

Abstract
Exome sequencing offers the potential to study the population-genomic variables that underlie patterns of deleterious variation. Runs of homozygosity (ROH) are long stretches of consecutive homozygous genotypes probably reflecting segments shared identically by descent as the result of processes such as consanguinity, population size reduction, and natural selection. The relationship between ROH and patterns of predicted deleterious variation can provide insight into the way in which these processes contribute to the maintenance of deleterious variants. Here, we use exome sequencing to examine ROH in relation to the distribution of deleterious variation in 27 individuals of varying levels of apparent inbreeding from 6 human populations. A significantly greater fraction of all genome-wide predicted damaging homozygotes fall in ROH than would be expected from the corresponding fraction of nondamaging homozygotes in ROH (p < 0.001). This pattern is strongest for long ROH (p < 0.05). ROH, and especially long ROH, harbor disproportionately more deleterious homozygotes than would be expected on the basis of the total ROH coverage of the genome and the genomic distribution of nondamaging homozygotes. The results accord with a hypothesis that recent inbreeding, which generates long ROH, enables rare deleterious variants to exist in homozygous form. Thus, just as inbreeding can elevate the occurrence of rare recessive diseases that represent homozygotes for strongly deleterious mutations, inbreeding magnifies the occurrence of mildly deleterious variants as well.
Introduction
The study of deleterious variation in the genome has fundamental importance to evolutionary genetics.1–15 In humans, it has been argued that an individual genome can contain tens to hundreds of variants that would be lethal in homozygous form2,3 and hundreds to thousands of mildly deleterious variants,6–8,15–19 the accumulation of which could potentially have health consequences.20 Because the distribution of these variants across individuals and populations reflects the result of natural selection and other population-genomic processes, investigations of patterns of deleterious variation can contribute insights into human adaptation, evolution, and genetic disease.
As genomic data became available, initial studies relied on limited numbers of genes to make inferences about the accumulation, distribution, and effects of deleterious variation. For example, Eyre-Walker and Keightley5 analyzed 46 genes by using sequences of human, chimpanzee, and the gene-specific closest available primate species at the time of the study to estimate the deleterious mutation rate in humans. Fay et al.6 used single-nucleotide polymorphism and divergence data from >100 genes to estimate that 80% of amino acid mutations are deleterious and that each diploid genome possesses ∼300 deleterious variants.
With the widespread availability of next-generation sequencing technology, exome sequencing now allows for the simultaneous study of nearly all known protein-coding regions. Because nonsynonymous mutations within protein-coding regions are particularly likely to be disruptive—by altering the encoded amino acid sequence—relative to noncoding regions, exome sequences can be used together with computational tools that assess the functional impact of amino acid changes for studying the genomic distribution of potentially deleterious variation.10,21 For example, Lohmueller et al.8 examined exomic data on 20 European Americans and 15 African Americans, finding that a greater number of homozygous variants were predicted to be deleterious in the European Americans compared to the African Americans. Tennessen et al.15 sequenced the exomes of more than 2,000 individuals, arguing that a large fraction of coding variation is recent, rare, and deleterious. In a study of 69 genome sequences, Torkamani et al.17 extended the work of Lohmueller et al.8 to further characterize variation in the number of deleterious genotypes among individuals from different human populations.
Examining the relationships between patterns of deleterious variation and population-genomic variables enables assessments of evolutionary processes that shape deleterious variation. For example, by using whole-genome sequences, Lohmueller et al.11 studied correlations among a variety of genomic variables related to coding variation, suggesting that a positive correlation between neutral diversity and recombination rate is the result of negative selection acting on large numbers of weakly deleterious variants.
Recent progress in the study of runs of homozygosity (ROH)22–29 provides a new basis for assessing the mechanism by which selection produces patterns of deleterious alleles. ROH regions—long stretches of consecutive homozygous genotypes, probably resulting from identity by descent as the result of demographic processes that reduce population size and increase homozygosity, cultural practices that promote consanguineous marriages, and natural selection that purges deleterious variants or elevates the frequencies of haplotypes surrounding a favored allele—are known to contain recessive disease mutations, and they have been a central focus of homozygosity mapping studies of recessive diseases.30–33 Pemberton et al.29 recently characterized worldwide patterns of ROH across the genome, separating ROH into length classes designed to represent the outcomes of different evolutionary forces, and reporting a database for additional analysis.
In light of the importance of ROH regions for recessive disease, we aim to characterize patterns of deleterious variation occurring inside and outside ROH regions. Specifically, we examine two hypotheses about the processes that shape patterns of deleterious variation in the human genome. First, a diploid genome with an ROH region containing many deleterious variants would carry these variants as homozygotes and would probably show reduced fitness, especially if the variants interact synergistically. As a result, this genome would be less viable than a genome whose ROH carry fewer deleterious homozygotes, and it is less likely to be extant in a population. We might therefore expect that random healthy individuals will carry an underrepresentation of deleterious homozygotes within their ROH. We can thus propose hypothesis 1: counting deleterious and neutral variants inside and outside of ROH regions, we expect to observe a smaller fraction of all genome-wide deleterious homozygotes in ROH regions compared to the fraction of neutral homozygotes occurring in ROH regions. Although deleterious homozygotes occurring outside of ROH regions will also incur a fitness cost, under this hypothesis, we expect that selection would more effectively purge homozygous regions if they carry more deleterious homozygotes. Consequently, ROH would be likely to contain fewer deleterious homozygotes as a proportion of all genome-wide deleterious homozygotes compared to their corresponding proportion of neutral homozygotes. This hypothesis implicitly requires that the negative impact of the deleterious homozygotes be strong and immediate or that the ROH regions be sufficiently stable across generations to accumulate the effect of selection.
Our second hypothesis proposes that low-frequency variants are more likely to be deleterious than common variants15,19,34,35 and that ROH regions can present low-frequency variants in homozygous form at a higher rate than non-ROH regions. Consider a rare variant that has allele frequency p in a population. If this variant were to occur in a nonidentical-by-descent region of a genome, then it would be in homozygous form with probability p2. If it instead occurred in an identical-by-descent ROH region, it would be homozygous with probability p, which exceeds p2. When homozygous deleterious variants are not lethal and inbreeding is recent, selection will not have had enough time to eliminate deleterious variants in ROH regions. In this case, we expect that when sampling a random set of individuals, we will observe an overrepresentation of deleterious homozygotes inside of ROH and inside long ROH in particular. Therefore we form hypothesis 2: counting deleterious and neutral variants inside and outside of ROH regions, we expect to observe a larger fraction of deleterious homozygotes in ROH regions compared to the fraction of neutral homozygotes occurring in ROH regions. We further expect that longer ROH, made of newer haplotypes, might have a higher relative fraction of deleterious homozygotes than shorter ROH, made of older haplotypes. Note that hypothesis 2 predicts an opposite pattern to that predicted by hypothesis 1.
