

Abstract
Normal human diploid fibroblasts have limited life span in culture and undergo replicative senescence after 50–60 population doublings. On the contrary, cancer cells typically divide indefinitely and are immortal. Expression of SV40 large T and small t antigens in human fibroblasts transiently extends their life span by 20–30 population doublings and facilitates immortalization. We have identified a rearrangement in chromosome 6 shared by SV40-transformed human fibroblasts. Rearrangements involving chromosome 6 are among the most frequent in human carcinogenesis. In this paper, we extend analysis of the 6q26–q27 region, a putative site for a growth suppressor gene designated SEN6 involved in immortalization of SV40-transformed cells. Detailed molecular characterization of the rearranged chromosomes (6q*, normal appearing; and 6qt, translocated) in the SV40-immortalized cell line HALneo by isolating each of these 2 chromosomes in mouse/HAL somatic cell hybrids is presented. Analysis of these mouse/HAL somatic cell hybrids with polymorphic and nonpolymorphic markers revealed that the 6q* has undergone a chromosomal break in the MLLT4 gene (alias AF6). This result in conjunction with previous published observations leads us to conclude that SEN6 lies between MLLT4 and TBP at chromosomal region 6q27. Examination of different genes (MLLT4, DLL1, FAM120B, PHF10) located within this interval that are expressed in HS74 normal fibroblast cells reveals that overexpression of epitope-tagged truncated PHF10 cDNAs resulted in reduced cell proliferation in multiple cell lines. Paradoxically, down-regulation of PHF10 by RNAi also resulted in loss of cell proliferation in normal fibroblast cells, indicating PHF10 function is required for cell growth. Taken together, these observations suggest that decreased cell proliferation with epitope-tagged truncated PHF10 proteins may be due to dominant negative effects or due to unregulated expression of these mutant proteins. Hence we conclude that PHF10 is not SEN6 but is required for cell growth.
Key Words: Chromosome 6q27, Mouse/human hybrid, RNAi, SEN6, SV40
Introduction
The development of human cancer is a complex and multistep process. SV40-mediated immortalization has long been utilized as a model system for human carcinogenesis which commonly involves inactivation of pRb and p53 tumor suppressor pathways, telomere stabilization and other genetic alterations. Normal human diploid fibroblast cells have a limited life span in culture [Hayflick, 1965]. These cells normally undergo 50–60 cell divisions, after which they stop dividing and enter a state of ‘replicative senescence’, with arrest within the G1 stage of the cell cycle [Stein and Dulic, 1995; Smith and Pereira-Smith, 1996]. This growth arrest has been attributed to the shortening and/or dysfunction of telomeres [Blackburn, 2000; Rodier et al., 2005; Stewart and Weinberg, 2006] resulting in activation of pRb and p53 tumor and growth suppressor pathways through up-regulation of p16 and p21 [Shay et al., 1991; Alcorta et al., 1996]. However, cells transformed by SV40 DNA tumor virus or its large T antigen (Tag) and small t antigen genes will replicate for an additional 20–30 generations. This extension of life span is achieved by the inactivation of the tumor suppressor proteins, p53 and pRb, through binding to the Tag protein [Bryan and Reddel, 1994]. Inactivation of p53 releases cell cycle checkpoints [Schwartz and Rotter, 1998; Vaziri and Benchimol, 1999], while inactivation of pRb leads to activation of the E2F family of transcription factors that are required for cell cycle progression [Nevins, 1992; Weinberg, 1995] inter alia. Inactivation of p53 and further shortening of the telomeres caused by the extended life span leads to genetic instability that contributes to extensive genomic rearrangements and thus ultimately to massive cell death (due to apoptosis) termed crisis [Macera-Bloch et al., 2002]. However, a rare cell survives or bypasses crisis and continues to grow indefinitely as an immortal clone. Such immortal clones typically occur at a very low frequency (less than 10−6) and have stabilized their telomeres. The low frequency of immortalization indicates that large Tag is required but is not sufficient for immortalization [Radna et al., 1989; Wright et al., 1989; Jha et al., 1998] and that a mutation in additional cellular gene(s) is needed for a cell to become immortal.
