

Abstract
The release kinetics and stability of ovalbumin encapsulated into polyanhydride microspheres with varying chemistries were studied. Polymers based on the anhydride monomers sebacic acid (SA), 1,6-bis(p-carboxyphenoxy)hexane (CPH), and 1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane (CPTEG) were utilized. Microspheres were fabricated using two non-aqueous methods: a solid/oil/oil double emulsion technique and cryogenic atomization. The studies showed that the two fabrication methods did not significantly affect the release kinetics of ovalbumin, even though the burst release of the protein was a function of the fabrication method and the polymer chemistry. Antigenic stability of ovalbumin released from microspheres prepared by cryogenic atomization was studied by western blot analysis. These studies indicate that the amphiphilic CPTEG:CPH polyanhydrides preserved protein structure and enhanced protein stability by preserving the immunological epitopes of released protein.
Keywords: polyanhydrides, microspheres, cryogenic atomization, protein stability, release kinetics
INTRODUCTION
Biodegradable polymers have been used as carriers for the controlled delivery of drugs and proteins for over two decades1. These carriers have the advantages of providing sustained release over long periods of time, well-controlled release profiles, and biocompatibility2. The most common biodegradable polymers used in drug delivery applications are polyesters such as poly(D,L-lactide-co-glycolide) (PLGA), polyanhydrides, and poly(orthoesters). A potential drawback in using the bulk-erodible PLGA for protein delivery is that the degradation products are acidic; for example, a pH of less than 3 for degradation products3 and a pH of 2 inside a PLGA drug delivery device4 have been reported. Studies have shown that at these pH values, some proteins can become denatured and/or irreversibly aggregated5. For biologically active proteins (enzymes, cytokines), this is problematic because a loss in structure is detrimental to function. In comparison, the microenvironmental pH resulting from polyanhydride degradation is higher, notably 4.2 for sebacic acid (SA) and 5.5 for 1,6-bis(p-carboxyphenoxy)hexane (CPH)6 and potentially less detrimental to biologically active proteins. The hydrophobic nature of polyanhydrides also prevents water-induced covalent aggregation of proteins, as water penetration into the polymer is negligible; however, non-covalent aggregation due to hydrophobic interactions may result7,8. This has motivated the development of polyanhydrides that are less hydrophobic9,10. Recently, Torres and co-workers reported that the incorporation of oligomeric ethylene glycol units into the backbone of hydrophobic aromatic polyanhydrides, such as poly(CPH), results in amphiphilic copolymers with mixed erosion mechanisms. It was shown that such amphiphilic polymers result in a reduction of both covalent and non-covalent aggregation of encapsulated proteins9, suggesting their promise as protein carriers.
Polyanhydrides have been studied for drug delivery applications since 1983, when Langer and co-workers reported their potential for controlled drug delivery based on their biodegradable properties, and non-toxic and non-mutagenic nature11. Due to their surface erosion characteristics, polyanhydrides are attractive as drug delivery devices as they exhibit a predictable zero-order release rate12. In the presence of water, degradation of polyanhydrides occurs by base-catalyzed hydrolysis of anhydride linkages to form dicarboxylic acids. In addition, the rate of degradation depends upon the chemistry of the polymer13-15. For example, SA and CPH tablets would degrade in 54 days and 1 year, respectively16. In contrast, 80% of the ethylene glycol containing polyanhydride, poly(1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane) (poly(CPTEG)), degrades in 28 days9. Thus, by combining different ratios of anhydride monomers into copolymer formulations, degradation rates can be tailored for specific applications17.
Biodegradable and biocompatible polymers are also preferred for parenteral drug delivery systems as there is no need to remove them following implantation. A size of less than 125 μm in diameter is normally preferred for such applications; this size allows for delivery into the tissue by the use of a syringe and needle18. In this regard, microspheres and nanospheres of biodegradable polymers have been widely used as injectable delivery systems for drugs, proteins, and immunogens2,19,20. Typical methods for microsphere fabrication include hot melt microencapsulation21, double emulsion12,22-26, spray drying27,28, and cryogenic atomization24,29-32. In particular, previous research has shown that double emulsion methods in which water/organic interfaces are present are potentially detrimental for protein stabilization25,33-36. Thus, there has been an emphasis on developing non-aqueous methods for preparing protein-loaded microspheres. Two commonly used non-aqueous techniques for fabricating microspheres include solid-oil-oil (S/O/O) double emulsion and cryogenic atomization (CA); besides avoiding the water/organic interface, these techniques prevent hydrolysis of the polymer by eliminating water from the process37.
The objective of the present study is to systematically evaluate the effects of polymer chemistry and fabrication method on the release kinetics of proteins from polyanhydride microspheres and on epitope availability of the released protein. Polymer chemistries based on the anhydride monomers SA, CPH, and CPTEG were chosen (Fig. 1). Ovalbumin (Ova) was selected as the model protein. Protein-loaded microspheres were fabricated by both the S/O/O and CA methods.
Figure 1.

Chemical structures of polymers used, from top, left to right: poly(sebacic acid), poly(1,6-bis(p-carboxyphenoxy)hexane), and poly(1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane). Here n represents the number of repeating monomer units.
