. (Aii) For P4, WGA-enriched skeletal-muscle homogenates were used, and the immunoblot was probed with IIH6, α-DG core, and β-DG antibodies, as well as by laminin overlay. β-DG appears as a possible doublet in P1 and P4 (and P8, Figure S6).
(B) Immunoblot analysis of fibroblast protein lysate from P1, P2, P4, P5, and P6. The membrane was incubated with IIH6 and β-DG antibodies. α-DG glycosylation is reduced in fibroblasts of individuals with GMPPB mutations.
(C) Flow cytometry of fibroblasts revealed reduced α-DG glycosylation for P1, P2, P4, P5, P6, and P7. This histogram shows the MFI of IIH6 staining for a secondary-only control, P1, P2, P4, P5, P6, P7, and a control fibroblast cell line. The MFI of the populations positive for IIH6 were 88.95 for the control, 19.6 for P1, 30.15 for P2, 26.34 for P4, 14.60 for P5, 28.63 for P6, and 45.3 for P7 (n = 6 for all). There was a statistically significant (p < 0.05) reduction in α-DG glycosylation in fibroblasts from the six individuals tested compared to controls, as determined by the average MFI of IIH6. The percentage of fibroblasts (out of a minimum of 10,000) that were positive for the IIH6 epitope was 85% for the control, 27% for P1, 16% for P2, 45% for P4, 60% for P5, 50% for P6, and 65% for P7. A IIH6-positive gate was set up with a negative control (where primary antibody was omitted) for all fibroblast cell lines tested and the control.
Next, we transfected fibroblasts from P1 with wild-type GMPPB cDNA to determine whether the reduced amount of α-DG glycosylation of this individual was related to dysfunctional GMPPB. The amount of glycosylated α-DG was increased as determined by the average MFI of IIH6 with the use of flow cytometry (Figure S7 and Table S7), suggesting that wild-type GMPPB complemented the altered GMPPB. Similar to the wild-type healthy control fibroblasts, 23.5% of the total transfected population of fibroblasts had a higher MFI (76.2) than the untransfected fibroblasts. Transfection of the wild-type gene into control fibroblasts did not alter the MFI of IIH6 (data not shown). These results suggest that GMPPB mutations are the cause of reduced α-DG glycosylation for P1.
In view of the role of GMPPB in multiple glycosylation reactions (Figure 2), we tested N-glycosylation of serum transferrin, abnormal in congenital disorders of glycosylation (CDG). For all affected individuals, routine diagnostic screening for CDG by transferrin isoelectric focusing showed normal results, which was confirmed by highly sensitive mass spectrometry of intact transferrin for P6 (Figure S8).
Subcellular Localization of Wild-Type and Mutant GMPPB
To further elucidate the effects of the missense mutations found in P1–P8, we introduced the mutations c.1000G>A, c.64C>T, c.553C>T, c.95C>T, c.860G>A, c.79G>C, and c.988G>A into wild-type GMPPB and studied the localization of the resultant altered proteins in C2C12 myoblasts. Wild-type GMPPB, a soluble enzyme,49 localized to the cytoplasm (Figure 5). The mutations c.1000G>A (found in P1 and P2), c.95C>T and c.860G>A (both found in P5 and P6), and c.988G>A (found in P7) caused the protein to form aggregates within the cytoplasm (Figure 5). The mutation c.64C>T (found in P2) caused GMPPB to aggregate near membrane protrusions into the cytoplasm (Figure 5). The mutations c.553C>T (found in P3, P4, and P8) and c.79G>C (found in P7) caused GMPPB to remain evenly distributed throughout the cytoplasm and to have no discernible changes compared with wild-type GMPPB (Figure 5).
Figure 5.
