

Abstract
Class V myosins (MyoV), the most studied unconventional myosins, recognize numerous cargos mainly via the motor’s globular tail domain (GTD). Little is known regarding how MyoV-GTD recognizes such a diverse array of cargos specifically. Here, we solved the crystal structures of MyoVa-GTD in its apo-form and in complex with two distinct cargos, melanophilin and Rab interacting lysosomal protein-like 2. The apo-MyoVa-GTD structure indicates that most mutations found in patients with Griscelli syndrome, microvillus inclusion disease, or cancers or in “dilute” rodents likely impair the folding of GTD. The MyoVa-GTD/cargo complex structure reveals two distinct cargo-binding surfaces, one primarily via charge–charge interaction and the other mainly via hydrophobic interactions. Structural and biochemical analysis reveal the specific cargo-binding specificities of various isoforms of mammalian MyoV as well as very different cargo recognition mechanisms of MyoV between yeast and higher eukaryotes. The MyoVa-GTD structures resolved here provide a framework for future functional studies of vertebrate class V myosins.
Keywords: myosin V, myo5a, MLPH, RILPL2, granuphilin
Myosin V (MyoV) is one of the earliest identified (1–3) and best characterized unconventional myosins (reviewed in refs. 4, 5) and is ubiquitously expressed in various types of cells in all eukaryotes. Vertebrates usually contain three MyoV paralogs (MyoVa, Vb, and Vc), whereas the budding yeast Saccharomyces cerevisiae has two class V myosins (myo2p and myo4p). As essential cellular cargo transporters, MyoV possesses the amazing ability to recognize numerous, highly diverse cargos (e.g., membrane vesicles, organelles, protein complexes, mRNA) (5, 6). The long tail of MyoV contains several coiled-coil segments and a globular tail domain (GTD) in its C terminus (Fig. 1A). Accumulating evidences indicate that the GTD is the major cargo-binding region in MyoV. In S. cerevisiae, the myo2p-binding sites of organelle and vesicle-specific adaptors have been mapped onto the GTD region (7, 8). In mammals, MyoV-GTD directly interacts with several adaptor proteins, including melanophilin (MLPH) (9–11), Rab interacting lysosomal protein-like 2 (RILPL2) (12), and granuphilin (13) for MyoVa, and Rab11-family interacting protein 2 (14) for MyoVb. As Rab-binding proteins (15–18), these adaptors presumably function to link MyoV with specific Rab-attached membranous cargos for cargo loading and transport (to simplify, we use “cargos” instead of “cargo adaptors” here).
Fig. 1.

Structural characterization of cargo-free MyoVa-GTD. (A) The domain organizations of class V myosins. The color coding of the regions is applied in other panels of this figure. (B) Ribbon representation of the MyoVa-GTD structure. The disordered loop connecting α7 and α8 is indicated by a dotted line. (C) Structural comparisons of MyoVa-GTD with GTD of myo2p (Protein Data Base code 2F6H) and myo4p (3MMI). The N terminus, CC-loop interface, and the α5/α7 connecting region in MyoVa in B and corresponding regions in myo2p and myo4p are highlighted by dashed circles. (D) The molecular details of the N terminus and the CC-loop interface of MyoVa-GTD showing the tight packing via extensive hydrophobic interactions. I1510, M1513, and D1519 are within or close to the interface; mutations of these residues cause a dilute phenotype in mice.
MyoVa is highly expressed in brain as well as other tissues such as melanocytes in vertebrates (2, 19–21). Defective mutations of MyoVa lead to “dilute-lethal” and neurological impairments in rodents (2, 22–24) and a severe hereditary disease known as Griscelli syndrome (GS) in humans (25). Many of these mutation sites are located in MyoVa-GTD, supporting the critical roles of the GTD in MyoVa’s function. The high-resolution GTD structure of yeast myo2p and myo4p shows that the GTD adopts a compact all-helical conformation (7, 26), shedding light on the potential target-binding mechanism of yeast MyoV GTDs. However, the very low sequence similarities between class V myosins (as well as their cargoes) from mammals and yeast (15–25% sequence identity, Fig. S1) have made it difficult to rationalize the underlying molecular mechanisms by which the mutations of mammalian MyoVa-GTD would affect its structure and function based on the available GTD structures of myo2p and myo4p.
Here, we determined the crystal structures of apo MyoV-GTD and MyoVa-GTD in complex with its two cargos, RILPL2 and MLPH. These structures reveal the versatile cargo recognition mechanisms of the class V myosins at the atomic resolution. RILPL2 and MLPH bind to two distinct sites of MyoVa-GTD using largely different interacting modes. RILPL2 forms a homodimer with a four-helix bundle conformation to interact with MyoVa-GTD, whereas MLPH folds as an extended loop and binds to a charge-rich groove of MyoVa-GTD. We also identified the minimal binding domain of another MyoVa cargo, granuphilin, and found that granuphilin and MLPH share the overlapping binding surface on MyoVa-GTD.
