

Abstract
Brain injury induces phenotypic changes in astrocytes, known as reactive astrogliosis, which may influence neuronal survival. Here we show that brain injury induces inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ signaling in astrocytes, and that the Ca2+ signaling is required for astrogliosis. We found that type 2 IP3 receptor knockout (IP3R2KO) mice deficient in astrocytic Ca2+ signaling have impaired reactive astrogliosis and increased injury-associated neuronal death. We identified N-cadherin and pumilio 2 (Pum2) as downstream signaling molecules, and found that brain injury induces up-regulation of N-cadherin around the injured site. This effect is mediated by Ca2+-dependent down-regulation of Pum2, which in turn attenuates Pum2-dependent translational repression of N-cadherin. Furthermore, we show that astrocyte-specific knockout of N-cadherin results in impairment of astrogliosis and neuroprotection. Thus, astrocytic Ca2+ signaling and the downstream function of N-cadherin play indispensable roles in the cellular responses to brain injury. These findings define a previously unreported signaling axis required for reactive astrogliosis and neuroprotection following brain injury.
Keywords: calcium signal, reactive astrocyte, translational repressor, stab wound
Astrocytes, a major type of glial cell in the brain, play essential roles not only in physiological functions, such as synaptic plasticity and hemodynamic responses (1), but also in pathophysiological conditions, such as trauma, infection, ischemia, epileptic seizures and stroke. In response to brain injury, astrocytes undergo characteristic phenotypic changes known as reactive astrogliosis, which can exert both beneficial and detrimental effects on surrounding neurons (2–4). Despite the importance of this process, the molecular mechanisms governing astrogliosis and the role of reactive astrocytes require further clarification.
Injury to the brain mobilizes astrocyte-reactivating factors, including ATP, endothelin 1 (ET1), glutamate, and inflammatory cytokines, as well as mechanical stress. These factors have been shown to evoke astrocytic Ca2+ signals not only in culture, but also in acute brain slice preparations and in the brains of live animals (5–7). These findings raise the possibility that injury to the brain evokes astrocytic Ca2+ signals, which in turn regulate reactive astrogliosis. However, injury-induced Ca2+ signaling in astrocytes heretofore had not been observed in vivo, and the role of Ca2+ signaling in pathophysiological processes after brain injury was not established.
In this study, we investigated the involvement of astrocytic Ca2+ signals and underlying molecular mechanisms in reactive astrogliosis and neuroprotection after traumatic brain injury. We found that neocortical injury evokes astrocytic Ca2+ signals, which are required for reactive astrogliosis and neuronal protection. We identified N-cadherin and pumilio 2 (Pum2) as molecules acting downstream of astrocytic Ca2+ signals. Around the injury site, N-cadherin is up-regulated owing to reduced translational repression by Pum2, which is down-regulated in a Ca2+-dependent manner. Furthermore, we identified an indispensable role of N-cadherin in astrogliosis and neuroprotection in mice with astrocyte-specific disruption of the N-cadherin gene. These results demonstrate that Ca2+-dependent translational regulation of N-cadherin expression in astrocytes is involved in reactive astrogliosis and neuroprotection.
Results
Requirement of Astrocytic Ca2+ Signaling for Injury-Induced Cellular Responses.
To gain insight to the potential involvement of astrocytic Ca2+ signaling in astrogliosis, we first examined whether injury induces astrocytic Ca2+ signaling in vivo. We performed laser ablation injury to the neocortex of mice during live Ca2+ imaging, and found that this treatment triggered robust Ca2+ signals in neighboring cells loaded with Oregon Green 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA)-1 acetoxymethyl (AM) ester, most of which were astrocytes (Fig. 1A, Fig. S1A, and SI Materials and Methods) (8). We also found that the injury-induced astrocytic Ca2+ signals persisted for more than 3 h (Fig. S1B). We then studied the source of injury-induced astrocytic Ca2+ signaling. Type 2 inositol 1,4,5-trisphosphate (IP3) receptor (IP3R2) is the dominant intracellular Ca2+ release channel in astrocytes (9, 10), and agonist-induced Ca2+ signals were absent in IP3R2KO neocortical astrocytes in acute slice preparations (Fig. S1C). Thus, we examined the injury-induced Ca2+ signal in IP3R2KO mice (11) and found that it was abrogated (Fig. 1A), indicating that injury to the brain mobilizes Ca2+ via IP3R2 in astrocytes.
