

Abstract
We engineered short ORFs and used them to control the expression level of recombinant proteins. These short ORFs, encoding a two-amino acid peptide, were placed upstream of an ORF encoding a protein of interest. Insertion of these upstream ORFs (uORFs) resulted in suppression of protein expression. By varying the base sequence preceding the uORF, we sought to vary the translation initiation rate of the uORF and subsequently control the degree of this suppression. Using this strategy, we generated a library of RNA sequence elements that can specify protein expression over a broad range of levels. By also using multiple uORFs in series and non-AUG start codons, we were able to generate particularly low expression levels, allowing us to achieve expression levels spanning three orders of magnitude. Modeling supported a mechanism where uORFs shunt the flow of ribosomes away from the downstream protein-coding ORF. With a lower translation initiation rate at the uORF, more ribosomes “leak” past the uORF; consequently, more ribosomes are able to reach and translate the downstream ORF. We report expression control by engineering uORFs and translation initiation to be robust, predictable, and reproducible across all cell types tested. We propose control of translation initiation as a primary method of choice for tuning expression in mammalian systems.
Keywords: eukaryotic translation, translation initiation site, Kozak consensus sequence, p21, synthetic biology
Currently there are few systematic approaches to precisely control the translation levels of recombinant proteins in mammalian cells. However, precise expression of proteins could be crucial to investigating physiologically relevant levels or genetically programming cells for a desired application. We sought RNA sequence elements that could be used to control translation initiation. In bacteria, constitutive control of translation has been achieved by varying the sequence and position of the ribosome-binding site, where ribosomes bind and assemble a short distance from the start of an ORF (1). Eukaryotes, in most cases, do not use such sequences to recruit and assemble ribosomes at the start of ORFs (Fig. 1A). Instead, a ribosomal preinitiation complex (43S, composed of a 40S subunit, initiator tRNA, and eukaryotic initiation factors) typically binds at the methylguanosine-capped 5′ end of mRNA and scans in the 3′ direction (Fig. 1B); it scans until it reaches and recognizes a translation initiation site (TIS) comprised of the start codon and neighboring bases (Fig. 1C). Upon recognition of a TIS, the complex pauses to enable release of a phosphate generated by GTP hydrolysis, release of initiation factors, and proper pairing of the tRNA anticodon to the start codon. Subsequently, the 60S ribosomal subunit joins, and translation initiates (2, 3).
Fig. 1.

Schematic of translation and RNA elements. (A) An ORF is defined by a start codon and an in-frame stop codon. The 43S ribosomal preinitiation complex is (B) loaded and then scans for a (C) TIS, consisting of the start codon (AUG) and adjacent bases (Ns). Upon recognition of the TIS sequence, the 60S ribosomal subunit joins. Translation initiates and proceeds until the ribosome reaches the stop codon. (D) Upstream ORFs, also with a start codon and in-frame stop codon, can affect translation of downstream, primary ORFs. 5′m, 5′ methylated cap of mRNA; AAA, poly-A tail.
Kozak is credited with first identifying the TIS as bases −3 to +4 (where the +1 position is the first base of the start codon) (4). In addition, Kozak demonstrated that the consensus motif (A/G)CCAUGG (5) strongly favored translation initiation (6). Subsequently, genetic engineers have often used this sequence to generate high levels of recombinant protein expression. In the human genome, 11% of genes (based on our own analysis) use the consensus (A/G)CCAUGG. With 89% of human genes using TIS sequences that differ from the consensus, this finding suggested to us that the use of different TIS sequences could be a natural strategy for tuning protein expression levels.
Cells can also use short upstream ORFs (uORFs; Fig. 1D) to regulate protein expression. Recently, genome analysis revealed that half of all human genes contain one or more uORFs (7). These uORFs can be as short as three codons (i.e., two codons that encode amino acids plus a stop codon), and their presence was shown to reduce expression from downstream, protein-coding ORFs. To devise a method to control constitutive protein translation levels, we combined two approaches—manipulation of the TIS sequence and insertion of uORFs—and report a strategy that allows unprecedented control of expression over a broad range of levels.
Results
Variation of TIS Sequence.