Results
Overall Structure of MyoVa-GTD.
To elucidate the cargo recognition mechanisms of class V myosins in high eukaryotes, we first set out to determine the GTD structure of MyoVa. Using the boundary with residues 1,469–1,853 (mouse MyoVa), we obtained high-quality crystals diffracted to 2.5 Å. The crystal structure of MyoVa-GTD was solved by single-wavelength anomalous dispersion using selenomethionine derivatives (Table S1). Except for a flexible loop connecting α7 and α8 and a few residues at the N terminus, the electron densities of the rest of MyoVa-GTD are clearly assigned (Fig. 1B). Each asymmetric unit of the native crystal contains eight MyoVa-GTD molecules with almost identical conformations (rmsd 0.3–0.7 Å).
MyoVa-GTD contains two helical-bundled subdomains (I and II) connected by a long helix α7 (Fig. 1B), which is similar to the GTD structures of yeast myo2p (rmsd 2.5 Å) and myo4p (rmsd 3.2 Å) (7, 26). A long C-terminal loop extends along the concave surface of the entire GTD structure and closely interacts with both subdomains I and II (Fig. 1 B and C). Thus, the C-terminal half of the C-terminal loop (CC loop) is assigned as part of the subdomain I.
Although sharing the similar overall fold with the yeast homologs, MyoVa-GTD contains some unique structural features. In myo2p and myo4p, the two termini of GTD form α-helical structures (α1 and α16) and interact with each other via an antiparallel helical interaction (Fig. 1C and Fig. S2 A and B). In contrast, each of the termini of MyoVa-GTD folds into short β-strands (β1 and β2) and forms a small antiparallel β-sheet (Fig. 1B and Fig. S2 A and B). In addition, the CC loop tightly packs with the N-terminal helical region (including α1 and α3) through extensive hydrophobic interactions, forming part of the structural core of the subdomain I (Fig. 1D). The N- and C-termini interactions were further strengthened by hydrogen bonds. The C-terminal carboxyl group forms two hydrogen bonds with main-chain nitrogen atoms from the N-terminal β1-strand. The hydrophobic residues that are responsible for the interactions between the N terminal and the CC loop shown in Fig. 1D are highly conserved among the class V myosins from worm, fly, and vertebrates (Fig. S1), indicating that all MyoV-GTDs found in metazoans likely share the similar N terminus (including β1, α1, and α3)/CC-loop interaction mode observed in MyoVa-GTD. Another major structural difference between MyoVa-GTD and its yeast counterparts is located at α7 and the region connecting α7 and its preceding α-helix (α5 in MyoVa and α6 in myo2p/4p) (Fig. 1 B and C and Fig. S1). In myo2p and myo4p, two long helices, α6 and α7, are connected with a short, disordered linking sequence. In contrast, α7 of MyoVa is much shorter in length and the α5/α7 linker is considerably longer, containing a short α-helix (α6) and two flanking structured loops (Fig. S2 C and D). Because the N-terminal part of α7 and the residues in the α5/α7 linker of myo2p are known to be involved in cargo recognitions, the large structural differences in this part of the GTD between yeast myo2p/4p and MyoVa points to their distinct cargo-binding properties. Correspondingly, the distinct structural features observed between myo2p/4p-GTD and MyoVa-GTD highlight the importance of the MyoVa-GTD structure for understanding the cargo-binding mechanism and cargo-mediated motor activity regulations of the class V myosins in higher eukaryotes, including humans.
Functional Defective Mutations of MyoV-GTD.
Loss-of-function mutations have been identified in MyoVa from dilute mice, including three missense mutations (I1510N, M1513K, and D1519G) and a C-terminal truncation mutation (deletion of residues 1,841–1,853) in the GTD region. The mutant mice display various degrees of pigment dilutions and neurological impairments (23). All of these mutations are located in subdomain I (Fig. 1D), indicating that subdomain I is crucial for MyoVa’s function in melanosome transport and neuronal development. The truncation mutation removes the majority of the CC loop, resulting in impairments of subdomain I folding. The three missense mutations are clustered in α3, which constitutes the structural core of subdomain I. Among these three sites, I1510 and M1513 are involved in the hydrophobic interaction between the CC loop and the N-terminal helical region. Substitutions of these hydrophobic residues to hydrophilic (I1510N) or charged residues (M1513K) are likely to destabilize subdomain I. The D1519G mutation eliminates two hydrogen bonds connecting α3 to the α6/α7-loop and is expected to impair subdomain I folding. Consistent with the structural analysis, the three missense mutations were found to abrogate the binding of MyoVa-GTD to MLPH (27).
