

Abstract
Pseudorabies virus (PRV) and herpes simplex virus type 1 (HSV-1) are distantly related alphaherpesviruses whose natural hosts are pigs and humans, respectively. Adult infections of natural hosts are mild and rarely lethal. However, both viruses are also able to infect other hosts, often with lethal effects. In this report, we use the paradigm of infection of a common permissive cell type and microarray analysis to determine if these two diverse alphaherpesviruses engage similar or different cellular pathways to obtain a common outcome: productive infection. We compared cellular gene expression in growth-arrested, primary rat embryonic fibroblasts that were mock infected or infected with either purified PRV-Becker or HSV-1(F). Infections by either virus affect the transcription of more than 1,500 cellular genes by threefold or more. Few differences are detected early, and the majority of changes occur during the late stages of infection. Remarkably, the transcripts of about 500 genes are regulated in common, while the rest are regulated in a virus-specific manner. Genes whose expression is affected by infection fall into a diverse group of functional classes and cellular pathways. Furthermore, a comparison of the cellular response to HSV-1 infection of primary human and rat fibroblasts revealed unexpected diversity in the transcript profiles.
Pseudorabies virus (PRV) and herpes simplex virus type 1 (HSV-1) are distantly related members of the subfamily Alphaherpesvirinae. PRV belongs to the Varicellovirus genus, whereas HSV-1 belongs to the Simplexvirus genus of alphaherpesviruses; by sequence comparison, the two viruses are placed at distal branches of an alphaherpesvirus phylogenetic tree (35). By direct DNA-DNA hybridization, only 8% of the sequences of their genomes is homologous (7). Nevertheless, PRV and HSV-1 genes share many positional and functional homologues (29), and their replication strategies are remarkably similar (36). PRV and HSV-1 can infect some cells, such as rat fibroblasts and neurons, by using the same cellular receptors for entry (e.g., the HveC receptor) (40).
The natural hosts for PRV and HSV-1 are pigs (domestic or feral) and humans, respectively. Adult infections of natural hosts are mild and rarely lethal. HSV-1 cannot infect pigs and PRV cannot infect humans (38, 49, 50, 60), but both viruses can infect other hosts, often with lethal effects. For example, HSV-1 and PRV can infect and kill most rodents such as mice, rats, and hamsters (2, 9, 17-19, 22, 34, 41, 59). In these nonnatural hosts, HSV-1 and PRV invade the peripheral nervous system from the site of infection and spread into the central nervous system. This property has led neuroanatomists to exploit PRV as a tracer for mapping networks of synaptically connected neurons in rats and other species (reviewed in reference 16).
Rodent infection models have long been used to study the pathogenesis promoted by both viruses. PRV mutants attenuated in the natural host also exhibit reduced virulence in rodents, a finding supporting the validity of the approach (5, 6, 54). Similar observations have also been documented for HSV-1 infections (reviewed in reference 16). These common virulence phenotypes and pathogenic outcomes in diverse hosts such as pigs, humans, mice, and rats may reflect common molecular interfaces of host and viral gene products during infection. To test this hypothesis, we used microarray technology to determine if two diverse alphaherpesviruses engage similar or different cellular pathways during the productive infection of a common permissive cell type. In particular, we used primary cultures of rat cells that are equally susceptible and permissive to infection by either virus.
Considerable work using HSV-1 infection of cultured human or monkey cells to study the cellular response has been done. HSV-1 infection can modulate the apoptosis response (reviewed in reference 3) and has been found to activate NF-κB pathway via IκB kinase activation (1), the Jun N-terminal kinase/stress-activated protein kinase, and p38 mitogen-activated protein kinase (MAPK) cascades (61). In addition, HSV-1 genes modulate host gene expression by posttranscriptional mechanisms, altering mRNA stability, mRNA transport, and translation (reviewed in reference 46). For PRV, mRNA differential display has been used to evaluate the cellular response to infection (23); however, the cellular pathways affected by PRV infection have not been studied in detail.
In recent years, microarray technology has proven useful for assessing the cellular transcriptional response to viral infections (10, 24, 37, 51, 53). In the present study, we examined the cellular mRNA levels at several times after PRV or HSV-1 infection of primary cultures of rat embryonic fibroblasts (REF). Using Affymetrix microarrays, we identified genes whose expression increased or decreased more than threefold over that of the mock-infected group upon infection. We then annotated these genes, classified them into functional groups, and subsequently assigned a subset of these to cellular pathways. We found that most host transcriptional changes occurred late after infection by both viruses and that both common and virus-specific pathways could be identified. Additionally, comparison of data obtained from HSV-1 infection of REF cells and primary human fibroblasts indicated a diverse host-specific cellular response.
MATERIALS AND METHODS
Viruses and cells.
In order to obtain high virus yields, PRV-Becker (PRV-Be) and HSV-1 strain F [HSV-1(F)] stocks were grown on PK15 and Vero cells, respectively. For virus purification, typically 8 × 107 PK15 or Vero cells were infected at a multiplicity of infection of 10 PFU per cell with PRV-Be or HSV-1(F). When complete cytopathic effect was observed, the majority of HSV-1 virions were found to remain cell associated, whereas PRV virions were present in the extracellular medium. Accordingly, we collected either the cells and extracellular media (for HSV-1) or only the extracellular media (for PRV) at the time of complete cytopathic effect. The virus-containing medium was clarified in a clinical centrifuge (Sorvall H-1000B rotor) at 2,200 rpm for 10 min and layered onto a 6-ml 30% sucrose cushion; the virions were then pelleted at 23,500 rpm for 60 min in a Beckman SW28 rotor. The virion pellet was resuspended in TNE (50 mM Tris-HCl, 150 mM NaCl, 10 mM EDTA), layered onto 20 to 50% dipotassium tartrate step gradients (6 ml of 20% tartrate on top of 2.5 ml of 50% tartrate) formed in phosphate-buffered saline (PBS) and centrifuged for 90 min at 24,100 rpm in an SW41 rotor with slow braking. The virions formed a band at the interface of the 50 and 20% tartrate phases. The virion band was pulled by using a 22-gauge needle, diluted three- to fourfold with PAE (PBS with 2 μg of aprotinin per ml and 1 mM EDTA), and pelleted by centrifugation at 20,500 rpm for 60 min with an SW41 rotor. The virion pellet was layered onto a linear 10-ml 20 to 50% dipotassium tartrate gradient formed in PBS and spun at 24,100 rpm for 16 h in an SW41 rotor. The virions separated into a tight lower band (infectious virions) and a diffuse upper band (presumably L particles). The lower band was pulled, and virions were pelleted as described before. The virion pellet was resuspended in 150 μl of PAE and separated into aliquots for titration and Western blot analysis. No immature forms of PRV or HSV-1 glycoproteins were detected in our virion preparations by Western blotting, a finding attesting to their purity (data not shown).
REF cells were isolated from gestation day 13 rat embryonic tissue, expanded, and frozen using standard protocols by C. Paulus (21). Passage 12 cells were used for all experiments. Cells were grown to confluence in Dulbecco's modified Eagle's medium (supplemented with 10% fetal bovine serum) at 37°C in a 5% CO2 atmosphere in a humidified chamber; the cells were then growth arrested by maintaining in the same (spent) medium for at least 10 days postconfluence.
Growth-arrested cells were infected with purified virions diluted in spent medium to a multiplicity of infection of 5 PFU/cell or with an equivalent volume of PAE buffer diluted in spent medium (for the mock-infected group). After 1 h of adsorption at 37°C, the inocula were replaced with warm spent medium. At the indicated times after the addition of virus, the medium was removed, the cells were lysed with TRIzol (Invitrogen), and the lysates were stored at −80°C. For each time point, three independent infections were carried out.
Cell viability of infected, growth-arrested REF cells was tested by using the Cell Titer Aqueous One solution cell proliferation assay (Promega) and following the manufacturer's protocol.
RNA purification and microarray hybridization.
Total RNA was isolated from TRIzol lysates according to the manufacturer's instructions. cDNA, derived from 20 μg of total RNA, was used as the template for biotin-labeled cRNA synthesis as described in the Affymetrix technical manual. The labeled cRNA samples were hybridized to Affymetrix RGU34A arrays. For each time point, the cRNA samples from three independent infections were hybridized to three different sets of arrays. The hybridized arrays were stained and washed in GeneChip fluidics stations by using the EukGE-WS2 v4 protocol defined in MAS 5.0 (Affymetrix). The microarrays were scanned with a GeneArray scanner system.
Data analysis.
The expression data were subjected to global scaling in MAS 5.0, using a target intensity of 150. The metrics values were imported into GeneSpring (Silicon Genetics) and normalized to the average of the corresponding time-matched mock-infected samples. The data were filtered to exclude probe sets that were absent in all conditions and then to retain only those whose expression varied from the averaged mock value with a maximum t test P value of 0.05 for at least one condition.
It is important that although the RGU34A arrays contain 8,799 probe sets, a few genes are represented by more than one probe set. These arrays also contain some annotative redundancies (a single gene might have several names or GenBank accession numbers corresponding to it). These redundancies were removed when the genes affected by infection were classified into functional classes.
Real-time PCR.
cDNAs corresponding to mock-infected and infected samples were used for real-time PCR analysis; the time point chosen corresponded to that when the gene of interest displayed maximal change of expression in microarrays. Primers were designed by using the PrimerExpress 2.0 software (Applied Biosystems [ABI]). Reactions were set up in a 25-μl volume with 10-fold dilutions of cDNA, PCR primers (100 nM), and SYBR Green PCR master mix (ABI). An ABI PRISM 7900 sequence detection system was used for monitoring the level of SYBR green fluorescence over 40 cycles of PCR. At the end of the cycling phase, a dissociation curve was produced by slow denaturation of the PCR end products to ensure specificity of amplification. For each sample, the quantity of cDNA corresponding to the gene of interest was normalized to the quantity of 18S rRNA. Relative expression levels were calculated by using the 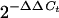 method (33) if the primer pair for the gene of interest passed a validation test described in the ABI user bulletin number 2 (http://docs.appliedbiosystems.com/pebiodocs/04303859.pdf). If the primer pair failed validation, a standard curve method described in the same publication was used.
method (33) if the primer pair for the gene of interest passed a validation test described in the ABI user bulletin number 2 (http://docs.appliedbiosystems.com/pebiodocs/04303859.pdf). If the primer pair failed validation, a standard curve method described in the same publication was used.
