

Abstract
The long terminal repeat (LTR) transcriptional promoters of different human immunodeficiency virus (HIV) type 1 subtypes were inserted into the LAI molecular clone of subtype B. The viral genotypes represent seven subtypes (A, B, C, D, E, F, and G) and one circulating recombinant form (AG). We performed replication studies with this isogenic set of viruses across six cellular environments. This approach revealed strong cellular environment effects, but the method was not sensitive enough to detect small differences in the replication rate between the subtypes. By conducting pairwise competition experiments between the virus variants in six cellular environments, we could demonstrate significant differences in the replication rates of the subtypes and that LTR-determined viral fitness depends both on the host cell type and the activation state of the cell. In addition, we determined the degree of conservation of the transcription factor-binding sites (TFBS) in the different-subtype LTRs by analyzing sequences from the HIV sequence database. The sequence analyses revealed subtype-specific conservation of certain TFBS. The results indicate that one should consider the possibility of subtype-specific viral replication rates in vivo, which are strongly influenced by the host environment. We argue that the multidimensional host environment may have shaped the genetic structures of the subtype LTRs.
The current human immunodeficiency virus type 1 (HIV-1) pandemic is caused by at least nine subtypes (termed A through K) and an increasing number of recombinant forms. The HIV-1 subtypes are not evenly distributed throughout the world but are clustered in certain areas. Genetic differences between subtypes can be up to 40% nucleotide sequence dissimilarity (e.g., in the V3 domain of the envelope protein), and even within subtypes these differences can exceed 20%. There has been a large skew in research focus on subtype B, which is the most prevalent subtype in the western world. Consequently, it is currently not clear whether the HIV-1 subtypes have different biological properties that cause differences in disease progression. Two studies have reported slower disease progression for subtype A (19, 20), and there may be differences in viral load at peak viremia shortly after infection and before the set point is reached in comparisons between subtypes B and C (46) and between subtypes B and E (16). Differences in transmission efficiency between subtypes have also been reported in some studies (8, 45). To what extent these differences are influenced by the interaction between the virus genotype and the host environment is not known. Here, we focused on the transcriptional promoter located in the long terminal repeat (LTR) of the HIV-1 genome, which is one of the most conserved regions among virus isolates and subtypes. This relative conservation indicates that the LTR is subject to strict constraints because it is important for viral gene expression and replication. The LTR is a major determinant for virulence in several animal retroviruses, and even minor changes or rearrangements within the transcription factor-binding sites (TFBS) can have a significant impact on cell tropism and pathogenicity (10, 33). For HIV-1 and its subtypes, it is largely unknown to what degree the promoter contributes to replication, infectivity, virulence, and viral fitness. We aimed to identify interactions between six cellular environments and nine viral genotypes with an isogenic set of HIV-1 molecular clones containing a subtype-specific LTR.
The LTR of HIV-1 can be subdivided into the U3, R, and U5 regions, the first two of which are essential for transcription. Upon transcription, the R region folds the TAR hairpin structure that interacts with the viral Tat protein to fully activate transcription. The U3 region contains the TATA box and several upstream TFBS that control transcription (Fig. 1). Most HIV-1 isolates have three SP1 sites and at least two NF-κB sites, although the upstream NF-κB site is changed into a GABP site in all subtype E isolates (51). There is considerable variation in the presence of other binding sites, such as AP1, NFAT, and USF, and some of these differences are subtype specific (18, 39). The TFBS exhibit distinct properties; SP1, for example, is required both for basal and induced transcription in the presence of the Tat-TAR complex (5, 6, 52). The downstream NF-κB site can make up for the absence of SP1 sites, depending on the T-cell line in which virus replication is tested (47). There are other examples where the lack of a binding site or the presence of a low-affinity site is compensated for by other regions in the LTR (17, 28, 42). This redundancy in transcription activation pathways and the presence of many different TFBS in HIV-1 may suggest a viral strategy to be productive across several cellular environments with large differences in the pool of transcription factors. There are more than 2,000 known transcription factors encoded by the human genome (23, 44). Differences in the presence or activation state of these transcription factors are due to the cell developmental program. This transcription factor pool can also change through exogenous influences (cytokines or coinfections) that trigger specific signaling pathways in the cell, which result in the activation of a new set of transcription factors (9). The presence of distinct TFBS in the HIV-1 subtypes and the temporal and/or spatial variation of transcription factors in the cellular environment may suggest that the phenotype or fitness of the subtypes depends on the environment. Recently, small differences in LTR transcriptional activity between subtypes were demonstrated (18, 36, 38, 44). Whether these differences in LTR activity result in subtype-specific differences in replication rate has not been addressed systematically thus far, and to what extent the cellular environment contributes to the viral replication rate is also unclear.
FIG. 1.

The HIV-1 LTR promoter in different subtypes. (A) Proviral genome of HIV-1 (not to scale). (B) A detailed picture of the subtype-specific TFBS in the U3 domain of the LTR promoter. Molecular clones were constructed through exchange of the U3 and R regions (nucleotides −147 to +63) of the subtype B molecular clone LAI with eight other subtype-specific promoter sequences. The U3 sequence changes do not affect the upstream Nef open reading frame. Because the variant sequences were introduced into the 3′ LTR of subtype B, the R element encoding TAR is inherited from the 5′ LTR. The asterisks beneath each TFBS indicate the predicted fit obtained by MatInspector, as follows: ***, good; **, average; *, poor. —, no prediction.
We inserted nine distinct subtype LTRs into the HIV-1 LAI molecular clone of subtype B. The viral genotypes represent seven subtypes (A, B, C, D, E, F, and G) and one circulating recombinant form (CRF02-AG). This approach allowed us to compare an isogenic set of virus variants that differ only in the viral promoter. We measured the replication rate of each subtype across six cellular environments, which revealed significant environmental effects. However, this approach was not sensitive enough to detect small differences in the replication rate between subtypes. By conducting pairwise competition experiments between all virus variants in six cellular environments, we were able to estimate the replication rate of every subtype in each environment. These results reveal significant differences in replication rate between the subtypes and show that the LTR-determined viral fitness depends on both the host cell type and the activation state. We also show that a few mutational differences are sufficient to cause a significant change in viral fitness and the way in which the virus reacts to changes in the environment.