To test these hypotheses, we perform whole-exome sequencing and computational prediction of deleteriousness, analyzing the predictions in conjunction with genomic ROH patterns previously estimated in the same individuals via SNP genotyping.29 We select 27 individuals from 6 populations, covering a wide range of genome-wide ROH coverage (4%–46%) and representing the extreme ends of the ROH distribution across the genome (Figure 1). The individuals are drawn from the HGDP-CEPH diversity panel of apparently healthy subjects collected for population-genetic studies.36 To predict whether a variant allele is deleterious, we use the PolyPhen2 program.37 For neutral variation, we consider both synonymous sites and missense sites predicted to be benign. Next, with the coordinates of called ROH regions,29 we count the number of predicted deleterious variants that lie in each individual’s ROH. Finally, we determine whether deleterious homozygotes occur within ROH more frequently than expected from the pattern of occurrence of neutral homozygotes, and we examine whether this pattern differs across ROH length classes chosen to largely reflect different population-genetic processes.
Figure 1.
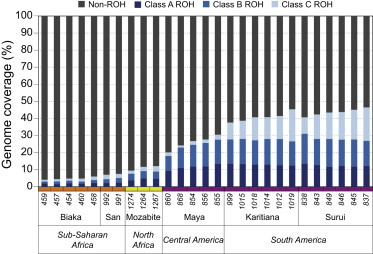
ROH Coverage across Individual Genomes
The y axis gives the percentage of individual genomes covered by short (class A), medium (class B), and long (class C) ROH. Numbers on the x axis represent identification numbers in the HGDP-CEPH diversity panel.36
Materials and Methods
We adapted a three-stage variant calling and quality control pipeline from DePristo et al.38 (Figure S1 available online). First, we mapped raw sequencing reads to the reference genome and performed quality control on the reads before single-nucleotide variant sites were called. Next, we called variant sites by using all samples jointly. Finally, we performed quality control at the site and genotype levels for all individuals. To assess whether a variant allele might have a deleterious effect, we computationally predicted deleteriousness with PolyPhen2.37 ROH data were taken directly from Pemberton et al.29
Raw Read Processing and Variant Calling
We performed Nimblegen SeqCap EZ v.1 (Roche Nimblegen) exome capture followed by sequencing with the Illumina Hiseq2000 system with one lane per sample. We aligned raw reads to the human reference sequence (assembly hg18; UCSC Genome Browser) with BWA39 and marked duplicate reads with Picard tools. We used the Genome Analysis Tool Kit (GATK) v.1.2-440 for lane-level local realignment around known and possible insertion-deletions (indels) and for lane-level recalibration of base quality scores. Finally, we called variants by using all samples jointly with the UnifiedGenotyper module of GATK with a minimum phred-scaled confidence score of 30 (minimum estimated error of 0.001). This analysis gave us a set of raw variant sites. In these analyses, we considered only biallelic single-nucleotide variant sites, and we excluded indels and multiallelic sites. The study was approved by the institutional review board of the University of Michigan Medical School; informed consent information appears in Cann et al.36
Quality Control of Called Variant Sites
The raw set of variant sites is expected to contain true variant sites but also to contain many false positives. We further filtered the initial set of variant calls to reduce false positives (Figure S1). The Nimblegen SeqCap EZ v.1 platform targets more than 175,000 coding exons with 100 bp padding into intronic segments flanking the targeted exons, and we retained only called variant sites that fell in the targeted regions or the padding. In principle, we expect a putative variant site that strongly deviates from the distribution of quality measures of known variant sites to be a likely false positive. We therefore utilized the variant quality score recalibrator module of GATK38 to build an adaptive error model using known variant sites that occur in our data set and their quality measure annotations (i.e., RMS Mapping Quality, Fisher’s exact test for strand bias, etc.). Utilizing the variant site quality measure from the joint variant calling step above, we estimated the probability that our called variant sites are true genetic variants.
The variant quality score recalibrator requires a set of likely true variant sites to train its error model. We considered two sets of likely true variant sites: called exome variant sites previously identified as HapMap 3.3 variant sites with a phred-scaled prior of 15 (96.84%), and called exome variant sites previously identified as Omni 2.5M HapMap variant sites with a phred-scaled prior of 12 (93.69%), as recommended by DePristo et al.38 Given this set of likely true variant sites, we trained the error model with the HaplotypeScore, HRun, MQRankSum, MQ, and FS quality score annotations.
After training the error model, all called variant sites in the data set were annotated with the variant quality score log-of-odds (VQSLOD), which represents the log odds of a site being a true variant versus a false positive. We considered the distribution of VQSLOD scores for called variant sites also found in HapMap 3.3 and chose a cut-off that returns 99% of these sites, as recommended by DePristo et al.38 After filtering sites below this cutoff, 96,797 remained (Ti/Tv = 2.9821). With dbSNP build 132 (excluding sites added after build 129), 53,286 were known (Ti/Tv = 3.1017) and 43,511 were novel (Ti/Tv = 2.8356).
Variant Classification by Predicted Functional Impact
Some of our variant sites might not be in the coding regions of the targeted genes because the NimbleGen platform pads the capture target by 100 bp on each side. We annotated the genomic location of each called variable site by using the MapSNPs algorithm provided with PolyPhen2.37 MapSNPs determined the genomic location of each site with respect to the Consensus CDS (CCDS) set of high-quality coding regions,41 and it successfully annotated 91,069 sites with 701 mapping to 2 CCDS regions (Figure S2). Any site that had a mutation classified as missense in one CCDS and as another type in another CCDS (e.g., synonymous) was considered only as a missense mutation for downstream analyses. Sites with a synonymous classification in one CCDS and a nonsense or UTR classification in another CCDS were removed. Sites with a missense classification in more than one CCDS were retained for further classification by PolyPhen2. If a missense mutation was classified by PolyPhen2 with respect to more than one CCDS, it was retained if the classifications were identical, and it was removed otherwise. After reconciling these double hits and removing sites that did not fall into a CCDS region, we were left with 26,776 missense sites and 29,914 synonymous sites.
We used PolyPhen2 to classify nonreference alleles that are missense changes. Given a set of missense mutations, PolyPhen2 predicts the potential disruption that the nonreference allele has on the encoded protein, incorporating knowledge of amino acid biochemistry, folded structure (if known), and conservation score. It does not use any population genetics information. PolyPhen2 categorizes missense mutations as “probably damaging,” “possibly damaging,” or “benign.” In some analyses, we combine synonymous sites with benign sites into a “nondamaging” superclass and possibly damaging and probably damaging sites into a “damaging” superclass. Although truly damaging variants might occur in the nondamaging class and nondamaging variants might occur in the damaging class, our concern is not with the prediction accuracy for any particular variant; rather, we aim to study genome-wide trends by generating classes that are separately enriched for damaging and nondamaging variants. Note that although the computational prediction of deleteriousness classifies the individual mutation that differs from the human genome reference sequence, for convenience we refer to a site as synonymous, probably damaging, possibly damaging, or benign if the nonreference allele at that site (also known as the “alternate” allele) has been classified as such. Reference alleles at damaging sites are not predicted to be damaging.