RESULTS
Infection of growth-arrested REF cells with PRV and HSV-1.
The standard method of infecting nonconfluent cells in various stages of the cell cycle failed to yield microarray hybridization data that were sufficiently reproducible (data not shown). Infection of growth-arrested REF cells increased the reproducibility of gene expression data among replicate samples. The quiescent cells were easily infected and produced high yields of infectious PRV or HSV-1 virions (Fig. 1A). Moreover, these cells were remarkably robust: infected cells displayed cytopathic effects (rounding up, nuclear distension) by 6 h postinfection (hpi) but maintained membrane integrity and remained attached to the plate beyond 12 hpi. Viability of REF cells infected with PRV and HSV-1 at 8 and 12 hpi was measured by examining the level of cellular metabolic activity, as determined by the bioreduction of a tetrazolium compound into a colored formazan product. As shown in Fig. 1B, the vast majority of cells were still metabolically active and intact 12 h after infection by either virus.
FIG. 1.

PRV and HSV-1 display similar growth kinetics in REF cells and keep these cells metabolically active for up to 12 hpi. (A) Single-step growth kinetics of PRV-Be (⋄) and HSV-1(F) (□) on growth-arrested REF cells. Scale on the y axis is logarithmic (n = 4). (B) Determination by colorimetric assay of the number of metabolically active PRV- and HSV-1-infected REF cells at 8 and 12 hpi, expressed as a percentage of mock-infected cells (n = 3). The values for mean ± standard error of the mean are plotted.
Differential cellular gene expression after PRV and HSV-1 infection.
Growth-arrested REF cells were mock infected or infected with PRV-Be or HSV-1(F) purified virions, and total RNA was isolated at 0, 1, 2, 3, 4, 5, 6, 8, and 12 hpi. The experiment was performed in triplicate (samples A, B, and C) for a total of 81 samples. Two of the chips (PRV-5hA and HSV-2hC) were found to have manufacturing defects after the initial scanning and were removed from subsequent analyses. Data from the remaining 79 samples can be found at http://www.herpes.lanl.gov. Expression data were imported into GeneSpring software, normalized, and filtered as described in Materials and Methods.
The filtered data set, consisting of 4,626 probe sets, was then used to deduce differences in gene expression that were effected by infection. Over the 12-h time course, 2,810 probe sets from the filtered data set did not change significantly (i.e., a more than threefold increase or decrease in mRNA levels).
Probe sets whose levels varied threefold or more were selected for further analysis. The distribution of 689 probe sets that increased mRNA levels by more than threefold after PRV or HSV-1 infection is shown in Fig. 2A. PRV and HSV-1 infection increased the mRNA levels of genes corresponding to 331 and 519 probe sets, respectively. Roughly 23% of the 689 total probe sets increased after infection with either virus. Figure 2B shows the distribution of 1,233 probe sets with reduced levels after infection by PRV or HSV-1. PRV and HSV-1 infection decreased the level of 878 and 695 probe sets, respectively. The expression of genes represented by 340 probe sets was reduced after infection with either virus. After removal of annotation- and probe set-related redundancies (see Materials and Methods), we found that the expression of 498 genes was regulated similarly by PRV and HSV-1, while 521 were affected only by PRV infection and 530 were altered only after HSV-1 infection.
FIG. 2.

Distribution of probe sets with more than threefold increased (A) or decreased (B) levels after PRV or HSV-1 infection. Depicted are Venn diagrams showing the number of probe sets that changed after infection by either or both viruses.
Temporal regulation of host gene expression after PRV and HSV-1 infection.
Distribution of the number of probe sets affected by PRV and HSV-1 infections at different times postinfection can be seen in Table 1. At early time points (0 to 3 hpi), PRV and HSV-1 infections increased the mRNA levels of 46 genes and decreased those of 83 genes by more than threefold (Table 2). However, the majority of transcriptional changes occurred late, between 8 and 12 hpi (Table 1). A similar study in human foreskin fibroblasts infected with HSV-1(F) found that while expression of 70 genes increased and 64 genes decreased more than threefold at 1 hpi, the majority of changes occurred later (450 genes increased and 310 decreased at 7 hpi) (51).
TABLE 1.
Number of probe sets affected by PRV-Be and HSV-1(F) infection
| Time (hpi) | No. of probe sets affected by infection with:
|
|||
|---|---|---|---|---|
| PRVa | HSV-1a | PRVb | HSV-1b | |
| 0 | 8 | 11 | 12 | 22 |
| 1 | 7 | 7 | 23 | 12 |
| 2 | 7 | 6 | 4 | 16 |
| 3 | 6 | 6 | 19 | 6 |
| 4 | 12 | 10 | 1 | 8 |
| 5 | 30 | 22 | 10 | 4 |
| 6 | 33 | 6 | 22 | 7 |
| 8 | 49 | 11 | 19 | 35 |
| 12 | 251 | 489 | 794 | 613 |
Threefold (or more) increase in mRNA levels.
Threefold (or more) decrease in mRNA levels.
TABLE 2.
Genes regulated early (0 to 3 hpi) by PRV and/or HSV-1 infection
| Gene
|
Data for PRV
|
Data for HSV
|
|||
|---|---|---|---|---|---|
| GenBank no. | Description | Maximum change (n-fold) | Time (h) | Maximum change (n-fold) | Time (h) |
| Threefold (or more) increase (0-3 hpi) | |||||
| AA892854 | ESTs, small inducible cytokine B13 precursor | 3.74 | 0 | —a | —a |
| AA892986 | EST | 3.66 | 0 | — | — |
| AI175900 | Ets1 | 3.41 | 0 | — | — |
| AB008521 | Dynein light intermediate chain 53/55 | 3.37 | 0 | — | — |
| L26110 | TGF-β type I | 3.02 | 0 | — | — |
| AA800851 | Carboxylesterase 3 | 4.48 | 1 | — | — |
| X07266 | Gene 33 polypeptide | 3.72, 4.23 | 1.3 | — | — |
| AA799593 | EST | 3.72 | 1 | — | — |
| AA800882 | EST | 3.59 | 1 | — | — |
| J04526 | Hexokinase M | 3.15 | 1 | — | — |
| M60786 | Endothelin receptor | 3.51 | 2 | — | — |
| U63318 | TGF-β type 1 receptor | 3.15 | 2 | — | — |
| J00712 | Casein gamma | 3.03 | 2 | — | — |
| X06769 | c-fos | 4.19 | 3 | — | — |
| U78102 | Krox20 (NGFI-B or Egr2) | 3.27 | 3 | — | — |
| X13722 | LDL receptor | —a | —a | 3.75 | 0 |
| M17527 | GTP-binding protein (G-alpha-i1) | — | — | 3.55 | 0 |
| AI639224 | Ribosomal protein S13 | — | — | 3.39 | 0 |
| U14647 | IL-1β converting enzyme (IL-1βCE) | — | — | 3.13 | 0 |
| AI014135 | Beta carotene 15, 15-dioxygenase | — | — | 3.03 | 0 |
| M31032 | Contiguous repeat polypeptides (CRP) | — | — | 4.24 | 1 |
| U04937 | Na-Ca exchanger isoform NACA1 | — | — | 3.78 | 1 |
| M35602 | Beta fibrinogen | — | — | 3.53 | 1 |
| AI639444 | Sarcosin (sarcomeric muscle protein) | — | — | 3.52 | 1 |
| AA893216 | Kidney-specific membrane protein | — | — | 3.23 | 2 |
| M57682 | Brain calcium channel alpha-1 subunit | — | — | 3.19 | 2 |
| AI058601 | Ankyrin 3 (G), Ank3 | — | — | 3.18 | 2 |
| H31479 | EST | — | — | 4.36 | 3 |
| AA800693 | EST | — | — | 3.42 | 3 |
| Z35138 | Fibroblast growth factor receptor 2b | — | — | 3.19 | 3 |
| AF036537 | Homocysteine-respondent protein HCYP2 | 4.93 | 2 | 3.05 | 1 |
| AA955554 | Splicing factor, arginine/serine-rich 5 | 3.74 | 2 | −4.20 | 1 |
| L26292 | Follicle-stimulating hormone-regulated protein (clone 59) | 5.38 | 3 | −3.16 | 2 |
| Z27513 | Carbamoylphosphate synthase I | −12.08 | 1 | 7.02, −9.43 | 0, 2 |
| AA875097 | EST, fibrinogen alpha/alpha-E chain precursor | −3.16 | 1 | 4.28, 3.07 | 0, 3 |