Rearrangement of human chromosome 6 has been a hallmark of many cancers [Devilee et al., 1991; Millikin et al., 1991; Morita et al., 1991; Gaidano et al., 1992; Saha et al., 1995; De Souza et al., 1995; Orphanos et al., 1995; Honchel et al., 1996; Tibiletti et al., 1996, 2000; Shridhar et al., 1999; Steinemann et al., 2003]. Karyotype analysis of matched sets of preimmortal and immortal cell lines by this laboratory and others has revealed nonrandom loss of chromosome 6q [Hubbard-Smith et al., 1992; Ray and Kraemer, 1992], suggesting that loss of gene(s) on chromosome 6q might be required for immortalization. Subsequent studies involving transfer of normal chromosome 6 or 6q into immortal cell lines exhibited growth suppression of immortal cells [Sandhu et al., 1994], further supporting the above hypothesis. More detailed molecular analysis of 17 different SV40-immortalized cell lines using microsatellite polymorphic markers has identified a minimum deletion region at 6q26–q27, suggesting that the gene responsible for immortalization (termed SEN6) resides within this region [Banga et al., 1997]. Unexpectedly, cytogenetic analysis of 2 immortal cell lines AR5 and HALneo that originated from the same primary SV40 transformant (SVtSA/HF-A) revealed that they are of independent origin. AR5 is hemizygous and therefore has retained only one chromosome 6, whereas HALneo has retained the other homolog of chromosome 6. In addition, FISH analysis revealed that HALneo appears to have retained a fragment of chromosome 6 that is derived from the 6q26–q27 region and is translocated to another unknown chromosome [Banga et al., 1997]. Further analysis of each of these 2 chromosomes of HALneo in mouse/HAL cell hybrids, however, demonstrated that the apparently intact chromosome 6 has a terminal deletion whereas the chromosome 6 fragment is derived from the region of chromosome 6 that is retained in AR5 [Banga et al., 1997]. Therefore, both these immortal sublines (AR5 and HALneo) in fact share the same region of 6q26–qter. In this report, we describe detailed molecular characterization of the normal appearing chromosome 6 (designated 6q*) and the fragment of chromosome 6 (designated 6qt) present in HALneo isolated in mouse/HAL cell hybrids using polymorphic and non-polymorphic markers. Analysis of these hybrids not only revealed integrity of these chromosomes with respect to their origin but also exposed a genetic rearrangement that would otherwise have gone undetected. Using this approach we were able to determine that chromosome 6q* has undergone one breakpoint in the MLLT4 genewhich is located at 6q27. Based upon these and previously published results, we have redefined the location of SEN6 to 6q27 between MLLT4 and TBP. Since several of the human tumors and SV40-transformed cells share loss of heterozygosity (LOH) in 6q26–qter [Saito et al., 1992; Foulkes et al., 1993; Banga et al., 1997; Tibiletti et al., 1998; Letessier et al., 2007], inactivation of SEN6 may be responsible for immortalization of these tumors as well. Overexpression studies involving different genes in this interval revealed epitope-tagged PHF10 cDNAs (a plant homeodomain-containing gene of unknown function in humans) resulted in growth suppression in multiple human cell lines. On the contrary, depletion of PHF10 in normal human fibroblast cells resulted in loss of cell proliferation by RNA interference (RNAi). Taken together, these observations suggest that decreased cell proliferation with epitope-tagged truncated PHF10 proteins may be due to dominant negative effects or due to unregulated expression of these mutant proteins. Hence we conclude that PHF10 is not SEN6 but rather is required for cell growth.