Defective mutations of mammalian MyoV lead to several genetic diseases, such as GS in humans (28) and a similar syndrome in horse MyoVa (29) and microvillus inclusion disease in human MyoVb (30, 31). Most of these changes are frame-shift or truncation mutations, which invariably result in truncated proteins lacking all or part of the GTD (Table S2). In addition, the GTD regions of the three human MyoV paralogs contain somatic mutations in various cancer samples (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) (Table S2). Mapping all of the missense mutations of the GTD structure reveals a surprisingly asymmetric distribution of the mutations found in MyoVa and Vc (Fig. 2). The mutations found in MyoVa are concentrated in subdomain I, whereas three MyoVc mutations are located in subdomain II, implying that the two subdomains play distinct roles in the two MyoV paralogs. Several missense mutations were found in both subdomains in MyoVb. Analyzing the mutation sites, we found that the majority of these residues contribute to the proper GTD folding (Table S2 and Fig. S3). Therefore, the majority of these identified frame-shift, nonsense, or missense mutations are expected to alter the GTD structure at different levels and thereby impair proper functions of MyoV.
Fig. 2.
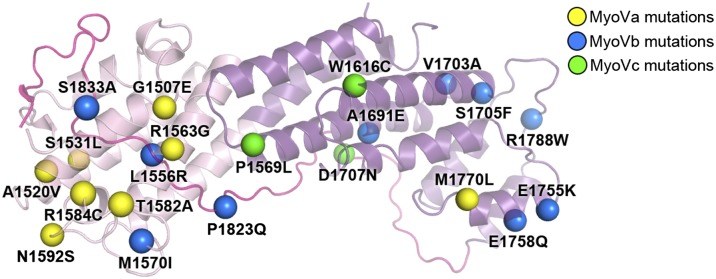
Disease-causing missense mutations of MyoV-GTD. The mutation sites (indicated by colored spheres) are mapped to the MyoVa-GTD structure (see Table S2 for the list of the mutations).
MyoVa-GTD Uses Different Sites to Interact with RILPL2 and MLPH.
We next characterized the interactions of two MyoVa cargos, RILPL2 and MLPH, with MyoVa-GTD in detail. RILPL2 contains two regions conserved in all RILP family members (including RILP, RILPL1, and RILPL2), referred to as RH1 and RH2 (Fig. 3A). Because the RH1 region of RILPL2 was recently shown to contain the MyoVa-GTD–binding domain (12), we characterized the interaction between MyoVa-GTD and RILPL2-RH1 in further detail. Analytical gel-filtration (Fig. 3B) and isothermal titration calorimetry (ITC)-based assays showed that MyoVa-GTD and RILPL2-RH1 interact with each other with high affinity (Kd ∼0.3 μM) (Fig. 3 B and C). The extension of RH1 to the full-length RILPL2 did not enhance its binding to MyoVa-GTD (Fig. S4A), indicating that the binding of MyoVa-GTD to RILPL2 only requires the RH1 region. MLPH contains two separate MyoVa binding sites, one interacting with exon F (referred to as EFBD) of the melanocyte-specific MyoVa isoform and the other interacting with the common MyoVa-GTD (referred to as GTBD; Fig. 3A) (10, 11, 27). The minimal MyoVa-GTD–binding region was further narrowed down to a 26-residue unstructured sequence (residues 176–201) in MLPH-GTBD by Geething et al. (32). We used a slightly extended fragment of MLPH-GTBD containing residues 170–208 (defined as GTBD hereafter) for subsequent biochemical and structural characterizations. Consistent with the results of Geething et al. (32), MyoVa-GTD binds to MLPH-GTBD with a submicromolar Kd (Fig. 3 D and E).
Fig. 3.

Biochemical characterizations of the interactions between MyoVa-GTD and its two cargos, RILPL2 and MLPH. (A) The domain diagrams of RILPL2 and MLPH. SHD, synaptotagmin-like protein homology domain; RBD, Rab-binding domain. The boundaries of the proteins used for the binding assays are indicated. (B, D, and F) Analytical gel filtration chromatography analysis of the MyoVa-GTD/cargo interactions. The profiles of the 1:1 mixtures of MyoVa-GTD/RILPL2-RH1 and MyoVa-GTD/MLPH-GTBD overlap in F as dashed curves. (C and E) The ITC curves showing the quantitative binding affinities between MyoVa-GTD and the two cargos.
Next, we asked whether RILP2 and MLPH compete with each other in binding to MyoVa-GTD. We noticed that the MyoVa-GTD/MLPH-GTBD interaction, but not the MyoVa-GTD/RILP2-RH1 interaction, is highly influenced by salt in the binding buffer (Fig. S4 B and C), indicating that MLPH-GTBD and RILP2-RH1 bind to MyoVa-GTD with different interaction modes. Consistent with our hypothesis, analytical gel-filtration chromatographic analysis of a 1:1:1 ratio mixture of MLPH-GTBD:RILP2-RH1:MyoVa-GTD showed that the three proteins were eluted together as a single peak with an elution volume larger than that of either one of the binary complexes (Fig. 3F), strongly indicating that MyoVa-GTD can interact with the two cargos simultaneously.
Overall Structure of the MyoVa-GTD/RILPL2-RH1/MLPH-GTBD Complex.