MATERIALS AND METHODS
Materials
Albumin from chicken egg white (ovalbumin/Ova), 1,6-dibromohexane, 4-hydroxybenzoic acid, 1-methyl-2-pyrrolidinone, sebacic acid (99%), monoclonal anti-chicken egg albumin (clone Ova-14), rabbit anti-chicken egg albumin, alkaline phosphatase conjugated goat anti-rabbit IgG, and tri-ethylene glycol were purchased from Sigma-Aldrich (St. Louis, MO). 4-p-fluorobenzonitrile was purchased from Apollo Scientific (Cheshire, UK). Acetic acid, acetic anhydride, acetone, acetonitrile, dimethyl formamide, ethyl ether, heptane, hexane, methylene chloride, petroleum ether, potassium carbonate, sodium hydroxide, sulfuric acid, and toluene were purchased from Fisher Scientific (Fairlaw, NJ). Dialysis cassettes, bicinchoninic acid (BCA) assay reagents, and GelCode blue were purchased from Pierce (Rockford, IL). Low molecular mass protein standards were purchased from BioRad (Hercules, CA). 12% tris-glycine PAGE Duramide Precast Gels were purchased from Lonza Bioscience (Basel, Switzerland). Dow Corning oil, ethanol, and liquid nitrogen were obtained from in-house bulk chemical supplies.
Monomer/polymer Synthesis
To synthesize the CPH monomer, the method described by Conix38 for synthesizing 1,3-bis(p-carboxyphenoxy)propane was altered, using 1,6-dibromohexane instead of 1,3-dibromopropane. Prepolymers for both CPH and SA were synthesized using a method outlined by Shen et al.39; CPH:SA copolymers of various compositions and poly(SA) were synthesized by melt polycondensation using a procedure outlined by Kipper and coworkers20. The CPTEG monomer and CPTEG:CPH copolymers were produced using a technique described by Torres et al9. The polymers, pre-polymers, and diacids were characterized by 1H NMR, using a Varian VXR-300 NMR (Palo Alto, CA), to ensure purity; a Waters GPC (Milford, MA) was used to measure the polymer molecular weight.
Protein Preparation
Ovalbumin particles were created prior to use by preparing Ova solutions (50 mg) in 10 mL of 50 mM ammonium bicarbonate and pumping the solution over 400 mL of liquid nitrogen. The liquid nitrogen was allowed to boil off, and the remaining protein was placed in a dryer oven at room temperature overnight; under these conditions, it is well known that there is no denaturation of freeze-thawed Ova40.
Contact Angle Measurements
To characterize the relative hydrophobicity of the polymers, contact angle measurements were carried out. Polymers were dissolved at a concentration of 2.5 % w/v in tetrahydrofuran (for poly(CPTEG) and CPTEG-containing copolymers) or methylene chloride (for poly(CPH), poly(SA), and their copolymers). After filtering solutions with 0.2 μm filters, the solutions were pipetted onto separate round glass cover slides. After the solvent dried, more solution was added until a suitable polymer thickness was obtained. To measure the contact angle, a water droplet was carefully placed on the surface of the polymer film immediately prior to imaging with a CCD camera. Image J (NIH, Bethesda, MD) was used to measure the contact angle. These analyses were performed in triplicate. A Student-t test (α=0.05) was performed with the statistical analysis software JMP® 6 (Cary, NC).
Solid-Oil-Oil (S/O/O) Double Emulsion
This method was modified from a previously published procedure25. Briefly, 6 mg of Ova were suspended in 100 mg of polymer that was already dissolved in methylene chloride. A Tissue-Tearor™ homogenizer (Biospec Products Inc., Bartlesville, OK) was used to homogenize the solution for one minute. For the second emulsion, Dow Corning oil and methylene chloride were added and homogenized at 10,000 rpm for one minute, to allow for thorough mixing during addition. The parameters used for each emulsion step for the different polymer chemistries are shown in Table 1. The solution was added drop-wise to a beaker containing 200 mL of heptane immersed in an ice bath (4 °C) and stirred at 300 rpm for two hours using a Caframo overhead stirrer (Wiarton, Ontario, Canada). Finally, the microspheres were filtered and placed in a vacuum oven overnight to eliminate any residual heptane.
Table 1.
Parameters for S/O/O double emulsion
| Methylene chloride for inner emulsion |
Rate for inner emulsion homogenization |
Rate for outer emulsion homogenization |
Diameter Range (Mean ± SD)* |
|
|---|---|---|---|---|
| 20:80 CPTEG:CPH |
2 mL | 20,000 rpm 3 min |
3mL oil/4 mL MeCl2 20,000 rpm, 3 min |
18 ± 11 μm |
| Poly(CPTEG) and 50:50 CPTEG:CPH |
2 mL | 20,000 rpm 3 min |
4mL oil/6 mL MeCl2 20,000 rpm, 3 min |
15 ± 11 μm 13 ± 9 μm |
| Poly(SA) | 3 mL | 30,000 rpm 1 min |
3mL oil/4 mL MeCl2 20000 rpm, 1 min |
0.3 ± 0.2 μm |
| 20:80 CPH:SA | 2 mL | 30000 rpm 1 min |
3mL oil/4 mL MeCl2 30000 rpm, 1 min |
10 ± 6 μm |
| 50:50 CPH:SA | 2 mL | 20000 rpm 1 min |
3mL oil/4 mL MeCl2 30000 rpm, 1 min |
10 ± 6 μm |
SD=Standard deviation of over 500 microspheres
Cryogenic Atomization (CA)
CA was also modified from previously published work25. Briefly, 100 mg of each polymer was dissolved in methylene chloride to which 6 mg of lyophilized Ova was added. Using a glass syringe, 20 gauge capillary tube, and programmable syringe pump (KD Scientific, Holliston, MA), the polymer solution was pumped over 200 mL of 200 proof ethanol (cooled by liquid nitrogen), leaving a small layer of liquid nitrogen overlaying the ethanol. The atomizing mist was provided by an ultrasonic atomizing nozzle (SonoTek Corporation, Milton, NY). After generating the microspheres, the beakers of ethanol were placed in a −80 °C freezer for three days to allow the liquid nitrogen to evaporate, the ethanol to warm up, and the methylene chloride to slowly be extracted. Afterwards, the microspheres were filtered and placed in a vacuum oven to dry overnight. Table 2 summarizes the operating parameters for this method for each of the polymer chemistries studied.