We attempted to control protein expression by varying the TIS sequence. We varied the three bases (positions −3, −2, and −1) preceding the start codon of an ORF encoding GFP (Fig. 2A, construct 1). We chose not to vary the +4 position, which remained a G, to avoid changes to the amino acid sequence. On our expression vectors, GFP was followed by an internal ribosome entry site (IRES) and a red fluorescent protein (RFP). Because ribosomes are loaded at the IRES and subsequently translate RFP in a manner that should be independent of GFP translation, RFP was used as a reference protein. Because GFP and RFP are expressed from the same transcript, normalization of GFP fluorescence intensity with that of RFP (GFP/RFP) allowed us to minimize reporter noise caused by variations in transcription and mRNA decay. Thus, the GFP/RFP value provided a better indicator for translation than GFP alone. We tested several different TIS sequences (Fig. 2B) and confirmed that the known Kozak consensus sequences with purines at the −3 position (i.e., GCCAUGG, ACCAUGG) produced high levels of translation. Other sequences that deviated from the consensus yielded lower translation levels, although even the weaker TIS sequences still produced relatively high translation levels. The weakest TIS that we evaluated, UUUAUGG, initiated translation at a level half that of the ACCAUGG Kozak consensus. Thus, for the TIS sequences we evaluated, we were not able to specify low expression levels over a sufficiently broad range.
Fig. 2.

Synthetic uORFs specify and tune protein expression levels. (A) Schematic of engineered mRNA elements and fluorescent protein reporter construct. 5′m, 5′ methylated cap of mRNA; AAA, poly-A tail; NNNG, bases preceding the GFP ORF; (NNN)S, non-AUG start codon; NNNu, bases preceding uORF; uORF, upstream ORF with sequence AUGGGUUGA where AUG and UGA are start and stop codons, respectively. All ORFs shown contain a G at the +4 position. (B–F) In PD31 cells, GFP/RFP levels as a measure of translation. (B) Effect of different TIS sequences without use of uORFs (construct 1). The sequence GCCACCAUGG (positions −6 to +4) was used by one construct, represented by GCCACC. One construct, RFP only, contained no GFP gene. (C) Effect of different numbers of uORFs (constructs 1–4). (D) Effect of distance (n) between upstream and GFP ORFs, where n is the number of bases after the uORF stop codon and before the GFP start codon (construct 5). (E) Variation of the three bases preceding the uORF and GFP ORF (construct 6) where GFP uses an AUG start codon. (F) Use of non-AUG start codons (in parentheses) to express GFP, where uORFs and bases preceding uORFs were varied (constructs 7 and 8). (G) GFP expression (not normalized to RFP) in different cell lines. Except for the transiently transfected 293T cells in G, all expression constructs were stably integrated into the genome. Unless noted, a five-base spacer exists between the uORF stop codon and GFP start codon, i.e., when n = 5 in D. Error bars represent SD from three experimental replicates.
Tuning Translation Levels with Synthetic uORFs.
Our next objective was to achieve a comprehensive range of lower expression levels. We evaluated synthetic uORFs for their ability to tune translation. We introduced a two-amino acid uORF with a strong TIS sequence (ACCAUGG) upstream of GFP. The uORF and GFP ORF were separated by five bases (Fig. 2A, construct 2), which led to 85% suppression of GFP translation. We also evaluated constructs with multiple uORFs (Fig. 2A, constructs 3 and 4), and for every additional uORF we achieved a lower level of translation (Fig. 2C). We also sought to tune expression levels by varying the distance between a uORF and the primary GFP ORF (Fig. 2A, construct 5). We found that spacer distances between three and nine bases all produced the same level of translation (Fig. 2D), although we cannot rule out that longer distances could have affected GFP translation.
Next we evaluated variation of TIS sequences and utilization of uORFs concurrently (Fig. 2A, construct 6). By tuning the TIS of both a single uORF and a downstream primary ORF, we were able to achieve a nearly continuous range of translation from 0.05 to 0.6 relative units, where 1.0 was expression from the ACCAUGG TIS without any uORF (Fig. 2E). In general, we found that the stronger the TIS of the uORF, the greater the suppression of the downstream ORF. Furthermore, we showed that inserting uORFs and varying TIS sequences could be used to tune expression of genes with AUG start codons, and also those with non-AUG start codons like ACG [Fig. 2 A (constructs 7 and 8) and F]. By both manipulating TIS sequences and/or uORFs, we were able to specify expression levels over a range of greater than three orders of magnitude (Fig. 2G; Fig. S1A). Furthermore, we found that use of uORFs and TIS RNA elements to tune translation achieved reproducible expression levels across different cell types: mouse pre-B lymphocytes (PD31), mouse plasmacytoma (MPC11), human chronic myelogenous leukemia (K562), human colon cancer (HCT-116), human embryonic kidney (293T), and Chinese hamster ovary (CHO-K1; Fig. 2G; Fig. S1A).