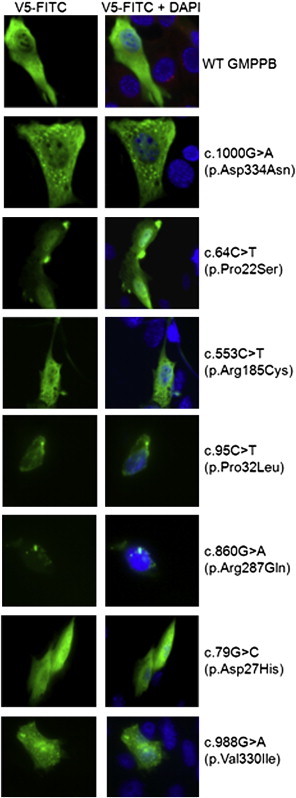
GMPPB Mutations Can Cause Mislocalization of GMPPB in Cultured Myoblasts
C2C12 myoblasts were transfected with GMPPB pcDNA 3.1 V5/HIS-TOPO that was either wild-type or had the missense mutation c.1000G>A (p.Asp334Asn), c.64C>T (p.Pro22Ser), c.553C>T (p.Arg185Cys), c.95C>T (p.Pro32Leu), c.860G>A (p.Arg287Gln), c.79G>C (p.Asp27His), or c.988G>A (p.Val330Ile). Compared to the wild-type, the mutations c.1000G>A, c.64C>T, c.95C>T, c.860G>A, and c.988G>A caused the protein to localize differently and aggregate, whereas the mutations c.553C>T and c.79G>C did not visibly alter the subcellular localization of GMPPB and the enzyme remained distributed evenly in the cytoplasm.
Knockdown of the GMPPB Ortholog in Zebrafish Embryos Recapitulates Features Characteristic of Dystroglycanopathies
In order to gather additional evidence of the role of GMPPB in vivo, we studied zebrafish (Danio rerio) gmppb. There is high sequence homology between the proteins encoded by these genes (81.4% identical; Table S6). We confirmed by RT-PCR that zebrafish gmppb is expressed throughout early embryonic development (Figure S9). To knock down zebrafish gmppb, we used a splice-blocking MO that targets the intron 4-exon 5 boundary within the nucleotidyl transferase domain (Figure S10).
Subsequently, we extracted RNA from wild-type and MO-injected embryos and performed RT-PCR with primers flanking the MO binding site (Figure S10). Compared to wild-type embryos, gmppb-MO-injected embryos (morphants) showed a clear reduction of the normal gmppb transcripts, whereas the amount of a housekeeping gene (actb1) remained roughly equivalent (Figure S10). This suggests that the gmppb MO specifically knocks down gmppb.
Morphologically, gmppb morphants were shorter than wild-type uninjected embryos at 48 hr postfertilization (hpf) and often had bent tails. Other phenotypes included hypopigmentation, micropthalmia, hydrocephalus, and reduced motility (Figure 6, Figure S11, and data not shown). The difference in diameter between the eyes of wild-type and gmppb morphant embryos was statistically significant (p < 1 × 10−7; Figure S11). Although none of our cases reported micropthalmia, this is a phenotype that is common in individuals with severe forms of CMD, such as WWS and MEB.3,4,20,23–25
Figure 6.
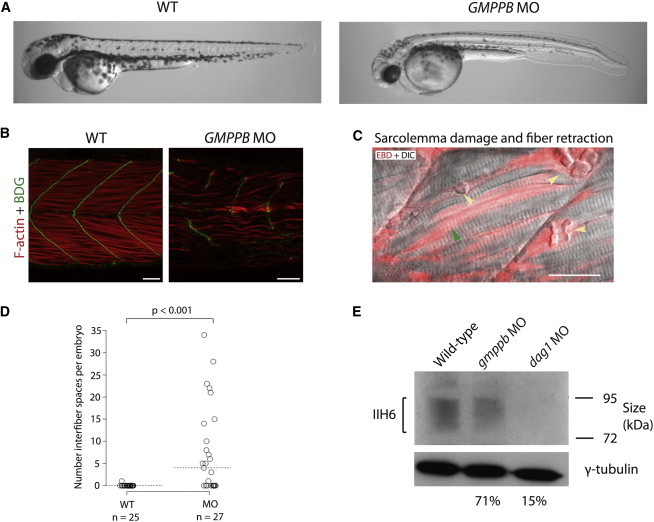
Zebrafish Embryos with gmppb Knockdown Have Morphological Defects, Damaged Muscle, and Hypoglycosylated α-DG at 48 hpf
(A) Bright-field microscopy of live embryos shows morphological defects of the gmppb-MO-injected embryos (injected with gmppb splice-blocking MO 3ng + p53 MO 6ng) as compared to uninjected wild-type embryos.