We determined the crystal structure of the MyoVa-GTD/RILPL2-RH1/MLPH-GTBD complex at the resolution of 2.4 Å (Table S1). In the complex structure, the three proteins form a 2:2:2 hexamer comprising two nearly identical MyoVa-GTD/RILPL2-RH1/MLPH-GTBD trimers related by a pseudo two-fold symmetry (Fig. 4A). The two trimers form the hexamer via homodimerization of RILPL2-RH1. In each trimer, MyoVa-GTD interacts with one RILPL2-RH1 and one MLPH-GTBD using two distinct binding sites at the opposite sides of the subdomain I (Fig. 4A), which is fully consistent with the noncompetitive bindings of the two cargos to MyoVa-GTD shown in Fig. 3. The conformation of MyoVa-GTD in the cargo-bound form and in its cargo-free form are essentially identical (rmsd <1 Å), indicating that the cargo binding does not induce obvious conformational changes to MyoVa-GTD.
Fig. 4.

The MyoVa-GTD/RILPL2-RH1/MLPH-GTBD complex structure. (A) Ribbon representation of the 2:2:2 hexamer structure of the MyoVa-GTD/RILPL2-RH1/MLPH-GTBD complex. MyoVa-GTD, RILPL2-RH1, and MLPH-GTBD in one 1:1:1 trimer are colored in purple, green, and cyan, with those in the other identical trimer colored in corresponding lighter colors. This color-coding scheme is used hereafter except as otherwise indicated. (B and C) The detailed interactions between MyoVa-GTD and RILPL2-RH1 (B) and between MyoVa-GTD and MLPH-GTBD (C) in the corresponding regions boxed in A. The disordered N- and C-termini of MLPH-GTBD are indicated by dotted lines. Hydrogen bonds and salt bridges are indicated by dashed lines.
Except for a highly flexible, 13-residue N-terminal fragment, RILPL2-RH1 adopts an all-helical structure. The RILPL2-RH1 homodimer is mainly mediated by a four-helix bundle formed by two antiparallel helices from each RH1 and further strengthened by a coiled coil formed by the C-terminal half of the α3-helix (α3C, Fig. 5A). The binding of RILPL2-RH1 to MyoVa-GTD is exclusively mediated by the four-helix bundle part of RH1, which buries ∼850 Å2 of solvent-accessible surface area on each MyoVa-GTD. Unlike RILPL2-RH1, MLPH-GTBD uses a much shorter region (14 residues in total, Fig. S5) to bind to MyoVa-GTD, and adopts an extended conformation (Fig. 4 A and C). The buried surface area on each MyoVa-GTD by MLPH-GTBD is ∼800 Å2.
Fig. 5.

Characterizations of the MyoVa-GTD/RILPL2-RH1 interaction. (A) The sequence alignment of the RH1 regions of the RILP family members. Dm, Drosophila melanogaster; Dr, Danio rerio; Hs, human; Mm, mouse; Xt, Xenopus tropicalis. Residues that are identical and highly similar are shown in red and yellow boxes, respectively. The secondary structural elements of RILPL2-RH1 are labeled above the alignment. The residues forming RILPL2 dimer interfaces in the helical bundle and coiled-coil regions are indicated by solid and open circles, respectively. The residues involved in the MyoVa-GTD/RILPL2-RH1 interaction are indicated by solid stars. (B and D) The MyoVa-GTD/RILPL2-RH1 interaction is mainly mediated by hydrophobic interactions. Two hydrophobic residues in α2MyoVa, L1502 and V1498, interact with the hydrophobic pocket of RILPL2-RH1 (B). Meanwhile, F56 in α3RILPL2 inserts its aromatic ring into the hydrophobic pocket of MyoVa (D). (C and E) Two sets of analytical gel filtration analysis were used for verification of the structural findings shown in B and D, respectively. (F) The structural basis of the four-helix, bundle-mediated dimer formation of RILPL2. α2MyoVa was shown to indicate the MyoVa binding site on RILPL2. (G) ITC-based analysis showing that truncation of the coiled-coil of RILPL2-RH1 (RH1_1-83) did not affect the MyoVa-GTD/RILPL2-RH1 interaction, whereas disruption of the RILPL2 homodimer (RH1_V61E) largely diminished this interaction.
The MyoVa-GTD/RILPL2-RH1 Interaction.