Fig. 1.
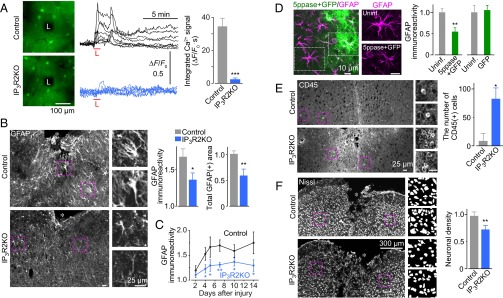
Injury-induced astrocytic Ca2+ signaling in astrogliosis and neuroprotection. (A) (Left) Neocortical astrocytic Ca2+ signals in vivo evoked by laser ablation injury (black areas marked with “L”). (Center) Ca2+ signals from individual astrocytes in IP3R2KO and control littermates (n = 8 each). (Right) Time integral of Ca2+ signals (for 120 s after injury). n = 47 for control, n = 53 for IP3R2KO, from three or four animals each. (B) (Left) Analysis of reactive astrogliosis by GFAP immunostaining. Magnified images correspond to boxed regions. Asterisks designate the injury site. (Right) Graphs showing the fold increase of GFAP immunoreactivity compared with the contralateral uninjured site and the total GFAP-positive area around the injury site, normalized to the average value in controls. n = 5–7. (C) Time course of reactive astrogliosis around the injury site, normalized to the intensity of the contralateral uninjured site. n = 4–9. (D) (Left) Effect of inhibition of astrocytic Ca2+ signals on reactive astrocytes after SWI. Adenovirus encoding 5ppase-IRES-GFP was injected. To the right are magnifications of boxed regions. (Right) Graph showing quantification of GFAP expression level in adenovirus-infected (5ppase + GFP) and uninfected (Uninf.) astrocytes around the injury site, normalized to uninfected astrocytes, along with results of a control experiment using adenovirus encoding GFP. n = 8–11. (E) Inflammatory cell invasion after SWI. CD45+ cells in the 0.5-mm2 area around the SWI site were counted. (Left) Representative images of CD45 immunostaining, with magnifications of boxed regions. (Right) Graph showing quantification of CD45+ cells. n = 6. (F) (Left) Density of surviving neurons around the injury site, with magnifications of binarized images of boxed regions. (Right) Graph showing quantification, normalized to that on the uninjured contralateral side. n = 6. *P < 0.05; **P < 0.01; ***P < 0.0001. The size of scale bars are common in each panel.
We then analyzed the role of Ca2+ signals in astrogliosis in the IP3R2KO mice after a neocortical stab wound injury (SWI), produced by a 0.8- to 1.3-mm-long and 0.5- to 0.8-mm-deep incision (12, 13). SWI is expected to generate Ca2+ signals in a larger population of astrocytes compared with a small laser ablation injury, making biochemical and histochemical analyses around the injury site more feasible. At 5 d after SWI, we tested for expression levels of glial fibrillary acidic protein (GFAP), an astrocytic marker that is up-regulated in reactive astrocytes, in astrocytes surrounding the injury site in control animals and in IP3R2KO mice. In the control animals, increased levels of GFAP expression were detected, often accompanied by polarized processes directed toward the injury site. In IP3R2KO mice, these changes associated with reactive astrogliosis were reduced (Fig. 1B and Fig. S2). The attenuation of SWI-induced GFAP up-regulation in IP3R2KO mice continued over a 14-d period (Fig. 1C). We confirmed these results using an alternative method to suppress Ca2+ signal by the IP3-hydrolyzing enzyme IP3 5-phosphatase (5ppase), which has been shown to abrogate astrocytic Ca2+ signals (14, 15). (See also Fig. S4C.) In neocortical astrocytes expressing 5ppase from an adenovirus vector, we found weak SWI-induced GFAP up-regulation, confirming our findings in the IP3R2KO mice (Fig. 1D).