(B) Phalloidin staining of filamentous actin (red) and immunostaining with an antibody against β-DG (green).
(C) Live gmppb-MO-injected embryos injected with EBD (red) and imaged by confocal microscopy. Some fibers showed EBD infiltration, indicating damage to the sarcolemma (green arrow), and other fibers detached from the myosepta and retracted (yellow arrow) and thus left a space. The following abbreviation is used: DIC, differential interference contrast. The scale bar represents 25 μm.
(D) gmppb morphants have significantly more interfiber spaces than do wild-type uninjected embryos. The horizontal dotted line shows the median.
(E) An immunoblot shows a reduction in α-DG glycosylation. Percentage figures indicate the intensity of morphant bands relative to that of the wild-type and are adjusted for the γ-tubulin loading control. “gmppb MO” indicates embryos injected with gmppb MO 3ng + p53 MO 6ng, and “dag1 MO” indicates embryos injected with dag1 translation-blocking morpholino (5 ng).
To characterize muscle defects in gmppb-knockdown zebrafish embryos, we used phalloidin to label filamentous actin, along with immunostaining with antibodies against β-DG (which localizes to the myosepta, the connective tissue to which muscle fibers anchor). We observed that the muscle fibers in gmppb morphants were sparse and disordered. Furthermore, fibers were frequently observed to span two somites, indicating damage or incomplete development of the myosepta (Figure 6).
To further explore the muscle phenotypes in gmppb morphants, we injected EBD into the pericardium of embryos at 2 days postfertilization. EBD is an azo dye that binds to proteins such as albumin and is transported in the serum. It fluoresces upon protein binding and infiltrates muscle where there are lesions between muscle fibers (interfiber spaces) or where there is sarcolemmal damage.50 Compared with uninjected control embryos, gmppb morphants had significantly more EBD accumulation within interfiber spaces (p < 0.001; Figure 6). In addition, EBD infiltrated both retracted and some intact muscle fibers in gmppb-knockdown embryos, suggesting that sarcolemma integrity was compromised prior to muscle-fiber breakdown (Figure 6).
Next, we investigated whether the laminin-binding glycan on α-DG is reduced in gmppb morphants. To do this, we performed immunoblots with the IIH6 antibody on membrane proteins enriched from wild-type embryos and gmppb morphants, as well as dag1 morphants as a negative control. After normalization to γ-tubulin loading control, gmppb morphants showed a slight but clear reduction in IIH6 levels (71% of that of the wild-type embryos) and dag1 morphants showed a strong reduction in IIH6 (15% of that of the wild-type embryos) (Figure 6). To confirm this finding, we performed double immunostaining with the IIH6 antibody and an antibody against laminins. In wild-type embryos, laminin and glycosylated α-DG colocalized at the myosepta. In gmppb morphants, the IIH6 staining was severely reduced and laminin staining revealed widened myosepta, indicating a reduction in glycosylation of α-DG associated with abnormal basement-membrane structure (Figure S12).