The MyoVa-GTD/RILPL2-RH1 interaction is mainly mediated by hydrophobic interactions (Fig. 4B), explaining why the high concentration of salt in the binding buffer had little impact on the interaction (Fig. S4C). The hydrophobic residues in the MyoVa-GTD/RILPL2-RH1 interface clusters into two continuous parts. In one part, V1498 and L1502 from α2MyoVa interact with a hydrophobic patch formed by the residues from α2RILPL2 and α3RILPL2 (Figs. 4B and 5B). In the other part, F56 from α3RILPL2 inserts its bulky side chain into a hydrophobic pocket formed by the residues mainly from α2MyoVa, α3MyoVa, and part of the CC loop (Figs. 4B and 5D). Consistently, substitution of V59, which is involved in the formation of the hydrophobic patch on RILPL2-RH1, with Gln totally abolished the MyoVa-GTD/RILPL2-RH1 interaction (Fig. 5C). Most of these interacting residues are highly conserved among the RILP family members (Fig. 5A), arguing that RILP or RILPL1 might also interact with MyoVa-GTD. Curiously and converse to our structure-based prediction, the N-terminal half of RILP (RILP-N), including the RH1 region, did not show detectable binding to MyoVa-GTD (Fig. S6A), indicating the exquisite specificity of MyoVa-GTD for RILPL2 instead of highly homologous RILP and RILPL1. Careful analysis of their sequences reveals that F56RILPL2 is replaced by a Pro in other RILP family members (e.g., P55 in mouse RILP) (Fig. 5A). This replacement presumably weakens the contact observed between F56RILPL2 and the hydrophobic pocket in MyoVa-GTD shown in Fig. 5D. Additionally, P55RILP may bend the α3RILP helix by introducing steric hindrances and thus alter its MyoVa-GTD binding. Consistent with this analysis, substituting F56RILPL2 with Pro led to the disruption of the interaction between RILPL2-RH1 and MyoVa-GTD (Fig. 5E). Conversely, replacing P55 in RILP-N with Phe rendered the mutant RILP-N capable of binding to MyoVa-GTD with an even higher binding affinity than that of RILPL2-RH1 (Kd of ∼0.05 μM vs. 0.3 μM; Figs. 3C and 6B). Despite of their overall high-sequence identities (up to 70%), the residues corresponding to α2MyoVa in MyoVb-GTD (and invertebrate MyoV-GTD) differ significantly from those of MyoVa-GTD (Fig. S1). Because most of the residues from α2MyoVa are involved in the binding to RILPL2-RH1, MyoVb is unlikely to be able to bind to RILPL2-RH1. Indeed, our analytical gel filtration–based assay could not detect direct interaction between MyoVb-GTD and RILPL2-RH1 (Fig. S6C).
Fig. 6.

Characterizations of the MyoVa-GTD/MLPH-GTBD interaction. (A) The multisequence alignment of MLPH-GTBD. Bt, bovine; Gg, chicken. The residues involved in the MyoVa-GTD/MLPH-GTBD interaction are indicated by solid stars. The negatively charged residues in the MLPH-GTBD C terminus may be involved in the charge–charge interaction with K1540, K1543, and K1544 (Fig. 4C) and are indicated by open stars. (B) The ITC-derived dissociation constants of the bindings between MyoVa-GTD and MLPH-GTBD. (C) Structural comparison of the MLPH-GTBD binding surface (cyan) on MyoVa-GTD and the Vac17/Mmr1-binding surface (blue) on myo2p-GTD. The four overlapped binding residues in MyoVa (purple) and myo2p (gray) are labeled.
The dimer formation is an important feature of RILPL2-RH1 (Fig. S6E). The RILPL2-RH1 dimer is structurally separated into two parts, the N-terminal four-helix bundle formed by α2 and α3N (Fig. 5F) and the C-terminal coiled coil formed by α3C (Fig. S6D). The four-helix bundle in the RILPL2-RH1 dimer is mainly stabilized by forming a hydrophobic core. The N-terminal small helix (α1RILPL2) and its following loop pack on α2RILPL2 from the same RILPL2 molecule and α3RILPL2 from the neighboring RILPL2 molecule and thus contribute to the bundle stability (Fig. 5F). The C-terminal parallel coiled coil is the asymmetric part in the hexamer because one of α3RILPL2 contains a short break in the middle (specifically, 72GS73 ; Fig. 4A, Lower). This coiled coil is dispensable for the RILPL2-RH1/MyoVa-GTD interaction because truncation of a large part of α3C (residues 84–97), which presumably disrupts the coiled-coil structure, had little impact on its binding to MyoVa-GTD (Fig. 5G). In contrast, the formation of the four-helix bundle RILPL2-RH1 dimer is critical for the RILPL2-RH1/MyoVa-GTD interaction because a monomeric mutant of RILPL2-RH1 (Fig. S6G), of which V61 (located in the helical bundle core but away from its MyoVa-GTD contact site) was substituted with Glu, displayed diminished binding to MyoVa-GTD (Fig. 5G).
The MyoVa-GTD/MLPH-GTBD Interaction.