Reactive astrogliosis is thought to enhance neuroprotection and prevent inflammatory cell infiltration into the neural parenchyma after injury (3, 12). To test whether injury-induced astrocytic Ca2+ signals are important for these functions of reactive astrocytes, we measured the abundance of these cells in the SWI model. We found significantly more infiltrating leukocytes, observed as CD45+ round cells (12), and fewer surviving neurons around the injury site in the IP3R2KO mice compared with control mice (Fig. 1 E and F). We confirmed that the IP3R2KO mice had no overt abnormalities in cortical histology, consistent with previous reports (10). Before injury, GFAP immunoreactivity and the density of CD45+ leukocytes were comparable in the IP3R2KO and control littermate mice (Fig. S3). Similarly, IP3R2 gene disruption caused no significant alteration in the immunoreactivity of ionized calcium-binding adapter molecule 1 (Iba1), a marker for microglia, which is implicated in reactive astrogliosis (16) (Fig. S3). These results suggest the involvement of injury-induced astrocytic Ca2+ signals in the cellular functions of reactive astrogliosis.
Pum2 and N-Cadherin Act Downstream of Astrocytic Ca2+ Signaling.
We next explored the mechanisms of Ca2+-dependent reactive astrogliosis. We first compared gene expression profiles between Ca2+ signal-silent and -active astrocytes in culture (SI Materials and Methods). Among highly down-regulated genes in Ca2+ signal-active astrocytes (Table S1), Pum2 drew our immediate attention because of its capacity as a translational repressor (17, 18). The decrease in Pum2 expression level in Ca2+ signal-active astrocytes was confirmed by quantitative PCR analysis (29.8 ± 5.1% compared with control) and Western blot analysis (Fig. S4A). Proteins whose translation is regulated by Pum2 are expected to be Ca2+-dependently up-regulated. We focused on N-cadherin as a potential target of Pum2 for several reasons. First, we previously found that N-cadherin is Ca2+-dependently up-regulated in astrocytes (14). Second, there is an evolutionarily conserved potential Pum2-binding sequence (17) in the 3′ UTR of N-cadherin mRNA (Fig. S4B). Third, N-cadherin is up-regulated in reactive astrocytes (19, 20), although its role in reactive astrogliosis remains elusive. In line with this notion, we found that Ca2+-mobilizing agonists ATP and ET1 induced down-regulation of Pum2 and up-regulation of N-cadherin in cultured astrocytes (Fig. 2 A and B and Fig. S4 C and D).
Fig. 2.

Effect of astrocytic Ca2+ signals on N-cadherin expression level as mediated by the translational repressor Pum2. (A) Agonist [30 µM ATP (Left) or 100 nM ET1 (Right)]-induced Ca2+ signals in cultured astrocytes. Ten representative traces are superimposed. (B) (Left) Pum2 and N-cadherin protein levels after incubation with 30 µM ATP or 100 nM ET1 for 15−18 h. (Right) Graphs showing signal intensity normalized to that of GAPDH (GAP). n = 5. (C) (Left) Pum2-induced down-regulation of N-cadherin in cultured astrocytes. RFP-Pum2 or mutant Pum2 without the RNA-binding domain (RFP-Pum2∆) was transfected. (Right) Graph showing quantification of N-cadherin expression normalized to that of adjacent untransfected cells. n = 21–25. (D) Results of Pum2 activity reporter assay in cultured astrocytes showing the effect of RFP-Pum2 or Pum2 without the RNA-binding region on reporters carrying N-cadherin 3′ UTR (GFP-N-UTR), N-cadherin 3′ UTR without the Pum2-binding sequence (GFP-N-UTRΔ), or E-cadherin 3′ UTR (GFP-E-UTR), which has no Pum2-binding sequence. The fluorescence intensity of GFP in cells expressing RFP-Pum2 was normalized to that in cells expressing RFP-Pum2∆. n = 22–84. *P < 0.05; **P < 0.01.