Table 2.
Parameters for CA
| Methylene chloride |
Flow rate | Wattage | Diameter (Mean ± SD)* |
|
|---|---|---|---|---|
| 20:80 CPTEG:CPH, 50:50 CPTEG:CPH, and Poly(CPTEG) |
7 mL | 3 mL/min | 1.5 W | 16 ± 8 μm 13 ± 8 μm 14 ± 8 μm |
| 50:50 CPH:SA | 3 mL | 1.5 mL/min | 2.5 W | 10 ± 5 μm |
| Poly(SA) and 20:80 CPH:SA |
3 mL | 3 mL/min | 1.5 W | 14 ± 8 μm 13 ± 8 μm |
SD=Standard deviation of over 500 microspheres
Size Distribution of Microspheres
A JEOL 840A scanning electron microscope (SEM) was used to determine the size and shape of the microspheres. Microspheres were smeared onto carbon stubs, sputter coated with 200 Å of gold, and imaged. Size distribution analysis was performed using Image J.
Ova Release Kinetics
Polyanhydride microspheres (10 mg) fabricated by S/O/O or CA were suspended in 1 mL of phosphate buffered saline (PBS, pH 7.4) containing 0.01% sodium azide and placed in an incubator at 37 °C while stirring at 100 rpm. Samples of the buffer were collected two hours later, daily for one week, and every other day for 30 days. An aliquot of 750 μL was sampled each time and subsequently replaced with 750 μL of fresh PBS to ensure perfect sink conditions; the microfuge tubes containing the microsphere suspensions were centrifuged before sampling to ensure that no microspheres were removed from along with the sampled PBS. In order to quantify the amount of protein released, a bicinchoninic acid (BCA) assay was run on each sample, in triplicate, as described by the manufacturer (Pierce).
After one month of release, the samples were added to dialysis cassettes (10 kDa molecular mass cut-off) to determine the amount of protein remaining inside the microspheres. The microspheres that had not completely eroded were suspended in 3 mL of 17 mM NaOH and sonicated to allow for complete degradation of the remaining polymer27. Each release sample was added to dialysis cassettes and incubated for one week at 40 °C and 100 rpm. A BCA assay was run on each sample in triplicate. The total protein loaded into the microspheres was calculated by adding the protein that was released in one month to the protein extracted from the remaining microspheres. The release data is presented as cumulative fraction of protein released, in which the amount of protein released is normalized by the total protein loaded into the microspheres. The total protein loaded into the microspheres was also used to evaluate the encapsulation efficiency of the protein into the microspheres by comparing the total protein loaded to the initial amount of protein.
Western Blot Analysis
Briefly, protein encapsulated in Ova-loaded microspheres (5% w/w) was quantified after incubation in dialysis cassettes as explained above. Prior to addition to the dialysis cassette, Ova-loaded microspheres were incubated in 1 mL of phosphate buffer solution (pH 7.4) with 0.01% sodium azide for 1h to ensure that all analyses were performed on released protein and not on protein adsorbed to the surface of the microsphere. Using the concentration from the BCA assay, 2 μg of protein from each sample were prepared under reducing conditions and analyzed by sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The gels were run at a constant voltage of 120 V. Subsequently, the gels were transferred into PVDF membranes for 3 hours at a constant current of 70mA. The PVDF membranes were blocked with TBST (0.1 mM tris buffered saline with 0.05% Tween, pH 7.6) containing 2.5% casein overnight. The following day, the membranes were rinsed thrice in TBST and incubated with primary antibody (rabbit anti-Ova, Sigma) diluted in TBST (1:1000). After 4 h of incubation and three washing steps, alkaline phosphatase conjugated goat anti-rabbit IgG diluted in TBST (1:1000) was added. After 2 h, the membranes were removed and rinsed thrice with TBST. A colorimetric detection method using 2 μg/mL naphthol phosphate and 2 μg/mL fast red solution in distilled water was used to reveal bands.
Unencapsulated Ova was used as a control or reference sample for any alterations to Ova structure during incubation at pH 10, denaturation and heating in the presence of SDS. Therefore, the western blot analysis of the Ova recovered after release from the microspheres was used to compare the epitope availability of the Ova structure following release from the different polymer chemistries.
RESULTS
Polymer Hydrophobicity
In order to compare the relative hydrophobicity of the various polymers, their contact angles were measured (Figure 2). As expected, poly(CPTEG), the polymer with the fastest degradation profile (within weeks9), had the smallest contact angle (29°) of all the polymer chemistries studied. Thus, poly(CPTEG) was significantly less hydrophobic than all the other polyanhydride chemistries analyzed (p ≤ 0.05). In contrast, poly(CPH), which is the most hydrophobic polymer, and would take ~ one year to completely degrade41, had the largest contact angle (60°). As Figure 2 demonstrates, an increase in CPH content within the CPTEG:CPH copolymer system results in an increase in contact angle, which is consistent with an increase in hydrophobicity. Copolymer compositions in the CPTEG:CPH system that are rich in CPTEG exhibit bulk erosion characteristics while compositions rich in CPH exhibit surface erosion characteristics, making this system versatile9. In the surface erodible CPH:SA system, since both CPH and SA are hydrophobic, their copolymers have relatively similar hydrophobicities, as evidenced by contact angles that are statistically indistinguishable from each other.