MLPH-GTBD contains a highly charged sequence with five continuous lysine or arginine and several negatively charged residues in its N and C terminus, respectively. These charged residues sandwich a few hydrophobic residues in the middle of MLPH-GTBD (Fig. 6A). Most of these residues are highly conserved, suggesting their potential roles in the MyoVa-GTD/MLPH-GTBD interaction. Consistently, these conserved residues form hydrophobic interaction, salt bridges, or hydrogen bonds with corresponding surface residues from α4, α7, and the α6/α7 loop of MyoVa-GTD (Fig. 4C). Compared with the MyoVa-GTD/RILPL2-RH1 interaction, the binding of MLPH-GTBD to MyoVa-GTD includes less hydrophobic interaction but more hydrogen bonds and salt bridges, explaining the salt-dependent binding between MLPH-GTBD and MyoVa-GTD (Fig. S4B). In the ITC-based assay, the alanine substitution mutants of charged residues in the binding interface reduced MLPH-GTBD binding to MyoVa-GTD by several fold (Fig. 6B). Similarly, introducing a negative charge in the interface by mutating K1539MyoVa to Glu disrupts the MyoVa-GTD/MLPH-GTBD interaction. As a control, the substitution of D193MLPH, which is not in the binding interface, with Ala had no detectable impact on the binding. Notably, substitution of either one of the two Phe residues in the MLPH-GTBD (F191 and F196) with Gln severely affected the binding of MLPH to MyoVa-GTD (Fig. 6B), indicating that the hydrophobic interaction is also crucial for the interaction. Correspondingly, the I1535E mutation in MyoVa decreased the binding affinity by ∼40-fold (Fig. 6B).
Except for MyoVb, the residues from MyoVa that are responsible for binding to MLPH-GTBD are not conserved in MyoVc and invertebrate MyoV (Fig. S1), suggesting that these MyoV homologs/paralogs are unlikely to bind to MLPH. Intriguingly, except for R1258, which is a His in MyoVb, all of the residues responsible for MyoVa binding to MLPH can be found in MyoVb (Fig. S1). However, MyoVb-GTD shows largely diminished binding to MLPH-GTBD (Fig. S7). Based on our structure, R1528 is intimately involved in the binding of MyoVa-GTD to MLPH-GTBD by forming two hydrogen bonds with a main-chain carbonyl group and a cation–π interaction with F191MLPH (Fig. 4C). Substitution of R1528 with His indeed weakened MyoVa-GTD’s binding to MLPH-GTBD but only by approximately fourfold (Fig. 6B), suggesting residues outside the interface may also contribute to the different binding affinities of MyoVa and MyoVb to MLPH through still-unknown mechanisms.
Several cargo-binding sites have been identified in yeast myo2p via structure-based, mutagenesis-based experiments (7, 8). Interestingly, the cargo-binding surface in subdomain I of myo2p, which is critical for binding to vacuole- or mitochondrial-specific cargos, partially overlaps with the corresponding MLPH-binding surface on MyoVa (Fig. 6C). Additionally, the cargo-binding sites in subdomain I of myo2p and MyoVa share several key cargo-recognizing residues (Fig. 6C), suggesting that at least that part of the cargo-binding functions of the class V myosins are evolutionarily linked.
Granuphilin Shares Its Binding Site with MLPH in MyoVa-GTD.
Very recently, granuphilin (Gran) was found to be an adaptor connecting secretory granules to MyoVa via binding to the motor’s GTD (13). We mapped the minimal MyoVa-GTD–binding region of Gran to its 141–350 fragment using pull-down assays (Fig. S8 A and B). Although sharing little amino acid sequence similarity with either RILPL2-RH1 or MLPH-GTBD, the MyoVa-GTD–binding region of Gran is predicted to be highly disordered and contains charge-rich sequences, suggesting that Gran may bind to MyoVa-GTD with a mode similar to that of MLPH. To test this hypothesis, we set up a competitive displacement assay by mixing the MyoVa-GTD/Gran complex with increasing amounts of the MLPH peptide. The result showed that the MLPH peptide effectively competes with Gran for binding to MyoVa-GTD (Fig. S8C). Additionally, we found that the MLPH-binding defective mutants (I1535E and K1539E) of MyoVa-GTD are also defective in binding to Gran (Fig. S7D, Upper), indicating Gran and MLPH share overlapping binding surfaces on MyoVa-GTD.
Discussion
Despite many years of extensive studies, it remains largely a mystery how MyoV-GTD specifically recognizes its broad ranges of cargos. Additionally, the underlying molecular bases of numerous loss-of-function mutations of MyoV-GTD found in patients or animals are poorly understood. Because of the very low amino acid sequence identity between GTDs of yeast and mammalian class V myosins, it has been difficult to translate knowledge obtained from the previously solved structures of yeast MyoV GTDs (7, 26) directly to mammalian class V myosins. The atomic structures of MyoVa-GTD both in its apo- and cargo-bound forms described in this work provide major advancements in these areas. The structure of apo-MyoVa-GTD reveals that the majority of the missense mutations found in MyoVa are concentrated within subdomain I and that these mutations are likely to perturb the overall folding of GTD, thereby indirectly affecting its cargo bindings (Fig. 2). Currently known loss-of-function missense mutations of MyoVb GTD are distributed in both subdomains I and II (Fig. 2), suggesting that both subdomains of MyoVb GTD are involved in cargo recognitions.