Because the 3′ UTR of N-cadherin mRNA harbors the potential Pum2-binding sequence, we examined the possibility that Pum2-dependent translational repression is involved in the regulation of N-cadherin expression in cultured astrocytes. Expression of red fluorescent protein (RFP)-tagged Pum2 (RFP-Pum2) reduced the basal expression level of N-cadherin, whereas Pum2 without its RNA-binding domain (RFP-Pum2∆) had no effect (Fig. 2C). Moreover, the expression of Pum2-activity reporter, consisting of a GFP coding sequence fused to the N-cadherin 3′ UTR sequence, was significantly reduced in astrocytes expressing RFP-Pum2 (Fig. 2D). The suppressive effect of Pum2 was abrogated by removal of the Pum2-binding sequence from the reporter or by replacement of the 3′ UTR sequence with that of E-cadherin mRNA (Fig. 2D). The expression of RFP-Pum2∆ had no effect on the expression level of all reporters. These results indicate that interaction between Pum2 and the Pum2-binding sequence in the 3′ UTR of N-cadherin mRNA mediates down-regulation of N-cadherin.
Injury-Induced Astrocytic Ca2+ Signals Regulate Expression Levels of Pum2 and N-Cadherin.
To study the role of Pum2 and N-cadherin in brain injury, we examined the expression levels of these molecules in vivo in the SWI model. Before injury, the adult neocortex expressed a significant amount of Pum2, whereas N-cadherin expression was barely detectable in both control and IP3R2KO mice (Fig. 3A). After SWI, Pum2 expression was significantly reduced and N-cadherin expression was markedly increased around the injury site in control mice, whereas these changes were significantly attenuated in IP3R2KO mice (Fig. 3 A and B). These SWI-induced changes in Pum2 and N-cadherin expression were confirmed by Western blot analysis using tissue samples collected within 0.5–1.0 mm of the SWI site (Fig. 3C). After SWI, Pum2 expression levels were 57 ± 9% of the contralateral expression level in control mice and 85 ± 8% of that in IP3R2KO mice, and respective N-cadherin expression levels were 289 ± 9% and 147 ± 13%.
Fig. 3.

Dependence of injury-induced Pum2 and N-cadherin expression on astrocytic Ca2+ signals. (A) (Left) Injury-induced changes in Pum2 and N-cadherin expression levels in IP3R2KO mice. (Right) Graph showing quantification of immunofluorescence intensity normalized to the average value in the uninjured site (for Pum2) or the injured site (for N-cadherin) in control mice. n = 5–7. (B) Time course of Pum2 (Left) and N-cadherin (Right) expression around the injury site, normalized to the intensity of the contralateral uninjured site. n = 3–7. (C) Western blot analysis of SWI-induced changes in Pum2 and N-cadherin expression levels. At 4 d after SWI, the brain region around the SWI was analyzed. SWI(−) was sampled from the contralateral side as an internal control. (D) (Left) Pum2-dependent suppression of SWI-induced N-cadherin up-regulation, analyzed at 5 d after injury. Arrowheads indicate N-cadherin signals on infected astrocytes. (Right) Graph showing quantified N-cadherin expression in RFP-Pum2–expressing cells, normalized to the average in RFP-expressing cells. n = 7. *P < 0.05; **P < 0.01. The size of scale bars are common in each panel.
To verify the responsibility of Pum2 for N-cadherin up-regulation in reactive astrocytes, we extrinsically expressed Pum2 in neocortical astrocytes using an adenoviral vector and then applied the SWI. Astrocytes overexpressing RFP-Pum2 showed significantly less N-cadherin up-regulation after SWI compared with control astrocytes transduced with a virus encoding RFP alone (Fig. 3D). These findings suggest that Pum2 keeps N-cadherin expression at a low level in quiescent astrocytes, and that after SWI, astrocytic Ca2+ signals down-regulate Pum2, which in turn leads to the up-regulation of N-cadherin through attenuation of Pum2-mediated translational repression.