Discussion
Here, we report the identification of GMPPB mutations in eight individuals with dystroglycanopathy by exome and Sanger sequencing. Immunohistochemistry showed reduced glycosylation of α-DG in muscle biopsies from affected individuals, and this finding was confirmed in available fibroblasts with the use of immunoblotting and flow cytometry, which revealed a reduction in the IIH6 signal. Transfection of an affected individual’s fibroblasts with wild-type GMPPB increased α-DG glycosylation, providing additional evidence that the GMPPB mutations carried by this individual are pathogenic and cause dystroglycanopathy.
Subcellular-localization studies indicated that GMPPB localizes to the cytosol, in agreement with its role in cytosolic synthesis of nucleotide sugars. This is consistent with the known localization of GDP-MP, the GMPPB ortholog in the protozoa L. mexicana.49 Assessment of the localization of altered GMPPB suggested that several of the altered enzymes formed aggregates within the cytoplasm or close to membrane protrusions into the cytoplasm and presumably interfered with the enzymatic function. Knocking down zebrafish gmppb recapitulated several aspects of the human phenotypes, including muscular dystrophy. Aspects of the phenotype were similar to those seen when other dystroglycanopathy genes are knocked down in zebrafish.20,24,50
Recent studies have revealed new details about α-DG glycosylation. For example, a highly specific O-mannosylated structure required for α-DG binding to extracellular ligands has been identified.51,52 Moreover, phosphorylation of an O-mannosyl glycan in the mucin-like domain of α-DG is required for its proper function.53 Recent mass-spectrometry studies have revealed the heterogeneity and complexity of α-DG O-mannosylation,54,55 a rare type of glycosylation that is particularly abundant on α-DG and clearly and specifically disrupted by mutations in well-characterized O-mannosyltransferase-encoding genes like POMT1, POMT2, and POMGNT1.3,4,12,13 More recently, mutations in genes encoding upstream components of the endoplasmic reticulum (ER) glycosylation pathway have been demonstrated to also cause dystroglycanopathies.14,15
GMPPB catalyzes the synthesis of GDP-mannose from GTP and mannose-1-phosphate. GDP-mannose is the substrate of cytosolic mannosyltransferases required for the synthesis of the core N-glycan structure, and it is required for the synthesis of Dol-P-Man in the ER membrane. Dol-P-Man synthesis is catalyzed by the DPM synthase complex, consisting of DPM1, DPM2, and DPM3. Dol-P-Man is the mannose donor required for all four mannosylation reactions that occur in the ER: O-mannosylation, C-mannosylation, N-glycosylation, and glycosylphosphatidylinositol-anchor formation.56 Substitutions in proteins required for the synthesis of Dol-P-Man could therefore theoretically perturb any of these pathways. Indeed, individuals with mutations in DPM2 and DPM3 have, in addition to a dystroglycanopathy, features evocative of a defect in N-glycosylation, such as abnormal glycosylation of transferrin, which is characteristic of CDG.14,15 However, in all our cases of mutant GMPPB, we found no evidence of perturbed transferrin glycoforms.
There are a number of possible explanations for this observation. For example, Dol-P-Man might be used to different extents in the N-glycosylation and O-mannosylation pathways. Previous research has shown that two enzymes in the glycosylation pathway compete for a common substrate and can use their substrates differentially, supporting this hypothesis.57 Alternatively, N-glycosylation could occur before O-mannosylation and the amount of Dol-P-Man would therefore be depleted by N-glycosylation before O-mannosylation starts.
Given what we know about the importance of glycosylation for α-DG function, it appears likely that the pathogenic mutations we have identified in GMPPB impair GMPPB function and thereby reduce the amount of GDP-mannose available for O-mannosylation of α-DG and ultimately cause the CMD phenotype. Although the full spectrum of clinical phenotypes associated with mutations in GMPPB has yet to be seen, the eight individuals described here demonstrate a wide range of muscle weakness, from a mild LGMD-like phenotype to a severe MEB- and/or FCMD-like phenotype, as well as cardiac involvement in the oldest surviving individuals; this phenotype range resembles the spectrum of phenotypes described for mutations in FKRP, FKTN, and ISPD. Features that were relatively common in individuals with GMPPB mutations but rare in other genotypes relate to the ophthalmologic finding of cataracts, observed not only in individuals with MEB- and/or FCMD-like phenotypes but also in individuals with CMD-MR (CMD with mental retardation) and LGMD-MR (LGMD with mental retardation) phenotypes.