We investigated other sequence-related mechanisms that could have affected expression levels. It was possible that differences in expression could be significantly affected by the sequence of GFP. However, when we expressed blue fluorescent protein (BFP), which has a different sequence than GFP and comes from a different organism, we found that our synthetic leaders tuned translation levels in a largely reproducible manner (Fig. 3A). It was possible that the different sequence elements caused changes in mRNA secondary structure, thus potentially affecting expression levels. However, we found changes in local mRNA folding energies had a minimal correlation with translation levels (Fig. 3B). It was also possible that the different sequence elements could have affected mRNA levels. However, we found that mRNA levels were largely unaffected by the different sequence elements (Fig. 3C). In further support of this, these sequence elements upstream of GFP did little to change the IRES-mediated expression of RFP (which in our constructs should have reflected mRNA abundance; Fig. S1B). Thus, for the TIS and uORF sequences that we evaluated, we deemed that translation levels were tuned in a manner largely independent of these other sequence-related issues.
Fig. 3.

Effect of ORF sequence, mRNA folding energy, and mRNA abundance. Normalized GFP expression plotted against (A) expression of BFP, which has a sequence unrelated to GFP; (B) the local ensemble mRNA folding energy; and (C) mRNA level. GFP expression tuned by varying TIS sequences with uORFs (●) or without uORFs (○). Fluorescent protein expression is reported as fluorescence intensity normalized to RFP fluorescence intensity. Error bars represent SD from three experimental replicates.
Tuning of Expression Abides by a Leaky Scanning Model.
We developed a model to describe the translation suppression of a downstream gene of interest (GOI) ORF by a uORF (Fig. 4; mathematical model described in SI Text). With this model, the uORF acts to shunt ribosomes (8–10) so that fewer ribosomes reach the downstream ORF (which in our experiments was GFP; Fig. 4A; Fig. S2). When each ribosome reaches the TIS of the uORF, it makes a probabilistic decision to initiate or not initiate translation of the uORF. We assumed that the decision to initiate translation is governed by a probability determined by the TIS sequence. If a ribosome initiates at the uORF, then after translating it and reaching its stop codon, it disassembles and eventually detaches from the mRNA. If a ribosome does not initiate at the uORF, it proceeds to the downstream ORF; upon reaching the TIS of the downstream ORF, it again makes a decision to initiate translation (or not). Again the probability of initiation is specified by the TIS sequence. Because there is a continuous procession of scanning ribosomes, the translation level of the downstream ORF is reflected by the product of a ribosomal flux and the probability of initiation. Because translation of the uORF decreases the flux reaching the downstream ORF, the translation level of the downstream ORF is decreased.
Fig. 4.
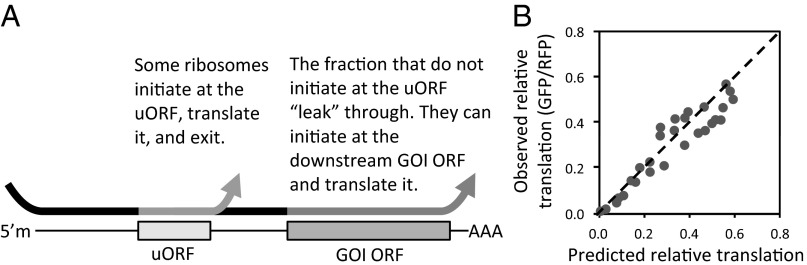
Effect of uORFs on protein translation described by a probabilistic leaky scanning model. (A) Schematic of leaky scanning mechanism. 5′m, 5′ methylated RNA cap; AAA, poly-A tail. In experiments, the GOI was GFP. Arrows indicate path of ribosomes. (B) Model prediction based on a probabilistic leaky scanning mechanism plotted against experimentally observed GFP translation levels. If model and predicted values agreed perfectly, data points would fall on the dotted line. The R2 correlation value was 0.92.