The final set of missense mutations classified by PolyPhen2 appears in Figure S3. Because we aim to examine both deleterious and nondeleterious variation, the final coding variation data set used in downstream analyses consists of the PolyPhen2-classified missense sites and the synonymous sites. The 979 missense sites for which PolyPhen2 was unable to predict a functional effect were removed from the data set.
Genotype-level Quality Control
Although site-level quality control generates a set of sites that are likely to be truly variable, specific genotypes might have poor quality. Therefore, we performed a final round of quality control per individual genotype on the remaining 29,914 synonymous and 25,797 missense sites. We assessed concordance with known genotypes for all 27 sampled individuals by comparing called genotypes at 6,180 variant sites previously studied via Illumina SNP genotyping.42 The percentage of called exome genotypes that agreed with the SNP genotypes was 99.2%; the concordance was 99.3% for called nonreference homozygotes, 99.5% for called reference homozygotes, and 98.7% for called heterozygotes (Table S1). Conversely, the percentage of SNP heterozygotes that were called as heterozygotes in the exome data was 98.4%. Considering these concordance levels, we chose a filter for homozygous genotypes of DP < 3, where DP is the read depth for the sample at that site. Applying this filter gave a new concordance rate of 99.6% for nonreference homozygous genotypes, while removing 42.6% of mismatches and only 1.1% of matches.
To filter heterozygous genotypes to achieve a similar concordance rate, we considered the distribution of called heterozygotes as a function of both DP and nonreference allele frequency, choosing, by hand, a progressive filter based on nonreference allele frequency as a function of DP. The cutoff is more permissive at lower DP and more restrictive at higher DP (Figure S4). Applying this filter produced a new concordance rate of 99.6% for heterozygous genotypes, removing 71.7% of mismatches and 0.5% of matches. Conversely, after filtering, 99.1% of Illumina heterozygotes were called as heterozygotes in the sequencing data. After filtering, 64 former variant sites did not have variant calls for any individual (all genotypes missing), and 1,288 were monomorphic. These sites were removed from the data set. After this final genotype filtering step, the data set has 54,359 sites (Figure 2). The mean coverage ranges from 38× and 81× across individuals, and the percentage of sites with ≥20× coverage ranges from 62% to 90% (Table S2).
Figure 2.
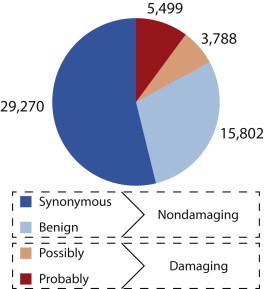
PolyPhen2 Classification of the Final Set of 54,359 Variants after All Filtering
Nonsense Variants
We also analyzed two data sets of nonsense mutations in our set of 27 individuals. The 264 sites with nonsense mutations in our data were subjected to the same quality control filtering as synonymous and missense mutations above, and 7 failed. Our first data set consists of all 257 nonsense sites that passed quality control. Our second data set consists of 66 nonsense sites that lie in the intersection of all 257 nonsense sites with the list of validated loss-of-function mutations from MacArthur et al.14
Runs of Homozygosity
Pemberton et al.29 characterized worldwide patterns of runs of homozygosity in 1,839 human individuals across 64 populations by an autozygosity-based LOD score method. They further classified these ROH into three categories designed to correspond to ROH that arose largely from different processes. Short ROH (class A) are tens of kilobases in size and reflect homozygosity of ancient haplotypes that predate continental migrations. Medium ROH (class B) are hundreds of kilobases to a few megabases long and mostly arise from background relatedness within populations. Finally, long ROH (class C) are several megabases long and probably result from recent parental relatedness. For the 27 individuals in our exome sequencing data set, we took the coordinates defining the ROH regions as well as the ROH size class boundary values so that we could identify a given ROH segment as belonging to a particular size class. With this information, we calculated
| (Equation 1) |
This quantity represents the total fraction of the genome of individual i covered by any ROH region (j = R) or the total fraction of the genome covered by a specific ROH class (; Table S3). With this information, for each individual, we mapped each variant site from Figure 2 to a specific ROH segment.
Results
Data Set
We sequenced the exomes of 27 individuals to an average read depth of 38×–81×. After variant calling and filtering, our data consist of 54,359 single-nucleotide sites for which at least 1 of the 27 individuals had a high-confidence nonreference allele called (Figure 2). At each site, every individual’s genotype is called, and low-confidence calls are considered missing genotypes. The per-individual missing data rate has a mean of 3.3% and a maximum of 10.6%. The concordance rate with SNP genotype data on the same samples, across 6,180 sites that overlap between the sequencing-based variant calls and genotype positions, treating diploid genotypes as concordant if they are identical and discordant otherwise, is 99.6% for both heterozygotes and nonreference allele homozygotes and 99.7% for reference homozygotes.
Heterozygous Genotypes in Different ROH Size Classes
We partitioned the genotypes in our data set into those occurring at damaging versus nondamaging sites and also those occurring outside ROH regions or inside ROH of a specific size class. An individual’s genotype at a site can be either homozygous for the reference allele (0/0), heterozygous (0/1), or homozygous for the alternate allele (1/1), and the alternate allele can be classified as damaging or nondamaging. For individual i, across all sites we denote by and the total number of sites with alternate alleles at nondamaging and damaging sites, respectively. For individual i, and represent the total number of sites with alternate alleles falling in ROH class at nondamaging and damaging sites, respectively. A, B, and C indicate the ROH classes of Pemberton et al.,29 R is the union of all three ROH classes, and N represents sites located outside of any ROH region. Thus,
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
By definition of ROH, heterozygotes occur less often within ROH than outside ROH. To account for possible genotyping errors and recent mutations, the approach of Pemberton et al.29 allows a nonzero number of heterozygotes to lie in an ROH. We expect to observe the fewest heterozygotes in long class C ROH, because these are the most confidently identified ROH, and the haplotypes that form these ROH have had the shortest length of time in which to develop mutations. Conversely, we expect to see an enrichment of heterozygotes in non-ROH regions relative to the genome-wide prevalence. To examine these expectations, we calculate the genome-wide fraction of heterozygotes in individual i as
| (Equation 6) |
Similarly, we calculate
| (Equation 7) |
representing the fraction of genotypes that are heterozygotes in individual i that do not occur in an ROH region (j = N), that occur in any ROH region (j = R), or that occur in an ROH region of a particular size class ().