MATERIALS AND METHODS
Constructs.
Molecular HIV-1 clones with a subtype-specific LTR were derived from the eight pBlue3′LTR constructs (A, C1, C2, D, E, F, G, and AG) described previously (18). Additional subtype C LTR clones from Ethiopia and subtype E LTR clones from Helsinki, Finland, will be described elsewhere. To obtain the molecular clones, a 1.7-kb XhoI-BglI fragment of each subtype was inserted into the molecular clone pLAI of subtype B. All molecular clones therefore had a pLAI backbone and carried a subtype-specific LTR from position −147 to + 63, stretching from the noncoding part of U3 to the R region, including the complete TAR hairpin (Fig. 1). The original pLAI molecular clone was used as the subtype B prototype (40). The subtype-specific LTR fragment was inserted into the 3′ LTR of pLAI. During reverse transcription, the U3 sequences will be inherited in both LTRs of the viral progeny. The R region (including the TAR element), however, is inherited from the 5′ LTR sequence. Thus, all nine viruses contain a subtype-specific LTR fragment from position −147 to −1 and a subtype B TAR element after the first round of replication.
Cell lines.
Human T-lymphocyte cell lines SupT1 and MT2 (49) were cultured in RPMI 1640 (Gibco BRL) supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 U/ml). The cervical carcinoma cell line C33A (ATCC HTB31) (3) was cultured in Dulbecco's modified Eagle's medium (Gibco BRL) with the same supplements. All cell lines were kept at 37°C with 5% CO2.
Transfection, virus replication, enzyme-linked immunosorbent assay, and TCID50 determination.
C33A cells were calcium phosphate transfected with 5 μg of plasmid DNA to produce virus stocks as described previously (12). Virus stock concentrations were determined by measuring CA-p24 by an enzyme-linked immunosorbent assay as described previously (18). For replication curves, SupT1 and MT2 cells (1.25 × 106/5 ml) were infected with virus stock (20 ng of CA-p24), and virus replication was monitored by CA-p24 production. Some cultures were supplemented with tumor necrosis factor alpha (TNF-α) (50 ng/ml) or phytohemagglutinin (PHA) (2.5 μg/ml). These factors were added to the relevant cultures every 3 days. We also measured the 50% tissue culture infectious dose (TCID50) of the input virus on SupT1 cells with or without TNF-α (2).
Competition experiments.
Competition experiments were performed at least twice from independent plasmid isolations and virus stocks to determine the ranking of the nine virus variants and their relative fitnesses. Cells (0.75 × 106) were infected with virus stocks of two different genotypes (10 ng of CA-p24 each). Infections were monitored by microscopic inspection. The peak of infection was reached between 6 and 15 days, depending on the environment, as judged by the presence of syncytia. At peak infection a small sample of cell-free supernatant (1 μl containing approximately 1 ng of CA-p24) was passaged onto fresh cells to continue the competition. Cells were harvested at two to five time points, total cellular DNA was isolated, and the frequencies of the two viruses were determined by sequencing (see below). Competition experiments were performed under six different conditions, which are referred to as cellular environments throughout this paper. Two environments are the T-cell lines SupT1 and MT2, which were cultured as described above, and the other four environments consist of one of these cell lines supplemented with either TNF-α or PHA.
Relative fitness.
For each pairwise competition experiment we computed the relative pairwise fitness Wij by comparing the initial and final viral genotype ratios and viral expansion, without having to specify the underlying growth process (27, 32), as follows:
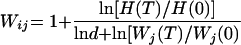 |
in which H(T) and H(0) are the genotype ratios at the end and at the start of the competition, respectively; d is the dilution factor or represents viral expansion; and Wj(T) and Wj(0) are the frequencies of virus j at the end and start of the competition, respectively.
Next, we introduce the relative fitness wi of each subtype within a certain environment, which is scaled to unity, so that 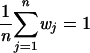 . Substituting wj = wiWji, we can estimate wi from all competitions involving subtype i (note that this method does not guarantee the scaling to unity; this could be corrected by a scaling factor, but in practice the error is very small):
. Substituting wj = wiWji, we can estimate wi from all competitions involving subtype i (note that this method does not guarantee the scaling to unity; this could be corrected by a scaling factor, but in practice the error is very small): 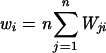 . Finally, we estimate the relative fitness Wi [Wij] for the individual competition experiments: Wi[Wij] = wjWij. From the competition experiments we can thus calculate n × (n − 1) × 2 (replicates) = 144 Wis per environment, which are tested by analysis of variance for significant differences in fitness.
. Finally, we estimate the relative fitness Wi [Wij] for the individual competition experiments: Wi[Wij] = wjWij. From the competition experiments we can thus calculate n × (n − 1) × 2 (replicates) = 144 Wis per environment, which are tested by analysis of variance for significant differences in fitness.
DNA isolation and sequencing.
Total cell DNA was isolated from approximately 0.25 × 106 cells. Cells were lysed in 500 μl of cell lysis buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 0.5% Tween 20 and 5 ng of proteinase K per ml) for 1 h at 56°C and 10 min at 95°C. Proviral LTR sequences were PCR amplified by using the sense primer T7-5′LAIxba (5′-TAA TAC GAC TCA CTA TAG GGT GGA AGG GCT AAT TCA CTC CC-3′) and the antisense primer Sp6-Lys21 (5′-ATT TAG GTG ACA CTA TAG GTT CAG GGA CAA GCC CGC GGT-3′). Population sequencing of proviral LTR DNA was performed with a T7 DYEnamic Direct cycle sequencing kit (Amersham) on an ABI 377 sequencer (Applied Biosystems).
TFBS predictions and sequence variation.
The web-based program MatInspector (43) was used to predict the presence of TFBS. We used the Kimura two-parameter model in the MEGA2 version 2.1 software package (24) to calculate sequence variation within the region stretching from nucleotide −147 to +63 in the LTR and within the individual TFBS.
RESULTS
Genetic structure of the LTR and relationships among the HIV-1 subtypes.