Materials and Methods
Cell Lines and Culture Conditions
SCSV3hygro is an SV40-immortalized mouse cell line that is deficient in double-strand break repair [Banga et al., 1994] and has been stably transfected with a selectable marker that provides resistance to hygromycin (pCMVHygtk). HALneo is a human fibroblast cell line that is immortalized with a temperature-sensitive SV40 T antigen [Hubbard-Smith et al., 1992]and has been stably transfected with a selectable marker neo (pRSVneo) conferring resistance to G418. SCSV3hygro and HALneo cells were grown at 7.5% CO2 in a medium supplemented with 10% fetal calf serum, penicillin and streptomycin. SCSV3hygro cells were maintained in medium containing 200 μg/ml of hygromycin at 37°C and HALneo cells were maintained in medium containing 150 μg of G418 at 35°C. HSF43 is a human foreskin fibroblast cell line and CT10-2A is an immortal cell line derived from HSF43 by SV40 transformation [Ray and Kraemer, 1992]. HS74, the fetal human bone marrow stromal cell line, which was used as the parent of the SV40-transformed cells generated in this laboratory, has been maintained as previously described [Small et al., 1982]. Other non-immortal and immortal SV40-transformed human cell lines including HALneo were maintained as previously described [Neufeld et al., 1987; Banga et al., 1997]. Non-immortal cell lines which were used to generate immortal derivatives were also termed ‘preimmortal’ cell lines. The SV40-transformed immortal cl39T-Tet-On cell line stably expressing rtTA (reverse transactivator) was generated by transfection of pTet-On plasmid (Clontech). The cl39T-Tet-On cell line was isolated and maintained under tetracycline repressed conditions.
Fluorescence in situ Hybridization
Metaphase preparations were hybridized with whole-chromosome-6-specific painting probe (SpectrumGreen) according to the instructions provided by the supplier (Vysis Inc.). Chromosomes were stained with propidium iodide. Fluorescent signals were detected by Olympus Fluorescent microscope and photographs were taken with a B20 camera using 400 ASA Kodak film.
Mouse/HAL Somatic Cell Hybrids
To obtain mouse/HAL somatic cell hybrids between SCSV3hygro and HALneo cells, 1.5 × 106 cells of SCSV3hygro and 2 × 106 cells of HALneo were grown together without selection in a 10-cm petri dish for 14 h at 35°C. The cells were fused using polyethylene glycol (PEG, BMB) for 2 min at 37°C followed by extensive washes with serum-free medium. Cells were then grown in non-selective medium for 22 h at 35°C in a 7.5% CO2 humidified chamber. After trypsinization, cells were subcultured into 10-cm dishes in culture medium containing 200 μg/ml of hygromycin and 400 μg/ml of G418 to select hybrid cells. Cells were then incubated for 10–12 days at 37°C. Discrete clones were picked and plated at low density in a 10-cm dish. Subclones were picked from 10-cm petri dishes and replated in 24-well plates individually. When cells in a well reached 50–90% confluency, 80–90% of cells were harvested from each well into 1.5-ml Eppendorf tubes for rapid DNA isolation. Remaining cells in each well were re-fed with selection medium to continue culture for additional isolation of DNA and for long-term storage.
Rapid Isolation of DNA from Somatic Cell Hybrids
Cells from each hybrid were washed twice with PBS and then resuspended in 50 μl of buffer containing 10 mM Tris Cl pH 8.3, 1 mM EDTA pH 8.0, 25 mM NaCl and 200 μg/ml proteinase K. After brief vortexing, cells were then incubated at 37°C for 20–30 min. The Eppendorf tubes containing DNA were incubated at 95°C for 8–10 min to denature proteinase K as well as DNA and cooled immediately on ice. The DNA samples were kept at −20°C until further analysis. Using this method one can isolate DNA for PCR analysis from a number of clones at a time. Once the hybrids containing a single copy of human 6q were identified, more DNA was isolated by following standard procedures.
Primers and Sequencing
Primers used in the study were either synthesized at the Molecular Resource Facility (New Jersey Medical School) or purchased from Research GeneticsInc. Primer sequence data for many genes, expressed sequence tag (est), sequence tag site (sts) and markers that are not provided in the manuscript can be obtained from the authors. To sequence the cDNA of MLLT4 in different immortal cell lines, we performed RT-PCR using total RNA of each cell line with primer pairs that were generated from the sequence of the MLLT4 cDNA clone [Prasad et al., 1993] provided by R. Dalla-Favera (New York). Additional exons present in longer splice variants were directly sequenced from respective genomic DNAs by designing primers from the sequence information available in the public databases.