Based on this model (SI Text), we projected expression of the downstream GOI ORF (XG) regulated by a uORF based on the TIS sequences of both ORFs according to the equation XG = (1 − kSu)(SG). The (1 − kSu) term represents the fraction of ribosomes that “leak” past the uORF and reach the downstream ORF. The parameter S represents the relative level of translation initiation associated with a TIS sequence. Previously, “good context” and “poor context” have been used to describe TIS sequences associated with high or low initiation rates, respectively. Thus, in our model, S represents the degree to which a TIS is a good or poor context sequence. Values for S (Su for the uORF and SG for the downstream GOI ORF) were determined from the experimentally observed expression levels (Fig. 2B) for GFP alone without any uORF. The constant k relates the experimentally observed initiation level S to a probability of initiation (SI Text). We then fit our experimental data (Fig. 2 B and E) to the model equation. Because the model values correlated relatively well (R2 = 0.92) to the observed uORF-regulated expression levels of GFP (Fig. 4B), we assert that the uORF-mediated suppression of GFP (the downstream GOI ORF) in our system can be largely modeled by a leaky scanning mechanism.
Tuning Cell-Cycle Activity by Controlling Translation of p21.
To demonstrate that our approach had utility beyond fluorescent reporter proteins, we attempted to tune cell-cycle activity by varying the translation level of ectopic protein p21 (CIP1/WAF1). Endogenous p21 can be transcriptionally activated by p53 in response to DNA damage or oncogenic stress (11, 12). By binding and inhibiting the complexes of cyclin-dependent kinases and cyclins, p21 can induce cell-cycle arrest (12, 13). In biotechnology applications, ectopic expression of p21 has been proposed as a means to slow the proliferation of recombinant CHO cells after reaching a high density and, as a result, increase recombinant protein production titers (14). We fused p21 with GFP and an estrogen receptor domain (GFP-p21-ER), which allowed posttranslational induction of activity upon addition of 4-hydroxytamoxifen (4-OHT). Using a subset of our leader sequences, we then expressed the fusion protein in HCT-116 cells deficient in endogenous p21. After addition of 4-OHT, we observed that we were able to tune GFP-p21-ER expression ranging from levels greater than the wild-type endogenous level induced by DNA damage [10 Gy ionizing radiation (IR)] down to subendogenous levels three orders of magnitude lower (Fig. 5 A and B). In the absence of exogenous DNA damage, though expression of GFP-ER did not significantly affect cell-cycle activity, we observed a shift in cell-cycle activity over a range of GFP-p21-ER expression (Fig. 5C). p21 is perhaps most well known for its ability to induce G1 arrest, inhibiting G1/S progression by binding cyclin-dependent kinase 2 complexed with cyclin E (Cdk2-CycE) and Cdk1/2-CycA (cyclin-dependent kinases 1 or 2 complexed with cyclin A) (12). In line with this notion, we observed predominant G1 arrest at high p21 levels close to or greater than the endogenous p21 levels induced by IR. At levels between 36- and 900-fold less, cell-cycle progression (i.e., cells in S phase) was still detected, but instead of G1 accumulation, we observed an increased accumulation of cells in G2/M. In this case, very low levels of p21 might still slow cell-cycle progression by binding to cyclin-dependent kinase 1 complexed with cyclin B (Cdk1/CycB), a known p21 binding partner that mediates G2/M progression (12); our report notes that the differences in cell-cycle distribution are a function of ectopic p21 dosage. More importantly, though other commonly used expression systems might have subjected cells to supraphysiological levels of p21, our RNA leaders were capable of reproducing ectopic expression levels over a broad, physiologically relevant range.
Fig. 5.

Effect of p21 dosage on cell-cycle distribution. (A) Immunoblot of HCT-116 p21−/− cells expressing GFP-p21-ER through use of different translation initiation leader sequences. Endogenous p21 from WT cells with and without IR. (B) Ectopic expression levels in p21−/− normalized to GAPDH measured by densitometry of immunoblot. Sequence notation is same as that of Fig. 2. (C) Cell-cycle population distribution vs. ectopic expression levels for GFP-ER (Upper) and GFP-p21-ER (Lower) in p21−/− cells. ER, estrogen-receptor domain, which allowed induction of activity upon addition of 4-OHT. Error bars represent SD from three experimental replicates.