Up-Regulation of Astrocytic N-Cadherin Is Indispensable for Neuroprotection.
We next examined the possibility that Ca2+-dependent N-cadherin up-regulation has a role in astrogliosis and neuroprotection. To study this possibility, we generated astrocyte-specific N-cadherin knockout (aNcadKO) mice, crossing N-cadherin floxed mice (21) and tamoxifen (Tx)-inducible Cre recombinase-expressing mice that is driven by the astrocyte specific L-glutamate/L-aspartate transporter promoter (GLAST-CreERT2) (22), and activated Cre recombinase by administration of Tx to adult mice. After SWI, GFAP immunoreactivity and the extent of morphological changes were significantly reduced in aNcadKO mice compared with control mice (Tx-administrated N-cadherin floxed mice without the GLAST-CreERT2 gene) (Fig. 4A and Fig. S5). We observed no significant effect of Tx administration on GFAP immunoreactivity or neuronal density before SWI (Fig. S6A). In addition, N-cadherin expression level before SWI was not significantly different in control and aNcadKO mice (Fig. S6B), suggesting that neuronal N-cadherin expression in aNcadKO mice is not down-regulated. However, on injury, N-cadherin up-regulation was markedly inhibited in aNcadKO mice (Fig. S6B). The slight up-regulation of N-cadherin in aNcadKO mice after SWI is likely produced by the population of astrocytes that escaped Cre-dependent recombination; recombination efficiency in these mice is ∼60% (22). Taken together, these results indicate that N-cadherin up-regulation is required for reactive astrogliosis after SWI. In support of this notion, we found that SWI-induced down-regulation of Pum2 in aNcadKO mice was comparable to that in control littermate mice (Fig. S6B).
Fig. 4.

Effects of injury-induced up-regulation of astrocytic N-cadherin on reactive astrogliosis and neuroprotection. (A) (Left) Analyses of reactive astrogliosis performed at 5 d after SWI in aNcadKO mice. Magnified images correspond to boxed regions. Asterisks indicate the injury site. (Right) Graphs showing quantification of GFAP immunoreactivity around the injury site. (Upper) Fold increase of GFAP immunoreactivity compared with the contralateral uninjured site. (Lower) Total GFAP-positive area normalized to the average value in control littermates (Tx-treated N-cadherin floxed homozygous mice without the GLAST-CreERT2 gene). n = 6. (B) Inflammatory cell invasion after SWI. CD45+ cells in a 0.5-mm2 area surrounding the site of SWI were counted. (Left) Representative images of CD45 immunostaining, with magnification of boxed regions. (Right) Graph showing quantification of CD45+ cells. n = 4. (C) (Left) Density of surviving neurons around the injury site, with magnification of binarized images in the boxed regions. (Right) Graph showing quantification, normalized to that on the uninjured contralateral side. n = 7. *P < 0.05; **P < 0.01. The size of scale bars are common in each panel.
We next performed an in vitro scratch wound assay (23, 24) to study the role of N-cadherin in reactive astrogliosis. Scratch wound-induced migration of astrocytes is considered an important in vitro model of reactive astrogliosis, because astrocytic migration toward the injury core site is an essential process during reactive astrogliosis (25). We found a more significant reduction in the distance of migration in N-cadherin–deficient astrocytes compared with control astrocytes (Fig. S7).
Finally, we studied the effect of astrocyte-specific N-cadherin deficiency on leukocyte infiltration and neuroprotection. In aNcadKO mice, the number of CD45+ cells around the injury site was significantly increased, whereas the density of surviving neurons was significantly decreased (Fig. 4 B and C). These phenotypes observed in aNcadKO mice are similar to those observed in IP3R2KO mice (compare Figs. 1 B, E, and F with Fig. 4). Astrocyte-specific N-cadherin deficiency had no effect on the resting activity and SWI-induced activation of microglia (Fig. S8). Thus, we conclude that Ca2+ signal-dependent up-regulation of N-cadherin in astrocytes regulates reactive astrogliosis and neuroprotection following brain injury.