| AF083331 | Kinesin-like protein KIF1B | −4.38 | 1 | 3.24 | 0 |
| AA875123 | EST | −5.06 | 1 | 3.84 | 2 |
| AA800456 | EST | −4.14, −3.06 | 1, 2 | 3.47 | 3 |
| AA875390 | Thioredoxin-like protein | 8.29 | 0 | 4.48 | 0 |
| H31859 | ESTs | 3.91 | 0 | 8.13 | 0 |
| AI175764 | Stearoyl-coenzyme A desaturase 1 (Scd1) | 3.46, −4.41, −3.11 | 0, 1, 3 | 9.32, −3.82 | 0, 3 |
| AI639318 | Ret proto-oncogene | −3.38, 4.06 | 0, 1 | 6.45 | 1 |
| AA800908 | EST | 3.91 | 1 | 4.26 | 1 |
| AA946542 | Prolactin-like protein D | −10.61, 4.33 | 1, 2 | −4.67, 4.31 | 1, 2 |
| AI639233 | Decorin | 3.1, −4.23 | 2, 3 | −3.75, 3.15 | 0, 2 |
| AF030088 | Activity and neurotransmitter-induced gene (ania-3) | 3.67 | 3 | 3.63 | 3 |
| Threefold (or more) decrease (0-3 hpi) | |||||
| U25282 | Brush border myosin (BBMI) | −5.50 | 0 | — | — |
| U59801 | Integrin alpha-M (Itgam) | −5.02 | 0 | — | — |
| M22926 | Muscarinic acetylcholine receptor M5 | −3.60 | 0 | — | — |
| M82845 | Peptidylglycine alpha-amidating monooxygenase-5 | −3.59, −4.28 | 0, 3 | — | — |
| AI639244 | EST | −3.47, −3.29 | 0, 3 | — | — |
| AI176191 | Vomeronasal neurons pheromone receptor V2R2 | −3.29 | 0 | — | — |
| L06804 | LIM homeodomain protein (LH-2) | −3.24 | 0 | — | — |
| AI639125 | EST | −3.09 | 0 | — | — |
| AI639244 | Neonatal submandibular gland protein B, Smgb | −7.30 | 1 | — | — |
| X06107 | IGF-I | −6.48, −3.02 | 1, 2 | — | — |
| Z83757 | Growth hormone receptor | −4.86 | 1 | — | — |
| AF039832 | Homeobox protein (rPtx2) | −4.49 | 1 | — | — |
| AI228110 | UDP-glucuronosyltransferase 8 (Ugt8) | −4.08 | 1 | — | — |
| AI639446 | EST | −4.00 | 1 | — | — |
| L22655 | Anti-acetylcholine receptor Abb gene, kappa chain | −3.93 | 1 | — | — |
| L39018 | Sodium channel protein 6 (SCP6) | −3.84 | 1 | — | — |
| AF034753 | DC16 mRNA | −3.57 | 1 | — | — |
| AA892240 | ESTs, 2008109A set gene (weakly similar) | −3.32 | 1 | — | — /PICK> |
| AI176460 | EST | −3.14 | 1 | — | — |
| M86758 | Estrogen sulfotransferase | −6.54 | 3 | — | — |
| M31788 | Phosphoglycerate kinase | −6.82 | 3 | — | — |
| M88709 | Cell adhesion-like molecule | −5.48 | 3 | — | — |
| AA926193 | Sulfotransferase family 1C, member 2, Sult1c2 | −5.15 | 3 | — | — |
| D50664 | Oligopeptide transporter | −4.82 | 3 | — | — |
| U32681 | Ebnerin | −3.67 | 3 | — | — |
| AI136175 | Rab38 | −3.65 | 3 | — | — |
| D31734 | Distal-less 3 (Dlx-3) homeobox protein | −3.63 | 3 | — | — |
| U95157 | Ryanodine receptor type II | −3.30 | 3 | — | — |
| AI639466 | EST | −3.23 | 3 | — | — |
| M58287 | Nonspecific lipid transfer protein (nsL-TP) | −3.12 | 3 | — | — |
| J05189 | Neuromedin K receptor | −3.10 | 3 | — | — |
| AI639358 | EST | −3.08 | 3 | — | — |
| AI639263 | EST | −3.05 | 3 | — | — |
| AA800930 | EST | — | — | −6.67 | 0 |
| AA799488 | EST | — | — | −6.16 | 0 |
| L24207 | Testosterone 6-beta-hydroxylase (CYP3A1) | — | — | −4.44 | 0 |
| L07315 | Dipeptidase (dpep1) | — | — | −4.44 | 0 |
| AI639315 | EST | — | — | −4.43 | 0 |
| AA799488 | Secretogranin II | — | — | −4.40 | 0 |
| AI030997 | Toll-like receptor 4 | — | — | −4.36 | 0 |
| AF043642 | Matrin cyclophilin | — | — | −4.04 | 0 |
| AA799538 | ESTs, cold-inducible RNA-binding protein | — | — | −4.03 | 0 |
| AI639301 | EST | — | — | −4.00 | 0 |
| M64867 | Serotonin receptor | — | — | −3.75 | 0 |
| D38629 | Adenomatosis polyposis coli protein | — | — | −3.65 | 0 |
| M36419 | Glutamate receptor (GluR-B) | — | — | −3.64 | 0 |
| AA818240 | Nuclear pore complex protein | — | — | −3.46 | 0 |
| AF055714 | Hypertension-regulated vascular factor-1C-4 | — | — | −3.44 | 0 |
| AA891650 | EST | — | — | −3.30 | 0 |
| AI639155 | EST | — | — | −3.25 | 0 |
| M31837 | Insulin-like growth factor binding protein 3, IGF-BP3 | — | — | −3.18 | 0 |
| D84418 | Chromosomal protein HMG2 | — | — | −3.15 | 0 |
| AF020046 | Integrin alpha OX-62 | — | — | −3.01 | 0 |
| AI639311 | EST | — | — | −7.37 | 1 |
| Z56277 | CLC-5 chloride channel | — | — | −4.20 | 1 |
| AF000901 | p23 (p58/p45) mRNA | — | — | −3.85 | 1 |
| M25892 | Murine IL-4 | −3.22 | 0 | −3.65, −3.33 | 1, 2 |
| AI639468 | EST | — | — | −3.11 | 1 |
| U17837 | Retinoblastoma-interacting zinc finger protein | — | — | −3.06 | 1 |
| X03369 | Beta-tubulin T beta15 | — | — | −7.93 | 2 |
| AF093267 | Homer-1b | — | — | −5.60 | 2 |
| M60811 | LINE 1 repeat element, ORFc II | — | — | −4.57 | 2 |
| M58495 | NAD(P)H: quinone reductase | — | — | −4.12 | 2 |
| AF065433 | Bcl-2-related ovarian death gene BOD-L | — | — | −3.86 | 2 |
| AF087945 | Nitzin mRNA | — | — | −3.80 | 2 |
| AF065431 | Bcl-2-related ovarian death gene BOD | — | — | −3.27 | 2 |
| AA799475 | EST | — | — | −3.18 | 2 |
| L14680 | Bcl-2 | — | — | −3.15 | 2 |
| E00444 | DNA coding for gamma interferon | — | — | −3.12 | 2 |
| AA893242 | Fatty acid coenzyme A ligase, long chain 2 | — | — | −3.10 | 2 |
| U78517 | Cyclic AMP-GEFII | — | — | −3.00 | 2 |
| J00777 | Fat prostatic steroid-binding protein, C3 peptide | — | — | −3.72 | 3 |
| L27059 | Putative phosphodiesterase | — | — | −3.71 | 3 |
| AA860015 | EST | — | — | −3.24 | 3 |
| AI639355 | EST | — | — | −3.12 | 3 |
| AA893870 | EST | — | — | −3.01 | 3 |
| U10303 | Edg-1 orphan receptor | −7.73 | 0 | −3.32 | 1 |
| M14775 | Cytochrome P-450 | −3.98 | 0 | −4.60 | 1 |
| AA892799 | ESTs, 3-phosphoglycerate dehydrogenase | −3.68 | 1 | −3.37 | 1 |
| X51615 | Connexin protein Cx26 | −4.07 | 1 | −4.18 | 1 |
| U52950 | Microtubule-associated protein 1B | −4.88 | 2 | −4.24 | 2 |
| D29646 | ADP-ribosyl cyclase (CD38) | −4.46 | 3 | −4.30 | 0 |
—, no change or absent.
Ab, antibody.
ORF, open reading frame.
Independent confirmation of microarray data.
We selected 15 genes whose expression was identified in the microarray analysis to be significantly affected by virus infection. Two of these genes (Arc and Btg2) were also tested for both PRV and HSV-1 infections, so that a total of 17 real-time PCR analyses were completed. The genes were chosen to represent a wide range of expression patterns and n-fold-change values. Of the 17 microarray data points tested using real-time PCR, 13 (76.5%) verified the array data, while 4 samples were discordant (Table 3). Our findings are comparable to those observed in similar microarray analyses of HSV-1-induced changes in host gene expression in infected primary human fibroblasts (51).
TABLE 3.
Validation of array data by real-time PCR
| Gene for: | Microarray data for:
|
RT-PCR data for:
|
||||
|---|---|---|---|---|---|---|
| PRV
|
HSV
|
PRV
|
HSV
|
|||
| Maximum change (n-fold) | Time (hpi) | Maximum change (n-fold) | Time (hpi) | Change (n-fold) | Change (n-fold) | |
| Amd1a | —a | — | 5.13 | 12 | NDb | −1.3 |
| Arc | 6.41 | 6 | 7.98 | 12 | 31.65 | 4.16 |
| Btg2 | 4.88 | 6 | 4.63 | 12 | 6.06 | −2.35 |
| Casp-3 | −7.88 | 12 | — | — | −1.65 | ND |
| Cox-2 | 11.21 | 8 | 3.41 | 12 | 60.7 | ND |
| Egr-1 | 9.38 | 8 | — | — | 13.8 | ND |
| Gene33 | 41.49 | 8 | — | — | 11.1 | ND |
| HO-1 | 4.06 | 8 | — | — | 2 | ND |
| IL-15 | −3.83 | 12 | — | — | −10.5 | ND |
| Lnk-1 | 4.06 | 8 | — | — | 7.5 | ND |
| MKP-3 | 10.92 | 8 | — | — | 4 | ND |
| Odc-1 | — | — | 14.81 | 12 | ND | −1.8 |
| PRG-1 | 3.18 | 8 | — | — | −1.7 | ND |
| SGK | 5.93 | 8 | — | — | 15.2 | ND |
| TagE4 | 3.25 | 8 | — | — | 3.7 | ND |
—, Not changed or absent.
ND, not done.
Genes regulated by PRV and HSV-1 infection belong to diverse functional classes.