Discussion
To develop a systematic approach for engineering protein translation levels, we identified RNA sequence elements that can be inserted immediately upstream of the ORFs of genes of interest. Varying TIS positions −3, −2, and −1 (bases preceding the AUG start codon) produced a range of primarily high expression levels; this alone would not be adequate as a general, all-purpose approach, because cellular proteins could have optimal or physiological activity at low levels. To achieve lower expression levels, we used short, dipeptide-encoding uORFs to decrease translation of the ORF of interest. To tune the degree of suppression by the uORF, we varied the TIS sequences of the uORFs. By varying both TIS sequences and using uORFs, we achieved a full range of protein expression spanning three orders of magnitude. For those who wish to use our RNA leader sequences, we recommend that they start with those that we used to assess the p21 dose–response (Fig. 5); because they all used strong TIS sequences at the downstream ORFs, there should be minimal leakage of ribosomes to internal AUG codons that could initiate translation of truncated or out-of-frame proteins. In addition, to best replicate the levels of expression reported here, users should also use a +4 G in engineered TIS sequences, although the translation control principles described here certainly do not require this. However, if a gene of interest normally uses a weaker TIS sequence (e.g., those with U at the −3 position) to purposefully promote internal initiation, then it may be prudent to use the identical or similar TIS sequence naturally associated with the gene.
We believe that our system abides by the leaky scanning mechanism proposed by Kozak (4, 15, 16), where either the uORF or the downstream ORF is translated. In addition to leaky initiation, Kozak observed that when there was an adequate distance between the uORF and downstream ORF, translation could occur at the uORF and then also reinitiate at the downstream ORF (16); she proposed that the 40S small ribosomal subunit could continue along the mRNA for a limited period after translation ends. Given an ample travel distance, the ribosome would have an opportunity to reload with initiation factors and reinitiate at the second downstream ORF. However, Kozak observed that reinitiation occurred when there were relatively large distances (specifically 41, 76, and 144 bases) between uORF and downstream ORF, but was highly inefficient when ORFs overlapped or were close (i.e., within eight bases) (16). In our system the ORFs were separated by only five bases. With such a short distance, we believe that translation of the downstream ORF is not likely to result from significant amounts of reinitiation. In support of this notion, our leaky initiation model was able to fit our experimental data without accounting for reinitiation. Thus, by using synthetic uORFs as a translational detour, we could precisely control the flux of ribosomes that reach and translate a downstream ORF of interest.
Although RNA secondary structures have been shown to affect translation levels (17–19), our results (Fig. 3B) suggested that our synthetic RNA elements tuned translation in a manner largely independent of secondary structure. In eukaryotes, it is believed that many secondary structures do not greatly affect translation because they are unwound by RNA helicases directly associated with scanning ribosomes (i.e., the 43S preinitiation complex) (2). However, secondary structures have been shown to suppress translation when intentionally placed close (within ∼10 bases) to the 5′ methylated cap (17, 18). On the other hand, when placed after the TIS so that they promote pausing of the ribosomes at the start codon, secondary structures can enhance the level of translation (19). Taking these points into consideration, our RNA elements (placed 510 bases from the 5′ methylated cap with no sequence elements added after the TIS) would not be highly likely to affect translation levels.
Previously, we (20–22) and others (23) have attempted to control constitutive expression levels through use of different promoters that mediate different levels of transcription. For higher eukaryotes, we believe that use of different RNA sequence elements to achieve constitutive control is superior to the use of different promoters. First, because cell type-specific expression is controlled in part through cell type-specific transcription factors, in synthetic applications different promoters, even those considered constitutive, can generate starkly different expression levels in different cell types (20). In contrast, we found that our translation control method performed similarly and reproducibly in a variety of different cell types. This reproducibility is likely because the translational machinery is conserved between cell types. Second, use of RNA leader sequences enables multicistronic gene expression (i.e., genes on a single transcript separated by IRESs) where different genes can be expressed at independently specified levels. In contrast, in eukaryotes independent control of individual genes on a cistron is inherently impossible through use of synthetic promoters, because the expression level of all genes will be dependent on the one transcription level associated with a chosen promoter. Third, when expression vectors are stably integrated into the genome, the transcription level will be determined not only by the vector’s promoter, but also by local enhancer elements and chromatin states in the genome. However, because our system uses RNA elements, the effect of different genomic integration sites will not affect the level of translation per mRNA transcript. It is common practice to express transgenes and an antibiotic resistance gene from the same transcript (where the resistance gene is preceded by an IRES) so that epigenetic silencing can be selected against by addition of antibiotics. However, if one uses a low-strength promoter to mediate low expression of a transgene, then the expression of the resistance gene will also be low and potentially inadequate for selection. By using RNA elements to independently control protein expression of both genes, transgene expression can be dialed to any level, and the resistance gene can be maintained at high levels capable of selecting cells that have not silenced the expression vector. This banal but practical point is one of the reasons all members of our laboratory have migrated to use of translation control elements.
