

Abstract
MicroRNAs (miRNAs), small noncoding RNAs that regulate target gene mRNAs, are known to contribute to pathogenesis of cancers. Acute myeloid leukemia (AML) is a group of heterogeneous hematopoietic malignancies with various chromosomal and/or molecular abnormalities. AML with chromosomal translocations involving the mixed lineage leukemia (MLL) gene are usually associated with poor survival. In the present study, through a large-scale, genomewide miRNA expression assay, we show that microRNA-9 (miR-9) is the most specifically up-regulated miRNA in MLL-rearranged AML compared with both normal control and non–MLL-rearranged AML. We demonstrate that miR-9 is a direct target of MLL fusion proteins and can be significantly up-regulated in expression by the latter in human and mouse hematopoietic stem/progenitor cells. Depletion of endogenous miR-9 expression by an appropriate antagomiR can significantly inhibit cell growth/viability and promote apoptosis in human MLL-rearranged AML cells, and the opposite is true when expression of miR-9 is forced. Blocking endogenous miR-9 function by anti-miRNA sponge can significantly inhibit, whereas forced expression of miR-9 can significantly promote, MLL fusion–induced immortalization/transformation of normal mouse bone marrow progenitor cells in vitro. Furthermore, forced expression of miR-9 can significantly promote MLL fusion–mediated leukemogenesis in vivo. In addition, a group of putative target genes of miR-9 exhibited a significant inverse correlation of expression with miR-9 in a series of leukemia sample sets, suggesting that they are potential targets of miR-9 in MLL-rearranged AML. Collectively, our data demonstrate that miR-9 is a critical oncomiR in MLL-rearranged AML and can serve as a potential therapeutic target to treat this dismal disease.
MicroRNAs (miRNAs) are regulatory RNAs, ∼22 nucleotides in length, which serve to posttranscriptionally regulate expression of target genes (1). Binding of target genes most frequently happens at the 3′-UTRs and induces mRNA translational repression and, particularly, degradation of the target mRNAs (1–3). Evidence is emerging that miRNAs play critical regulatory roles in virtually all bioprocesses in both normal development and pathogenesis of disease (1–4).
Leukemia is a cancer arisen from uncontrolled proliferation in the hematopoietic lineage. Acute myeloid leukemia (AML) accounts for roughly 70–80% of all adult acute leukemias and 20% of all childhood acute leukemias (5). Approximately 40–60% of AML patients contain chromosomal abnormalities with varying prognosis. Specifically, translocations involving the mixed lineage leukemia (MLL) gene account for roughly 10% of AML (2, 6, 7). Patients with MLL-rearranged AML are often associated with poor prognosis, and effective targeted therapies are not available (2, 6, 7). Dysregulation of miRNAs has been frequently observed in AML, including those carrying MLL rearrangements (2). We and others have shown that MLL translocations induce aberrant overexpression of a set of miRNAs such as individual miRNAs in the miR-17–92 cluster and miR-196b (8–13).
In the present study, through a large-scale, genomewide miRNA expression profiling assay of 85 human AML cases (including 10 MLL rearranged and 75 others), along with 15 normal controls, we show that besides the aforementioned individual miRNAs, a number of other miRNAs are also significantly overexpressed in MLL-rearranged AML relative to normal controls and non–MLL-rearranged AML. In particular, miR-9 appears to be the most specifically and consistently overexpressed miRNA in MLL-rearranged AML. We then show that miR-9 is a direct target of MLL fusions and plays an essential oncogenic role in MLL-rearranged leukemia.
Results
miR-9 Is the Most Specifically and Consistently Overexpressed miRNA in MLL-Rearranged AML.