The 1,549 genes whose expression is regulated by PRV and/or HSV-1 infection were classified by hand into 24 functional classes (Table 4). Of the 1,549 genes, 419 were expressed sequence tags (ESTs) and 117 were not easily assigned. The two largest classes of genes were involved in metabolism and signaling, followed by genes involved in cell adhesion, cellular transport (including genes encoding cytoskeletal components), and transcription factors. Several genes involved in stress response, immunity, and apoptosis were also affected by both virus infections. The complete list of functionally classified genes regulated by the two viruses can be found in the supplemental information (available at http://www.molbio.princeton.edu/labs/enquist/SupplInfo.html).
TABLE 4.
Classes of genes regulated by PRV-Be and HSV-1(F) infection
| Functional class | No. of genes | PRV only | HSV only | PRV and HSV |
|---|---|---|---|---|
| Signaling | 146 | 47 | 58 | 41 |
| Metabolism | 138 | 46 | 49 | 43 |
| Cell adhesion | 96 | 11 | 38 | 47 |
| Cellular transport | 85 | 37 | 20 | 28 |
| Transcription factor | 55 | 28 | 12 | 15 |
| Neuronal | 53 | 16 | 22 | 15 |
| Stress response | 49 | 21 | 15 | 13 |
| Channels and transporters | 42 | 7 | 19 | 16 |
| Immunity | 41 | 9 | 17 | 15 |
| Kinase | 38 | 15 | 10 | 13 |
| Protein folding | 36 | 15 | 14 | 7 |
| Nucleus and nuclear matrix | 34 | 13 | 14 | 7 |
| Ribosomal proteins | 25 | 20 | 2 | 3 |
| Oncogenesis | 24 | 10 | 5 | 9 |
| Hormone related | 22 | 5 | 5 | 12 |
| Apoptosis | 20 | 8 | 8 | 4 |
| RNA-binding proteins | 18 | 9 | 3 | 6 |
| Cell cycle related | 17 | 4 | 7 | 6 |
| Cytokine or chemokine | 15 | 6 | 7 | 2 |
| Electron transport | 21 | 5 | 9 | 7 |
| Growth factor | 12 | 0 | 7 | 5 |
| Prostaglandin related | 12 | 2 | 5 | 5 |
| Peroxisome related | 8 | 3 | 3 | 2 |
| Translation | 6 | 2 | 2 | 2 |
| Unclassified | 117 | 24 | 42 | 51 |
| ESTs | 419 | 158 | 137 | 124 |
| Total | 1,549 | 521 | 530 | 498 |
Pathways affected by PRV and HSV-1 infection.
One indication that the observed transcript differences may have biological relevance is if sets of genes in known pathways show coordinated regulation. Accordingly, the functionally classified genes listed in the supplemental information were mapped to known cellular pathways. Table 5 shows a list of genes that were assigned to these cellular pathways, and some of these are discussed in detail below.
TABLE 5.
Pathways of genes regulated upon infection with PRV-Be and HSV-1(F)
| Gene
|
Data for PRV
|
Data for HSV
|
|||
|---|---|---|---|---|---|
| GenBank no. | Description | Maximum change (n-fold) | Time (hpi) | Maximum change (n-fold) | Time (hpi) |
| TGF-β | |||||
| M81225 | Famesyltransferase alpha subunit | −7.96 | 12 | —a | —a |
| U08136 | Golgi protein MG-160 | −6.98 | 12 | — | — |
| AI172476 | TIEG, TGF-β-inducible early growth response | −5.89 | 12 | — | — |
| AF067727 | Smad1 protein | −3.38 | 12 | — | — |
| M31076 | TGF-α | −3.29 | 12 | — | — |
| M24067 | PAI-1, plasminogen activator inhibitor 1 | 6.22 | 8 | — | — |
| AF001417 | Kruppel-like transcription factor Zf9/COPEB | 4.27 | 8 | — | — |
| S98336 | Mullerian inhibiting substance | 4.37 | 12 | — | — |
| AA875033 | Fibulin 5 | —a | —a | −9.26 | 12 |
| U03491 | TGF-β3 | — | — | −8.40 | 12 |
| X52498 | TGF-β1 | — | — | −6.79 | 12 |
| AB017912 | Smad2 protein | — | — | −4.63 | 12 |
| AA849769 | Follistatin-related protein | — | — | −3.94 | 12 |
| L25785 | TSC-22, TGF-β-stimulated clone 22 | −12.66 | 12 | −6.14 | 12 |
| Y12760 | Latent TGF-β binding protein 2 | −10.1 | 12 | −31.25 | 12 |
| AF014503 | p8 mRNA | −6.49 | 12 | −6.99 | 12 |
| L26110 | TGF-β type I receptor | 8.87 | 12 | 6.94 | 12 |
| MAPK | |||||
| U73142 | p38 MAPK | −4.98 | 12 | — | — |
| S81478 | MKP-1/CL 100 | 3.14, −3.58 | 8, 12 | — | — |
| J02722 | HO-1, heme oxygenase 1 | 4.06 | 8 | — | — |
| X13411 | Elk protein | 3.47 | 12 | — | — |
| L04485 | MAPKK | — | — | −5.63 | 12 |
| AI231354 | MAPK9 | — | — | −3.69 | 12 |
| AI007614 | ESTs, MAPKKKK-4 (highly similar) | — | — | −3.39 | 12 |
| U37462 | MAP kinase/ERK kinase 5 (MEK5) | — | — | 3.83 | 12 |
| AF013144 | Dual-specificity phosphatase, cpg21 | — | — | 3.64 | 12 |
| AA963674 | MAPKK2 | 3.21 | 12 | −8.38 | 12 |
| X94185 | Dual-specificity phosphatase (MKP-3) | 10.92, −3.46 | 8, 12 | −3.04 | 12 |
| AA800613 | Zfp36/Tis11 | 5.69, −6.85 | 8, 12 | 4.69 | 12 |
| M64301 | MAPK6 (ERK3) | −6.37 | 12 | −3.83 | 12 |
| JAK/STAT | |||||
| AJ000556 | JAK1 | −6.04 | 12 | — | — |
| U57391 | SH2-B PH containing signal mediator 1 | −5.76 | 12 | — | — |
| X91810 | STAT3 | −5.75 | 12 | — | — |
| X98490 | p32 subunit of replication protein A | −3.24 | 12 | — | — |
| U02320 | Neu differentiation factor | — | — | 5.65 | 12 |
| X91988 | STAT5B | 4.05 | 12 | 5.97 | 12 |
| Insulin-like growth factor | |||||
| U59809 | IGF-II receptor | — | — | −14.51 | 12 |
| M91595 | IGF binding protein 2 | — | — | −5.50 | 12 |
| A09811 | BRL-3A binding protein (IGF-BP) | — | — | −5.12 | 12 |
| M31837 | IGF binding protein 3 | — | — | 8.17 | 12 |
| AA924289 | IGF binding protein | — | — | 3.77 | 12 |
| X06107 | IGF-1 | −6.48, −3.02, 3.58, 3.43 | 1, 2, 8, 12 | −5.69 | 12 |
| AI029920 | IGF binding protein 5 | −9.09 | 12 | −3.06 | 12 |
| X17012 | IGF-II | 4.1 | 12 | 13.08 | 12 |
| S46785 | IGF-BP complex acid-labile subunit | 3.11 | 12 | 3.42 | 12 |
| PI-3K/Akt | |||||
| J05210 | ATP citrate-lyase | −6.96 | 12 | — | — |
| Y15748 | PDK1 | −6.87 | 12 | — | — |
| L13193 | Brain factor 3 (HFH-BF-3) | 3.27 | 8 | — | — |
| D30040 | RAC protein kinase alpha (Akt) | — | — | −6.45 | 12 |
| S55223 | 14-3-3 protein beta | — | — | −5.08 | 12 |
| U01146 | Nuclear orphan receptor HZF-3 | — | — | 6.36 | 12 |
| L13201 | HNF-3/forkhead homologue 1 (HFH-1) | — | — | 5.22 | 12 |
| U50412 | PI3K regulatory subunit, p85 alpha | — | — | 5.08 | 12 |
| M87634 | Brain factor 1 (HFH-BF-1) | — | — | 4.37 | 12 |
| AJ006710 | Phosphatidylinositol 3-kinase (PI3K) | −8.13 | 12 | −5.68 | 12 |
| AA942751 | 14-3-3 protein, theta polypeptide | −7.63 | 12 | −4.24 | 12 |
| AI105076 | Murine thymoma viral (v-akt) oncogene 2 | −4.1 | 12 | −3.06 | 12 |
| Z46614 | Caveolin | −3.55 | 12 | −10.75 | 12 |
| A1180424 | 14-3-3 protein, zeta polypeptide | −3.35 | 12 | −6.49 | 12 |
| L01624 | SGK, serum-glucocorticoid-regulated kinase | 5.93 | 8 | 8.21 | 12/PICK> |
| Apoptosis: Bcl-2 family | |||||
| U49930 | Caspase 3 | −7.88 | 12 | — | — |
| AF025670 | Caspase 6 | −4.71 | 12 | — | — |
| L14680 | Bcl-2 | — | — | −3.15 | 2 |
| AA818072 | BAD, Bcl-2-associated death agonist | — | — | −12.16 | 12 |
| AF065433 | BimL | −3.48 | 5 | −3.86, 5.48 | 2, 12 |
| AF065431 | BimL | −3.09 | 6 | 7.56 | 12 |
| U49729 | rBax alpha | −4.7 | 12 | −5.35 | 12 |
| X94185 | Dual-specificity phosphatase (MKP-3) | 10.92, −3.46 | 8, 12 | −3.04 | 12 |
| Notch signaling | |||||
| L38483 | Jagged-1 (Notch ligand) | −21.13 | 12 | — | — |
| D13417 | Transcriptional repressor HES-1 | −8.15 | 12 | — | — |
| AF009329 | SHARP-1 | −4.84 | 12 | — | — |
| X99267 | Presenilin-2 | −3.25 | 12 | — | — |
| AI101320 | Jagged-2, Jag2 | 4.45 | 12 | 3.15 | 12 |
| Insulin | |||||
| AB017596 | Plasma cell membrane glycoprotein, PC1 | −8.75 | 12 | — | — |