We constructed an isogenic set of nine viruses, representing seven subtypes (A, B, C [two strains], D, E, F, and G) and one recombinant form (AG). The subtype-specific U3-R region stretching from nucleotide −147 to +63 was inserted into the 3′ LTR of the molecular clone pLAI from subtype B. The U3 region will be inherited in both LTRs of the viral progeny after a single round of replication. The R region (including the TAR element) is inherited from the 5′ LTR sequence (22); therefore, all viruses contain a subtype B TAR element. Figure 1A depicts the proviral genome of HIV-1, and Fig. 1B shows the positions of known motifs in the exchanged part of the transcriptional promoter. Indicated are the TFBS for each HIV-1 subtype that was used in this study and the predicted fit of these binding sites as determined by the web-based program MatInspector (43). This analysis reveals some small differences from the TFBS previously proposed by Jeeninga et al. (18). For instance, we now predict at least one AP1 site for all viruses, and a single ETS-1 site is present in most variants, except for subtype E.
We determined how well the selected sequences represent the subtypes that have been submitted to the HIV sequence database. For this, we compared the TFBS and their predicted fit with entries from the database. Table 1 lists the most prevalent TFBS in each subtype across a set of LTR sequences, with the predicted fit calculated by MatInspector and the amount of sequence diversity in each binding site within a subtype. Little sequence variation and a good fit indicate strong conservation of a particular TFBS within a certain HIV-1 subtype, suggesting that there may have been positive selection for an optimal binding site for a particular transcription factor. Table 1 also shows that the presence of some well-known TFBS is less obvious than generally assumed. For example, the downstream SP1-I site has a poor MatInspector fit for most subtypes, and the amount of sequence variation is comparable to that of the large LTR segment. In contrast, the NF-κB sites in all subtypes and the GABP site in subtype E have good predictions and low sequence variability. Thus, some TFBS are highly conserved and might have a more important role than others, and some of these effects are subtype specific. Comparing the MatInspector fit in Table 1 with Fig. 1 indicates that the selected LTRs are good representatives of the subtypes.
TABLE 1.
Sequence diversity and presence of several TFBS differ among the HIV-1 subtypes
| Subtype (no. of se- quences) | Fita and sequence diversity of TFBS:
|
Sequence diversity (mean ± SE) of LTR segment from position −147 to +63 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ETS1 | API-III | API-II | NF-κB-III | NF-κB-II | NF-κB-I | SP1-III | SP1-II | SP1-I | ||
| A (10) | ***, 0.067b | ***, 0.020 | ***, 0.061 | — | ***, 0.050 | ***, 0 | *, 0.074 | ***, 0.075 | *, 0.033 | 0.054 ± 0.011 |
| B (33) | ***, 0.067 | ***, 0.100 | — | — | ***, 0.007 | ***, 0.025 | **, 0.083 | ***, 0.048 | *, 0.059 | 0.054 ± 0.008 |
| C (21) | ***, 0.130 | ***, 0.020 | — | **/***, 0.021 | ***, 0 | ***, 0.053 | *, 0.093 | ***, 0.080 | *, 0.139 | 0.054 ± 0.009 |
| D (11) | ***, 0.069 | ***, 0.093 | — | — | ***, 0 | ***, 0.020 | **, 0.164 | ***, 0.148 | *, 0.031 | 0.071 ± 0.009 |
| E (15) | *, 0.110 | ***, 0.041 | — | — | ***, (GABP), 0.012 | ***, 0 | *, 0 | ***, 0.046 | *, 0.029 | 0.029 ± 0.006 |
| F (9) | ***, 0.060 | ***, 0.124 | ***, 0.038 | — | ***, 0 | ***, 0.024 | **, 0.342 | ***, 0.165 | *, 0.090 | 0.076 ± 0.015 |
| G (9) | ***, 0.080 | ***, 0.022 | ***, 0.087 | — | ***, 0.025 | ***, 0 | **, 0.268 | *, 0.337 | *, 0.095 | 0.086 ± 0.013 |
| AG (11) | ***, 0.138 | ***, 0.039 | ***, 0.022 | — | ***, 0.020 | ***, 0 | *, 0.097 | **, 0.112 | *, 0.049 | 0.068 ± 0.012 |
Fit determined by MatInspector. ***, good; **, average; *, poor; —, no prediction.
Sequence diversity of a particular TFBS.
Replication experiments in six different cellular environments.
Recent studies have reported small LTR-directed differences in transcriptional activity of the HIV-1 subtypes (18, 36, 38). However, the effect on viral replication was not accurately assessed. We performed at least two replication assays with the SupT1 T-cell line by measuring CA-p24 production over time. Figure 2A shows a representative replication curve for each subtype, which reveals small differences in replication rate between the subtypes in this particular cell type. We proceeded by performing replication experiments in five further cellular environments, i.e., a second T-cell line (MT2) and both T-cell lines supplemented with TNF-α or PHA. We assume that each cellular environment represents a different nuclear pool of transcription factors. For instance, SupT1 cells contain the NF-κB transcription factors p50 and p65 in an inactive form in the cytoplasm, and TNF-α triggers their transport to the nucleus (44, 51). PHA is a lectin that binds nonselectively to glycosylated receptors, thereby activating several signaling cascades that affect the nuclear pool of transcription factors.
FIG. 2.

The subtype LTR directs differential viral replication. Virus replication was monitored by measuring CA-p24 production. (A) Replication curves of the nine virus variants in three different SupT1 cellular environments. (B) Replication in three different MT2 cellular environments.
Figure 2A demonstrates that such a change in the cellular environment can have a strong effect on the replication rate of HIV-1. Addition of TNF-α to the SupT1 T-cell line has a positive effect on the replication rates of all subtypes. The peak of infection is reached approximately 2 days earlier, and this result was confirmed by the observation of syncytia. In contrast, addition of PHA to the SupT1 T-cell line has a negative effect on the replication rates of all subtypes; the peak of infection is delayed by approximately 4 days (results are summarized in Table 2). We confirmed these results in a TCID50 assay, in which a much lower viral input (approximately 10-fold lower) was required to initiate a productive infection in TNF-α-treated SupT1 cells compared to the untreated control cells (data not shown).
TABLE 2.
Manipulation of the cellular environment affects HIV-1 replication
| Environment | Peak of infection (days)a | P value |
|---|---|---|
| SupT1 | After 9 | |
| SupT1 + TNF-α | −2 | <0.0001 |
| SupT1 + PHA | +4-6 | <0.001 |
| MT2 | After 6 | <0.0001 |
| MT2 + TNF-α | 0 | NSb |
| MT2 + PHA | +1 | <0.001 |
−, earlier peak; +, delayed peak; 0, no change in peak.