We performed a large-scale, genomewide miRNA expression profiling assay of 85 AML primary patient samples (including 10 MLL rearranged and 75 others; Table S1), along with 15 normal control [6 CD34+ hematopoietic stem/progenitor cell, 5 CD33+ myeloid progenitor cell, and 4 mononuclear cell (MNC)] samples, by use of Exiqon miRCURY LNA arrays (v10.0; covering 757 human miRNAs). As shown in Fig. 1A, 29 miRNAs are expressed at a significantly higher level [significant analysis of microarrays (SAM), q < 0.05; false discovery rate (FDR) < 0.05] in MLL-rearranged AML than in both normal controls and non–MLL-rearranged AML; in contrast, the opposite pattern was observed for only 1 miRNA (i.e., miR-495) (14). Consistent with our previous miRNA expression profiling assay using a bead-based method (8, 13), we found in this new profiling assay that individual miRNAs in the miR-17–92 cluster (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92) or its paralogous clusters (i.e., miR-106a-363 and miR-106b-25) are highly expressed in MLL-rearranged AML (Fig. 1A). Similarly, overexpression of miR-196a/b (8–10) was also confirmed in this new assay (Fig. 1A). However, among the 29 overexpressed miRNAs, miR-9 appears to be the most specifically and consistently overexpressed one in MLL-rearranged AML (Fig. 1 A and B). The significant up-regulation of miR-9 was also observed in various leukemia cell lines (including MONOMAC-6, THP-1, ML-2, and KOCL-48) carrying MLL rearrangements but not in those without MLL translocations (e.g., HL-60 and U937), compared with normal controls (Fig. S1).
Fig. 1.

MLL fusion proteins directly up-regulate expression of miR-9. (A) Expression profiles of 30 miRNAs that are significantly (q < 0.05; FDR < 0.05; SAM) up-regulated (29 miRNAs) or down-regulated (1 miRNA, i.e., miR-495) in MLL-rearranged AML (n = 10) compared with both normal controls [n = 15, including 6 CD34+ hematopoietic stem/progenitor, 5 CD33+ myeloid progenitor, and 4 MNC cells] and non–MLL-rearranged AML (n = 75). Expression data are mean centered, and the relative value for each sample is represented by a color: high expression is represented with red and low expression is represented with green (scale shown in the upper left). (B) Relative expression levels of miR-9 in 10 human MLL-rearranged AML (i.e., MLL), 75 non–MLL-rearranged AML (i.e., Non-MLL), and 15 normal control (i.e., NC) samples, as detected by Exiqon miRNA microarray assays. (C) qPCR analysis of miR-9 expression level in human cord blood CD34+ cells that were retrovirally transduced with MSCV-MLL-AF9 (MA9-1, -2, and -3), MSCV-AML-ETO (AE-1, -2, and -3), or empty vector (NC-1, -2, and -3). (D) qPCR analysis of miR-9 expression level in colony cells derived from mouse BM progenitor cells retrovirally transduced with MLL-AF9 (MLL-AF9-MSCVneo+MSCVpig), HOXA9+MEIS1 (HOXA9-MSCVpig+MEIS1-MSCVneo), or empty vector (MSCVneo+MSCVpig). (E) ChIP assay of miR-9 promoter regions using anti-MLL (N-terminal), anti-IgG, and anti-H3K79me2 (H3K79 di-methylation) antibodies in MONOMAC-6 cell line. (F) Withdrawal of 4-hydroxy-tamoxifen (4-OHT) (day 7 is shown) results in a significant decrease of miR-9 expression in MLL-ENL-ERtm cell line. *P < 0.05, two-tailed t test.
miR-9 Is a Direct Downstream Target of MLL Fusion Proteins.
We then investigated whether increased expression of miR-9 is directly attributed to MLL translocations. We found that miR-9 could be significantly up-regulated by retrovirally transduced MLL-AF9 [resulting from t(9;11)(p22;q23), the most frequent MLL rearrangement observed in AML], but not by AML1-ETO [resulting from t(8;21)], in human hematopoietic stem/progenitor cells (15) (Fig. 1C). Similarly, MLL-AF9 could also significantly up-regulate miR-9 expression in normal mouse bone marrow (BM) progenitor cells (Fig. 1D). In contrast, we could not observe a significant up-regulation of miR-9 expression in mouse BM progenitor cells transduced with HOAX9 and MEIS1 (Fig. 1D), which are two critical downstream targets of MLL fusion proteins, and their aberrant overexpression is required for the pathogenesis of MLL-rearranged AML (16–18). Thus, our data suggest that MLL fusion proteins may directly up-regulate expression of miR-9 and not through downstream mediators (e.g., HOXA9 and MEIS1).