GMPPB orthologs have been knocked down in various species, including Saccharomyces cerevisiae, Aspergillus fumigatus, Arabidopsis thaliana, Solanum tuberosum, Trypanosoma brucei, and Leishmania mexicana.49,58–63 This caused glycosylation defects and a range of pathogenic phenotypes from defective cell growth to lethality. This severity suggests that complete loss of function of GMPPB might be lethal. In this respect, it is interesting to note that we did not identify any case with two null alleles, and it is possible that some GMPPB function was retained in our cases.
In humans, GMPPB has a paralog, GMPPA. The proteins encoded by these genes are 30% identical to each other. In pigs, GMPPB and GMPPA act as a complex (GMPP) to catalyze the synthesis of both GDP-mannose and GDP-Glc (they have a higher affinity for synthesizing GDP-Glc). GMPPB alone has a very high affinity for synthesizing GDP-mannose and a low but detectible affinity for synthesizing GDP-Glc.47 Although to our knowledge the affinity of GMPPA for synthesizing GDP-Man has not been investigated, these results suggest some functional overlap between GMPPA and GMPPB. If this is true for humans, GMPPA might be able to synthesize some GDP-mannose in our CMD cases, which could contribute to the unexpectedly mild phenotype.
In summary, by using a combination of exome and Sanger sequencing, we have identified mutations in GMPPB in eight individuals with a wide spectrum of dystroglycanopathy phenotypes, ranging from CMD with structural brain involvement to LGMD. The spectrum and overall frequency of the conditions resulting from GMPPB mutations appear to be as wide as those reported to result from mutations in genes such as FKRP, FKTN, or ISPD.22,26 This brings the total number of genes in which mutations can cause a dystroglycanopathy to 15. Although many details of the complex glycosylation of α-DG are still unknown, the picture becomes a little more complete with each gene discovered.
Acknowledgments
This study makes use of data generated by the UK10K Consortium. A full list of the investigators who contributed to the generation of the data is available at http://www.uk10k.org/publications_and_posters.html. Funding for UK10K was provided by the Wellcome Trust under award WT091310. We are grateful to the UK10K consortium for making this study possible. Acknowledgments are continued in the Supplemental Data.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ImageJ, http://rsb.info.nih.gov/ij/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
xBrowse, http://atgu.mgh.harvard.edu/xbrowse
Accession Numbers
The European Genome-phenome Archive study ID for the P1 exome sequence data reported in this paper is EGAS00001000101.