Discussion
In this study, we have shown that injury-induced astrocytic Ca2+ signals, acting with downstream Pum2 and N-cadherin, regulate reactive astrogliosis and neuroprotection after brain injury (Fig. 5), thereby highlighting the importance of intracellular Ca2+ signaling in the pathophysiological function of astrocytes. The requirement of IP3R2 in this Ca2+-dependent mechanism suggests that IP3-generating agonists such as ATP, ET1, and glutamate, which are released from the injured tissue, are likely to be the initiation signal. We also found that Pum2 mediates the Ca2+-dependent regulation of N-cadherin expression level through its function as a translational repressor. The various changes observed after experimental N-cadherin dysregulation, including impaired GFAP up-regulation and cell hypertrophy, seem to be consistent with N-cadherin’s known regulation of various cellular processes (26–28).
Fig. 5.

Scheme of the signaling pathway underlying astrocytic Ca2+ signal-dependent neuroprotection on brain injury. Injury evokes Ca2+ signals in astrocytes via IP3R2 (1, 2), which lead to down-regulation of Pum2 (3). This in turn induces N-cadherin up-regulation (4) by relieving Pum2-mediated repression of N-cadherin translation. Up-regulated N-cadherin plays an essential role in reactive astrogliosis and neuroprotection (5).
Mechanism of Astrocytic Ca2+-Dependent Reactive Astrogliosis.
The present study clarifies the critical role of Pum2 in reactive astrogliosis by showing that Ca2+-dependent regulation of Pum2 expression occurs at the mRNA level, suggesting the involvement of Ca2+-dependent transcriptional factor or miRNA regulation. The precise nature of this regulation remains to be elucidated. Our data are consistent with a recent study showing that Ca2+ influx across the plasma membrane regulates Pum2 expression level in neurons (29). Thus, Ca2+ dependent regulation of Pum2 appears to be common to both neurons and glia.
The putative Pum2-binding sequence is present in an array of mRNAs, suggesting that Ca2+-dependent Pum2 signaling regulates multiple potential targets. However, our data suggest that the dominant target of Ca2+-Pum2 signaling is likely N-cadherin. Specifically, whereas the effect of Ca2+ signal disruption on both astrogliosis and neuroprotection in IP3R2KO mice was recapitulated in aNcadKO mice, the extent of injury-induced Pum2 down-regulation in aNcadKO mice was similar to that seen in control mice, suggesting that N-cadherin is the dominant target of Ca2+-Pum2 signaling in the experimental system presented here. Nonetheless, it will be important to explore other targets of Pum2 in various cellular functions. In addition to the Ca2+-dependent mechanism described in this study, other signaling cascades have been identified as mediators of reactive astrogliosis (3, 4, 30). How these pathways collaborate to regulate the complex and multistep cellular processes in reactive astrogliosis remains to be studied.
N-Cadherin Regulates Reactive Astrogliosis and Neuroprotection.
Although the function of N-cadherin in neurons has been studied extensively, its function in glial cells has remained elusive. We found that N-cadherin up-regulation in astrocytes is essential for reactive astrogliosis and neuroprotection after brain injury. In previous studies, N-cadherin and its family proteins were initially characterized by their function in cell–cell adhesion, but were subsequently found to regulate other cellular functions as well, including differentiation, proliferation, cell polarization, migration, and gene expression (26–28). These multiple functions of cadherin family proteins may underlie reactive astrogliosis.