| M81225 | Farnesyltransferase alpha subunit | −7.96 | 12 | — | — |
| X58375 | Insulin receptor (IRS-1) | −3.89 | 12 | — | — |
| U38481 | ROK-alpha | −3.02 | 12 | — | — |
| M64711 | Endothelin-1 | 9.13 | 12 | 4.97 | 12 |
| Endothelial differentiation | |||||
| U10699 | Edg5 | −4.2 | 12 | — | — |
| AA848831 | Edg2 | −3.72 | 8 | — | — |
| AA956930 | Endothelin-converting enzyme 1 | — | — | 4.54 | 12 |
| U93306 | VEGF receptor-2/FLK-1 | — | — | 3.46 | 12 |
| AA850734 | VEGF | — | — | 3.03 | 12 |
| U10303 | Edg-1 orphan receptor | −7.73 | 0 | −3.32 | 1 |
| AA891746 | ESTs, endothelial differentiation factor 1 | −3.37 | 12 | −4.98 | 12 |
| L20913 | VEGF-3 | −3.18 | 12 | 3.95 | 12 |
| M60786 | Endothelin receptor | 3.51, −4.72 | 2, 12 | −4.2 | 12 |
| M64711 | Endothelin-1 | 9.13 | 12 | 4.97 | 12 |
| IL-1 | |||||
| U14647 | IL-1β converting enzyme | — | — | 3.13, −3.92 | 0, 8 |
| U14010 | Soluble IL-1 receptor type I | — | — | −3.25 | 12 |
| U48592 | IL-1 receptor accessory protein | — | — | 7.39 | 12 |
| M98820 | IL-1β propeptide (active form) | — | — | 5.04 | 12 |
| Interferon related | |||||
| AA893384 | ESTs, IFN regulatory factor 3 (IRF-3) | — | — | −6.76 | 12 |
| X61381 | IFN-induced mRNA | — | — | −3.67 | 12 |
| M34253 | IFN regulatory factor 1 (IRF-1) | — | — | 10.68 | 12 |
| AI014163 | IFN-related development regulator 1 | — | — | 6.64 | 12 |
| D87919 | IFN-β | — | — | 3.59 | 12 |
| AA892259 | ESTs, IFN consensus sequence-BP | — | — | 3.5 | 12 |
| E00444 | DNA coding for IFN-γ | −3.8 | 6 | −3.12 | 2 |
| Prostaglandin synthesis | |||||
| U38376 | Cytosolic phospholipase A2 | −3.12 | 12 | — | — |
| U28966 | Prostacyclin receptor | 3.74 | 12 | — | — |
| S87522 | Leukotriene A4 hydrolase | — | — | −8.46 | 12 |
| AF051895 | Lipocortin V | — | — | −4.07 | 12 |
| AI171962 | Annexin 1 (p35) (Lipocortin 1) | — | — | −3.54 | 12 |
| AI170268 | Prostaglandin F receptor | — | — | −3.2 | 12 |
| AI169372 | Arachidonic acid epoxygenase | — | — | 7.32 | 12 |
| AI175764 | Scd1 | 3.46, −4.41, −3.11, −4.9 | 0, 1, 3, 8 | 9.32, −3.82, 4.05, −3.29 | 0, 3, 8, 12 |
| AF036761 | Scd2 | −10.53 | 12 | −7.04 | 12 |
| AI145502 | Prostaglandin F2 receptor-negative regulator | −4 | 12 | −20 | 12 |
| AF000901 | p23 (cytosolic PG-E synthase) | 3.6, −5.66 | 8, 12 | −3.85 | 1 |
| S67722 | Cox-2 | 11.21 | 8 | 3.92 | 12 |
| Heat shock | |||||
| AI104388 | Hsp27 | −9.1 | 12 | — | — |
| M11942 | Hsc70 | −4.97 | 12 | — | — |
| AA108277 | Hsp-E7I | −3.62 | 12 | — | — |
| AI170613 | Hsp10-1 | — | — | −3.60 | 12 |
| Z75029 | Hsp70.2 | — | — | 31.02 | 12 |
| Z27118 | Hsp70 | — | — | 26.34 | 12 |
| X15705 | Hst70 | −3.67 | 12 | 3.68, 7.27 | 8, 12 |
| AA818604 | Hsp 70-1 | −3.07 | 12 | 23.12 | 12 |
| AA875620 | Hsp70-3 | 4 | 12 | 3.54 | 12 |
| M14050 | Hsp70-5 | −18.87 | 12 | −3.91 | 12 |
| AI176546 | ESTs, Hsp90-β | 3.12 | 12 | — | — |
| Oxidative stress | |||||
| AI014169 | Vdup1 | −7.49 | 12 | — | — |
| AI138143 | GST-theta2 | −5.08 | 12 | — | — |
| AI233261 | Glutamate-cysteine ligase, regulatory | −4.45 | 12 | — | — |
| AI136977 | FK506 binding protein (BP) 4 | −3.39 | 12 | — | — |
| Z24721 | Superoxide dismutase 3 | −3.36 | 12 | — | — |
| J02722 | HO-1, heme oxygenase 1 | 4.06 | 8 | — | — |
| S81478 | MKP-1/CL100 | 3.14 | 8 | — | — |
| K01932 | GST-Yc | — | — | −8.37 | 12 |
| AI010083 | Peroxiredoxin 1 | — | — | −3.25 | 12 |
| AI235747 | GST-alpha | — | — | 7.06 | 12 |
| AA926149 | Catalase | — | — | 3.93 | 12 |
| M62642 | Hemopexin | — | — | 3.78 | 12 |
| AF058787 | HO-3, heme oxygenase 3 | — | — | 3.58 | 12 |
| AA875390 | Thioredoxin-like protein | 8.29 | 0 | 4.48, 3.9, −3.13, 4.95 | 0, 5, 6, 12 |
| AA799650 | Peroxiredoxin 3 | −3.6 | 12 | −4.33 | 12 |
| AI228738 | FK506-binding protein (BP) 1 (12 kDa) | −6.76 | 12 | −3.04 | 12 |
| Fibrinolysis | |||||
| M23697 | Tissue plasminogen activator (t-PA) | −9.41 | 12 | — | — |
| M24067 | PAI-1, Plasminogen activator inhibitor 1 | 6.22 | 8 | — | — |
| X71898 | uPAR1, Urinary plasminogen activator receptor | 5.25 | 8 | — | — |
| AF001417 | Kruppel-like transcription factor Zf9/COPEB | 4.27 | 8 | — | — |
| M35602 | Beta fibrinogen | 3.53, −3.41 | 1, 8 | ||
| M81642 | G-protein coupled thrombin receptor | — | — | −5.69 | 12 |
| M26247 | Factor IX | 3.48 | 12 | ||
| X05861 | Gamma fibrinogen | 3.22 | 12 | ||
| AA875097 | EST, fibrinogen alpha | −3.16, 8.7 | 1, 5 | 5.08 | 5 |
| X53565 | TGN38 (upregulates PAI-1 secretion) | −16.95 | 12 | −5.21 | 12 |
—, no change or absent.
TGF-β signaling.
The expression of many genes belonging to the transforming growth factor β (TGF-β) signaling pathway was decreased at 12 hpi by PRV infection. These include farnesyltransferase alpha (an insulin target that interacts with the TGF-β receptor [48, 56]), Golgi protein MG-160 (latent TGF-β complex protein 1 [25, 39]), TIEG, Smad1, and TGF-α. HSV-1 infection also resulted in decreased expression of TGF-β signaling genes, such as TGF-β3, TGF-β1, Smad2, follistatin-related protein (upregulated by TGF-β1 [62]), and fibulin 5 (induced by TGF-β [45]). Moreover, genes such as TSC-22, p8 mRNA (a TGF-β-responsive gene that enhances Smad transcriptional activity [20]), and latent TGF-β-BP2 (an extracellular matrix protein that targets TGF-β action [43]) also displayed reduced expression after both infections. In addition, the Kruppel-like transcription factor Zf9/COPEB (induces uPA expression and activates latent TGF-β1 [30]) and PAI-1 (a TGF-β1-responsive gene that detaches cells from the extracellular matrix by inactivating integrins [14]) were induced at 8 hpi by PRV. PRV and HSV-1 infections also increased the expression of the TGF-β type I receptor gene at 12 hpi.
MAPK signaling genes.
MAPK signaling genes were expressed to various levels following infection by PRV and HSV-1. PRV infection reduced expression of p38 MAPK at 12 hpi and increased the expression of MKP-1 (oxidative stress-inducible protein tyrosine phosphatase) and heme oxygenase 1 (oxidative stress-inducible via MAPKs of Jun N-terminal kinase and p38 pathways) at 8 hpi. PRV infection also reduced the expression of Elk protein (downstream target of p38 MAPK) and MAPKK2 at 12 hpi. HSV-1 infection reduced expression of MAPKK2, MAPKK, MAPK9, and MAPKKKK4 and increased expression of MEK5 and of the dual-specificity phosphatase (cpg21) at 12 hpi. Zn finger protein (Zfp36/Tis11/TTP) expression increased 12 h after PRV infection and after HSV-1 infection at 8 hpi, whereas MKP-3 (which dephosphorylates ERK1/2 [42]) expression increased 8 h after PRV infection and decreased 12 h after infection by either PRV or HSV-1. Infection by either virus reduced the levels of MAPK6 at 12 hpi.
JAK/STAT pathway.
JAK/STAT pathway genes such as JAK1, STAT3, the p32 subunit of replication protein A (which regulates STAT3 transcription [26]) and SH-2 pleckstrin homology containing signaling mediator 1 (JAK2 substrate) were expressed to lower levels 12 h after infection by PRV. HSV-1 infection increased expression of neu differentiation factor (i.e., neuregulin, which activates the JAK/STAT pathway [32]) at 12 hpi. STAT5B expression was also increased by both infections at 12 hpi.
IFN- and IL-1-related pathways.