We propose that RNA leaders using engineered uORFs and TISs serve as the primary method of choice for tuning constitutive expression in mammalian cells. The mechanism of control will not interfere with most genetic programming methods, e.g., transcriptional activators, repressors, inducible promoters, inducible protein stability domains, and secondary RNA structures. Genetic circuits can be first “wired” with these other components and then optimized by tuning translation initiation. We anticipate that our sequence elements will perform successfully not only in any mammalian system but any vertebrate system. In principle, our system can be adapted to control translation in other eukaryotes, including plant, yeast, and insect cells.
Materials and Methods
Vector Construction.
To generate pCru5-GFP-IRES-mCherry, mCherry was PCR-amplified from pCru5-mCherry-IRES-Blast (24) and inserted in place of the puromycin resistance gene in the retroviral expression vector pCru5-GFP*-IRES-Puro (25) using standard plasmid construction methods; GFP* was then replaced with monomeric GFP (EGFP A207K, here referred to as GFP), which was derived and PCR-amplified from pEGFP-N1 (Clontech Laboratories Inc.). Sequences to control translation initiation were added to GFP by PCR amplification. GFP was amplified using the forward primer 5′CATCCTCTAGACTGCCGGATCTCGAGTAACTAACTAA(NNN)G(NNN)SGGCGAATTCAGCAAGGGCGAGGAGCTGTTC3′ for leader sequences with variable TIS only or 5′CATCCTCTAGACTGCCGGATCTCGAGTAACTAACTAA(NNN)uATGGGTTGA(T)n-3(NNN)G(NNN)SGGCGAATTCAGCAAGGGCGAGGAGCTGTTC3′ for leader sequences with variable TISs and synthetic uORFs, where the varied nucleotides are indicated by Ns and n and follow the notation of Fig. 2. For leader sequences with multiple uORFs, GFP was amplified with the forward primer 5′CATCCTCTAGACTGCCGGATCTCGAGTAACTAACTAA(ACCATGGGTTGATT)nACCATGGGCGAATTCAGCAAGGGCGAGGAGCTGTTC3′, where n was 1, 2, or 3. All reactions used the reverse primer 5′CGGAATTGGCCGCCCTAGATGCATGCTTATTCGAACTTGTACAGCTCGTCCATGCCGA3′ (Integrated DNA Technologies). The GFP-amplified product was then inserted into the retroviral expression plasmid pCru5-GFP-IRES-mCherry (where GFP was monomeric EGFP) at XhoI and EcoRI restriction sites. In a subset of plasmids, mCherry was replaced with the puromycin resistance gene from pCru5-GFP-IRES-Puro (25). The p21 gene was PCR-amplified from human cDNA. Estrogen receptor domain was PCR-amplified from pBabe-Puro-OmoMyc-ER (a gift from G. Evan, University of California, San Francisco, CA). GFP-ER and GFP-p21-ER fusions (where GFP was monomeric EGFP) were generated by standard molecular biology techniques and then inserted into the retroviral expression vector pCru5-(UUU)GGFP-IRES-Puro at the EcoRI and NsiI sites. Different RNA leader sequences were then substituted into this vector by inserting the EcoRI-NotI fragments from these vectors into the EcoRI-NotI sites on the pCru5-GFP-IRES-mCherry plasmid variants. In a subset of plasmids, monomeric BFP, mTagBFP, was PCR-amplified from pCru5-Puro-CMV-BFP-HRasG12V (21) and inserted in place of GFP in the pCru5-GFP-IRES-mCherry plasmid variants.
Cell Culture.