NS, not significant.
The MT2 T-cell line had a strong positive effect on the replication rates of all subtypes compared to the SupT1 T-cell line, and the peak of infection was reached 3 days earlier (Fig. 2B) (results are summarized in Table 2). TNF-α did not significantly affect the rate of virus replication in MT2 cells. PHA again had a negative influence on the replication rate and delayed the peak of infection by approximately 1 day. Although the replication experiments point towards differences in the growth rates of the different subtypes, the variation in growth rate in the cell cultures was too large for accurate quantification. To reveal differences in relative fitness, we performed pairwise competition experiments between subtypes. Although conditions between competition experiments may vary slightly, within each experiment both viral subtypes face exactly the same conditions.
Differences in replication rate among HIV-1 subtypes.
The SupT1 T-cell line was infected with equal amounts of two viruses, and the frequencies of the viral genotypes were assessed at two to five time points by population sequencing of the proviral LTR. Two representative competitions, between subtypes A and B and between subtypes A and E, are shown in Fig. 3. The ranking and relative fitnesses of the nine viral genotypes were determined by performing 36 pairwise competitions in SupT1 T-cells (Table 3). We performed each competition at least twice with independent virus stocks and obtained the same result. Subtype E had the strongest competitive ability and always out-competed all other viruses. Variant C2 was the second best and was followed by A, C1, G, D, AG, B, and F. This dominance in ranking is very robust and absolute; i.e., the higher-ranked subtype always wins the pairwise competition. Furthermore, the difference in relative fitness between two subtypes is a good indicator of the competition rate; i.e., the larger the fitness difference between two genotypes, the faster that outgrowth of the fittest variant is observed.
FIG. 3.

Virus competition. The percentage of each virus in the competition was determined at five time points. SupT1 cells were infected with equal amounts of subtypes A and B (▵) or of subtypes A and E (▴). Only the change in the percentage of subtype A is shown.
TABLE 3.
Relative fitness and ranking of HIV-1 subtypes in three SupT1 and three MT2 environments
| Cell line | Viral geno- type | Mean fitness ± SE (ranking)a in cells with:
|
||
|---|---|---|---|---|
| No addition | TNF-α | PHA | ||
| SupT1 | A | 1.04 ± 0.011a (3) | 1.00 ± 0.009b (4) | 1.01 ± 0.026a,b,c (3) |
| B | 0.94 ± 0.019b,d (8) | 1.03 ± 0.010a (3) | 1.01 ± 0.023b,c (4) | |
| C1 | 0.99 ± 0.010b,c (4) | 1.05 ± 0.011a (2) | 1.00 ± 0.022c (5) | |
| C2 | 1.07 ± 0.012a (2) | 1.11 ± 0.009 (1) | 1.07 ± 0.016a (1) | |
| D | 0.96 ± 0.014b (6) | 0.99 ± 0.009b (5) | 0.92 ± 0.016d (9) | |
| E | 1.13 ± 0.016 (1) | 0.92 ± 0.008 (9) | 1.07 ± 0.019a,b (2) | |
| F | 0.92 ± 0.010d (9) | 0.95 ± 0.007c (7) | 0.96 ± 0.020c,d (7) | |
| G | 0.99 ± 0.013c (5) | 0.98 ± 0.008b (6) | 1.00 ± 0.021c (6) | |
| AG | 0.96 ± 0.014b,c (7) | 0.95 ± 0.009c (8) | 0.96 ± 0.021c,d (8) | |
| MT2 | A | 1.02 ± 0.007a (3) | 1.02 ± 0.008b (3) | 1.02 ± 0.005a,b (3) |
| B | 0.96 ± 0.008c (9) | 0.98 ± 0.008d,e (7) | 0.96 ± 0.004f (9) | |
| C1 | 0.99 ± 0.007b (5) | 1.01 ± 0.006b,c (5) | 0.99 ± 0.004c (5) | |
| C2 | 1.05 ± 0.009 (1) | 1.07 ± 0.012a (1) | 1.06 ± 0.006 (1) | |
| D | 0.98 ± 0.004b,c (8) | 0.98 ± 0.008d,e (8) | 0.97 ± 0.004d,e (7) | |
| E | 1.02 ± 0.007a (2) | 1.05 ± 0.006a (2) | 1.03 ± 0.005a (2) | |
| F | 1.01 ± 0.005a (4) | 1.01 ± 0.008b,c (4) | 1.01 ± 0.004b (4) | |
| G | 0.99 ± 0.006b (6) | 0.99 ± 0.008c,d (6) | 0.99 ± 0.007c,d (6) | |
| AG | 0.98 ± 0.006b (7) | 0.96 ± 0.012e (9) | 0.97 ± 0.004e,f (8) | |
Values with the same letter indicate that the subtypes have similar relative fitnesses within an environment; however, in such cases the ranking remains dominant.
Subtype fitness depends on the cellular environment.
Next, all pairwise competitions were performed in duplicate in the five additional cellular environments. We were interested whether different cellular environments could affect the replication rate in a subtype-specific manner. An environmentally induced change in fitness of a particular subtype is best illustrated by a change in the ranking of viral fitness. Figure 4 depicts the competitive ability or relative fitness of each subtype in a particular cellular environment and the effect of a change in cellular environment, and Table 3 lists the relative fitness values.
FIG. 4.

Change in viral fitness as determined by the environment. Shown are the reaction norms for each subtype, which indicate the change in relative fitness in a changing environment. The relative fitness values are depicted in Table 3. ▪, subtype A; ▴, subtype B; ×, subtype C1; ◊, subtype C2; □, subtype D; ▵, subtype E; •, subtype F; ○, subtype G; ⧫, subtype AG.