MLL fusion proteins can bind to the promoter regions of their critical target genes and promote their expression through recruiting DOT1L-mediated methylation of histone H3 lysine 79 (10, 13, 19–21). To determine whether MLL fusion proteins also directly bind to the miR-9 promoter regions, we performed a ChIP assay in the MONOMAC-6 cell line that carries t(9;11)/MLL-AF9 (Fig. 1E). miR-9 has three loci, miR-9-1, miR-9-2, and miR-9-3, which all produce an identical mature miRNA product (22). We observed that MLL fusion proteins were significantly (P < 0.05) enriched on the promoter regions of miR-9-1, -2, and -3 loci, associated with enrichment of H3K79me2, a mark for active transcription (20). To determine whether ovrexpression of miR-9 is dependent on the presence of MLL fusions, we used MLL-ENL-ERtm, a mouse myeloid cell line that expresses a stable derivative of MLL-ENL in the presence of 4-hydroxytamoxifen (4-OHT), as a model (16, 23). We found that depletion of MLL-ENL following withdrawal of 4-OHT led to a significant decrease (P < 0.05) in miR-9 expression (Fig. 1F). Together, our data indicate that miR-9 is a direct target of MLL fusion proteins.
miR-9 Promotes Cell Survival and Blocks Apoptosis in Human MLL-Rearranged AML Cells.
To determine the role of miR-9 in AML, we performed both loss- and gain-of-function studies in human MLL-rearranged AML cells. Transfection of miR-9 inhibitors (i.e., antagomiR oligos) into MONOMAC-6 cells led to a significant decrease in cell viability and an increase in apoptosis (Fig. 2A), as well as a significant decrease in cell growth/proliferation (Fig. 2B). Similar phenomena were observed in THP-1, another AML cell line containing t(9;11) (Fig. S2). Conversely, we found that forced expression of miR-9 caused a significant increase in cell viability and a decrease in apoptosis (Fig. 2C). In addition, in a model of monocytic differentiation of THP-1 cells induced by phorbol-12-myristate-13-acetate (PMA) (24), we found that miR-9 expression was significantly down-regulated during the differentiation (Fig. S3), implying that miR-9 may also play a role in cell differentiation.
Fig. 2.
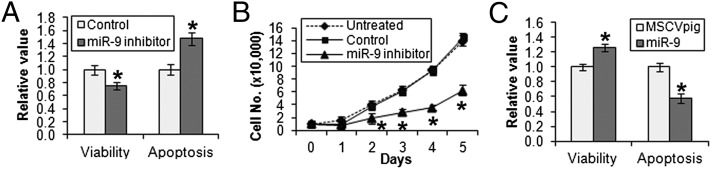
Effects of miR-9 on cell viability, proliferation, and apoptosis of human MLL-rearranged AML cells in vitro. (A) Transfection of miR-9 inhibitor (i.e., anti-miR-9 antagomiR oligos) into MONOMAC-6 cells results in a significant decrease of cell viability and an increase in apoptosis compared with transfection of antagomiR scrambled control oligos (as control). (B) Inhibition of miR-9 by miR-9 inhibitor also significantly suppresses cell growth and proliferation of MONOMAC-6 compared with control and untreated MONOMAC-6 cells. Cell numbers were counted daily. (C) Forced expression of miR-9 (via transfection with MSCVpig-miR-9) significantly increases cell viability and decreases apoptosis of MONOMAC-6 cells compared with transfection with MSCVpig empty vector control. Cell viability and apoptosis were evaluated 48 h after transfection. *P < 0.05, two-tailed t test.
miR-9 Plays a Critical Oncogenic Role in MLL Fusion–Mediated Cell Transformation In Vitro.
Through in vitro colony-forming/replating assays, we show that, although forced expression of miR-9 alone is not sufficient to transform normal hematopoietic progenitor cells, miR-9 can significantly enhance MLL-AF9–mediated cell transformation (Fig. 3 A and B). Conversely, cotransduction of MLL-AF9 and miR-9 sponge [competitive inhibitors of miR-9 (22)] result in a significant decrease of colony numbers (Fig. 3C) and a dramatic reduction of blast cell proportion in colony cells (Fig. 3D) compared with cotransduction of MLL-AF9 and sponge control. Notably, we also found a synergistic effect in cell transformation between HOXA9 and miR-9 (Fig. S4) but not between HOXA9 and miR-196b [an miRNA that regulate both oncogenic and tumor-suppressor targets (10)]. Thus, miR-9 plays an important oncogenic role in cell transformation mediated by MLL fusions, probably through a synergetic effect with other oncogenic downstream targets (e.g., HOXA9) of MLL fusions.
Fig. 3.