We observe, as expected, that the percentage of heterozygotes in any ROH region is substantially lower than genome-wide and in non-ROH regions (Table 1). As we move from short to long ROH, the heterozygote percentage drops dramatically. This result is consistent with the view that short (class A) ROH are made of older haplotypes, which have had time to accumulate more mutations, and long (class C) ROH are made of younger haplotypes, which have accumulated fewer mutations. In these analyses, we consider only the 54,359 sites polymorphic in our 27 individuals, so that the denominator in heterozygote percentages does not include the far greater number of sequenced sites at which all 27 samples showed the homozygous reference genotype. Absolute heterozygote frequencies are therefore much lower than the values in Table 1.
Table 1.
Percentage of All Polymorphic Exon Variants that Are Heterozygous in a Given Region of an Individual’s Genome
| Population | Individual ID | Genome-wide (%,) | Non-ROH (%,) | Any ROH (%,) | Class A (%,) | Class B (%,) | Class C (%,) |
|---|---|---|---|---|---|---|---|
| San | 991 | 17.7 | 18.8 | 4.2 | 6.4 | 4.7 | 0.6 |
| 992 | 17.4 | 18.5 | 2.6 | 5.8 | 2.8 | 0.4 | |
| Biaka | 454 | 17.9 | 18.5 | 2.6 | 3.8 | 2.7 | 0.2 |
| 457 | 18.0 | 18.6 | 3.3 | 5.5 | 2.6 | 2.2 | |
| 458 | 17.2 | 18.1 | 2.1 | 4.7 | 1.8 | 0.2 | |
| 459 | 17.9 | 18.6 | 1.7 | 3.4 | 1.1 | 0.0 | |
| 460 | 17.6 | 18.1 | 4.6 | 5.7 | 7.6 | 1.0 | |
| Mozabite | 1264 | 14.6 | 15.9 | 2.3 | 2.6 | 2.9 | 1.2 |
| 1267 | 14.7 | 16.1 | 2.4 | 4.1 | 2.2 | 0.8 | |
| 1274 | 15.6 | 16.8 | 2.4 | 3.4 | 2.5 | 0.1 | |
| Maya | 854 | 11.4 | 14.6 | 2.6 | 2.7 | 2.6 | 1.0 |
| 855 | 10.8 | 14.6 | 2.2 | 2.8 | 2.0 | 1.1 | |
| 856 | 11.2 | 14.3 | 2.3 | 2.9 | 1.9 | 1.7 | |
| 860 | 12.8 | 15.0 | 2.7 | 3.4 | 2.2 | 1.9 | |
| 868 | 12.0 | 14.9 | 2.5 | 2.8 | 2.3 | 1.5 | |
| Karitiana | 999 | 9.1 | 14.3 | 1.9 | 2.9 | 2.3 | 0.4 |
| 1012 | 8.8 | 14.2 | 1.8 | 3.0 | 2.6 | 0.3 | |
| 1014 | 9.4 | 14.2 | 1.8 | 2.6 | 2.2 | 0.3 | |
| 1015 | 9.1 | 14.2 | 1.7 | 2.6 | 2.2 | 0.4 | |
| 1018 | 8.1 | 13.7 | 1.4 | 2.9 | 2.0 | 0.6 | |
| 1019 | 9.9 | 14.2 | 2.1 | 3.0 | 1.8 | 0.3 | |
| Surui | 837 | 8.3 | 14.6 | 1.5 | 2.8 | 2.1 | 0.3 |
| 838 | 9.6 | 14.6 | 2.3 | 3.6 | 2.4 | 0.7 | |
| 843 | 8.6 | 13.7 | 1.7 | 3.1 | 1.6 | 0.6 | |
| 845 | 8.7 | 14.2 | 1.9 | 3.2 | 2.3 | 0.7 | |
| 846 | 9.1 | 14.8 | 1.8 | 3.3 | 2.2 | 0.4 | |
| 849 | 8.8 | 14.5 | 1.6 | 2.9 | 1.8 | 0.5 |
Number of Damaging Homozygous Genotypes in ROH
Tables S4 and S5 report the counts for reference homozygotes (0/0), heterozygotes (0/1), and nonreference homozygotes (1/1) at damaging and nondamaging sites, respectively, that fall into ROH regions and non-ROH regions (all and ). For nondamaging sites, the ordering across populations by number of homozygotes per individual genome is consistent with known levels of genetic diversity in these populations,42–44 with the African populations having higher diversity and fewer homozygotes than the Native American populations. This trend also holds for nonreference homozygotes that are predicted to be damaging, with a large share of those genotypes falling in ROH regions.
These results underscore the substantial mutational burden many individuals are carrying, particularly the individuals with a high genomic ROH content. For instance, Surui individual 837 has the highest ROH coverage (46.4% of the genome) and carries a total of 357 predicted damaging variants in homozygous form (189 probably damaging and 168 possibly damaging). By contrast, Biaka individual 459 has the lowest ROH coverage (4.0% of the genome) and has 212 predicted damaging variants in homozygous form (109 probably damaging and 103 possibly damaging).
For each individual, Figure 3 shows the total number of damaging nonreference homozygotes (1/1) as a function of the total fraction of the genome covered by ROH (, Equation 1). The red points represent damaging homozygotes that occur within ROH (), and the black points represent damaging homozygotes that occur outside ROH (). As the genome is increasingly covered by more ROH and longer ROH (high values of ), we expect a greater number of homozygotes (damaging or not) to fall within ROH. We indeed see a strong linear relationship between the number of damaging homozygotes and genomic ROH fraction (Pearson r = 0.9897, slope 584.3, intercept −9.2). Similarly, we expect the number of homozygotes occurring outside ROH to decrease with genomic ROH fraction, because the genome simply contains fewer ROH-free regions. As expected, we see a strong negative correlation of damaging homozygotes outside ROH with genomic ROH fraction (Pearson r = −0.8378, slope −139.0, intercept 181.3). The decreasing slope for non-ROH regions is shallower than the increasing slope for ROH regions, however, indicating that the rise in damaging homozygotes in ROH regions outpaces the decline of damaging homozygotes in non-ROH regions. The fitted lines predict that an average noninbred individual () carries approximately 181 damaging variants in homozygous form. Increasing the ROH coverage of the genome by 10% results in a mean increase of damaging homozygotes in ROH regions by 58 and a mean decrease of damaging homozygotes in non-ROH regions by 14, for a net increase of 44.
Figure 3.
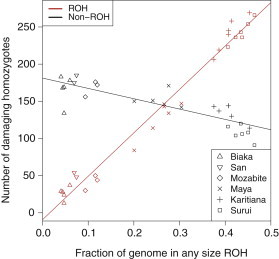
The Number of Damaging Nonreference Homozygotes versus the Fraction of the Genome Covered by ROH for Each Individual
Red points represent the number of damaging homozygotes falling within ROH regions, and black points represent the number of damaging homozygotes falling outside ROH regions.