Most strikingly, subtype E changed from being the strongest competitor with the highest replication rate in SupT1 cells to being the worst competitor with the lowest replication rate in the SupT1-TNF-α environment (Fig. 4). Other prominent changes across these two cellular environments include the increase in the competitive ability of viral genotypes C1 and B, which moved up in ranking from the fourth and eighth positions in SupT1 cells to the second and third positions, respectively, in the SupT1-TNF-α environment. It has been shown that LTR-directed transcription in a TNF-α-stimulated SupT1 T-cell environment is positively correlated with the number of NF-κB sites (18, 36, 37, 44). This is most likely due to the elevated level of activated transcription factors p50 and p65 in the nucleus. Our experiments show that the replication rate is also positively correlated with the number of NF-κB sites in the LTR. The two C genotypes, with three NF-κB sites, are most fit in the SupT1-TNF-α environment, and subtype E, with a single NF-κB site, is the least fit genotype. In the SupT1 T-cell environment, in which a low level of p50 and p65 transcription factors is present in the nucleus, there is no correlation between the number of NF-κB sites and fitness. Above we showed that addition of PHA to the SupT1 cells has a negative influence on the replication rates of all subtypes. Figure 4 shows that the HIV-1 subtypes react differently to the PHA signal and that the PHA effect differs from the TNF-α response. Thus, subtypes C2 and E were most fit and had comparable fitness in the SupT1-PHA environment, followed by subtypes A, G, B, C1, F, AG, and D.
A change in host cell type can have a strong and subtype-specific effect on viral fitness. For instance, when SupT1 and MT2 cells are compared, subtype F increases from the ninth to the fourth fitness rank, and the relative fitness of subtype E decreases strongly (Fig. 4). In general, fitness differences between the subtypes are smaller in the MT2 cell line, as indicated by the twofold-lower range of fitness values (Fig. 4). This also applies to the effect of TNF-α or PHA stimulation in these cells. Because the replication rate is higher in MT2 cells, it is possible that a host cell factor becomes limiting, thus imposing an upper limit on virus replication. Nevertheless, there are minor but significant differences across the three MT2 environments, which are illustrated by two general features. The first feature is the dominance of the ranking. For instance, recombinant AG becomes the worst competitor in the MT2-TNF-α environment, compared to the seventh fitness rank in the MT2 environment. The second feature is the magnitudes of the slopes of the reaction norms in Fig. 4. For instance, competition between subtypes E and A proceeds slightly faster in the MT2-TNF-α environment than in the MT2 environment, which is due to increased fitness of subtype E in the former environment.
Fitness properties are conserved within a subtype.
We described rather significant differences among the LTR promoters of the different HIV-1 subtypes, in terms of both viral fitness and their response to environmental changes. We already raised the issue of intrasubtype variation and conservation of the LTR sequences with respect to the present TFBS and their MatInspector fit. Intrasubtype variation raises the possibility that the observed subtype-specific effects are merely anecdotal. To test whether the observed expression patterns are a more general property of a particular subtype, we analyzed additional viral strains. We selected two subtype E isolates (E-Fin24 and E-Fin4) from a recent miniepidemic among drug users in Helsinki, Finland, and two subtype C isolates from Ethiopia (C-Eth26 and C-Eth9) which closely resemble the C2 variant that was included in the initial analysis. All sequences possess the characteristic subtype signature; the two additional subtype E sequences have a GABP site instead of the NF-κB II site, and the subtype C sequences possess three NF-κB sites. We measured virus fitness in SupT1 cells and determined their reaction norms upon addition of TNF-α. The results are summarized in Fig. 5 and strongly argue that the LTR-imposed effect on virus replication in different cellular milieus is indeed a property that holds for multiple isolates within a subtype.
FIG. 5.

Conserved fitness and environmental responsiveness in different subtype C and E strains. The change in relative fitness was determined for two additional subtype E (E-Fin24 and E-Fin4) and two subtype C (C-Eth26 and C-Eth9) strains in SupT1 cells and SupT1 cells plus TNF-α. Relative fitness and the responsiveness to a changing environment are very similar for the genotypes belonging to the same subtype. ⧫, C2; ▪, C-Eth26; ▴, C-Eth9; ▵, E; ◊, E-Fin24; □, E-Fin4.
In summary, the competition assay is very robust in revealing small replication differences among viral genotypes. The competition assay provides strong evidence for the presence of interactions between the viral genotype and the cellular environment (30). This means that viral fitness based on the LTR promoter is strongly influenced by the environment, which in turn is determined by the pool of nuclear transcription factors present in the host cell.
DISCUSSION
It is unclear whether the HIV-1 subtypes exhibit different virus replication properties in vivo and thereby cause subtype-specific disease characteristics. Moreover, the role of the host cell environment in the performance of the different subtypes is currently unknown. In this in vitro study, we showed that there are subtype-specific replication rates that are dictated by differences in the TFBS composition of the LTR promoter. In addition, we demonstrated, by means of replication studies and competition experiments, a profound influence of the cellular environment on viral fitness. We also showed that a small number of LTR changes can have a major impact on these genotype-environment interactions. Below we discuss the possible origin of these interactions and the consequences for the genetic structure of the virus.
Previous studies have reported small differences in the transcriptional activities of the subtype LTR promoters (18, 36, 38, 44). Whether these differences result in subtype-specific replication rates has not been systematically addressed. One study did report differences in the replication rates of HIV-1 subtypes B and C, but the viral isolates used in that study differed to a large and unknown extent, leaving the cause for the differences unclear (4). The subtypes that we constructed differ exclusively in the U3 part of the LTR promoter. The selected LTR segments encode TFBS similar to those of viral isolates in the HIV database and are thus good representatives of the different subtypes. For example, the selected LTR segment and all subtype E isolates possess an inactivating one-nucleotide deletion in the upstream NF-κB site, which is converted into a GABP site (51). This conversion seems to have a strong positive fitness effect in the SupT1 environment but a negative fitness effect in the SupT1-TNF-α environment, indicating a possible evolutionary trade-off. Because the LTR structure within a subtype is relatively conserved in terms of the number and MatInspector fit of TFBS, it is likely that the LTR-directed fitness and environmental influences are general properties of most isolates within a subtype. This was confirmed by the inclusion of additional viral strains, to give a total of four subtype C genotypes (C1, C2, C-Eth26, and C-Eth9) and three subtype E genotypes (E, E-Fin4, and E-Fin24). The isolates of a certain subtype exhibit almost identical fitness, and they also react similarly to the environmental change that is induced by addition of TNF-α to SupT1 cells. The E genotypes are the best competitors in the SupT1 environment but become the worst competitors in the SupT1-TNF-α milieu. The four C genotypes are the best competitors upon addition of TNF-α to the SupT1 cells. Genotype C1 is a special case; it was originally chosen because it represents a different phylogenetic branch within subtype C (18). Genotype C1 is different in that it has no predicted SP1-III site, which may explain why it has a significantly lower fitness than the three other C genotypes across all environments.