Our structural and biochemical studies described in this work demonstrated that MyoVa-GTD can use its subdomain I to interact with its multiple cargos by distinct binding regions and interaction modes. In one site, MyoVa-GTD employs the α2-helix and its vicinity region to bind to RILPL2-RH1 mainly via hydrophobic interactions (Fig. 4B). The other cargo-binding site (“site 2”) is located at the opposite side of the RILPL2-binding site and enriched with charged residues (Fig. 4C). Matching with the surface properties of site 2, the MyoVa-binding regions of MLPH and Gran are rich in charged amino acid residues (Fig. 6 and Fig. S8). Previous studies of yeast myo2p suggested the site 2 in yeast MyoV is involved in binding to several target proteins; many of these proteins do not have corresponding counterparts in mammals (7, 8), indicating that both MyoV-GTD and its cargos undergo extensive changes during evolution. Despite of the high-sequence similarity between MyoVa and Vb, neither RILPL2 nor MLPH shows specific binding to MyoVb-GTD, indicating exquisite cargo-binding specificities of the closely related MyoV paralogs. Our structural studies also showed that both RILPL2 and MLPH bind to subdomain I of MyoVa-GTD, leaving subdomain II completely free. Nevertheless, it is likely that subdomain II is also involved in recognitions of other cargos. Finally, it is known that tissue-specific alternative splicings of MyoV’s tail regions N terminus to GTD also directly regulate cargo bindings (9, 11, 33), adding further complexities to cargo recognitions for class V myosins.
Materials and Methods
All proteins used in this study was expressed in Escherichia coli BL21 (DE3) and purified by Ni2+-NTA affinity chromatography followed by size-exclusion chromatography. Crystals were obtained by hanging drop vapor diffusion method at 16 °C. An extended method describing protein preparation, crystallization, structure determination, and biochemical assays can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the BL17U1 beamline team of the Shanghai Synchrotron Radiation Facility for the beamline time. This work was supported by the Research Grants Council of Hong Kong Grants 663610, 663811, 663812, Hong Kong University of Science and Technology (HKUST)6/CRF/10, SEG_HKUST06, AoE/M-04/04, and T13-607/12R (to M.Z.) and 662710 (to Z.W.). Z.W. is an Institute for Advanced Study Tin Ka Ping Fellow at HKUST, and M.Z. is a Kerry Holdings Professor in Science and a Senior Fellow of IAS at HKUST.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 3WB8 (apo MyoV-GTD) and 4KP3 (cargo bound MyoVa-GTD)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306768110/-/DCSupplemental.
References
- 1.Johnston GC, Prendergast JA, Singer RA. The Saccharomyces cerevisiae MYO2 gene encodes an essential myosin for vectorial transport of vesicles. J Cell Biol. 1991;113(3):539–551. doi: 10.1083/jcb.113.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mercer JA, Seperack PK, Strobel MC, Copeland NG, Jenkins NA. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature. 1991;349(6311):709–713. doi: 10.1038/349709a0. [DOI] [PubMed] [Google Scholar]
- 3.Espreafico EM, et al. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J Cell Biol. 1992;119(6):1541–1557. doi: 10.1083/jcb.119.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammer JA, 3rd, Sellers JR. Walking to work: Roles for class V myosins as cargo transporters. Nat Rev Mol Cell Biol. 2012;13(1):13–26. doi: 10.1038/nrm3248. [DOI] [PubMed] [Google Scholar]
- 5.Sellers JR, Weisman LS. Myosin V. In: Coluccio LM, editor. Myosins, a Superfamily of Molecular Motors. Vol 9. Dordrecht: Springer; 2008. pp. 289–323. [Google Scholar]
- 6.Reck-Peterson SL, Provance DW, Jr, Mooseker MS, Mercer JA. Class V myosins. Biochim Biophys Acta. 2000;1496(1):36–51. doi: 10.1016/s0167-4889(00)00007-0. [DOI] [PubMed] [Google Scholar]
- 7.Pashkova N, Jin Y, Ramaswamy S, Weisman LS. Structural basis for myosin V discrimination between distinct cargoes. EMBO J. 2006;25(4):693–700. doi: 10.1038/sj.emboj.7600965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eves PT, Jin Y, Brunner M, Weisman LS. Overlap of cargo binding sites on myosin V coordinates the inheritance of diverse cargoes. J Cell Biol. 2012;198(1):69–85. doi: 10.1083/jcb.201201024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Au JS, Huang JD. A tissue-specific exon of myosin Va is responsible for selective cargo binding in melanocytes. Cell Motil Cytoskeleton. 2002;53(2):89–102. doi: 10.1002/cm.10061. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda M, Kuroda TS, Mikoshiba K. Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: Implications of a tripartite protein complex for melanosome transport. J Biol Chem. 2002;277(14):12432–12436. doi: 10.1074/jbc.C200005200. [DOI] [PubMed] [Google Scholar]
- 11.Wu XS, et al. Identification of an organelle receptor for myosin-Va. Nat Cell Biol. 2002;4(4):271–278. doi: 10.1038/ncb760. [DOI] [PubMed] [Google Scholar]