Figure 2.

Contact angle (in degrees) of polyanhydride films. Error bars represent standard deviations of the mean generated from three separate experiments. A Student-t test was performed and significance defined as p < 0.05 (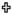 = statistically different from CPH and SA homopolymers ,
= statistically different from CPH and SA homopolymers , 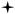 =statistically different from CPTEG homopolymer).
=statistically different from CPTEG homopolymer).
Ova Release Kinetics
Scanning electron photomicrographs of 50:50 CPH:SA and 20:80 CPTEG:CPH microspheres fabricated by S/O/O and CA methods are shown in Figures 3 and 4. In general, the mean diameter of the CPTEG-containing particles ranged from 4-29 μm (Table 1) when prepared by S/O/O, similar to the mean diameter of microspheres (5-24 μm) prepared by CA (Table 2). As expected, the majority of the microspheres (>50%) were below 10 μm, as previously described24. For the CPH:SA compositions, the mean diameter of the microspheres ranged between 0.1-16 μm and 5-22 μm for S/O/O and CA respectively. Noticeably, Poly(SA) microspheres fabricated by S/O/O were considerably smaller than all the other microspheres fabricated.
Figure 3.

50:50 CPH:SA microspheres fabricated by S/O/O (left) and CA (right) methods. Scale bars represent 50 μm.
Figure 4.

20:80 CPTEG:CPH microspheres fabricated by S/O/O (left) and CA (right) methods. Scale bars represent 20 μm.
Both the S/O/O and the CA methods resulted in ~100% encapsulation efficiency of the protein, which is consistent with previous work on these methods25. In Figure 5, the release profiles of Ova from poly(SA), 20:80 CPH:SA and 50:50 CPH:SA copolymer microspheres fabricated by S/O/O and CA methods are shown. All these chemistries exhibit sustained release kinetics after the initial burst of protein, which is consistent with previous work25. Each polyanhydride chemistry exhibited a different release rate consistent with the hydrophobicity of the polymer, but upon comparing the two fabrication methods, this was found to be unrelated to the fabrication method; for example, poly(SA) microspheres fabricated by both S/O/O and CA released 90% of the encapsulated Ova within 30 days. In the CPH:SA system, as the polymer hydrophobicity increased, the release rate of the protein decreased. The fabrication method did influence the size of the initial burst of protein release. Microspheres produced by the S/O/O technique demonstrated a smaller initial burst of protein than those produced by CA. This is attributed to the interplay between two phenomena: the rate at which the polymer precipitates during microsphere formation and the rate at which the methylene chloride is extracted into the non-solvent to form the microspheres. In addition, polymer hydrophobicity appears to have an influence on the burst effect, as a greater difference in initial bursts was noted for the less hydrophobic poly(SA) than the CPH:SA copolymers.
Figure 5.

Fractional release kinetics of ovalbumin (Ova) released from poly(SA) and CPH:SA copolymer microspheres prepared by solid/oil/oil/ (S/O/O) (left) or cryogenic atomization (CA) (right) fabrication techniques. Error bars indicate standard deviation of the mean generated from three separate experiments.
The Ova release profiles from poly(CPTEG), 20:80 CPTEG:CPH, and 50:50 CPTEG:CPH copolymer microspheres fabricated by S/O/O and CA methods are shown in Fig. 6. Because poly(CPTEG) is bulk-eroding, the protein release kinetics does not mimic the polymer degradation kinetics, but rather depends upon a combination of degradation, water swelling, and diffusion9. While one would expect the protein released from poly(CPTEG) microspheres to have the fastest release profile, the 50:50 CPTEG:CPH copolymer releases protein at the same rate (~90% for CA) or slightly faster (90% vs. 80% for S/O/O) than poly(CPTEG). Previous work has shown that even though the mass loss (i.e., erosion) of the microspheres was consistent with the hydrophobicities of poly(CPTEG) and 50:50 CPTEG:CPH copolymer, the water penetration and polymer degradation rates of both chemistries were very similar9. Our protein release data is consistent with these observations. On the other hand, the 20:80 CPTEG:CPH microspheres released Ova at a slower rate, and both fabrication methods were comparable in their sustained release profiles by releasing ~50% of protein in one month. Once again, the only variation of protein release kinetics as a result of the fabrication methods was that the initial burst release of Ova in the S/O/O method was smaller. Furthermore, poly(CPTEG) microspheres demonstrated the largest difference in amount of Ova released during the initial burst (8% in S/O/O vs. 42% in CA). This is consistent with the observations reported for the CPH:SA system.
Figure 6.

Fractional release kinetics of ovalbumin (Ova) released from poly(CPTEG) and CPTEG:CPH copolymer microspheres prepared by solid/oil/oil (S/O/O) (left) and cryogenic atomization (CA) (right) fabrication techniques. Error bars indicate standard deviation of the mean generated from three separate experiments.
Stability of Released Ova
Because the studies evaluating the release kinetics did not show significant differences between the S/O/O and CA microspheres, microspheres fabricated by cryogenic atomization were used for the protein stability studies due to ease of scale-up and simplicity of fabrication. Additionally, in vaccine delivery applications, it would be advantageous to have a high initial burst of antigen release in order to more rapidly and vigorously induce a robust primary immune response26.