The results of this study confirm that a small number of mutations or even a single point mutation can significantly affect fitness (10, 33, 51). Moreover, our data suggest that a mutation can have a positive fitness effect in one cellular environment and a negative fitness effect in another. One of the most fundamental consequences of this study is that it may not be possible to assign a single fitness value to a particular subtype. We show that viral fitness is not a rigid value but is largely influenced by environmental factors. A single replication or competition experiment may therefore not provide an accurate fitness value, because fitness, as determined by the LTR promoter, depends entirely on the tight interplay between the genetic structure of the virus and the cellular environment. It is therefore not possible to tell which subtype is better, but only which subtype is better in a particular environment.
The origin of these subtype-specific genotype-environment interactions in the HIV-1 LTR is unknown. Following the neutral theory of evolution (21), HIV-1 genomes do not evolve but rather float freely through sequence space, and genetic drift may be the most important force that shapes the genetic structure (14, 48). However, HIV-1 will adapt to the host environment in the presence of a strong environmental cue. For instance, this has been shown for the evolution of drug resistance (26) and escape from neutralizing antibodies (41). Inspection of the TFBS composition in Table 1 may suggest a role of selection and drift in shaping of the transcriptional promoter. First, there are TFBS with a good fit and low sequence variation compared to the complete LTR segment, for example, NF-κB-I in all subtypes and NF-κB-II in all subtypes except subtype E, which instead has a conserved GABP site. Second, there are TFBS with a poor fit and low sequence diversity, for example, SP1-I in recombinant AG and subtypes G and D. Third, there are TFBS with a poor fit and high sequence diversity, such as SP1-II in subtype G. The first pattern may be explained by strong purifying selection for a particular TFBS to retain a certain level of gene expression. The second pattern may be caused by the presence of overlapping TFBS at the same location. For instance, MatInspector predicts binding of LYF-1, a T-cell-specific transcription factor, to sequences overlapping the SP1-I site in subtypes D and G and recombinant AG. The third pattern could be explained by redundancy in TFBS, which may allow some elements to mutate and to drift freely through sequence space because other TFBS compensate to maintain a certain LTR activity. Through this turnover scenario, a new TFBS identity may evolve. New TFBS are frequently generated through duplication of existing sites, in particular when the starting virus is a poor replicator due to mutations that affect viral gene expression (7, 51). Sequences from HIV-1-infected patients confirm that new sites can be generated by duplication of preexisting TFBS (13, 23, 53). These results suggest that there is also a large adaptive potential in the LTR, despite the conservation of the overall genetic make-up.
The selectional cue for the transcriptional promoter of HIV-1 comes from the activated pool of transcription factors in the cellular environment. It is estimated that more than 2,000 transcription factors are encoded in the human genome (25, 50). These factors are not equally expressed in all cells, and many are induced at specific states of cellular development. With respect to HIV-infected individuals, differences in the pool of activated transcription factors may be found in different cell types (e.g., macrophages and T-helper cells), the differential activation state of cells (e.g., Th1 and Th2 states of T cells and naive and memory T cells), different body compartments (e.g., lymphocytes, spleen, and brain), the occurrence of viral and bacterial coinfections (e.g., hepatitis B [15], tuberculosis [11], or Epstein-Barr virus [31]), or genetic polymorphisms of the human host. The local environment of the HIV-1 LTR and other promoters is therefore multidimensional, as the pool of transcription factors varies both temporally and spatially. The principle of allocation predicts that high fitness in a specific environment can be attained only at the expense of average fitness in other environments (29). This means that specialization and maximization of fitness in a particular environment will be at the expense of fitness in another environment. The alternative is to optimize fitness across several environments, which will be at the expense of high fitness in a specific environment. Between these two extremes of the adaptational spectrum lie intermediate solutions with characteristics of both strategies, depending on the variability of the environment in time and space (35). We and others have shown that the subtype-specific LTRs of HIV-1 possess the signatures of both the generalist and specialist strategies. On the one hand, there is redundancy and overlap in the TFBS function that ensures replication in several cellular environments. On the other hand, the LTR also contains sites that enhance replication in specific cellular environments (1, 28, 34, 42, 47, 51). It seems likely that the subtype-specific variation in HIV-1 represents different adaptational solutions as a result of a changing and/or fluctuating environment. Therefore, the subtypes are different solutions in sequence space and are positioned onto different and distant fitness peaks in the adaptive landscape. These different solutions in the LTR may have been explored shortly after the zoonotic transmission from apes to humans, when there was room for increased replication because the simian-adapted virus was not optimized for replication in the new human host.
In this study, we showed that the subtype-specific LTRs dictate different HIV-1 replication rates. Fitness is not a constant parameter but is strongly influenced by the environment. A few mutational differences between viral genotypes can change fitness and the way in which the virus reacts upon environmental changes. Under the influence of the variability of the host environment, a trade-off seems to have arisen with respect to the number and type of TFBS in the subtype-specific LTRs and the degree of specialization. There have been several reports of subtype-specific differences in disease characteristics and transmission. Our study confirms that biological differences between subtypes exist, but the impact on viral pathogenesis and transmission remains to be determined.
Acknowledgments
We thank Marijn van Ballegooijen, Mark Geels, Sara Magalhaes, Bill Paxton, Tony de Ronde, Andre de Roos, Maurice Sabelis, and Nadine Vastenhouw for discussions and comments on the manuscript.
This research was supported by NWO-ALW project number 811.35.001 and by a fellowship from the Royal Netherlands Academy of Arts and Sciences to M.C.B.
REFERENCES
- 1.Ait-Khaled, M., J. E. McLaughlin, M. A. Johnson, and V. C. Emery. 1995. Distinct HIV-1 long terminal repeat quasispecies present in nervous tissues compared to that in lung, blood and lymphoid tissues of an AIDS patient. AIDS 9:675-683. [DOI] [PubMed] [Google Scholar]
- 2.Aldovini, A., and B. D. Walker. 1990. Techniques in HIV research. Stockton Press, New York, N.Y.