Interferon (IFN)- and interleukin 1 (IL-1)-related genes were regulated primarily by HSV-1 but not by PRV infection. Expression of IFN regulatory factor 3 and IFN-induced mRNA was reduced, while that of IFN-β, IFN regulatory factor 1, IFN-related developmental regulator 1, and IFN consensus sequence binding protein was induced at 12 hpi by HSV-1 infection. However, HSV-1 and PRV infections decreased expression of the IFN-γ gene at 2 and 6 hpi, respectively. IL-1β-converting enzyme expression increased early and decreased late after HSV-1 infection. Soluble IL-1 receptor type 1 expression was reduced, while that of IL-1 receptor accessory protein and IL-1β propeptide was induced at 12 hpi by HSV-1 infection.
IGF pathway.
The insulin-like growth factor (IGF) pathway was affected by PRV and HSV-1 infections. Infection by either virus results in increased expression of IGF-II and of the acid-labile subunit of the IGF-binding protein (IGF-BP) complex as well as decreased expression of IGF-BP5 at 12 hpi. The IGF-II receptor, IGF-BP3, and the BRL-3A binding protein were expressed to lower levels, whereas IGF-BP2 and IGF-BP were expressed to higher levels 12 h following infection by HSV-1. The expression of IGF-I was reduced early after PRV infection but increased late (8 and 12 hpi). HSV-1 infection caused a reduction in IGF-I expression at 12 hpi.
PI3K/Akt survival pathway.
Expression of genes in the PI3K/Akt survival pathway was reduced by PRV and HSV-1 infection. In both infections, the mRNA levels of phosphatidylinositol 3-kinase (PI3K), 14-3-3-theta and -zeta (protein kinase C regulatory proteins; PI3K target), murine thymoma viral (v-akt) oncogene homolog 2, and caveolin (which induces the PI3K/Akt signaling pathway [47]) were found to be decreased at 12 hpi. In addition, PRV infection decreased expression of ATP citrate-lyase (Akt substrate [8]) and PDK1 (which phosphorylates Akt and is inhibited by binding 14-3-3 [44]), while HSV-1 infection reduced expression of RAC protein kinase alpha (Akt) and 14-3-3-beta at 12 hpi. Transcript levels of SGK (a target of PI3K that phosphorylates forkhead transcription factor FOXO3a [12]) and forkhead transcription factors such as brain factor 3, HZF-3, and HFH-1 increased late after infection by either virus.
Apoptotic pathway.
Genes belonging to the Bcl-2 family of pro- and antiapoptotic proteins were altered upon PRV and HSV-1 infection. Caspase 3 and caspase 6 mRNA levels were decreased after PRV infection, whereas infection with HSV-1 reduced the expression of Bcl-2 (antiapoptotic) and Bad (a Bcl-2-associated death agonist). rBax alpha (proapoptotic) mRNA levels were reduced after infection with either virus. BimL (proapoptotic) levels were reduced by PRV infection and induced by HSV-1 infection, and MKP-3 (which causes apoptosis by degradation of Bcl-2) mRNA levels increased after PRV infection at 8 hpi and decreased at 12 hpi after infection by either PRV or HSV-1.
Notch signaling pathway.
Notch signaling pathway genes such as jagged-1 (notch ligand [58]), transcriptional repressor HES-1 (notch effector), SHARP-1 (a component of the histone deacetylase corepressor that represses notch target genes in the absence of activated Notch [4]), and presenilin 2 (a component of gamma-secretase that cleaves notch and APP [27, 28]) decreased 12 h after infection by PRV. Infection by either virus increased expression of jagged-2 at 12 hpi.
Heat shock and oxidative stress pathways.
Infection by both viruses affected the expression of genes stimulated by heat shock and oxidative stress late after infection. Expression of Hsp27, Hsc70, and Hsp-E71 was reduced, and Hsp90-beta expression was induced by PRV infection. Infection by HSV-1 decreased levels of Hsp10-1 expression but increased those of Hsp70 and Hsp70-2. Infection by both viruses increased Hsp70-3 and decreased Hsp70-5 expression. However, HSV-1 infection induced the expression of Hsp70-1 and Hst70, while PRV infection repressed these genes. Both viruses reduced expression of the oxidative stress-related genes peroxiredoxin 3 and FK506-BP1. Expression of Vdup1 (which is suppressed by H2O2-induced oxidative stress [57]), glutamate-cysteine ligase (a key enzyme in glutathione synthesis), glutathione S-transferase (GST)-theta2 (glutathione synthesis), FK506-BP4, and superoxide dismutase 3 was decreased at 12 hpi, while that of the oxidative stress-inducible genes heme oxygenase 1 and MKP-1 was increased at 8 hpi by PRV infection. Infection by HSV-1 reduced mRNA levels of GST-Yc and peroxiredoxin and induced those of GST-alpha, catalase (marker of oxidative stress), hemopexin (an antioxidant that binds and transports heme to prevent heme-mediated oxidative stress [55]), and heme oxygenase 3.
DISCUSSION
We used a comparative virology approach to guide our analysis of the cellular response to infection. The rationale was that despite their low sequence similarity and distinct natural hosts, PRV and HSV-1 display remarkable similarities in their genome structure, gene conservation, virion structure, and replication cycle. Remarkably, of all the cellular genes significantly affected by infection in our study, only 32% (498 out of 1,549) are common to both viruses (Table 4). Moreover, the diversity of gene functions that are regulated after infection by both viruses was unexpected. For most functional classes, the numbers of PRV-specific and HSV-1-specific genes were about equal. In other cases, a functional class was affected upon infection by one virus but not the other. For example, the cellular transport-cytoskeleton class, transcription factors, stress response genes, ribosomal proteins, oncogenesis, and RNA binding protein are more affected by PRV infection than by HSV-1 infection. Similarly, HSV-1 infection affects expression of genes in the cell adhesion, immunity, channels and transporters, and growth factor classes more than does PRV infection. Such variations in the number of PRV- and HSV-1-specific genes belonging to the different classes are not predictable.
After classifying genes into functional groups, we assigned them to various cellular pathways (Table 5). This classification revealed that the transcription of several genes belonging to the PI3K/Akt signaling and other related pathways changed after infection by PRV and HSV-1. Trophic factors (such as IGF-I) have been shown to play a role in promoting cell survival via the PI3K/c-Akt pathway. Phospholipids generated by PI3K act by multiple mechanisms that cooperate to regulate Akt kinase activity (and also that of SGK and some protein kinase C isoforms). One mechanism involves phospholipid binding to Akt, resulting in its relocalization to the plasma membrane, bringing Akt in proximity to regulatory kinases (such as PDK1) and causing its subsequent activation by phosphorylation (reviewed in reference 15).
Targets of activated Akt include the proapoptotic protein Bad, which is sequestered in its phosphorylated form by the 14-3-3 protein (thereby promoting cell survival) and by forkhead transcription factors that are retained in the cytoplasm when phosphorylated, thereby preventing transcription of forkhead target genes which include Fas ligand and insulin response sequences in the IGF-BP1 promoter (11, 31, 52). The transcription of genes such as IGF-I, IGF-II, IGF-II receptor, IGF-BPs, PI3K, PDK1, Akt, SGK, 14-3-3 protein, forkhead transcription factors (HFH-1, HFH-BF3, HZF-3), and Bcl-2 family genes including Bad were found to change upon infection by PRV and/or HSV-1 (Table 5). At 12 hpi, the expression of some of these genes changed in a proapoptotic direction, while others did so to promote cell survival, suggesting the existence of a fine balance between the two pathways in infected cells at late time points.
IFN- and IL-1-related genes were regulated primarily by HSV-1 infection and not by PRV infection. Our failure to see more IFN- and IL-1-related genes affected by PRV infection is both surprising and intriguing. Future studies might shed light on the significance of these differences in the IFN- and IL-1-stimulated responses elicited by the two virus infections and clarify whether they might underlie differences in pathogenicity in rodent infections by these viruses.
Many oxidative stress-stimulated genes were regulated by PRV and HSV-1 infections. Perhaps this result stems from a cytoprotective response effected by either the host or possibly the virus to counter hypoxic stress induced by infection. The antioxidant glutathione has recently been shown to inhibit the growth of influenza virus (13), a finding that underscores the importance of regulating oxidation during a productive viral infection.
We have assumed that the coordinate regulation of transcripts in a pathway is an indication that the proteins are actually produced and are functional. This assumption must be tempered with the knowledge that both HSV and PRV express proteins that affect mRNA stability, transport, and translation. We have verified that at least two transcripts induced late after infection by PRV and HSV infection from the cox-1 and cox-2 genes are functional, yielding increased amounts of Cox-1 and -2 proteins that are easily detected by Western blot analysis. Moreover, inhibitors of Cox-1 and -2 enzyme activity significantly reduce the yield of both viruses in several cell types (our unpublished results).
The virion host shutoff (Vhs) protein, which enters the cells as part of the virion tegument, stimulates nonspecific degradation of cellular mRNAs early in PRV and HSV-1 infection. In view of this activity, our observations and those of Taddeo et al. (51) that relatively few genes displayed reduced expression early after infection may seem contradictory. However, the modest early host shutoff observed may be linked to the growth-arrested state of the primary fibroblasts used in both studies. It is conceivable that compared to cycling cells, which have been used to demonstrate Vhs activity, growth-arrested cells might be less transcriptionally active and therefore display a less dramatic Vhs-induced decrease in mRNA levels—at least early in infection. At late times postinfection with either PRV or HSV-1, the expression of a large number of genes decreased more than threefold from that of the mock-infected group. As suggested by Taddeo et al., this finding could be the result of delayed Vhs activity or RNA decay in the absence of de novo transcription (51). Another reason might be that the newly synthesized Vhs protein is more abundant than that found immediately after infection. At late times postinfection, we also observed the increased expression of a large number of genes for both PRV and HSV-1. These transcripts increased in the face of Vhs activity and must be either induced to high levels by cellular pathways or are resistant to Vhs action and subsequent degradation.