Ova has a tendency to form moisture-induced covalent aggregates42, which is shown by the presence of characteristic bands between 54 and 97 kDa (lane 2), in addition to the major Ova band at 48 kDa (Figure 7). Strongly antigenically reactive bands are visible for both states, which was comparable to the unencapsulated Ova, for the protein released from each of the CPTEG-containing polymers (lanes 6-8), indicating that the antigenic epitopes of Ova were conserved. This data also indicated that the primary structure of Ova was not altered by encapsulation and release from the CPTEG-containing polymers. Encapsulation and release from 50:50 CPH:SA microspheres also appeared to preserve antigenic epitopes of the 45 kDa Ova, however, only faint bands for the aggregated forms of Ova were detectable. Poly(SA), due to the acidic nature of sebacic acid6, appeared to degrade Ova below the limit of detection by immunoblot analysis; 20:80 CPH:SA also showed a similar effect.
Figure 7.

Western blot analysis of ovalbumin (Ova) released from polyanhydride microspheres. Lane 1 – molecular size standards; lane 2 – Ova incubated at pH 10; lane 3 – Ova released from poly(SA); lane 4 – Ova released from 20:80 CPH:SA; lane 5 – Ova released from 50:50 CPH:SA; lane 6 – Ova released from poly(CPTEG); lane 7 – Ova released from 20: 80 CPTEG:CPH; and lane 8 – Ova released from 50:50 CPTEG:CPH. The Ova was recovered following a two-week period of protein release from the microspheres and the immunoblots were developed using a polyclonal anti-Ova antisera as described in Materials and Methods.
DISCUSSION
The present work provided insights into the effect of polymer chemistry and two non-aqueous microsphere fabrication methods on protein release kinetics and stability of the protein upon release. As expected, the more hydrophobic polymers released the protein more slowly. In these CPH-rich polymers, the rate at which water penetrates into the bulk slows in correlation with the amount of CPH43. Both microsphere fabrication methods yielded consistent results for the release kinetics of Ova. CA is the preferred method of preparing microspheres, due to its ease of scale up and increased protein encapsulation efficiency25.
The burst profiles correlated with the polymer hydrophobicity, as the more hydrophobic CPH:SA microspheres displayed the larger bursts regardless of the fabrication method. This may be attributable to the incompatibility of the protein with the more hydrophobic polymers. Therefore, even though the actual amount of protein released at the start of the degradation/erosion cycle may vary, the degree of hydrophobicity positively correlated with the observed burst effect. When a drug or a protein is incorporated into a microsphere, the drug/protein molecules may be non-uniformly distributed due to thermodynamic incompatibility with the polymer carrier. Therefore, when drug-loaded microspheres are immersed into a solution, the drug that is closer to the surface immediately escapes into the bulk solution, resulting in a large, instantaneous release of the drug12. Microspheres fabricated with S/O/O method exhibited different burst characteristics than CA, with higher initial bursts resulting from the cryogenic atomized microspheres. This could be attributed to the differences in polymer precipitation during the process of microsphere formation and the subsequent solvent extraction rate for each method. For CA, the polymer solution is sprayed into ultra-cold ethanol (-180 °C) hardening the polymer microspheres instantaneously. If the protein is not homogenously distributed in the polymer solutions it will be released faster, leading to larger initial bursts of protein release. In addition, processes that require an extended length of time to extract the solvent also affect the initial burst release. Due to the slow rate kinetics of the methylene chloride extraction during the fabrication of microspheres during CA, the protein can be displaced to the surface of the microspheres instead of being evenly dispersed. This will also contribute to a greater burst effect. In contrast, microspheres made by the S/O/O method may have more uniformly distributed protein within the polymer matrix because the process is conducted at 4 °C and occurs over 2 h. In addition, polymer chemistry affected the release kinetics in both fabrication methods; the more hydrophobic the polyanhydride, the smaller is the effect on the initial burst.
Overall, the present studies indicate that the chemistry of the carrier has a significant effect on protein stability. Even though equivalent amounts of protein were loaded on to each SDS-PAGE gel, the data indicated that polyclonal antibody was unable to detect Ova that had been encapsulated into poly(SA) and 20:80 CPH:SA copolymer suggesting that the protein had been degraded (Figure 7). Based on the immunoblot analysis, there was less degradation of Ova that was encapsulated into the 50:50 CPH:SA microspheres and the antigenic recognition was similar to that of un-encapsulated Ova. It is likely that the observed degradation was related to the acidic microenvironment resulting from the degradation of poly(SA) and 20:80 CPH:SA. This is consistent with previously reported results which demonstrated that acidic environments generated by degradation products are detrimental to protein stability and that the low pH produced inside bulk eroding microspheres exacerbates the loss of protein stability44. Among the degradation products studied, SA is the most acidic and, therefore, SA-rich polymers may be less favorable to protein stability. Nevertheless, it is important to mention that the acidity of SA is not as harsh as that caused by the well-studied PLGA6. On the other hand, epitope integrity was better maintained in CPTEG-containing microspheres regardless of composition or fabrication method. This is likely due to the amphiphilic nature of the CPTEG-containing copolymers and the lower acidity of the resultant degradation products as previously reported24,45.