In vitro colony-forming/replating assays. (A) Forced expression of miR-9 promoted MLL-AF9–mediated cell transformation. Normal mouse BM progenitor cells were retrovirally transduced with MSCVneo+MSCVpig (i.e., Control), MSCVneo+MSCVpig-miR-9 (i.e., miR-9), MSCVneo-MLL-AF9+MSCVpig (i.e., MLL-AF9), or MSCVneo-MLL-AF9+MSCVpig-miR-9 (i.e., MLL-AF9+miR-9) and then plated into methylcellulose medium under double selection of puromycin and G418 to form colonies. The colony cells were replated every 7 d for up to six passages. Mean± SD values of colony numbers are shown. (B) Cytospin morphology analysis of first passage of colony cells (see Fig. 3A) via Wright-giemsa staining. (C) Block of miR-9 function inhibited MLL-AF9–mediated cell transformation. Normal mouse BM progenitor cells were retrovirally transduced with MSCVneo+pBABE-puro-scrambled sponge (i.e., Control), MSCVneo+pBABE-puro-miR-9 sponge (i.e., miR-9 sponge), MSCVneo-MLL-AF9+pBABE-puro-scrambled sponge (i.e., MLL-AF9), or MSCVneo-MLL-AF9+pBABE-puro-miR-9 sponge (i.e., MLL-AF9+miR-9 sponge), and colony forming and replating were conducted as described above for up to three passages. Mean ± SD values of colony numbers are shown. (D) Cytospin morphology analysis of first passage of colony cells (see Fig. 3C) via Wright-giemsa staining. The length of bars in B and D represents 10 μm. *P < 0.05; **P < 0.01; two-tailed t test.
miR-9 Enhances MLL Fusion–Mediated Leukemogenesis In Vivo.
To investigate in vivo pathological function of miR-9, we retrovirally cotransduced BM progenitor cells (CD45.2) with MSCVneo+MSCVpig (control), MSCVneo+MSCVpig-miR-9 (miR-9), MSCVneo-MLL-AF9+MSCVpig (MLL-AF9), and MSCVneo-MLL-AF9+MSCVpig-miR-9 (MLL-AF9+miR-9) and then cultured them on methylcellulose medium for 7 d to form colonies to select transduction double-positive cells. Colony cells were then transplanted into lethally irradiated CD45.1 mice. As expected, although forced expression of miR-9 alone could not induce leukemia in transplanted mice, recipient mice transplanted with both miR-9 and MLL-AF9 developed AML (Fig. S5A) with a significantly shorter latency compared with the mice transplanted with MLL-AF9 alone (median overall survival: 64 vs. 79 d; P = 0.001; Fig. 4A). Furthermore, all mice in the MLL-AF9+miR-9 group exhibited a remarkable increase in the proportion of c-Kit+ blast cells in the BM, spleen, and peripheral blood compared with MLL-AF9 mice (Fig. 4B; Fig. S5B; Table S2). Morphologically, MLL-AF9+miR-9 leukemic BM cells exhibited a higher degree of immature/blast counts than MLL-AF9 leukemic BM cells (Fig. 4C). Histological analysis also showed a greater degree of leukemic infiltration of spleen and liver in MLL-AF9+miR-9 mice compared with MLL-AF9 mice (Fig. 4D). Collectively, MLL-AF9+miR-9 mice exhibited a much more aggressive leukemic phenotype than MLL-AF9 mice, highlighting the important oncogenic role of miR-9 in the pathogenesis of MLL-rearranged leukemia.
Fig. 4.

miR-9 promotes MLL-AF9–mediated leukemogenesis in vivo. (A) Kaplan-Meier survival curves of lethally irradiated CD45.1 recipient mice, which were reconstituted with colony cells derived from CD45.2 mouse BM progenitor transduced with MSCVneo+MSCVpig (control, n = 5), MSCVneo+MSCVpig-miR-9 (miR-9; n = 5), MSCVneo-MLL-AF9+MSCVpig (MLL-AF9; n = 8), or MSCVneo-MLL-AF9+MSCVpig-miR-9 (MLL-AF9+miR-9; n = 5). Overexpression of miR-9 accelerates MLL-AF9–mediated leukemia (P = 0.001, log-rank test) compared with the MLL-AF9 alone group. (B) Flow cytometric analysis of bone marrow (BM), spleen (SP), and peripheral blood (PB) from MLL-AF9 and MLL-AF+miR-9 representative mice stained for c-Kit/Mac-1. (C) Wright-Giemsa–stained BM cytospin. (D) H&E-stained SP and liver sections showing massive leukemic infiltration.
Identification of Potential Target Genes of miR-9 in MLL-Rearranged AML.