PD31 cells were cultured in RPMI-1640 medium (Life Technologies) with 10% (vol/vol) FBS (Gemini Bio-Products), 2 mM glutamine, 1 mM sodium pyruvate, and 0.05 mM 2-mercaptoethanol. K562 cells were cultured in RPMI-1640 with 5% FBS and 2 mM glutamine. The 293T human embryonic kidney cells were cultured in DMEM (Life Technologies) with 10% FBS, 4.5 g/mL glucose, and 2 mM glutamine. CHO-K1 cells were cultured in F-12 Kaighn's Modification media (HyClone Laboratories, Inc.) with 10% FBS and 2 mM glutamine. HCT-116 cells were cultured in McCoy's 5A media (Thermo Fisher Scientific) with 10% FBS and 2 mM glutamine. For experiments involving stably transduced GFP-ER and GFP-p21-ER and measurement of mRNA levels by quantitative real-time PCR, puromycin selection was performed. In these cases, PD31 cells and HCT-116 cells were cultured with 1 μg/mL puromycin. GFP-ER and GFP-p21-ER activity was induced by supplementing cells with 0.5 µM 4-OHT 24 h before analysis. All cells were cultured with 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C with 5% CO2.
To evaluate RNA leader sequences by transient transfection, expression vectors were introduced to 293T cells using the calcium phosphate precipitation method (CalPhos Mammalian Transfection Kit; Clontech Laboratories, Inc.). To evaluate the leader sequences in stable cell lines, cells were transduced with retroviral vectors. Retroviral particles were produced by cotransfecting the retroviral expression vectors with either pCL-Eco (ecotropic pseudotyping for PD31 and CHO-K1) or pCL-Ampho (amphotropic pseudotyping for MPC11, K562, HCT-116, and HCT-116 p21−/−) (26) into 293T using the calcium phosphate precipitation method. Virus-containing supernatant was harvested and added with 3 μg/mL (PD31, MPC11, K562) or 8 μg/mL (HCT-116, CHO-K1) Polybrene (hexadimethrine bromide) to cells. CHO-K1 cells were also supplemented with 0.4 μg/mL tunicamycin 18 h before infection. Virus was titered so that transduced cells received a single copy of the vectors.
Quantitative Real-Time PCR.
Total mRNA was extracted from MPC11 cells (QIAzol extraction reagent; Qiagen) and used to generate cDNA (QuantiTect Reverse Transcription Kit; Qiagen). cDNA was measured by quantitative real-time PCR (TaqMan Fast Advanced Master Mix; Life Technologies) to detect GFP (forward primer, 5′-CTGCTGCCCGACAACCA-3′; probe, 5′-FAM-TACCTGAGCACCCAGTCCGCCCT-Iowa Black FQ-3′; reverse primer, 5′-TGTGATCGCGCTTCTCGTT-3′; Integrated DNA Technologies) and GAPDH as a reference (4352339E, VIC-labeled; Life Technologies). All reactions were performed in triplicate on a StepOnePlus real-time PCR machine (Life Technologies).
Analysis of RNA Folding Energy.
The ensemble RNA folding energies of sequences containing the engineered RNA elements were calculated using the method previously used by Hofacker (27), available as part of the Vienna RNA package (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). For the constructs with only variation at the TIS and not including uORFs, the folding energy was calculated for the sequence starting 40 bases before the first base of the start codon of GFP and ending 32 bases afterward. For constructs that also contained uORFs, the analyzed sequences started 40 bases before the first base of the start codon of the first uORF and ended 32 bases after the first base of the GFP start codon.
Immunoblotting.
Immunoblotting was performed using standard methods. Electrophoresis was performed using 10% Mini-PROTEAN TGX gels (Bio-Rad). p21 was detected using a primary anti-p21 rabbit monoclonal antibody (2947; Cell Signaling) and a secondary anti-rabbit IgG conjugated to HRP (7074; Cell Signaling). GAPDH was detected using a rabbit anti-GAPDH antibody conjugated to HRP (3683; Cell Signaling). HRP activity was detected using the WesternBright ECL HRP substrate (K-12045-D50; Advansta).
Flow Cytometry.
An LSRII flow cytometer (BD Biosciences) was used for all analyses. GFP and RFP mCherry levels were quantified by measuring fluorescence intensities by flow cytometry. The rate of translation was gauged by computing the quotient GFP divided by RFP levels. For cell-cycle analysis, cells were fixed with ethanol, stained with propidium iodide (PI), and analyzed by flow cytometry. Flow cytometry data were analyzed with FlowJo software (Tree Star). For cells expressing GFP-ER or GFP-p21-ER, GFP-positive cells were gated using the software before analysis of PI staining.