References
- 1.Holt K.H., Crosbie R.H., Venzke D.P., Campbell K.P. Biosynthesis of dystroglycan: processing of a precursor propeptide. FEBS Lett. 2000;468:79–83. doi: 10.1016/s0014-5793(00)01195-9. [DOI] [PubMed] [Google Scholar]
- 2.Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 3.Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W.B. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Reeuwijk J., Janssen M., van den Elzen C., Beltran-Valero de Bernabé D., Sabatelli P., Merlini L., Boon M., Scheffer H., Brockington M., Muntoni F. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright K.M., Lyon K.A., Leung H., Leahy D.J., Ma L., Ginty D.D. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron. 2012;76:931–944. doi: 10.1016/j.neuron.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durbeej M., Henry M.D., Ferletta M., Campbell K.P., Ekblom P. Distribution of dystroglycan in normal adult mouse tissues. J. Histochem. Cytochem. 1998;46:449–457. doi: 10.1177/002215549804600404. [DOI] [PubMed] [Google Scholar]
- 7.Durbeej M., Larsson E., Ibraghimov-Beskrovnaya O., Roberds S.L., Campbell K.P., Ekblom P. Non-muscle alpha-dystroglycan is involved in epithelial development. J. Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herzog C., Has C., Franzke C.W., Echtermeyer F.G., Schlötzer-Schrehardt U., Kröger S., Gustafsson E., Fässler R., Bruckner-Tuderman L. Dystroglycan in skin and cutaneous cells: beta-subunit is shed from the cell surface. J. Invest. Dermatol. 2004;122:1372–1380. doi: 10.1111/j.0022-202X.2004.22605.x. [DOI] [PubMed] [Google Scholar]
- 9.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 10.Michele D.E., Campbell K.P. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 11.Hara Y., Balci-Hayta B., Yoshida-Moriguchi T., Kanagawa M., Beltrán-Valero de Bernabé D., Gündeşli H., Willer T., Satz J.S., Crawford R.W., Burden S.J. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med. 2011;364:939–946. doi: 10.1056/NEJMoa1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 13.Clement E.M., Godfrey C., Tan J., Brockington M., Torelli S., Feng L., Brown S.C., Jimenez-Mallebrera C., Sewry C.A., Longman C. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch. Neurol. 2008;65:137–141. doi: 10.1001/archneurol.2007.2. [DOI] [PubMed] [Google Scholar]
- 14.Barone R., Aiello C., Race V., Morava E., Foulquier F., Riemersma M., Passarelli C., Concolino D., Carella M., Santorelli F. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann. Neurol. 2012;72:550–558. doi: 10.1002/ana.23632. [DOI] [PubMed] [Google Scholar]
- 15.Lefeber D.J., Schönberger J., Morava E., Guillard M., Huyben K.M., Verrijp K., Grafakou O., Evangeliou A., Preijers F.W., Manta P. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 2009;85:76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lefeber D.J., de Brouwer A.P., Morava E., Riemersma M., Schuurs-Hoeijmakers J.H., Absmanner B., Verrijp K., van den Akker W.M., Huijben K., Steenbergen G. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011;7:e1002427. doi: 10.1371/journal.pgen.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longman C., Brockington M., Torelli S., Jimenez-Mallebrera C., Kennedy C., Khalil N., Feng L., Saran R.K., Voit T., Merlini L. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum. Mol. Genet. 2003;12:2853–2861. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- 18.Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M.E., Yu L., Campbell K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buysse K., Riemersma M., Powell G., van Reeuwijk J., Chitayat D., Roscioli T., Kamsteeg E.J., van den Elzen C., van Beusekom E., Blaser S. Missense mutations in β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet. 2013;22:1746–1754. doi: 10.1093/hmg/ddt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens E., Carss K.J., Cirak S., Foley A.R., Torelli S., Willer T., Tambunan D.E., Yau S., Brodd L., Sewry C.A., UK10K Consortium Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am. J. Hum. Genet. 2013;92:354–365. doi: 10.1016/j.ajhg.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 22.Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manzini M.C., Tambunan D.E., Hill R.S., Yu T.W., Maynard T.M., Heinzen E.L., Shianna K.V., Stevens C.R., Partlow J.N., Barry B.J. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 2012;91:541–547. doi: 10.1016/j.ajhg.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roscioli T., Kamsteeg E.J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L.E. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 2012;44:581–585. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D.B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012;44:575–580. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cirak S., Foley A.R., Herrmann R., Willer T., Yau S., Stevens E., Torelli S., Brodd L., Kamynina A., Vondracek P., UK10K Consortium ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain. 2013;136:269–281. doi: 10.1093/brain/aws312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercuri E., Messina S., Bruno C., Mora M., Pegoraro E., Comi G.P., D’Amico A., Aiello C., Biancheri R., Berardinelli A. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology. 2009;72:1802–1809. doi: 10.1212/01.wnl.0000346518.68110.60. [DOI] [PubMed] [Google Scholar]
- 28.Jae L.T., Raaben M., Riemersma M., van Beusekom E., Blomen V.A., Velds A., Kerkhoven R.M., Carette J.E., Topaloglu H., Meinecke P. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bouchet C., Gonzales M., Vuillaumier-Barrot S., Devisme L., Lebizec C., Alanio E., Bazin A., Bessières-Grattagliano B., Bigi N., Blanchet P. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum. Mutat. 2007;28:1020–1027. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- 30.Manzini M.C., Gleason D., Chang B.S., Hill R.S., Barry B.J., Partlow J.N., Poduri A., Currier S., Galvin-Parton P., Shapiro L.R. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum. Mutat. 2008;29:E231–E241. doi: 10.1002/humu.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godfrey C., Clement E., Mein R., Brockington M., Smith J., Talim B., Straub V., Robb S., Quinlivan R., Feng L. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 32.Messina S., Tortorella G., Concolino D., Spanò M., D’Amico A., Bruno C., Santorelli F.M., Mercuri E., Bertini E. Congenital muscular dystrophy with defective alpha-dystroglycan, cerebellar hypoplasia, and epilepsy. Neurology. 2009;73:1599–1601. doi: 10.1212/WNL.0b013e3181c0d47a. [DOI] [PubMed] [Google Scholar]
- 33.Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Timal S., Hoischen A., Lehle L., Adamowicz M., Huijben K., Sykut-Cegielska J., Paprocka J., Jamroz E., van Spronsen F.J., Körner C. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 2012;21:4151–4161. doi: 10.1093/hmg/dds123. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 36.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dubowitz V., Sewry C.A., Oldfors A. Saunders Elsevier; Oxford: 2013. Muscle Biopsy: A practical approach. [Google Scholar]
- 40.Schuler F., Sorokin L.M. Expression of laminin isoforms in mouse myogenic cells in vitro and in vivo. J. Cell Sci. 1995;108:3795–3805. doi: 10.1242/jcs.108.12.3795. [DOI] [PubMed] [Google Scholar]
- 41.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 42.Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.F., Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., van der Meulen J. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langheinrich U., Hennen E., Stott G., Vacun G. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr. Biol. 2002;12:2023–2028. doi: 10.1016/s0960-9822(02)01319-2. [DOI] [PubMed] [Google Scholar]
- 44.Parsons M.J., Campos I., Hirst E.M., Stemple D.L. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development. 2002;129:3505–3512. doi: 10.1242/dev.129.14.3505. [DOI] [PubMed] [Google Scholar]
- 45.Abramoff M.D., Magalhaes P.J., Ram S.J. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 46.Rafiq M.A., Kuss A.W., Puettmann L., Noor A., Ramiah A., Ali G., Hu H., Kerio N.A., Xiang Y., Garshasbi M. Mutations in the alpha 1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2011;89:176–182. doi: 10.1016/j.ajhg.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ning B., Elbein A.D. Cloning, expression and characterization of the pig liver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glc pyrophosphorylases are different proteins. Eur. J. Biochem. 2000;267:6866–6874. doi: 10.1046/j.1432-1033.2000.01781.x. [DOI] [PubMed] [Google Scholar]