We analyzed four major miRNA target prediction programs including TragetScan, PITA, miRanda, and miRBase Targets and identified a total of 4,941 genes as putative targets of miR-9 in both human and mouse genomes as predicted by at least one of the four programs. To identify potential target genes of miR-9 in AML, we also performed mRNA expression profiling of 79 of the 100 human samples used in the miRNA expression profiling, including 70 AML (composed of 9 MLL- and 61 non–MLL-rearranged AML cases) and 9 normal controls (composed of 3 CD34+, 2 CD33+, and 4 MNC cell samples) by use of an Agilent custom-design microarray platform (14). Through correlation of expression of miR-9 with that of its putative target genes across the 79 samples, we found that 170 putative targets exhibited a significantly inverse correlation of expression (r < −0.2, P < 0.05, Pearson correlation) with miR-9 (Table S3). Of the 170 genes, 80 also exhibited a significant inverse correlation of expression (r < −0.4, P < 0.05, Pearson correlation) with miR-9 across the 18 samples of MLL-rearranged AML (n = 9) and normal controls (n = 9). In addition, we also performed Affymetrix exon arrays of 13 human MLL-rearranged AML samples and 9 normal controls (including 3 each of CD34+, CD33+, and MNC samples) (10). We found that of these 80 potential target genes, 31 exhibited a significant down-regulation in MLL-rearranged AML samples compared with the normal controls (Table S3). Moreover, in analysis of Affymetrix gene arrays of nine mouse MLL-AF9 leukemic samples and six control samples (10, 14, 21), we found that 17 of the 31 candidate targets were also significantly down-regulated in mouse MLL-AF9 leukemic samples relative to the normal controls (Table S3). Thus, these 17 genes (including CPEB4, CYFIP2, ENDOD1, HBP1, JAK1, KLF6, LHFPL2, MAP3K8, RAB8B, RHAG, RHOH, RYBP, SERPINB9, TAL1, TFRC, TRAK2, and VAMP5) are highly likely targets of miR-9 in MLL-rearranged AML (see Fig. 5 for their expression profiles).
Fig. 5.

Expression profiles of 17 candidate target genes of miR-9 in MLL-rearranged AML. Expression data were mean centered, and the relative value for each sample is represented by a color, with red representing high expression and green representing low expression (scale shown in the upper left). (A) Expression profiles of miR-9 and its 17 candidate target genes in the 79 human sample set (including 9 MLL-rearranged AML, 61 non–MLL-rearranged AML, and 9 normal control samples) and their expressional correlations are shown. (B) Expression profiles of the 17 candidate target genes in the 22 human sample set including 13 MLL-rearranged AML and 9 normal control samples. (C) Expression profiles of the 17 candidate target genes in the 15 mouse sample set including 9 MLL-rearranged AML and 6 normal control samples. The fold changes and corresponding q values (as detected by SAM) of the 17 target genes in MLL-rearranged AML samples compared with normal controls are shown in B and C.
We then chose RHOH and RYBP, two genes that show the greatest inverse correlation of expression with miR-9 in the set of 79 samples (Fig. 5A), as candidate targets for further validation. As expected, both genes are significantly down-regulated in human hematopoietic stem/progenitor cells transduced with MLL-AF9 (Fig. 6A), in a manner opposite to miR-9 (Fig. 1C). In the presence of MLL-AF9, forced expression of miR-9 can cause a further significant repression of Rhoh and Rybp expression in mouse BM progenitor cells (Fig. 6B), and a similar pattern was observed in human MONOMAC-6 cells (Fig. 6C). Thus, these results supported the likelihood that miR-9 negatively regulates the expression of these potential targets.
Fig. 6.
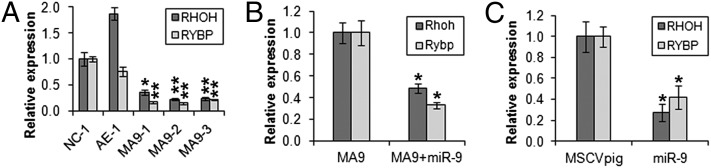
RHOH and RYBP are potential direct targets of miR-9. (A) Relative expression of RHOH and RYBP in normal human cord blood CD34+ cells transduced with MSCV-MLL-AF9 (MA9-1, -2, and -3), MSCV-AML1-ETO (AE-1), or empty vector (NC-1) (15). (B) Relative expression of Rhoh and Rybp in normal mouse BM progenitor cells transducted with MSCVneo-MLL-AF9+MSCVpig (MA9) or MSCVneo-MLL-AF9+MSCVpig-miR-9 (MA9+miR-9). Cells used for the analysis are passage II colony cells from the in vitro colony-forming/replating assay shown in Fig. 3A. (C) Relative expression of RHOH and RYBP in human MONOMAC-6 cells 48 h after transfection with empty vector (MSCVpig) or miR-9 (MSCVpig-miR-9). Expression level of each target gene in the control group in each plot was set to 1 for comparison and statistical analysis. *P < 0.05; **P < 0.01; two-tailed t test.