Supplementary Material
Acknowledgments
We thank Roosmery Yang for analysis of RNA folding energies; Goutam Nistala for help with quantitative real-time PCR; and Bill Noderer, Stacey Shiigi, and Kunal Mehta for helpful discussions. We thank Gerard Evan (University of California, San Francisco) for pBabe-Puro-OmoMycER and Bert Vogelstein (Johns Hopkins University) for the HCT-116 lines. This work was supported by National Science Foundation CAREER Award 0846392 and Ellison Medical Foundation Grant AG-NS-0550-09.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305590110/-/DCSupplemental.
References
- 1.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27(10):946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinnebusch AG. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev. 2011;75(3):434–467. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11(2):113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozak M. Possible role of flanking nucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes. Nucleic Acids Res. 1981;9(20):5233–5252. doi: 10.1093/nar/9.20.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12(2):857–872. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44(2):283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 7.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA. 2009;106(18):7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Racine T, Duncan R. Facilitated leaky scanning and atypical ribosome shunting direct downstream translation initiation on the tricistronic S1 mRNA of avian reovirus. Nucleic Acids Res. 2010;38(20):7260–7272. doi: 10.1093/nar/gkq611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stacey SN, et al. Leaky scanning is the predominant mechanism for translation of human papillomavirus type 16 E7 oncoprotein from E6/E7 bicistronic mRNA. J Virol. 2000;74(16):7284–7297. doi: 10.1128/jvi.74.16.7284-7297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozak M. Emerging links between initiation of translation and human diseases. Mamm Genome. 2002;13(8):401–410. doi: 10.1007/s00335-002-4002-5. [DOI] [PubMed] [Google Scholar]
- 11.el-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 12.Abbas T, Dutta A. p21 in cancer: Intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 14.Fussenegger M, Schlatter S, Dätwyler D, Mazur X, Bailey JE. Controlled proliferation by multigene metabolic engineering enhances the productivity of Chinese hamster ovary cells. Nat Biotechnol. 1998;16(5):468–472. doi: 10.1038/nbt0598-468. [DOI] [PubMed] [Google Scholar]
- 15.Kozak M. Selection of initiation sites by eucaryotic ribosomes: Effect of inserting AUG triplets upstream from the coding sequence for preproinsulin. Nucleic Acids Res. 1984;12(9):3873–3893. doi: 10.1093/nar/12.9.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozak M. Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol. 1987;7(10):3438–3445. doi: 10.1128/mcb.7.10.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol. 1989;9(11):5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA. 2006;12(5):851–861. doi: 10.1261/rna.2309906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozak M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci USA. 1990;87(21):8301–8305. doi: 10.1073/pnas.87.21.8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira JP, Peacock RWS, Lawhorn IEB, Wang CL. Modulating ectopic gene expression levels by using retroviral vectors equipped with synthetic promoters. Syst Synth Biol. 2011;5(3-4):131–138. doi: 10.1007/s11693-011-9089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreira JP, Lawhorn IE, Peacock RW, Wang CL. Quantitative assessment of Ras over-expression via shotgun deployment of vectors utilizing synthetic promoters. Integr Biol (Camb) 2012;4(1):108–114. doi: 10.1039/c1ib00082a. [DOI] [PubMed] [Google Scholar]
- 22.Peacock RW, Lawhorn IE, Ferreira JP, Wang CL. Flow cytometry of v-Abl transformed pre-B cells heterogeneous in ectopic expression levels reveals Ras dose-response. J Immunol Methods. 2012;384(1-2):177–183. doi: 10.1016/j.jim.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 23.Jensen PR, Hammer K. Artificial promoters for metabolic optimization. Biotechnol Bioeng. 1998;58(2-3):191–195. [PubMed] [Google Scholar]
- 24.Peacock RW, Wang CL. A genetic reporter system to gauge cell proliferation rate. Biotechnol Bioeng. 2011;108(9):2003–2010. doi: 10.1002/bit.23163. [DOI] [PubMed] [Google Scholar]
- 25.Wang CL, Harper RA, Wabl M. Genome-wide somatic hypermutation. Proc Natl Acad Sci USA. 2004;101(19):7352–7356. doi: 10.1073/pnas.0402009101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70(8):5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofacker IL. (2004) RNA secondary structure analysis using the Vienna RNA package. Curr Protoc Bioinformatics Chap 12:Unit 12.2, 10.1002/0471250953.bi1202s04. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.