Damaging and Nondamaging Homozygotes in ROH of Any Size
We next turn to testing the two hypotheses regarding the role of selection on patterns of deleterious variation. Recall that our expectations center around comparing the fraction of damaging homozygotes inside and outside of ROH regions to the corresponding fraction of nondamaging homozygotes. Under hypothesis 1, damaging homozygotes occur more often in non-ROH regions relative to the proportion of genome-wide nondamaging homozygotes occurring in non-ROH regions. Under hypothesis 2, damaging homozygotes occur more often in ROH regions relative to the proportion of genome-wide nondamaging homozygotes occurring in ROH regions. Hypothesis 2 additionally predicts an effect of ROH size class, with long ROH having the greatest enrichment of damaging homozygotes.
To test these hypotheses, we compute
| (Equation 8) |
where is the fraction of nondamaging 1/1 homozygotes in individual i that fall in any ROH region. These numbers represent the baseline distribution for nondamaging homozygotes with which we compare the distribution of damaging homozygotes. Similarly, we compute
| (Equation 9) |
where is the fraction of damaging 1/1 homozygotes in individual i that fall in any ROH region. Under hypothesis 1, we expect , whereas under hypothesis 2, .
Figure 4A plots and versus total genomic ROH coverage (). Both the fraction of nondamaging homozygous genotypes in ROH and the fraction of damaging homozygous genotypes in ROH are positively correlated with total genomic ROH coverage (nondamaging Pearson r = 0.9983, damaging Pearson r = 0.9938). The correlations are expected, given that we expect a larger fraction of homozygous genotypes to occur in ROH as ROH comprise increasingly more of the genome. In accordance with hypothesis 2, the fraction of genome-wide damaging homozygotes in ROH consistently exceeds the fraction of genome-wide nondamaging homozygotes in ROH.
Figure 4.
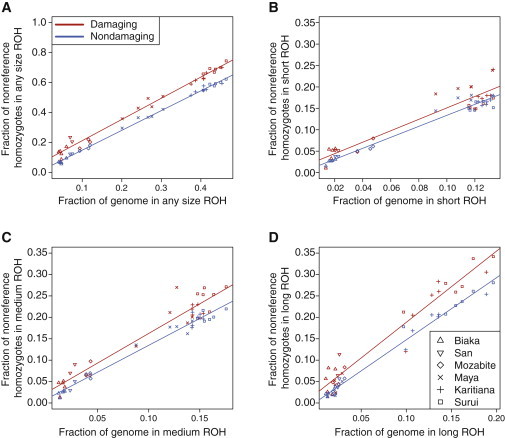
The Fraction of All Genome-wide Nonreference Homozygotes Falling in ROH Regions versus the Fraction of the Genome Covered by ROH, for Each Individual
(A) Any ROH region.
(B) Short (class A) ROH regions.
(C) Medium (class B) ROH regions.
(D) Long (class C) ROH regions.
Red points represent damaging homozygotes, and blue points represent nondamaging homozygotes.
To assess the statistical significance of the two linear regressions on total genomic ROH coverage for the damaging and nondamaging genotypes, we fit a linear model,
| (Equation 10) |
where is a vector of length 54 containing, for all individuals, the fraction of genome-wide damaging homozygotes in any ROH region () and the fraction of genome-wide nondamaging homozygotes in any ROH region (). is the fraction of the genome covered by ROH of any size for individual i, as given in Equation 1, and is an indicator variable, taking a value of 1 if the observed response is of damaging homozygotes and a value of 0 for nondamaging homozygotes. A statistically significant (two-tailed t test) indicates a difference in the intercepts of separate regressions for damaging and nondamaging homozygotes, and a statistically significant (two-tailed t test) indicates a difference in the regression slopes.
We find (p = 8.591 × 10−6) and (p = 8.394 × 10−3), indicating significantly different intercepts and slopes between the regressions in Figure 4A. Thus, as predicted by hypothesis 2, damaging homozygotes occur more often in ROH than expected on the basis of nondamaging homozygotes.
Damaging and Nondamaging Homozygotes by ROH Size Class
Under hypothesis 2, we expect to observe an excess of damaging homozygotes compared to nondamaging homozygotes specifically in long (class C) ROH, and an excess of damaging homozygotes in long (class C) ROH versus damaging homozygotes in short (class A) ROH. To examine these predictions, we separately consider each ROH size class. For homozygous genotypes falling in ROH of size class j, we calculate
| (Equation 11) |
| (Equation 12) |
for damaging and nondamaging 1/1 homozygotes, respectively. Because we investigate the same number of data points for each size class—27 individuals, each with a value of and a value of —statistical tests for each size class are equally powered.
Figure 4B plots and versus total genomic coverage for class A ROH (). Both the fraction of nondamaging homozygous genotypes in class A ROH and the fraction of damaging homozygous genotypes in class A ROH are positively correlated with class A genomic coverage (nondamaging Pearson r = 0.9829, damaging Pearson r = 0.9365), though the two regressions have no significant difference in either the intercept (, p = 0.3027) or the slope (, p = 0.6466). Figure 4C plots and versus total genomic coverage by class B ROH (). The regressions for nondamaging (r = 0.9892) and damaging (r = 0.9629) homozygotes have smaller p values than in the case of class A, but again with no significant difference in either the intercept (, p = 0.1312) or the slope (, p = 0.1425). Figure 4D plots and versus total genomic coverage by class C ROH () and the regressions for nondamaging (r = 0.9921) and damaging (r = 0.9727) homozygotes. We now find a significant difference in both the intercept (, p = 0.03679) and slope (, p = 0.01863). These results are consistent with hypothesis 2, supporting the view that inbreeding that generates long ROH is driving the differences in the proportions of damaging and nondamaging homozygotes in ROH regions.
Under hypothesis 2, we expect damaging homozygotes to occur more frequently in class C ROH than in class A ROH. We compare the fraction of damaging homozygotes falling in class C ROH () to the fraction of damaging homozygotes falling in class A ROH (). We can see in Figure 5 that the high-ROH-coverage individuals have a substantially higher fraction of genome-wide damaging homozygotes occurring in class C versus class A. We test the statistical significance of the difference of these regressions with a linear model analogous to Equation 10. Here, however, we are concerned with distinguishing the distributions of damaging homozygotes in ROH of classes C and A. The regression model now becomes
| (Equation 13) |
where is a vector of length 54 containing, for all individuals, the fractions of genome-wide damaging homozygotes in class C ROH () and class A ROH (). is the fraction of the genome covered by either class C () or class A () ROH for individual i, and is an indicator variable, taking a value of 1 if the observed response is of damaging homozygotes in class C ROH and a value of 0 if the observed response is of damaging homozygotes in class A ROH. Although the intercepts of the regressions are not significantly different (, p = 0.7278), the slopes are significantly different (, p = 0.01389). This result suggests that the increase in the fraction of damaging homozygotes is higher per unit increase in ROH coverage for class C ROH versus class A ROH, consistent with hypothesis 2.