EVI1 Has No Apparent Effect on Expression of miR-9 in MLL-Rearranged AML.
Senyuk et al. reported recently that ecotropic viral integration site 1 (EVI1) could repress the expression of miR-9, with antagonistic effects on myelopoiesis and EVI1-induced leukemogenesis (25). Interestingly, previously studies indicate that Evi1 is a transcriptional target of MLL fusion proteins (26). Consistent with a previous report that EVI1 was aberrantly overexpressed in ∼40–50% of MLL-rearranged AML patients (27), we found that EVI1 was expressed at a higher level in four (44%) of our nine patients with MLL-rearranged AML than its average level in the nine patients (Fig. S6A). We classified these four samples as “EVI1-high” and the remaining five samples as “EVI1-low.” As expected, EVI1 is expressed at a significantly higher level in the EVI1-high group than in both the EVI1-low group and the normal control group, but there is no significant difference between the latter two groups (Fig. S6 A and B). Notably, there is a significantly positive correlation of expression (r = 0.29, P = 0.01; Pearson correlation) between miR-9 and EVI1 across our 79 human samples (Fig. S6A). This positive correlation is largely attributed to the co-overexpression of miR-9 and EVI1 in the subset of MLL-rearranged AML, and when MLL-rearranged AML samples were excluded from the correlation analysis, no significance (r = −0.028, P = 0.81; Pearson correlation) was observed across the remaining 70 samples. In contrast to the previous report that forced expression of EVI1 could significantly repress expression of miR-9 in normal hematopoietic progenitor cells (25), we show here that in MLL-rearranged AML, miR-9 expression level in the EVI1-high group is even higher than in the EVI1-low group, although the difference is not statistically significant (Fig. S6 C and D). Thus, our data indicate that MLL fusion–mediated up-regulation of miR-9 expression likely overrides EVI1-mediated suppression in MLL-rearranged AML cells.
Discussion
Here we show that as many as 29 miRNAs are significantly up-regulated, whereas only 1 miRNA (i.e., miR-495) is significantly down-regulated, in MLL-rearranged AML compared with normal controls and other AMLs. It should not be a surprise that the miRNAs specifically dysregulated in MLL-rearranged AML are predominantly up-regulated ones, because previous studies from us and others have shown that MLL fusion proteins function predominantly as transcriptional activators, rather than inhibitors, in regulating expression of downstream targets (10, 13, 16, 19, 28). In particular, miR-9 is the most specifically and consistently up-regulated miRNA in MLL-rearranged AML compared with normal controls and other AMLs. Similar to miR-17-92 and miR-196b (10, 13), we demonstrate that miR-9 is also a direct target of MLL fusion proteins.
Both tumor suppressor and oncogenic roles of miR-9 have been reported in various solid tumors (22, 29–31). However, its role in leukemia is unclear. Here we show that, although gain- or loss-of-function of miR-9 alone does not exhibit any significant effect on cell transformation, its forced expression can significantly enhance MLL-AF9– or HOXA9-mediated transformation of normal mouse BM progenitor cells in vitro. Depletion of miR-9 expression can significantly inhibit viability/growth and promote apoptosis of human MLL-rearranged AML cells, and the opposite is true when expression of miR-9 is forced. Moreover, forced expression of miR-9 can significantly accelerate MLL-AF9–mediated leukemogensis in transplanted mice, leading to a more aggressive leukemic phenotype. Thus, our data clearly indicate that miR-9 plays an essential oncogenic role in the pathogenesis of MLL-rearranged AML. Notably, a very recent study reported a potential tumor suppressor role for miR-9 in EVI1-mediated leukemogenesis (25). EVI1 is also a direct downstream target of MLL fusion proteins (26). We show that co-overexpression of miR-9 and EVI1 occurs in around half of the MLL-rearranged AML cases, and the expression level of miR-9 in the EVI1-high subgroup is even relatively higher than that in the EVI1-low subgroup of MLL-rearranged AML, suggesting that EVI1-mediated repression is overridden by MLL fusion–mediated up-regulation of miR-9 expression in MLL-rearranged AML. Therefore, those data together suggest that miR-9’s role in tumorigenesis is context dependent.