The results of these studies are of particular importance when developing protocols for the design of protein delivery devices. As discussed earlier, polymer chemistry plays an important role that can be beneficial or detrimental for protein stability and biological function. A balance between hydrophobic and hydrophilic microenvironment (i.e., amphiphilic) is necessary to ensure protein stability as discussed elsewhere45. Drugs such as insulin have been shown to be structurally altered following release from PLGA microspheres because of the acidic nature of the polymer degradation products 46. In addition, the inability to exclude water from bulk eroding microspheres has been shown to affect protein stability. For example, insulin can undergo both covalent and non-covalent aggregation when introduced to moisture-rich environments, thus, affecting biological function of the encapsulated protein47. Since proteins rely on the preservation of their hierarchical structure for functional activity, their integrity must be maintained following encapsulation; thus, it is imperative that the delivery device and the fabrication methodology must not induce perturbations to protein structure6,24,25. Maintaining immunogenic epitopes is also crucial for the design of efficacious vaccination regimen. This would be particularly important for vaccines containing multiple protective epitopes that are essential for inducing immunity against diseases such as cancer48 and AIDS49. Furthermore, epitopes recognized by antibodies (B cells) can be linear or conformational within a folded protein. It is known that the immunogenicity of linear B cell epitopes is enhanced when protein conformation is retained49. The Towbin buffer used in the transfer phase of the western blot experiments does not contain SDS and allows proteins to refold into their native state. As shown in Figure 7, the antigenic structure (i.e., epitopes) of Ova was preserved upon release from the CPTEG:CPH containing copolymers. Maintaining the immunogenicity of multiple epitopes would improve vaccine efficacy as many disease-causing agents evade host immune mechanisms by altering dominant epitopes50. Based on the studies presented, polyanhydride delivery vehicles represent a promising platform that is capable of preserving the availability of antigenic epitopes. Thus, the choice of both polymer chemistry and the method of fabrication can significantly affect protein stability and the resultant efficacy of a vaccine delivery system.
CONCLUSIONS
These studies established that polyanhydride chemistry and microsphere fabrication methods significantly affect epitope stability of the protein and its release kinetics. The two fabrication methods studied in this work (S/O/O and CA) did not significantly affect the release kinetics of ovalbumin, even though the burst release of the protein was a function of the fabrication method and the polymer chemistry. The antigenic stability studies revealed that amphiphilic polyanhydrides (i.e., CPTEG:CPH copolymers) conserved protein structure and enhanced protein stability by preserving the immunological epitopes of released protein. These studies add to the growing literature on the importance of amphiphilic polymer carriers for protein stabilization and suggest a rational approach to designing protein carriers for both therapeutic and prophylactic applications.
ACKNOWLEDGMENTS
The authors would like to thank ISU undergraduate students Kristina Staley for help with polymer synthesis and release studies and Andrew Glowacki for help with size distribution analysis. The authors would like to acknowledge funding from the US Department of Defense – Office of Naval Research (ONR Award # N00014-06-1-1176), the Hunke Fellowship (to S.K.L), and NIH (Ruth Kirschstein Fellowship to M.P.T).
REFERENCES
- 1.Peppas NA, Langer R. New challenges in biomaterials. Science. 1994;263(5154):1715–20. doi: 10.1126/science.8134835. [DOI] [PubMed] [Google Scholar]
- 2.Sinha VR, Trehan A. Biodegradable microspheres for parenteral delivery. Crit Rev Ther Drug Carrier Syst. 2005;22(6):535–602. doi: 10.1615/critrevtherdrugcarriersyst.v22.i6.20. [DOI] [PubMed] [Google Scholar]
- 3.Zhu G, Schwendeman SP. Stabilization of proteins encapsulated in cylindrical poly(lactide-co-glycolide) implants: mechanism of stabilization by basic additives. Pharm Res. 2000;17(3):351–7. doi: 10.1023/a:1007513425337. [DOI] [PubMed] [Google Scholar]
- 4.Herrlinger M. Polymerabbau un Wirksttoffreigabe von Poly-DL-Lakid-Formlingen. University of Heidelberg; Germany: 1994. [Google Scholar]
- 5.Jaenicke R. Stability and stabilization of globular proteins in solution. J Biotechnol. 2000;79(3):193–203. doi: 10.1016/s0168-1656(00)00236-4. [DOI] [PubMed] [Google Scholar]