Figure 5.
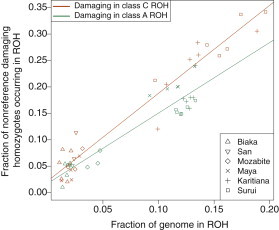
The Fraction of All Genome-wide Nonreference Homozygotes Falling in Different Sized ROH versus the Fraction of the Genome Covered by ROH, for Each Individual
Orange points represent damaging homozygotes in long (class C) ROH regions, and green points represent damaging homozygotes in short (class A) ROH regions.
To assess the robustness of these results, we repeated the analysis with SIFT, an alternative program for predicting deleterious alleles.45 SIFT generates two classifications, tolerated and damaging, analogous to our use of nondamaging and damaging classes with PolyPhen2. In general, SIFT produces the same patterns observed with PolyPhen2 (Figures S5 and S6). With PolyPhen2, we found significant differences between the nondamaging and damaging intercepts and slopes when considering all ROH regions and class C ROH regions, and we further found a significant difference in the slopes when comparing damaging homozygotes in class A versus class C ROH. All of these results are recapitulated with SIFT, with the exception that for the comparison of intercept terms for nondamaging and damaging variants, PolyPhen2 produced a result significant at the 0.05 level (p = 0.03679) whereas with SIFT, the test is not significant (p = 0.06029).
The divergence in slopes in Figure 5 might be driven partly by an excess of damaging variants in Native American populations as a function of all Native American variants, compared to the analogous computation in African populations. To examine this possibility, for each population in our data set, we consider the fraction of population-specific (private) alternate alleles that are predicted to be damaging. Here, we call a site private if the alternate allele at that locus is found in only one of the populations in our sample. For each population, we calculate the proportion of alleles private to the population that are of a particular predicted functional class by computing
| (Equation 14) |
where is the fraction of alleles private to population p that have predicted functional class {synonymous, benign, probably damaging, possibly damaging}, is the number of alleles private to population p of predicted functional class s, and is the total number of alleles private to population p. Figure 6 depicts this fraction, illustrating that the proportion of private variants that are predicted to be damaging is lowest in sub-Saharan African populations (∼20%) and highest in the Native American populations (>30%). The Native American populations have the highest levels of genomic ROH coverage, and long ROH, produced by younger haplotypes, are likely to possess a disproportionate number of young and potentially private variants (Figure S7). Thus, the combination of a high proportion of private damaging alleles and high homozygosity is probably contributing to the significant divergence in slopes observed in Figure 5.
Figure 6.
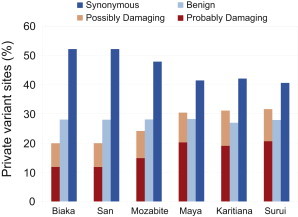
The Fraction of All Private Nonreference Variants that Are Synonymous or Missense Variants
Private variants are defined as variants for which the nonreference allele appears only in a single population in our sample. Missense variants are further split by PolyPhen2 into predicted benign, predicted possibly damaging, and predicted probably damaging classes.
Nonsense Variants and ROH
The patterns we have observed thus far consider homozygotes from two predicted classes: damaging and nondamaging. Although we expect the damaging class of variants to be enriched for variants that have deleterious effects, it is useful to study a subset of variants with an even higher likelihood of being deleterious. First, when we consider probably damaging, possibly damaging, benign, and synonymous homozygotes as four separate classes rather than combining benign and synonymous homozygotes and probably damaging and possibly damaging homozygotes, the observed patterns are similar to those observed in the combined analysis, with the probably damaging homozygotes having the highest fraction within ROH and the benign and synonymous homozygotes having nearly identical fractions (Figure S8). Next, noting that nonsense mutations create stop codons in the reading frame and are a priori more likely to interfere with the proper functioning of a protein than are missense mutations, we examined the placement of two nested sets of nonsense mutations in relation to ROH. The first is a set of 257 nonsense mutations that passed our quality-control procedures, and the second is a subset of those 257 sites consisting of 66 nonsense mutations that are verified loss-of-function (LoF) variants as identified by MacArthur et al.14 Tables S6 and S7 report at nonsense and LoF nonsense sites, respectively, the counts for reference homozygotes (0/0), heterozygotes (0/1), and nonreference homozygotes (1/1) that fall into ROH regions and non-ROH regions.
Because these data sets are substantially smaller than the full sets of damaging and nondamaging sites, instead of considering individuals separately, we combine individuals into two groups: “low-ROH” individuals and “high-ROH” individuals. For comparisons involving all ROH regions (Figure 7A), individuals with less than 20% genomic ROH coverage were classified as low ROH, and those with more than 20% ROH coverage were classified as high ROH. For comparisons involving specific ROH size classes (Figures 7B–7D), individuals with less than 5% genomic ROH coverage of the respective size class were classified as low ROH, and the remaining individuals were regarded as high ROH.
Figure 7.
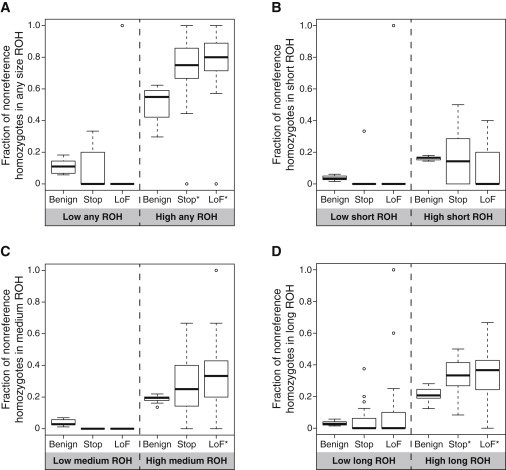
The Fraction of All Genome-wide Nonreference Homozygotes Falling in ROH Regions for Nondamaging Variants, Nonsense Variants, and LoF Nonsense Variants versus the Fraction of the Genome Covered by ROH, for Individuals Grouped into “Low-ROH” and “High-ROH” Groups
(A) Any ROH region.
(B) Short (class A) ROH regions.
(C) Medium (class B) ROH regions.
(D) Long (class C) ROH regions.
Means that exceed the mean for benign sites for the same ROH coverage class at the p < 0.05 significance level are indicated by asterisks.