N-cadherin in cultured astrocytes is known to regulate cell polarity, the essential determinant of the cell morphology and direction of migration (23, 31). In line with this notion, aNcadKO astrocytes demonstrated defects in cell migration and morphological change after injury (Fig. 4A and Figs. S5 and S7). N-cadherin binds the fibroblast growth factor receptor (FGFR) and activates the FGFR-dependent signaling cascade, which can enhance GFAP expression in cultured astrocytes (4, 26, 32). This regulatory mechanism may underlie the defect of GFAP up-regulation in IP3R2KO, 5ppase-expressing, and aNcadKO astrocytes (Figs. 1B and D and 4A). Further work is needed to clarify the molecular mechanism of N-cadherin in reactive astrogliosis.
Pathophysiological Functions of the Astrocytic Ca2+ Signal.
Recently reported results indicate that astrocytic Ca2+ signaling underlies physiological functions, including gliotransmitter release, that may regulate synaptic transmission, synaptic plasticity, and local blood flow in the brain (1). In the present study, we focused on pathophysiological functions of astrocytes and found a significant phenotype of IP3R2KO mice regarding astrogliosis. Pathophysiological Ca2+ signaling in neurons often has a deleterious effect on neurons and results in neuronal cell death (33, 34). On the other hand, our present findings indicate that brain injury-induced Ca2+ signaling in astrocytes drives astrocytic reactivation, which has beneficial effects on neurons around the injury site. In addition, microglial Ca2+ signaling after brain injury mediates the clearance of dying cells (35). Thus, in glial cells in the adult central nervous system, Ca2+ signaling may have a tissue-protective role after brain injury.
In conclusion, the signaling pathway identified in this study highlights the importance of astrocytic intracellular Ca2+ signaling in the beneficial effect of reactive astrocytes under pathophysiological conditions, and provides a platform to further characterize the cellular and molecular basis of reactive astrogliosis.
Materials and Methods
All animal experiments were performed in accordance with the regulations and guidelines of the Institutional Animal Care and Use Committee of the University of Tokyo and with approval from the Institutional Review Board of the Graduate School of Medicine, University of Tokyo. In vivo Ca2+ imaging was performed using a two-photon laser-scanning microscope operating at 800 nm with a laser-scanning system and water-immersion objective lens. SWI was applied to IP3R2KO and aNcadKO mice and their control littermates. For SWI, skulls were exposed, and the somatosensory cortex was impaled with a scalpel (36). After 2–14 d, histochemistry was performed using antibodies for monoclonal N-cadherin, polyclonal GFAP, polyclonal Pum2, or NeuroTrace. Purified adenovirus encoding 5ppase-internal ribosome entry sites (IRES)-GFP, RFP-Pum2, RFP-IRES-Cre, GFP, or RFP (15) was injected into somatosensory cortex of anesthetized mice. The expression vectors for RFP-Pum2, RFP-Pum2∆, and Pum2-activity reporters were introduced into astrocytes prepared from the neocortices of rat embryo or mouse neonates. All data are expressed as mean ± SEM. Significance was determined using the Student t test or a one-way ANOVA with Tukey’s post hoc test. More detailed information is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Y. Kawashima, Y. Horiguchi, C. Fujita, and A. Fukasawa for technical assistance, and following researchers for providing transgenic mice: J. Chen and N. Tada (IP3R2KO); G. Radice and M. Takeichi (flox N-cad KO); and M. Götz, K. Tanaka, O. Komine, and T. Aida (GLAST-ERT2Cre). This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology and the Takeda Science Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.D.F. is a guest editor invited by the Editorial Board.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE39979).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300378110/-/DCSupplemental.