Our analysis provides an opportunity to compare cellular gene expression induced by HSV-1 infection in rat and human fibroblasts. As mentioned above, changes in cellular gene expression induced by HSV-1 infection in growth-arrested human foreskin fibroblast cells was reported recently (51). We compared this data set with ours to extract a set of 29 genes whose expression is increased more than threefold compared to that of the mock-infected group upon HSV-1 infection of rat and human fibroblasts (Table 6). A subset of these genes also exhibited increased expression (more than threefold) after PRV infection of REF cells and are indicated as such in the same table. Expression of these genes might constitute part of an alphaherpesvirus signature. The same comparative analysis also allowed us to prepare lists of genes that are induced by HSV-1 infection in a cell-type-specific manner (supplemental information is available at http://www.molbio.princeton.edu/labs/enquist/SupplInfo.html). The genes induced only in the human cells by HSV-1 infection may be part of the response that contains spread in the natural host so that infection is mild and rarely lethal. On the other hand, genes that are induced only in the rat cells may reflect the response of a nonnatural host to infection by a human virus and provide insight into the process of the acute and often lethal infection that HSV-1 causes in rodents.
TABLE 6.
Genes that increase more than threefold by HSV-1(F) infection of rat and human fibroblasts
| Gene
|
REF cells
|
HFFb cells
|
|||
|---|---|---|---|---|---|
| GenBank no. | Description | MFIa | Time (hpi) | MFI | Time (hpi) |
| X13722 | LDL receptor | 3.75 | 0 | 7.8 | 3 |
| X03369 | Beta-tubulin | p 4.71 | 5 | 4.1 | 12 |
| Z27118 | Hsp70 | 26.34 | 12 | 6.3 | 12 |
| AI176710 | Nuclear receptor 4A3 | p 14.42 | 12 | 10.1 | 1 |
| X17012 | IGF-II | p 13.08 | 12 | 6 | 12 |
| M34253 | IRF-1 | 10.68 | 12 | 7.2 | 7 |
| L01624 | SGK | p 8.21 | 12 | 4 | 7 |
| AI230256 | Id2 | p 8.13 | 12 | 33.2 | 7 |
| AF065431 | BimL | 7.56 | 12 | 14.2 | 7 |
| AI014163 | IFN-related development regulator 1 | 6.64 | 12 | 16 | 12 |
| X06769 | c-fos | p 6.4 | 12 | 35.3 | 1 |
| AI639441 | eIF-2 | 5.8 | 12 | 4.8 | 12 |
| X07320 | Phosphorylase kinase-gamma | p 5.59 | 12 | 11.7 | 12 |
| AI008131 | Amd1a | 5.13 | 12 | 5.6 | 12 |
| AA800613 | Zfp36/Tis11 | p 4.69 | 12 | 9.4 | 7 |
| M60921 | Btg2 | p 4.63 | 12 | 8 | 7 |
| L23148 | Id1 | 4.56 | 12 | 26.2 | 7 |
| J04197 | Fruct-2,6-bisphosphatase | p 4.48 | 12 | 42.3 | 3 |
| U64705 | eIF4A | 4.44 | 12 | 3.3 | 7 |
| U17013 | Oct1 | 4.3 | 12 | 4.2 | 7 |
| AA955859 | Splicing factor, Arg/Ser-rich 10 | 4.18 | 12 | 5.4 | 12 |
| AF005099 | Neuronal pentraxin receptor | 3.85 | 12 | 4.6 | 7 |
| AF000942 | Id3 | 3.78 | 12 | 3.8 | 7 |
| AA848218 | DNA topoisomerase 1 | 3.58 | 12 | 7.1 | 7 |
| AA875165 | EST, gamma-tubulin complex | 3.54 | 12 | 4.1 | 7 |
| X96437 | PRG1/IEX-1 | p 3.5 | 12 | 4.6 | 7 |
| AA850734 | VEGFc | 3.03 | 12 | 7.7 | 7 |
| U75397 | Krox24 (NGFI-A or Egr1) | p 3.03 | 12 | 3.3 | 12 |
| D26500 | Dynein-like protein 9 | 3.02 | 12 | 75 | 1 |
| D49708 | RNA-BP (transformer 2 like) | 3.01 | 12 | 8.4 | 12 |
MFI, maximum fold increase; “p” in front of the MFI indicates that the gene was also up-regulated in PRV-infected REF cells.
HFF, human foreskin fibroblast.
VEGF, vascular endothelial growth factor.
In summary, this work represents our first comparative analysis of the global cellular transcriptional changes induced in a common cell type by two alphaherpesviruses. We believe that these analyses may lay the foundation for larger comparative studies aimed at assessing the molecular signatures of herpesvirus infection in different cell types and hosts. PRV and HSV-1 are distantly related alphaherpesviruses that engage and modulate a multitude of cellular processes to achieve a productive infection. The primary question we asked was whether the two viruses engage the same or different pathways in a common cell type. Obviously, the outcome of infection in vivo depends on the response of infected cells, the singular event that triggers a cascade leading to spread or containment of the infection. A striking finding of our study was the large increase in cellular transcription changes late in infection. While we often consider the early modulation of host defenses essential to establishment of a productive infection, the cellular events that happen late in infection, when early cellular defenses have been breached, are not well understood. The late changes observed in stress response and heat shock genes might be involved in sending out general alarm signals to the immune system, reporting the imminent danger induced by virus infection. However, the late response is more than a general stress response because more than one-third of these late changes are virus specific. It is now of some interest to determine if these cellular transcripts are functional, if different cell types have similar or different responses, and if the resulting cellular proteins influence the infection in a given animal.
Acknowledgments
We thank C. Paulus for providing REF cells and constant technical guidance; T. delRio for assistance with developing the virion purification protocol; T. Shenk and colleagues for valuable discussions and suggestions; B. Roizman for providing HSV-1(F), advice, and unpublished information; S. Cadambi for assistance with PERL scripts; and C. Paulus, C. Hengartner, and A. Flood for critical reading of the manuscript.
This work was supported by NIH grant number 5P01 CA87661 to L. W. Enquist.
REFERENCES
- 1.Amici, C., G. Belardo, A. Rossi, and M. G. Santoro. 2001. Activation of Iκb kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J. Biol. Chem. 276:28759-28766. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, J. R., and H. J. Field. 1983. The distribution of herpes simplex type 1 antigen in mouse central nervous system after different routes of inoculation. J. Neurol. Sci. 60:181-195. [DOI] [PubMed] [Google Scholar]
- 3.Aubert, M., and J. A. Blaho. 2001. Modulation of apoptosis during herpes simplex virus infection in human cells. Microbes Infect. 3:859-866. [DOI] [PubMed] [Google Scholar]
- 4.Azmi, S., and R. Taneja. 2002. Embryonic expression of mSharp-1/mDEC2, which encodes a basic helix-loop-helix transcription factor. Mech. Dev. 114:181-185. [DOI] [PubMed] [Google Scholar]
- 5.Babic, N., B. Klupp, A. Brack, T. C. Mettenleiter, G. Ugolini, and A. Flamand. 1996. Deletion of glycoprotein gE reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology 219:279-284. [DOI] [PubMed] [Google Scholar]
- 6.Babic, N., T. C. Mettenleiter, A. Flamand, and G. Ugolini. 1993. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J. Virol. 67:4421-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Porat, T., R. A. Veach, and S. Ihara. 1983. Localization of the regions of homology between the genomes of herpes simplex virus, type 1, and pseudorabies virus. Virology 127:194-204. [DOI] [PubMed] [Google Scholar]
- 8.Berwick, D. C., I. Hers, K. J. Heesom, S. K. Moule, and J. M. Tavare. 2002. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 277:33895-33900. [DOI] [PubMed] [Google Scholar]
- 9.Bianchi, A. T., H. W. Moonen-Leusen, F. J. van Milligen, H. F. Savelkoul, R. J. Zwart, and T. G. Kimman. 1998. A mouse model to study immunity against pseudorabies virus infection: significance of CD4+ and CD8+ cells in protective immunity. Vaccine 16:1550-1558. [DOI] [PubMed] [Google Scholar]
- 10.Browne, E. P., B. Wing, D. Coleman, and T. Shenk. 2001. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 75:12319-12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. [DOI] [PubMed] [Google Scholar]
- 12.Brunet, A., J. Park, H. Tran, L. S. Hu, B. A. Hemmings, and M. E. Greenberg. 2001. Protein kinase SGK mediates survival signals by phosphorylating the Forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 21:952-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai, J., Y. Chen, S. Seth, S. Furukawa, R. W. Compans, and D. P. Jones. 2003. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 34:928-936. [DOI] [PubMed] [Google Scholar]
- 14.Czekay, R. P., K. Aertgeerts, S. A. Curriden, and D. J. Loskutoff. 2003. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J. Cell Biol. 160:781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905-2927. [DOI] [PubMed] [Google Scholar]
- 16.Enquist, L. W., P. J. Husak, B. W. Banfield, and G. A. Smith. 1998. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 51:237-347. [DOI] [PubMed] [Google Scholar]
- 17.Field, H. J., and T. J. Hill. 1974. The pathogenesis of pseudorabies in mice following peripheral inoculation. J. Gen. Virol. 23:145-157. [DOI] [PubMed] [Google Scholar]
- 18.Field, H. J., and T. J. Hill. 1975. The pathogenesis of pseudorabies in mice: virus replication at the inoculation site and axonal uptake. J. Gen. Virol. 26:145-148. [DOI] [PubMed] [Google Scholar]