- 12.Lisé MF, et al. Myosin-Va-interacting protein, RILPL2, controls cell shape and neuronal morphogenesis via Rac signaling. J Cell Sci. 2009;122(Pt 20):3810–3821. doi: 10.1242/jcs.050344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brozzi F, et al. Molecular mechanism of myosin Va recruitment to dense core secretory granules. Traffic. 2012;13(1):54–69. doi: 10.1111/j.1600-0854.2011.01301.x. [DOI] [PubMed] [Google Scholar]
- 14.Hales CM, Vaerman JP, Goldenring JR. Rab11 family interacting protein 2 associates with Myosin Vb and regulates plasma membrane recycling. J Biol Chem. 2002;277(52):50415–50421. doi: 10.1074/jbc.M209270200. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Takeuchi T, Yokota H, Izumi T. Novel rabphilin-3-like protein associates with insulin-containing granules in pancreatic beta cells. J Biol Chem. 1999;274(40):28542–28548. doi: 10.1074/jbc.274.40.28542. [DOI] [PubMed] [Google Scholar]
- 16.Matesic LE, et al. Mutations in Mlph, encoding a member of the Rab effector family, cause the melanosome transport defects observed in leaden mice. Proc Natl Acad Sci USA. 2001;98(18):10238–10243. doi: 10.1073/pnas.181336698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsui T, Ohbayashi N, Fukuda M. The Rab interacting lysosomal protein (RILP) homology domain functions as a novel effector domain for small GTPase Rab36: Rab36 regulates retrograde melanosome transport in melanocytes. J Biol Chem. 2012;287(34):28619–28631. doi: 10.1074/jbc.M112.370544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hales CM, et al. Identification and characterization of a family of Rab11-interacting proteins. J Biol Chem. 2001;276(42):39067–39075. doi: 10.1074/jbc.M104831200. [DOI] [PubMed] [Google Scholar]
- 19.Provance DW, Jr, Wei M, Ipe V, Mercer JA. Cultured melanocytes from dilute mutant mice exhibit dendritic morphology and altered melanosome distribution. Proc Natl Acad Sci USA. 1996;93(25):14554–14558. doi: 10.1073/pnas.93.25.14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nascimento AA, Amaral RG, Bizario JC, Larson RE, Espreafico EM. Subcellular localization of myosin-V in the B16 melanoma cells, a wild-type cell line for the dilute gene. Mol Biol Cell. 1997;8(10):1971–1988. doi: 10.1091/mbc.8.10.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rogers SL, et al. Regulation of melanosome movement in the cell cycle by reversible association with myosin V. J Cell Biol. 1999;146(6):1265–1276. doi: 10.1083/jcb.146.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang JD, et al. Molecular genetic dissection of mouse unconventional myosin-VA: Head region mutations. Genetics. 1998;148(4):1951–1961. doi: 10.1093/genetics/148.4.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang JD, et al. Molecular genetic dissection of mouse unconventional myosin-VA: Tail region mutations. Genetics. 1998;148(4):1963–1972. doi: 10.1093/genetics/148.4.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takagishi Y, et al. The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci Lett. 1996;215(3):169–172. doi: 10.1016/0304-3940(96)12967-0. [DOI] [PubMed] [Google Scholar]
- 25.Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: A model system to study vesicular trafficking. Pigment Cell Melanoma Res. 2009;22(3):268–282. doi: 10.1111/j.1755-148X.2009.00558.x. [DOI] [PubMed] [Google Scholar]
- 26.Heuck A, et al. The structure of the Myo4p globular tail and its function in ASH1 mRNA localization. J Cell Biol. 2010;189(3):497–510. doi: 10.1083/jcb.201002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuda M, Kuroda TS. Missense mutations in the globular tail of myosin-Va in dilute mice partially impair binding of Slac2-a/melanophilin. J Cell Sci. 2004;117(Pt 4):583–591. doi: 10.1242/jcs.00891. [DOI] [PubMed] [Google Scholar]
- 28.Pastural E, et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet. 1997;16(3):289–292. doi: 10.1038/ng0797-289. [DOI] [PubMed] [Google Scholar]
- 29.Brooks SA, et al. Whole-genome SNP association in the horse: Identification of a deletion in myosin Va responsible for Lavender Foal Syndrome. PLoS Genet. 2010;6(4):e1000909. doi: 10.1371/journal.pgen.1000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller T, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 31.Ruemmele FM, et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum Mutat. 2010;31(5):544–551. doi: 10.1002/humu.21224. [DOI] [PubMed] [Google Scholar]
- 32.Geething NC, Spudich JA. Identification of a minimal myosin Va binding site within an intrinsically unstructured domain of melanophilin. J Biol Chem. 2007;282(29):21518–21528. doi: 10.1074/jbc.M701932200. [DOI] [PubMed] [Google Scholar]
- 33.Roland JT, Lapierre LA, Goldenring JR. Alternative splicing in class V myosins determines association with Rab10. J Biol Chem. 2009;284(2):1213–1223. doi: 10.1074/jbc.M805957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.