In addition, we identified 17 potential targets of miR-9 through analyses of a series of human and mouse leukemia sample sets (Fig. 5; Table S3). The miR-9–mediated repression of two representative potential targets (i.e., RHOH and RYBP) has been validated. RYBP (RING1 and YY1-binding protein), a polycomb complex-associated protein, can stabilize p53 by modulating MDM2 and thus has tumor suppressor activity (32). In contrast, RHOH, encoding a hematopoietic-specific, GTPase-deficient member of the Rho GTPase protein family, was first identified as a partner of BCL6 in a fusion transcript resulting from t(3;4)(q27;p13) in a non-Hodgkin lymphoma cell line (33) and was aberrantly overexpressed in B-cell chronic lymphocytic leukemia (CLL), in which it played an essential oncogenic role (34). However, in AML, low expression of RHOH has been shown to be an independent unfavorable prognostic factor for both overall and disease-free survival of the patients and contributes to chemotherapy resistance in leukemia cells (35), implying a tumor suppressor role in AML. Similarly, TAL1 is a common target of chromosomal rearrangements in T-cell acute lymphoblastic leukemia (T-ALL) and is aberrantly expressed in up to 60% of pediatric T-ALL, in which it plays an oncogenic role (36); however, the down-regulation of TAL1 has been frequently observed in AML (37, 38). Thus, it is likely that both RHOH and TAL1 are oncogenes in lymphoblastic leukemia but serve as tumor suppressors in myeloid leukemia. Indeed, a recent study showed that Notch signaling, a main oncogenic trigger of T-ALL, plays an essential tumor suppressor role in myeloid leukemia (39). Several other potential targets such as HBP1, KLF6, and SERPINB9 are also tumor suppressors in myeloid leukemia (40–42). Therefore, it is quite possible that miR-9 plays a critical oncogenic role in the pathogenesis of MLL-rearranged AML through targeting a group of tumor suppressor genes.
Thus far, we showed that MLL fusion proteins could directly up-regulate expression of miR-9, miR-17-92 (8, 11, 13), and miR-196b (10), and all of them play oncogenic roles in the pathogenesis of MLL-rearranged leukemia. It would be important to study how these miRNAs work together in promoting leukemogenesis. These miRNAs may regulate different sets of critical targets in distinct pathways and thus contribute to leukemogenesis through different pathways and may also regulate some common target genes or different components in the same pathways to achieve a synergistic effect in inhibiting the corresponding pathways. Clearly, our studies demonstrate that miRNAs are functionally important mediators in the pathogenesis of MLL-rearranged leukemia. Comprehensive understanding of their roles and identification of their essential target genes would broaden and deepen our understanding of the complex molecular mechanisms underlying MLL fusion–mediated leukemogensis, which may lead to the development of effective targeted therapy strategies to treat this presently therapy-resistant disease.
Materials and Methods
Exiqon miRCURY LNA arrays, Agilent’s custom-design microarrays, and Affymetrix exon or gene arrays have been used for miRNA and mRNA expression profiling. ChIP, cell apoptosis and viability assays, colony-forming/replating assays, and bone marrow transplantation (BMT) were performed as described previously (8, 10, 13, 14, 21), with some modifications. Additional details can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Drs. Gregory Hannon, Scott Hammond, Lin He, and Scott Armstrong for providing retroviral constructs and Dr. Robert K. Slany for the MLL-ENL-ERtm cell line. This work was supported in part by National Institutes of Health Grants R01 CA127277 (to J.C.), F31 CA171702 (to C.P.), R01 CA118319 (to J.C.M.), and P01 CA40046 and P30 CA014599 (to M.M.L.B.), the Intramural Program of National Human Genome Research Institute (A.E. and P.P.L.), an American Cancer Society Research Scholar grant (to J.C.), the Leukemia and Lymphoma Society (translational research grant to J.D.R. and J.C., special fellowship to Z.L., and scholar award to J.C.M.), the University of Chicago Committee on Cancer Biology fellowship program (X.J.), and Gabrielle’s Angel Foundation for Cancer Research (J.C., X.J., Z.L., H.H., and G.M.H.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The microarray data sets have been submitted to the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession nos. GSE30258, GSE34184, and GSE34185).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1310144110/-/DCSupplemental.