Figure 7A examines the distribution of nonsense mutations across all ROH. For low-ROH individuals, no significant difference exists in the mean fraction of homozygotes falling in ROH between nondamaging and nonsense variants (p = 0.6788, one-tailed t test) or between nondamaging and LoF variants (p = 0.2581, one-tailed t test). For high-ROH individuals, however, the fraction of nondamaging homozygotes falling in ROH is significantly lower than those for nonsense (p = 2.368 × 10−3, one-tailed t test) and LoF (p = 3.160 × 10−4, one-tailed t test) homozygotes falling in ROH. Separately analyzing class A, class B, and class C ROH (Figures 7B–7D), for individuals with high genomic ROH coverage, the fraction of nonsense homozygotes in class C ROH is significantly greater than the fraction of nondamaging homozygotes in class C ROH (p = 2.862 × 10−3, one-tailed t test); the comparisons are not significant for class A (p = 0.4976, one-tailed t test) or class B (p = 0.05689, one-tailed t test) ROH. The fraction of nonsense LoF homozygotes significantly exceeds the fraction of nondamaging homozygotes in class B ROH (p = 0.01203, one-tailed t test) and class C ROH (p = 8.931 × 10−3, one-tailed t test) but not in class A ROH (p = 0.8964, one-tailed t test). These results are consistent with our hypothesis 2, in that high-ROH individuals are observed to have a higher fraction of damaging homozygotes (nonsense and LoF nonsense) occurring in ROH of any size (Figure 7A) and that the pattern is driven primarily by long (class C) ROH (Figure 7D).
Discussion
Through sequencing-based variant discovery efforts, it has been widely recognized that each human individual carries numerous deleterious variants.8,14,15,18,19 Our data set extends this observation by showing that many individuals can carry at least 147 and up to 357 such damaging variants in homozygous form (Table S4). Further, more than half the individuals in our sample (14) carry five or more homozygous verified LoF variants (Table S7). The fact that the combined presence of so many homozygous deleterious variants is compatible with life supports the view that most deleterious variants must have relatively small fitness effects. The numbers of predicted deleterious homozygotes generally accord with other recent studies, which have estimated values from a few dozen to a thousand or more.8,17,18 Because we selected samples for our analysis to extend across the range of genomic ROH coverage observed worldwide, we expect that the number of deleterious variants in individuals from populations that we have not included would be comparable to the values we report.
Our analysis of deleterious variation with respect to ROH was framed by two alternative hypotheses. Under hypothesis 1, because of more effective selection in ROH against deleterious variants, we might have expected the fraction of genome-wide damaging homozygotes occurring in ROH to be less than the corresponding fraction of genome-wide nondamaging homozygotes. In this case, the result would have been driven by the expectation that selection would purge haplotypes containing many deleterious recessive alleles. On the other hand, under hypothesis 2, we expected inbreeding to present an excess of low-frequency and damaging variants in homozygous form, with selection not having had sufficient time to eliminate them. Under this hypothesis, we expected ROH to consist of long IBD haplotypes that combine otherwise rare and probably deleterious variants into homozygotes. Because IBD regions present homozygotes at a higher frequency than would be predicted by Hardy-Weinberg equilibrium, we expected ROH to contain a higher fraction of damaging homozygotes than the corresponding fraction of nondamaging homozygotes, with class C ROH—the most recent in origin and the result of recent inbreeding—driving this difference.
As we saw in Figure 4A, the fraction of damaging homozygotes was significantly greater in ROH regions than the corresponding fraction of nondamaging homozygotes, consistent with hypothesis 2. The pattern was observed more strongly for medium (class B) ROH than for short (class A) ROH, and for long (class C) ROH more strongly than for medium (class B) ROH; for long ROH, the difference was significant. Each of these patterns was recapitulated by nonsense and LoF nonsense variants (Figure 7).
Our identification of an enrichment of deleterious variation in ROH accords with and extends recent related work. Studies beginning with Lohmueller et al.8 have found that lower-diversity populations carry an excess of recessive deleterious variants, presumably as the result of founding events that have inflated the frequencies of otherwise rare alleles. If the same founding events that have amplified recessive deleterious variants are responsible for ROH, it might be expected that those variants would lie within ROH regions that have resulted from such founder events—typically short and medium length ROH (classes A and B). Although we do see some evidence for an excess of deleterious variants in these ROH classes, the signal is strongest for long ROH (class C), indicating a role for both inbreeding and founder events in increasing the occurrence of recessive deleterious variants.
We note that the excess of recessive deleterious variants in ROH regions might also result partially from a scenario in which positive selection generating long haplotypes leads to production of ROH, and neighboring deleterious variants in the ROH surrounding the positively selected sites hitchhike to high frequencies. However, Pemberton et al.29 found that across the genome, the frequency at which ROH occur is only weakly correlated with haplotype-based scores for positive selection; thus, although we expect that hitchhiking might account for some deleterious variants in ROH, this result suggests that it is unlikely as a general explanation. Another possibility is that ROH preferentially cover gene regions of systematically lower genomic constraint and that deleterious alleles in ROH are occurring in these gene regions. It is difficult to assess this hypothesis, however, because the computational prediction algorithms we used to classify deleterious mutations rely on genomic constraint measures, so that we cannot easily separate measures of constraint from predictions of deleteriousness.
One of the limitations of this study was our reliance on predicted variant function. Computational prediction algorithms are concerned primarily with molecular functions of gene products and not with overall impact on organism-level disease risks or fitness. Thus, the PolyPhen2 prediction is only a proxy for the fitness consequence of a mutation. Although we do not expect the functional classification given by PolyPhen2 to be accurate for every missense variant, it is reasonable to consider the full sets of damaging and nondamaging variants as enriched for truly deleterious and benign mutations, respectively. Because our study deals with these variants in aggregate, prediction accuracy for individual variants is not as important as the observed general trends. Our claims are robust in that we reproduced our findings in two higher-confidence sets of deleterious variants: nonsense and verified loss-of-function variants. Furthermore, by using the program SIFT in place of PolyPhen2 to computationally predict functional effect, we reproduced our findings from Figures 4 and 5 in Figures S5 and S6.
The human genome contains a spectrum of variants with a rich gradation of functional impact. Our results suggest that inbreeding not only amplifies the occurrence of recessive genetic diseases of significant fitness effect, it also amplifies the burden of mildly deleterious homozygotes. Indeed, inbreeding has long been known to be deleterious to the health of offspring,2,46–51 and if a variant in a population is lethal in homozygous form, inbreeding will greatly increase the chance of generating a genome with the lethal genotype. Our work finds that inbreeding also has a more subtle effect, enabling the accumulation of mildly deleterious variants as well. If some of these variants act in synergistic fashion, then, as suggested by recent results implicating an excess of runs of homozygosity as a contributor to disease phenotypes52 and other traits,53 the simultaneous presence of multiple deleterious variants in homozygous form could systematically underlie an important component of complex human diseases.