- 19.Fraser, G., and S. P. Ramachandran. 1969. Studies on the virus of Aujeszky's disease. I. Pathogenicity for rats and mice. J. Comp. Pathol. 79:435-444. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Montero, A. C., S. Vasseur, L. E. Giono, E. Canepa, S. Moreno, J. C. Dagorn, and J. L. Iovanna. 2001. Transforming growth factor β1 enhances Smad transcriptional activity through activation of p8 gene expression. Biochem. J. 357:249-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldman, A. 1998. Isolation and culture of fibroblasts, p. 4.1-4.7. In D. L. Spector, R. D. Goldman, and L. A. Leinwand (ed.), Cells: a laboratory manual, vol. 1. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- 22.Hill, T. J., H. J. Field, and W. A. Blyth. 1975. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J. Gen. Virol. 28:341-353. [DOI] [PubMed] [Google Scholar]
- 23.Hsiang, C. Y., T. Y. Ho, C. H. Lin, K. Wu, and T. J. Chang. 1996. Analysis of upregulated cellular genes in pseudorabies virus infection: use of mRNA differential display. J. Virol. Methods 62:11-19. [DOI] [PubMed] [Google Scholar]
- 24.Jones, J. O., and A. M. Arvin. 2003. Microarray analysis of host cell gene transcription in response to varicella-zoster virus infection of human T cells and fibroblasts in vitro and SCIDhu skin xenografts in vivo. J. Virol. 77:1268-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawano, J., T. Nakayama, T. Kotani, H. Matsubayashi, M. T. Yamamoto, and T. Suganuma. 2002. Identification and characterization of an insect homologue of the vertebrate Golgi apparatus protein 1 (MG-160/cysteine-rich fibroblast growth factor receptor/E-selectin ligand-1/latent transforming growth factor-β complex protein-1) with a Golgi-specific monoclonal antibody. Histochem. Cell Biol. 117:381-389. [DOI] [PubMed] [Google Scholar]
- 26.Kim, J., D. Kim, and J. Chung. 2000. Replication protein a 32 kDa subunit (RPA p32) binds the SH2 domain of STAT3 and regulates its transcriptional activity. Cell Biol. Int. 24:467-473. [DOI] [PubMed] [Google Scholar]
- 27.Kimberly, W. T., W. P. Esler, W. Ye, B. L. Ostaszewski, J. Gao, T. Diehl, D. J. Selkoe, and M. S. Wolfe. 2003. Notch and the amyloid precursor protein are cleaved by similar γ-secretase(s). Biochemistry 42:137-144. [DOI] [PubMed] [Google Scholar]
- 28.Kimberly, W. T., M. J. LaVoie, B. L. Ostaszewski, W. Ye, M. S. Wolfe, and D. J. Selkoe. 2003. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 100:6382-6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klupp, B. G., C. J. Hengartner, T. C. Mettenleiter, and L. W. Enquist. 2004. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78:1683-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kojima, S., S. Hayashi, K. Shimokado, Y. Suzuki, J. Shimada, M. P. Crippa, and S. L. Friedman. 2000. Transcriptional activation of urokinase by the Kruppel-like factor Zf9/COPEB activates latent TGF-β1 in vascular endothelial cells. Blood 95:1309-1316. [PubMed] [Google Scholar]
- 31.Kops, G. J., N. D. de Ruiter, A. M. De Vries-Smits, D. R. Powell, J. L. Bos, and B. M. Burgering. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398:630-634. [DOI] [PubMed] [Google Scholar]
- 32.Liu, J., and J. A. Kern. 2002. Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am. J. Respir. Cell Mol. Biol. 27:306-313. [DOI] [PubMed] [Google Scholar]
- 33.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 34.McFerran, J. B., and C. Dow. 1970. Experimental Aujeszky's disease (pseudorabies) in rats. Br. Vet. J. 126:173-179. [DOI] [PubMed] [Google Scholar]
- 35.McGeoch, D. J., A. Dolan, and A. C. Ralph. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J. Virol. 74:10401-10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mettenleiter, T. C. 2000. Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesis—state of the art, June 1999. Vet. Res. 31:99-115. [DOI] [PubMed] [Google Scholar]
- 37.Mossman, K. L., P. F. Macgregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nara, P. L. 1985. Porcine herpesvirus 1, p. 89-113. In R. G. Olsen, S. Krakowka, and J. J. R. Blakeslee (ed.), Comparative pathobiology of viral diseases, vol. 1. CRC Press, Inc., Boca Raton, Fla. [Google Scholar]
- 39.Olofsson, A., U. Hellman, P. Ten Dijke, S. Grimsby, H. Ichijo, A. Moren, K. Miyazono, and C. H. Heldin. 1997. Latent transforming growth factor-β complex in Chinese hamster ovary cells contains the multifunctional cysteine-rich fibroblast growth factor receptor, also termed E-selectin-ligand or MG-160. Biochem J. 324(Pt. 2):427-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richart, S. M., S. A. Simpson, C. Krummenacher, J. C. Whitbeck, L. I. Pizer, G. H. Cohen, R. J. Eisenberg, and C. L. Wilcox. 2003. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 77:3307-3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roizman, B., and A. E. Sears. 1996. Herpes simplex viruses and their replication, p. 1043-1107. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fundamental virology, 3rd ed., vol. 1. Lippincott-Raven Publishers, Philadelphia, Pa. [Google Scholar]
- 42.Rossig, L., C. Hermann, J. Haendeler, B. Assmus, A. M. Zeiher, and S. Dimmeler. 2002. Angiotensin II-induced upregulation of MAP kinase phosphatase-3 mRNA levels mediates endothelial cell apoptosis. Basic Res. Cardiol. 97:1-8. [DOI] [PubMed] [Google Scholar]
- 43.Saharinen, J., M. Hyytiainen, J. Taipale, and J. Keski-Oja. 1999. Latent transforming growth factor-β binding proteins (LTBPs)—structural extracellular matrix proteins for targeting TGF-β action. Cytokine Growth Factor Rev. 10:99-117. [DOI] [PubMed] [Google Scholar]
- 44.Sato, S., N. Fujita, and T. Tsuruo. 2002. Regulation of kinase activity of 3-phosphoinositide-dependent protein kinase-1 by binding to 14-3-3. J. Biol. Chem. 277:39360-39367. [DOI] [PubMed] [Google Scholar]
- 45.Schiemann, W. P., G. C. Blobe, D. E. Kalume, A. Pandey, and H. F. Lodish. 2002. Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion. Fibulin-5 is induced by transforming growth factor-β and affects protein kinase cascades. J. Biol. Chem. 277:27367-27377. [DOI] [PubMed] [Google Scholar]
- 46.Schneider, R. J., and I. Mohr. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130-136. [DOI] [PubMed] [Google Scholar]
- 47.Shack, S., X. T. Wang, G. C. Kokkonen, M. Gorospe, D. L. Longo, and N. J. Holbrook. 2003. Caveolin-induced activation of the phosphatidylinositol 3-kinase/Akt pathway increases arsenite cytotoxicity. Mol. Cell. Biol. 23:2407-2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solomon, C. S., and M. L. Goalstone. 2001. Dominant negative farnesyltransferase α-subunit inhibits insulin mitogenic effects. Biochem. Biophys. Res. Commun. 285:161-166. [DOI] [PubMed] [Google Scholar]
- 49.Subramanian, G., R. A. LeBlanc, R. C. Wardley, and A. O. Fuller. 1995. Defective entry of herpes simplex virus types 1 and 2 into porcine cells and lack of infection in infant pigs indicate species tropism. J. Gen. Virol. 76(Pt. 9):2375-2379. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian, G., D. S. McClain, A. Perez, and A. O. Fuller. 1994. Swine testis cells contain functional heparan sulfate but are defective in entry of herpes simplex virus. J. Virol. 68:5667-5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taddeo, B., A. Esclatine, and B. Roizman. 2002. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. Proc. Natl. Acad. Sci. USA 99:17031-17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang, E. D., G. Nunez, F. G. Barr, and K. L. Guan. 1999. Negative regulation of the Forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274:16741-16746. [DOI] [PubMed] [Google Scholar]
- 53.Thomas, J. T., S. T. Oh, S. S. Terhune, and L. A. Laimins. 2001. Cellular changes induced by low-risk human papillomavirus type 11 in keratinocytes that stably maintain viral episomes. J. Virol. 75:7564-7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tirabassi, R. S., and L. W. Enquist. 1998. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 72:4571-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tolosano, E., and F. Altruda. 2002. Hemopexin: structure, function, and regulation. DNA Cell Biol. 21:297-306. [DOI] [PubMed] [Google Scholar]
- 56.Wang, T., P. D. Danielson, B. Y. Li, P. C. Shah, S. D. Kim, and P. K. Donahoe. 1996. The p21RAS farnesyltransferase alpha subunit in TGF-β and activin signaling. Science 271:1120-1122. [DOI] [PubMed] [Google Scholar]
- 57.Wang, Y., G. W. De Keulenaer, and R. T. Lee. 2002. Vitamin D3-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J. Biol. Chem. 277:26496-26500. [DOI] [PubMed] [Google Scholar]
- 58.Weijzen, S., M. P. Velders, A. G. Elmishad, P. E. Bacon, J. R. Panella, B. J. Nickoloff, L. Miele, and W. M. Kast. 2002. The Notch ligand Jagged-1 is able to induce maturation of monocyte-derived human dendritic cells. J. Immunol. 169:4273-4278. [DOI] [PubMed] [Google Scholar]
- 59.Whitley, R. J., D. W. Kimberlin, and B. Roizman. 1998. Herpes simplex viruses. Clin. Infect. Dis. 26:541-555. [DOI] [PubMed] [Google Scholar]
- 60.Wittmann, G., and H. J. Rziha. 1989. Aujeszky's disease (pseudorabies) in pigs., p. 230-333. In G. Wittmann (ed.), Herpesvirus diseases of cattle, horse and pigs. Kluwer Academic Publishers, Boston, Mass.
- 61.Zachos, G., B. Clements, and J. Conner. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097-5103. [DOI] [PubMed] [Google Scholar]
- 62.Zhang, G., Y. Ohsawa, S. Kametaka, M. Shibata, S. Waguri, and Y. Uchiyama. 2003. Regulation of FLRG expression in rat primary astroglial cells and injured brain tissue by transforming growth factor-β 1 (TGF-β 1). J. Neurosci. Res. 72:33-45. [DOI] [PubMed] [Google Scholar]