- 3.Auersperg, N. 1964. Long-term cultivation of hypodiploid human tumor cells. J. Natl. Cancer Inst. 32:135-163. [PubMed] [Google Scholar]
- 4.Ball, S. C., A. Abraha, K. R. Collins, A. J. Marozsan, H. Baird, M. E. Quinones-Mateu, A. Penn-Nicholson, M. Murray, N. Richard, M. Lobritz, P. A. Zimmerman, T. Kawamura, A. Blauvelt, and E. J. Arts. 2003. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J. Virol. 77:1021-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berkhout, B., A. Gatignol, A. B. Rabson, and K. T. Jeang. 1990. TAR-independent activation of the HIV-1 LTR: evidence that tat requires specific regions of the promoter. Cell 62:757-767. [DOI] [PubMed] [Google Scholar]
- 6.Berkhout, B., and K. T. Jeang. 1992. Functional roles for the TATA promoter and enhancers in basal and Tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 66:139-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkhout, B., K. Verhoef, J. L. van Wamel, and N. K. Back. 1999. Genetic instability of live, attenuated human immunodeficiency virus type 1 vaccine strains. J. Virol. 73:1138-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackard, J. T., B. Renjifo, W. Fawzi, E. Hertzmark, G. Msamanga, D. Mwakagile, D. Hunter, D. Spiegelman, N. Sharghi, C. Kagoma, and M. Essex. 2001. HIV-1 LTR subtype and perinatal transmission. Virology 287:261-265. [DOI] [PubMed] [Google Scholar]
- 9.Brivanlou, A. H., and J. E. Darnell, Jr. 2002. Signal transduction and the control of gene expression. Science 295:813-818. [DOI] [PubMed] [Google Scholar]
- 10.Carvalho, M., M. Kirkland, and D. Derse. 1993. Protein interactions with DNA elements in variant equine infectious anemia virus enhancers and their impact on transcriptional activity. J. Virol. 67:6586-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins, K. R., M. E. Quinones-Mateu, Z. Toossi, and E. J. Arts. 2002. Impact of tuberculosis on HIV-1 replication, diversity, and disease progression. AIDS Rev. 4:165-176. [PubMed] [Google Scholar]
- 12.Das, A. T., B. Klaver, and B. Berkhout. 1999. A hairpin structure in the R region of the human immunodeficiency virus type 1 RNA genome is instrumental in polyadenylation site selection. J. Virol. 73:81-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Estable, M. C., B. Bell, M. Hirst, and I. Sadowski. 1998. Naturally occurring human immunodeficiency virus type 1 long terminal repeats have a frequently observed duplication that binds RBF-2 and represses transcription. J. Virol. 72:6465-6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frost, S. D., M. J. Dumaurier, S. Wain-Hobson, and A. J. Brown. 2001. Genetic drift and within-host metapopulation dynamics of HIV-1 infection. Proc. Natl. Acad. Sci. USA 98:6975-6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Gonzalo, M., M. Carretero, J. Rullas, E. Lara-Pezzi, J. Aramburu, B. Berkhout, J. Alcami, and M. Lopez-Cabrera. 2001. The hepatitis B virus X protein induces HIV-1 replication and transcription in synergy with T-cell activation signals: functional roles of NF-kappaB/NF-AT and SP1-binding sites in the HIV-1 long terminal repeat promoter. J. Biol. Chem. 276:35435-35443. [DOI] [PubMed] [Google Scholar]
- 16.Hu, D. J., S. Vanichseni, T. D. Mastro, S. Raktham, N. L. Young, P. A. Mock, S. Subbarao, B. S. Parekh, L. Srisuwanvilai, R. Sutthent, C. Wasi, W. Heneine, and K. Choopanya. 2001. Viral load differences in early infection with two HIV-1 subtypes. AIDS 15:683-691. [DOI] [PubMed] [Google Scholar]
- 17.Ilyinskii, P. O., and R. C. Desrosiers. 1996. Efficient transcription and replication of simian immunodeficiency virus in the absence of NF-κB and Sp1 binding elements. J. Virol. 70:3118-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeeninga, R. E., M. Hoogenkamp, M. Armand-Ugon, M. de Baar, K. Verhoef, and B. Berkhout. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 74:3740-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaleebu, P., A. Ross, D. Morgan, D. Yirrell, J. Oram, A. Rutebemberwa, F. Lyagoba, L. Hamilton, B. Biryahwaho, and J. Whitworth. 2001. Relationship between HIV-1 Env subtypes A and D and disease progression in a rural Ugandan cohort. AIDS 15:293-299. [DOI] [PubMed] [Google Scholar]
- 20.Kanki, P. J., D. J. Hamel, J. L. Sankale, C. Hsieh, I. Thior, F. Barin, S. A. Woodcock, A. Gueye-Ndiaye, E. Zhang, M. Montano, T. Siby, R. Marlink, I. NDoye, M. E. Essex, and S. M. Boup. 1999. Human immunodeficiency virus type 1 subtypes differ in disease progression. J. Infect. Dis. 179:68-73. [DOI] [PubMed] [Google Scholar]
- 21.Kimura, M. 1968. Evolutionary rate at the molecular level. Nature 217:624-626. [DOI] [PubMed] [Google Scholar]
- 22.Klaver, B., and B. Berkhout. 1994. Premature strand transfer by the HIV-1 reverse transcriptase during strong-stop DNA synthesis. Nucleic Acids Res. 22:137-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koken, S. E., J. L. van Wamel, J. Goudsmit, B. Berkhout, and J. L. Geelen. 1992. Natural variants of the HIV-1 long terminal repeat: analysis of promoters with duplicated DNA regulatory motifs. Virology 191:968-972. [DOI] [PubMed] [Google Scholar]
- 24.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244-1245. [DOI] [PubMed] [Google Scholar]
- 25.Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921. [DOI] [PubMed] [Google Scholar]
- 26.Larder, B. A., G. Darby, and D. D. Richman. 1989. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science 243:1731-1734. [DOI] [PubMed] [Google Scholar]
- 27.Lenski, R. E., M. R. Rose, S. C. Simpson, and S. C. Tadler. 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat. 138:1315-1341. [Google Scholar]
- 28.Leonard, J., C. Parrott, A. J. Buckler-White, W. Turner, E. K. Ross, M. A. Martin, and A. B. Rabson. 1989. The NF-κB binding sites in the human immunodeficiency virus type 1 long terminal repeat are not required for virus infectivity. J. Virol. 63:4919-4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levins, R. 1968. Evolution in changing environments. Princeton University Press, Princeton, N.J.