- 6.Determan AS, Wilson JH, Kipper MJ, Wannemuehler MJ, Narasimhan B. Protein stability in the presence of polymer degradation products: consequences for controlled release formulations. Biomaterials. 2006;27(17):3312–20. doi: 10.1016/j.biomaterials.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 7.Ron E, Turek T, Mathiowitz E, Chasin M, Hageman M, Langer R. Controlled release of polypeptides from polyanhydrides. Proc Natl Acad Sci USA. 1993;90:4176–4180. doi: 10.1073/pnas.90.9.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabata Y, Gutta S, Langer R. Controlled delivery systems for proteins using polyanhydride microspheres. Pharm Res. 1993;10(4):487–96. doi: 10.1023/a:1018929531410. [DOI] [PubMed] [Google Scholar]
- 9.Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J Biomed Mater Res A. 2006;76(1):102–10. doi: 10.1002/jbm.a.30510. [DOI] [PubMed] [Google Scholar]
- 10.Vogel BM, Mallapragada SK. Synthesis of novel biodegradable polyanhydrides containing aromatic and glycol functionality for tailoring of hydrophilicity in controlled drug delivery devices. Biomaterials. 2005;26(7):721–8. doi: 10.1016/j.biomaterials.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Rosen H, Chang J, Wnek G, Linhardt R, Langer R. Bioerodible polyanhydrides for controlled drug delivery. Biomaterials. 1983;4:131–133. doi: 10.1016/0142-9612(83)90054-6. [DOI] [PubMed] [Google Scholar]
- 12.Determan AS, Trewyn BG, Lin VS, Nilsen-Hamilton M, Narasimhan B. Encapsulation, stabilization, and release of BSA-FITC from polyanhydride microspheres. J Control Release. 2004;100(1):97–109. doi: 10.1016/j.jconrel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Kumar N, Langer RS, Domb AJ. Polyanhydrides: an overview. Adv Drug Deliv Rev. 2002;54(7):889–910. doi: 10.1016/s0169-409x(02)00050-9. [DOI] [PubMed] [Google Scholar]
- 14.Leong KW, D’Amore PD, Marletta M, Langer R. Bioerodible polyanhydrides as drug-carrier matrices. II. Biocompatibility and chemical reactivity. J Biomed Mater Res. 1986;20(1):51–64. doi: 10.1002/jbm.820200106. [DOI] [PubMed] [Google Scholar]
- 15.Laurencin C, Peirrie-Jacques H, Langer R. Toxicology and biocompatibility considerations in the evaluation of polymeric materials for biomedical applications. Clin Lab Med. 1990;10:549–570. [PubMed] [Google Scholar]
- 16.Temenoff JS, Mikos AG. Injectable biodegradable materials for orthopedic tissue engineering. Biomaterials. 2000;21(23):2405–12. doi: 10.1016/s0142-9612(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 17.Eldridge JH, Staas JK, Meulbroek JA, McGhee JR, Tice TR, Gilley RM. Biodegradable microspheres as a vaccine delivery system. Mol Immunol. 1991;28(3):287–94. doi: 10.1016/0161-5890(91)90076-v. [DOI] [PubMed] [Google Scholar]
- 18.Tice TR, Tabibi E. Parenteral drug delivery: injectables. In: Kydonieus A, editor. Treatise on controlled drug delivery: fundamentals optimization, applications. Marcel Dekker; New York: 1991. pp. 315–339. [Google Scholar]
- 19.Hanes J, Cleland JL, Langer R. New advances in microsphere-based single dose vaccines. Advanced Drug Delivery Reviews. 1997;28:97–119. doi: 10.1016/s0169-409x(97)00053-7. [DOI] [PubMed] [Google Scholar]
- 20.Kipper MJ, Shen E, Determan A, Narasimhan B. Design of an injectable system based on bioerodible polyanhydride microspheres for sustained drug delivery. Biomaterials. 2002;23(22):4405–12. doi: 10.1016/s0142-9612(02)00181-3. [DOI] [PubMed] [Google Scholar]
- 21.Mathiowitz E, Langer R. Poly(anhydrides) microspheres as drug carriers. I. Hot-melt microencapsulation. J. Controlled Release. 1987;5:13–22. [Google Scholar]
- 22.Mathiowitz E, Amato C, Dor P, Langer R. Polyanhydride microspheres: 3. Morphology and characterization of systems made by solvent removal. Polymer. 1990;31:547–555. [Google Scholar]
- 23.Mathiowitz E, Saltzman W, Domb A, Dor P, Langer R. Polyanhydride Microspheres as Drug Carriers. II. Microencapsulation by Solvent Removal. J Applied Polymer Sciences. 1988;35:755–774. [Google Scholar]
- 24.Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials. 2007;28(1):108–16. doi: 10.1016/j.biomaterials.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Determan AS, Graham JR, Pfeiffer KA, Narasimhan B. The role of microsphere fabrication methods on the stability and release kinetics of ovalbumin encapsulated in polyanhydride microspheres. J Microencapsul. 2006;23(8):832–43. doi: 10.1080/02652040601033841. [DOI] [PubMed] [Google Scholar]
- 26.Kipper MJ, Wilson JH, Wannemuehler MJ, Narasimhan B. Single dose vaccine based on biodegradable polyanhydride microspheres can modulate immune response mechanism. J Biomed Mater Res A. 2006;76(4):798–810. doi: 10.1002/jbm.a.30545. [DOI] [PubMed] [Google Scholar]
- 27.Tamada J, Langer R. The development of polyanhydrides for drug delivery applications. J Biomater Sci Polym Ed. 1992;3(4):315–53. doi: 10.1163/156856292x00402. [DOI] [PubMed] [Google Scholar]
- 28.Mathiowitz E, Bernstein H, Giannos S, Dor P, Turek T, Langer R. Polyanhydride microspheres. IV. Morphology and characterization of systems made by spray drying. Journal of Applied Polymer Science. 1992;45(125-134) [Google Scholar]
- 29.Johnson OL, Cleland JL, Lee HJ, Charnis M, Duenas E, Jaworowicz W, Shepard D, Shahzamani A, Jones AJ, Putney SD. A month-long effect from a single injection of microencapsulated human growth hormone. Nat Med. 1996;2(7):795–9. doi: 10.1038/nm0796-795. [DOI] [PubMed] [Google Scholar]