Acknowledgments
The authors would like to thank Brendan Tarrier, Christine Brennan, and Robert Lyons of the University of Michigan DNA Sequencing Core. Funding for this research was provided by National Institutes of Health R01 GM081441 and R01 HG005855, an NARSAD Young Investigator Award, and a grant from the Burroughs Wellcome Fund.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Picard, http://picard.sourceforge.net/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Muller H.J. Our load of mutations. Am. J. Hum. Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
- 2.Morton N.E., Crow J.F., Muller H.J. An estimate of the mutational damage in man from data on consanguineous marriages. Proc. Natl. Acad. Sci. USA. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondrashov A.S. Contamination of the genome by very slightly deleterious mutations: why have we not died 100 times over? J. Theor. Biol. 1995;175:583–594. doi: 10.1006/jtbi.1995.0167. [DOI] [PubMed] [Google Scholar]
- 4.Charlesworth B., Charlesworth D. Some evolutionary consequences of deleterious mutations. Genetica. 1998;102-103:3–19. [PubMed] [Google Scholar]
- 5.Eyre-Walker A., Keightley P.D. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- 6.Fay J.C., Wyckoff G.J., Wu C.-I. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 8.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun S., Fay J.C. Evidence for hitchhiking of deleterious mutations within the human genome. PLoS Genet. 2011;7:e1002240. doi: 10.1371/journal.pgen.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper G.M., Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 11.Lohmueller K.E., Albrechtsen A., Li Y., Kim S.Y., Korneliussen T., Vinckenbosch N., Tian G., Huerta-Sanchez E., Feder A.F., Grarup N. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genet. 2011;7:e1002326. doi: 10.1371/journal.pgen.1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Necşulea A., Popa A., Cooper D.N., Stenson P.D., Mouchiroud D., Gautier C., Duret L. Meiotic recombination favors the spreading of deleterious mutations in human populations. Hum. Mutat. 2011;32:198–206. doi: 10.1002/humu.21407. [DOI] [PubMed] [Google Scholar]
- 13.Lesecque Y., Keightley P.D., Eyre-Walker A. A resolution of the mutation load paradox in humans. Genetics. 2012;191:1321–1330. doi: 10.1534/genetics.112.140343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torkamani A., Pham P., Libiger O., Bansal V., Zhang G., Scott-Van Zeeland A.A., Tewhey R., Topol E.J., Schork N.J. Clinical implications of human population differences in genome-wide rates of functional genotypes. Front. Genet. 2012;3:211. doi: 10.3389/fgene.2012.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., Tyler-Smith C., 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crow J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 21.Sunyaev S.R. Inferring causality and functional significance of human coding DNA variants. Hum. Mol. Genet. 2012;21(R1):R10–R17. doi: 10.1093/hmg/dds385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson J., Morton N.E., Collins A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 23.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Curtis D., Vine A.E., Knight J. Study of regions of extended homozygosity provides a powerful method to explore haplotype structure of human populations. Ann. Hum. Genet. 2008;72:261–278. doi: 10.1111/j.1469-1809.2007.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McQuillan R., Leutenegger A.-L., Abdel-Rahman R., Franklin C.S., Pericic M., Barac-Lauc L., Smolej-Narancic N., Janicijevic B., Polasek O., Tenesa A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auton A., Bryc K., Boyko A.R., Lohmueller K.E., Novembre J., Reynolds A., Indap A., Wright M.H., Degenhardt J.D., Gutenkunst R.N. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nothnagel M., Lu T.T., Kayser M., Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum. Mol. Genet. 2010;19:2927–2935. doi: 10.1093/hmg/ddq198. [DOI] [PubMed] [Google Scholar]
- 28.Kirin M., McQuillan R., Franklin C.S., Campbell H., McKeigue P.M., Wilson J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pemberton T.J., Absher D., Feldman M.W., Myers R.M., Rosenberg N.A., Li J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012;91:275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lander E.S., Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 31.Broman K.W., Weber J.L. Long homozygous chromosomal segments in reference families from the Centre d’Etude du Polymorphisme Humain. Am. J. Hum. Genet. 1999;65:1493–1500. doi: 10.1086/302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildebrandt F., Heeringa S.F., Rüschendorf F., Attanasio M., Nürnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S., Haynes C., Barany F., Ott J. Genome-wide autozygosity mapping in human populations. Genet. Epidemiol. 2009;33:172–180. doi: 10.1002/gepi.20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiezun A., Pulit S.L., Francioli L.C., van Dijk F., Swertz M., Boomsma D.I., van Duijn C.M., Slagboom P.E., van Ommen G.J.B., Wijmenga C., Genome of the Netherlands Consortium Deleterious alleles in the human genome are on average younger than neutral alleles of the same frequency. PLoS Genet. 2013;9:e1003301. doi: 10.1371/journal.pgen.1003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cann H.M., de Toma C., Cazes L., Legrand M.-F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 37.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pruitt K.D., Harrow J., Harte R.A., Wallin C., Diekhans M., Maglott D.R., Searle S., Farrell C.M., Loveland J.E., Ruef B.J. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 43.Ramachandran S., Deshpande O., Roseman C.C., Rosenberg N.A., Feldman M.W., Cavalli-Sforza L.L. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. USA. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.-C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 45.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Darwin C.R. John Murray; London: 1876. The Effects of Cross and Self Fertilisation in the Vegetable Kingdom. [Google Scholar]
- 47.Garrod A.E. The incidence of alkaptonuria: a study in chemical individuality. Lancet. 1902;160:1616–1620. [Google Scholar]
- 48.Bittles A.H., Neel J.V. The costs of human inbreeding and their implications for variations at the DNA level. Nat. Genet. 1994;8:117–121. doi: 10.1038/ng1094-117. [DOI] [PubMed] [Google Scholar]
- 49.Jorde L.B. Consanguinity and prereproductive mortality in the Utah Mormon population. Hum. Hered. 2001;52:61–65. doi: 10.1159/000053356. [DOI] [PubMed] [Google Scholar]
- 50.Rudan I., Rudan D., Campbell H., Carothers A., Wright A., Smolej-Narancic N., Janicijevic B., Jin L., Chakraborty R., Deka R., Rudan P. Inbreeding and risk of late onset complex disease. J. Med. Genet. 2003;40:925–932. doi: 10.1136/jmg.40.12.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Charlesworth D., Willis J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 52.Keller M.C., Simonson M.A., Ripke S., Neale B.M., Gejman P.V., Howrigan D.P., Lee S.H., Lencz T., Levinson D.F., Sullivan P.F., Schizophrenia Psychiatric Genome-Wide Association Study Consortium Runs of homozygosity implicate autozygosity as a schizophrenia risk factor. PLoS Genet. 2012;8:e1002656. doi: 10.1371/journal.pgen.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McQuillan R., Eklund N., Pirastu N., Kuningas M., McEvoy B.P., Esko T., Corre T., Davies G., Kaakinen M., Lyytikäinen L.-P., ROHgen Consortium Evidence of inbreeding depression on human height. PLoS Genet. 2012;8:e1002655. doi: 10.1371/journal.pgen.1002655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.