- 30.Lynch, M., and B. Walsh. 1998. Genetics and analysis of quantitative traits. Sinnauer Associates, Sunderland, Mass.
- 31.Mallon, R., J. Borkowski, R. Albin, S. Pepitoni, J. Schwartz, and E. Kieff. 1990. The Epstein-Barr virus BZLF1 gene product activates the human immunodeficiency virus type 1 5′ long terminal repeat. J. Virol. 64:6282-6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maree, A. F., W. Keulen, C. A. Boucher, and R. J. De Boer. 2000. Estimating relative fitness in viral competition experiments. J. Virol. 74:11067-11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maury, W. 1998. Regulation of equine infectious anemia virus expression. J. Biomed. Sci. 5:11-23. [DOI] [PubMed] [Google Scholar]
- 34.McAllister, J. J., D. Phillips, S. Millhouse, J. Conner, T. Hogan, H. L. Ross, and B. Wigdahl. 2000. Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: evidence for cell type-specific gene regulation and viral replication. Virology 274:262-277. [DOI] [PubMed] [Google Scholar]
- 35.Meyers, L. A., and J. J. Bull. 2002. Fighting change with change: adaptive variation in an uncertain world. Trends Ecol. E vol. 17:551-557. [Google Scholar]
- 36.Montano, M. A., C. P. Nixon, and M. Essex. 1998. Dysregulation through the NF-κB enhancer and TATA box of the human immunodeficiency virus type 1 subtype E promoter. J. Virol. 72:8446-8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montano, M. A., C. P. Nixon, T. Ndung'u, H. Bussmann, V. A. Novitsky, D. Dickman, and M. Essex. 2000. Elevated tumor necrosis factor-alpha activation of human immunodeficiency virus type 1 subtype C in Southern Africa is associated with an NF-kappaB enhancer gain-of-function. J. Infect. Dis. 181:76-81. [DOI] [PubMed] [Google Scholar]
- 38.Naghavi, M. H., M. C. Estable, S. Schwartz, R. G. Roeder, and A. Vahlne. 2001. Upstream stimulating factor affects human immunodeficiency virus type 1 (HIV-1) long terminal repeat-directed transcription in a cell-specific manner, independently of the HIV-1 subtype and the core-negative regulatory element. J. Gen. Virol. 82:547-559. [DOI] [PubMed] [Google Scholar]
- 39.Naghavi, M. H., S. Schwartz, A. Sonnerborg, and A. Vahlne. 1999. Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Res. Hum. Retroviruses 15:1293-1303. [DOI] [PubMed] [Google Scholar]
- 40.Peden, K., M. Emerman, and L. Montagnier. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185:661-672. [DOI] [PubMed] [Google Scholar]
- 41.Peterlin, B. M., and D. Trono. 2003. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat. Rev. Immunol. 3:97-107. [DOI] [PubMed] [Google Scholar]
- 42.Pohlmann, S., S. Floss, P. O. Ilyinskii, T. Stamminger, and F. Kirchhoff. 1998. Sequences just upstream of the simian immunodeficiency virus core enhancer allow efficient replication in the absence of NF-kappaB and Sp1 binding elements. J. Virol. 72:5589-5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quandt, K., K. Frech, H. Karas, E. Wingender, and T. Werner. 1995. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23:4878-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quivy, V., E. Adam, Y. Collette, D. Demonte, A. Chariot, C. Vanhulle, B. Berkhout, R. Castellano, Y. de Launoit, A. Burny, J. Piette, V. Bours, and C. Van Lint. 2002. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-κB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J. Virol. 76:11091-11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renjifo, B., W. Fawzi, D. Mwakagile, D. Hunter, G. Msamanga, D. Spiegelman, M. Garland, C. Kagoma, A. Kim, B. Chaplin, E. Hertzmark, and M. Essex. 2001. Differences in perinatal transmission among human immunodeficiency virus type 1 genotypes. J. Hum. Virol. 4:16-25. [PubMed] [Google Scholar]
- 46.Rinke de Wit, T. F., A. Tsegaye, D. Wolday, B. Hailu, M. Aklilu, E. Sanders, M. Hagos, A. Kliphuis, G. Pollakis, A. Krol, R. Geskus, F. Miedema, J. Goudsmit, R. Coutinho, and A. L. Fontanet. 2002. Primary HIV-1 subtype C infection in Ethiopia. J. Acquir. Immune Defic. Syndr. 30:463-470. [DOI] [PubMed] [Google Scholar]
- 47.Ross, E. K., A. J. Buckler-White, A. B. Rabson, G. Englund, and M. A. Martin. 1991. Contribution of NF-κB and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: distinct patterns of viral growth are determined by T-cell types. J. Virol. 65:4350-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sala, M., and S. Wain-Hobson. 2000. Are RNA viruses adapting or merely changing? J. Mol E vol. 51:12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith, C. D., M. Shatsky, P. S. Cohen, R. Warnke, M. P. Link, and B. E. Glader. 1984. Monoclonal antibody and enzymatic profiles of human malignant T-lymphoid cells and derived cell lines. Cancer Res. 44:5657-5662. [PubMed] [Google Scholar]
- 50.Venter, J. C., M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, et al. 2001. The sequence of the human genome. Science 291:1304-1351. [DOI] [PubMed] [Google Scholar]
- 51.Verhoef, K., R. W. Sanders, V. Fontaine, S. Kitajima, and B. Berkhout. 1999. Evolution of the human immunodeficiency virus type 1 long terminal repeat promoter by conversion of an NF-κB enhancer element into a GABP binding site. J. Virol. 73:1331-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yedavalli, V. S., M. Benkirane, and K. T. Jeang. 2003. Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J. Biol. Chem. 278:6404-6410. [DOI] [PubMed] [Google Scholar]
- 53.Zhang, L., Y. Huang, H. Yuan, B. K. Chen, J. Ip, and D. D. Ho. 1997. Identification of a replication-competent pathogenic human immunodeficiency virus type 1 with a duplication in the TCF-1α region but lacking NF-κB binding sites. J. Virol. 71:1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]