- 48.Mostacciuolo M.L., Miorin M., Martinello F., Angelini C., Perini P., Trevisan C.P. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum. Genet. 1996;97:277–279. doi: 10.1007/BF02185752. [DOI] [PubMed] [Google Scholar]
- 49.Davis A.J., Perugini M.A., Smith B.J., Stewart J.D., Ilg T., Hodder A.N., Handman E. Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana. J. Biol. Chem. 2004;279:12462–12468. doi: 10.1074/jbc.M312365200. [DOI] [PubMed] [Google Scholar]
- 50.Lin Y.Y., White R.J., Torelli S., Cirak S., Muntoni F., Stemple D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011;20:1763–1775. doi: 10.1093/hmg/ddr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Endo T., Toda T. Glycosylation in congenital muscular dystrophies. Biol. Pharm. Bull. 2003;26:1641–1647. doi: 10.1248/bpb.26.1641. [DOI] [PubMed] [Google Scholar]
- 52.Chiba A., Matsumura K., Yamada H., Inazu T., Shimizu T., Kusunoki S., Kanazawa I., Kobata A., Endo T. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J. Biol. Chem. 1997;272:2156–2162. doi: 10.1074/jbc.272.4.2156. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida-Moriguchi T., Yu L., Stalnaker S.H., Davis S., Kunz S., Madson M., Oldstone M.B., Schachter H., Wells L., Campbell K.P. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nilsson J., Nilsson J., Larson G., Grahn A. Characterization of site-specific O-glycan structures within the mucin-like domain of alpha-dystroglycan from human skeletal muscle. Glycobiology. 2010;20:1160–1169. doi: 10.1093/glycob/cwq082. [DOI] [PubMed] [Google Scholar]
- 55.Stalnaker S.H., Hashmi S., Lim J.M., Aoki K., Porterfield M., Gutierrez-Sanchez G., Wheeler J., Ervasti J.M., Bergmann C., Tiemeyer M., Wells L. Site mapping and characterization of O-glycan structures on alpha-dystroglycan isolated from rabbit skeletal muscle. J. Biol. Chem. 2010;285:24882–24891. doi: 10.1074/jbc.M110.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maeda Y., Kinoshita T. Dolichol-phosphate mannose synthase: structure, function and regulation. Biochim. Biophys. Acta. 2008;1780:861–868. doi: 10.1016/j.bbagen.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 57.Sharma V., Ichikawa M., He P., Scott D.A., Bravo Y., Dahl R., Ng B.G., Cosford N.D., Freeze H.H. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J. Biol. Chem. 2011;286:39431–39438. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warit S., Zhang N., Short A., Walmsley R.M., Oliver S.G., Stateva L.I. Glycosylation deficiency phenotypes resulting from depletion of GDP-mannose pyrophosphorylase in two yeast species. Mol. Microbiol. 2000;36:1156–1166. doi: 10.1046/j.1365-2958.2000.01944.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhang N., Gardner D.C., Oliver S.G., Stateva L.I. Down-regulation of the expression of PKC1 and SRB1/PSA1/VIG9, two genes involved in cell wall integrity in Saccharomyces cerevisiae, causes flocculation. Microbiology. 1999;145:309–316. doi: 10.1099/13500872-145-2-309. [DOI] [PubMed] [Google Scholar]
- 60.Jiang H., Ouyang H., Zhou H., Jin C. GDP-mannose pyrophosphorylase is essential for cell wall integrity, morphogenesis and viability of Aspergillus fumigatus. Microbiology. 2008;154:2730–2739. doi: 10.1099/mic.0.2008/019240-0. [DOI] [PubMed] [Google Scholar]
- 61.Qin C., Qian W., Wang W., Wu Y., Yu C., Jiang X., Wang D., Wu P. GDP-mannose pyrophosphorylase is a genetic determinant of ammonium sensitivity in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA. 2008;105:18308–18313. doi: 10.1073/pnas.0806168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keller R., Renz F.S., Kossmann J. Antisense inhibition of the GDP-mannose pyrophosphorylase reduces the ascorbate content in transgenic plants leading to developmental changes during senescence. Plant J. 1999;19:131–141. doi: 10.1046/j.1365-313x.1999.00507.x. [DOI] [PubMed] [Google Scholar]
- 63.Denton H., Fyffe S., Smith T.K. GDP-mannose pyrophosphorylase is essential in the bloodstream form of Trypanosoma brucei. Biochem. J. 2010;425:603–614. doi: 10.1042/BJ20090896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.