References
- 1.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86(3):1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- 2.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 3.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robel S, Berninger B, Götz M. The stem cell potential of glia: Lessons from reactive gliosis. Nat Rev Neurosci. 2011;12(2):88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- 5.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science. 1990;247(4941):470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 6.Duffy S, MacVicar BA. Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J Neurosci. 1995;15(8):5535–5550. doi: 10.1523/JNEUROSCI.15-08-05535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9(6):816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- 8.Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1(1):31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- 9.Holtzclaw LA, Pandhit S, Bare DJ, Mignery GA, Russell JT. Astrocytes in adult rat brain express type 2 inositol 1,4,5-trisphosphate receptors. Glia. 2002;39(1):69–84. doi: 10.1002/glia.10085. [DOI] [PubMed] [Google Scholar]
- 10.Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J Neurosci. 2008;28(19):4967–4973. doi: 10.1523/JNEUROSCI.5572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin-1–induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate (IP3)-receptor type 2-deficient mice. Circ Res. 2005;96(12):1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- 12.Bush TG, et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23(2):297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 13.Buffo A, et al. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci USA. 2008;105(9):3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanemaru K, Okubo Y, Hirose K, Iino M. Regulation of neurite growth by spontaneous Ca2+ oscillations in astrocytes. J Neurosci. 2007;27(33):8957–8966. doi: 10.1523/JNEUROSCI.2276-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mashimo M, et al. Inositol 1,4,5-trisphosphate signaling maintains the activity of glutamate uptake in Bergmann glia. Eur J Neurosci. 2010;32(10):1668–1677. doi: 10.1111/j.1460-9568.2010.07452.x. [DOI] [PubMed] [Google Scholar]
- 16.Hanisch UK, Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 17.White EK, Moore-Jarrett T, Ruley HE. PUM2, a novel murine puf protein, and its consensus RNA-binding site. RNA. 2001;7(12):1855–1866. [PMC free article] [PubMed] [Google Scholar]
- 18.Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3′ UTR regulation as a way of life. Trends Genet. 2002;18(3):150–157. doi: 10.1016/s0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- 19.Vázquez-Chona F, Geisert EE., Jr N-cadherin at the glial scar in the rat. Brain Res. 1999;838(1-2):45–50. doi: 10.1016/s0006-8993(99)01679-0. [DOI] [PubMed] [Google Scholar]
- 20.Tran MD, Wanner IB, Neary JT. Purinergic receptor signaling regulates N-cadherin expression in primary astrocyte cultures. J Neurochem. 2008;105(1):272–286. doi: 10.1111/j.1471-4159.2008.05214.x. [DOI] [PubMed] [Google Scholar]
- 21.Kostetskii I, et al. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96(3):346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- 22.Mori T, et al. Inducible gene deletion in astroglia and radial glia—a valuable tool for functional and lineage analysis. Glia. 2006;54(1):21–34. doi: 10.1002/glia.20350. [DOI] [PubMed] [Google Scholar]
- 23.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106(4):489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 24.Liang CC, Park AY, Guan JL. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 25.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12(7):829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 26.Cavallaro U, Dejana E. Adhesion molecule signalling: Not always a sticky business. Nat Rev Mol Cell Biol. 2011;12(3):189–197. doi: 10.1038/nrm3068. [DOI] [PubMed] [Google Scholar]
- 27.Harris TJ, Tepass U. Adherens junctions: From molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11(7):502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- 28.Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8(1):11–20. doi: 10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- 29.Fiore R, et al. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009;28(6):697–710. doi: 10.1038/emboj.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maragakis NJ, Rothstein JD. Mechanisms of disease: Astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2(12):679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 31.Dupin I, Camand E, Etienne-Manneville S. Classical cadherins control nucleus and centrosome position and cell polarity. J Cell Biol. 2009;185(5):779–786. doi: 10.1083/jcb.200812034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suyama K, Shapiro I, Guttman M, Hazan RB. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2(4):301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- 33.Narayanan R, Dougherty KJ, Johnston D. Calcium store depletion induces persistent perisomatic increases in the functional density of h channels in hippocampal pyramidal neurons. Neuron. 2010;68(5):921–935. doi: 10.1016/j.neuron.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt S, Ehrlich BE. Unloading intracellular calcium stores reveals regionally specific functions. Neuron. 2010;68(5):806–808. doi: 10.1016/j.neuron.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koizumi S, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446(7139):1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buffo A, et al. Expression pattern of the transcription factor Olig2 in response to brain injuries: Implications for neuronal repair. Proc Natl Acad Sci USA. 2005;102(50):18183–18188. doi: 10.1073/pnas.0506535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.