References
- 1.He L, Hannon GJ. MicroRNAs: Small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Odenike O, Rowley JD. Leukaemogenesis: More than mutant genes. Nat Rev Cancer. 2010;10(1):23–36. doi: 10.1038/nrc2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao C, Rajewsky K. MicroRNA control in the immune system: Basic principles. Cell. 2009;136(1):26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 5.Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341(14):1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 6.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 7.Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. doi: 10.1146/annurev-pathol-011811-132434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105(40):15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popovic R, et al. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood. 2009;113(14):3314–3322. doi: 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, et al. miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012;3:688. doi: 10.1038/ncomms1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, et al. Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res. 2009;69(3):1109–1116. doi: 10.1158/0008-5472.CAN-08-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong P, et al. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010;70(9):3833–3842. doi: 10.1158/0008-5472.CAN-09-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mi S, et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci USA. 2010;107(8):3710–3715. doi: 10.1073/pnas.0914900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang X, et al. MiR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2012;109(47):19397–19402. doi: 10.1073/pnas.1217519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei J, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13(6):483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeisig BB, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004;24(2):617–628. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21(21):2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faber J, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2009;113(11):2375–2385. doi: 10.1182/blood-2007-09-113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milne TA, et al. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102(41):14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22(4):524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mueller D, et al. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7(11):e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuchiya S, et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982;42(4):1530–1536. [PubMed] [Google Scholar]
- 25.Senyuk V, et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc Natl Acad Sci USA. 2013;110(14):5594–5599. doi: 10.1073/pnas.1302645110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arai S, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117(23):6304–6314. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 27.Gröschel S, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: A study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J Clin Oncol. 2013;31(1):95–103. doi: 10.1200/JCO.2011.41.5505. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121(8):1422–1431. doi: 10.1182/blood-2012-07-442004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiao CH, et al. High incidence of CD56 expression and relapse rate in acute myeloid leukemia patients with t(8;21) in Taiwan. J Formos Med Assoc. 2002;101(6):393–398. [PubMed] [Google Scholar]
- 30.Gharib TG, et al. Proteomic analysis of cytokeratin isoforms uncovers association with survival in lung adenocarcinoma. Neoplasia. 2002;4(5):440–448. doi: 10.1038/sj.neo.7900257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz DR, et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res. 2002;62(16):4722–4729. [PubMed] [Google Scholar]
- 32.Chen D, et al. RYBP stabilizes p53 by modulating MDM2. EMBO Rep. 2009;10(2):166–172. doi: 10.1038/embor.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dallery E, et al. TTF, a gene encoding a novel small G protein, fuses to the lymphoma-associated LAZ3 gene by t(3;4) chromosomal translocation. Oncogene. 1995;10(11):2171–2178. [PubMed] [Google Scholar]
- 34.Sanchez-Aguilera A, et al. Involvement of RhoH GTPase in the development of B-cell chronic lymphocytic leukemia. Leukemia. 2010;24(1):97–104. doi: 10.1038/leu.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwasaki T, et al. Prognostic implication and biological roles of RhoH in acute myeloid leukaemia. Eur J Haematol. 2008;81(6):454–460. doi: 10.1111/j.1600-0609.2008.01132.x. [DOI] [PubMed] [Google Scholar]
- 36.Begley CG, Green AR. The SCL gene: From case report to critical hematopoietic regulator. Blood. 1999;93(9):2760–2770. [PubMed] [Google Scholar]
- 37.Larson RA, Le Beau MM. Therapy-related myeloid leukaemia: A model for leukemogenesis in humans. Chem Biol Interact. 2005;153-154:187–195. doi: 10.1016/j.cbi.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 38.Whitman SP, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. Blood. 2010;116(18):3622–3626. doi: 10.1182/blood-2010-05-283648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klinakis A, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473(7346):230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao CJ, Works K, Romagnoli PA, Austin GE. Effects of overexpression of HBP1 upon growth and differentiation of leukemic myeloid cells. Leukemia. 2005;19(11):1958–1968. doi: 10.1038/sj.leu.2403918. [DOI] [PubMed] [Google Scholar]
- 41.Humbert M, et al. Deregulated expression of Kruppel-like factors in acute myeloid leukemia. Leuk Res. 2011;35(7):909–913. doi: 10.1016/j.leukres.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 42.Stirewalt DL, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47(1):8–20. doi: 10.1002/gcc.20500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.