- 30.Cleland JL, Jones AJ. Stable formulations of recombinant human growth hormone and interferon-gamma for microencapsulation in biodegradable microspheres. Pharm Res. 1996;13(10):1464–75. doi: 10.1023/a:1016063109373. [DOI] [PubMed] [Google Scholar]
- 31.Lam X, Duenas E, Daugherty A, Levin N, Cleland J. Sustained release of recombinant human insulin-like growth factor-I for treatment of diabetes. J Control Release. 2000;67:281–292. doi: 10.1016/s0168-3659(00)00224-8. [DOI] [PubMed] [Google Scholar]
- 32.Gombotz WR, Pankey SC, Bouchard LS, Phan DH, MacKenzie AP. Stability, characterization, formulation, and delivery system development for transforming growth factor-beta 1. Pharm Biotechnol. 1996;9:219–45. doi: 10.1007/0-306-47452-2_4. [DOI] [PubMed] [Google Scholar]
- 33.Meinel L, Illi O, Zapf J, Malfanti M, Merkle H, Gander B. Stabilizing insulin-like growth factor-I in poly(D,L-lactide-co-glycolide) microspheres. J Control Release. 2001;70:193–202. doi: 10.1016/s0168-3659(00)00352-7. [DOI] [PubMed] [Google Scholar]
- 34.Carrasquillo KG, Stanley AM, Aponte-Carro JC, De Jesus P, Costantino HR, Bosques CJ, Griebenow K. Non-aqueous encapsulation of excipient-stabilized spray-free dried BSA into poly(lactide-co-glycolide) microspheres results in release of native protein. J Control Release. 2001;76:199–208. doi: 10.1016/s0168-3659(01)00430-8. [DOI] [PubMed] [Google Scholar]
- 35.Sah H. Protein instability toward organic solvent/water emulsification: implications for protein microencapsulation into microspheres. PDA J Pharm Sci Technol. 1999;53(1):3–10. [PubMed] [Google Scholar]
- 36.Castellanos IJ, Cuadrado WO, Griebenow K. Prevention of structural perturbations and aggregation upon encapsulation of bovine serum albumin into poly(lactide-co-glycolide) microspheres using the solid-in-oil-in water technique. J Pharm Pharmacol. 2001;53(8):1099–107. doi: 10.1211/0022357011776487. [DOI] [PubMed] [Google Scholar]
- 37.Mathiowitz E, Langer R. Polyanhydride microspheres as drug delivery systems. In: Donbrow M, editor. Microcapsules and Nanoparticles in Medicine and Pharmacy. CRC Press; Boca Raton, FL: 1992. pp. 100–122. [Google Scholar]
- 38.Conix A. Poly[1,3-bis(p-carboxyphenoxy)-propane anhydride] Macromolecular Synthesis. 1966;2:95–98. [Google Scholar]
- 39.Shen E, Pizsczek R, Dziadul B, Narasimhan B. Microphase separation in bioerodible copolymers for drug delivery. Biomaterials. 2001;22(3):201–10. doi: 10.1016/s0142-9612(00)00175-7. [DOI] [PubMed] [Google Scholar]
- 40.Koseki T, Kitabatake N, Doi E. Freezing denaturation of ovalbumin at acid pH. J Biochem. 1990;107:389–394. doi: 10.1093/oxfordjournals.jbchem.a123055. [DOI] [PubMed] [Google Scholar]
- 41.Leong KW, Brott BC, Langer R. Bioerodible polyanhydrides as drug-carrier matrices. I: Characterization, degradation, and release characteristics. J Biomed Mater Res. 1985;19:941–955. doi: 10.1002/jbm.820190806. [DOI] [PubMed] [Google Scholar]
- 42.Stotz C, Winslow S, Houchin M, D’Souze A, Ji J, Topp E. Degradation pathways for lyophilized peptides and proteins. In: Costantino HR, Pikal M, editors. Lyophilization of Biopharmaceuticals. Springer; 2004. [Google Scholar]
- 43.Gopferich A. Mechanisms of polymer degradation and erosion. Biomaterials. 1996;17:103–114. doi: 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- 44.Mader K, Bittner B, Li Y, Wohlauf W, Kissel T. Monitoring microviscosity and microacidity of the albumin microenvironment inside degrading microparticles from poly(lactide-co-glycolide) (PLG) or ABA-triblock polymers containing hydrophobic poly(lactide-co-glycolide) A blocks and hydrophilic poly(ethylene oxide) B blocks. Pharm. Res. 1998;15:787–793. doi: 10.1023/a:1011939607573. [DOI] [PubMed] [Google Scholar]
- 45.Kissel T, Li Y, Volland C, Gorich S, Koneberg R. Parenteral protein delivery systems using biodegradable polyesters of ABA block structure, containing hydrophobic poly(lactide-co-glyocolide) A blocks and hydrophilic poly(ethylene oxide) B blocks. J Control Release. 1996;39:315–326. [Google Scholar]
- 46.Uchida T, Yagi A, Oda Y, Nakada Y, Goto S. Instability of bovine insulin in poly(lactide-co-glycolide) (PLGA) microspheres. Chem Pharm Bull (Tokyo) 1996;44(1):235–6. doi: 10.1248/cpb.44.235. [DOI] [PubMed] [Google Scholar]
- 47.Costantino HR, Langer R, Klibanov AM. Moisture-induced aggregation of lyophilized insulin. Pharm Res. 1994;11(1):21–9. doi: 10.1023/a:1018981208076. [DOI] [PubMed] [Google Scholar]
- 48.Harandi A. Immunoplacental therapy, a potential multi-epitope cancer vaccine. Med Hypotheses. 2006;66(6):1182–7. doi: 10.1016/j.mehy.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Lu Y, Xiao Y, Ding J, Dierich M, Chen Y. Multiepitope vaccines intensively increased levels of antibodies recognizing three neutralizing epitopes on human immunodeficiency virus-1 envelope protein. Scan. J. Immunol. 2000;51:497–501. doi: 10.1046/j.1365-3083.2000.00713.x. [DOI] [PubMed] [Google Scholar]
- 50.Xiao Y, Lu Y, Chen Y. Epitope-vaccine as a new strategy against HIV-1 mutation. Immunol. Lett. 2001;77:3–6. doi: 10.1016/s0165-2478(01)00187-0. [DOI] [PubMed] [Google Scholar]