Polymerase Chain Reaction
2 μl of DNA was taken from each mouse/HAL hybrid for PCR amplification in a reaction volume of 25 μl. A standard PCR reaction contained 1× Taq buffer (BMB), 200 μM dNTPs, 50 pM of each primer and 1 U of Taq polymerase (BMB). PCR conditions were as follows: 3 min at 94°C for 1 cycle, 1 min at 94°C, 1 min at 55–57°C (depending upon primer used), 1 min at 72°C for 35 cycles and 6 min at 72°C for 1 cycle. One tenth of the amplified products were separated in 4% agarose gels containing ethidium bromide to visualize amplified products.
RT-PCR and Real-Time Quantitative RT-PCR
Expression of each gene or EST from the 6q27 region was detected by RT-PCR using total RNA from a normal HS74 and a preimmortal SV40-transformed cell line (RT-PCR Kit, Invitrogen). Each experiment was repeated at least twice. For QRT-PCR of gene expression analysis of different transcripts of MLLT4, serial dilutions of input RNA (100–1.56 ng) were analyzed using the ABI PRISM 7500HT sequence detector system, primers, probes, and the TaqMan One-Step RT-PCR Master Mix Reagents kit (Applied Biosystems). Three distinct primer pairs were used to evaluate different MLLT4 splice variants (Applied Biosystems). The reaction mixture was held at 48°C for 30 min for reverse transcription of RNA into cDNA. This was followed by incubation at 95°C for 10 min to activate the Taq polymerase. PCR amplification of cDNA was performed for 40 cycles using the following cycling conditions: denaturing for 15 s at 95°C, and annealing and extending for 60 s at 60°C. All samples were tested in triplicate, and average values were used for quantitation. Analysis was performed using SDS v2.1 software (Applied Biosystems) according to the manufacturer's instructions. GAPDH was used to normalize RNA loading. Relative Standard Curve Method was used for quantitation of gene expression.
Plasmids and cDNA Cloning
cDNA of DLL1 (kindly provided by S. Atravanis-Tsakonas, Charleston, Mass.) was tagged with an HA epitope at the 3′ end by PCR and then cloned into tetracycline regulatable vector pTRE-Tight-PGK-Puro carrying the PAC gene for puromycin resistance. pTRE-Tight-PGK-Puro vector was constructed by cloning PGK-Puro into pTRE-Tight (Clontech). PHF10b variant 2 (NM_133325.1, Open Biosystems) was also tagged with an HA epitope at the 3′ end and then cloned into pTRE-Tight-PGK-Puro. This cDNA lacks exon 1 and 2 of the revised version NM_018288.3 by the NCBI (fig. 1). We tagged FAM120B (alias BC012177) cDNA (Open Biosystems) by PCR with a 5′ primer containing an HA tag and a 3′ primer containing 3× FLAG epitopes and cloned it into pTRE-Tight-PGK-Puro vector to generate pTRE-Tight · (HA) BamH1-FAM120B-BamH1(FLAG) · PGK-Puro. To facilitate the cloning of other cDNAs with HA and FLAG tags, we removed the FAM120B cDNA by digestion with BamH1 and religated the vector to generate pTRE-Tight · (HA)BamH1(FLAG) · PGK-Puro. A longer version of PHF10(PHF10L) cDNA (ORF 99 −1454 nt) lacking exon 1 of the revised version NM_018288.3 by NCBI (fig. 1)was cloned from normal HS74 cells by RT-PCR using the primer pair carrying BamH1 (underlined) sites (5′CTGGATCCATGGGCTCAGGAGATAGTTC 3′ and 5′ CTGGATCCTCCCTCTTTGCTGTTTTTCC 3′). This additional exon 2 adds 123 nt (41 aa) to the PHF10b variant 2 ORF. The isolation of PHF10L cDNA was based on ORF analysis of NM_133325.1 sequence and existence of a similar size ORF in Pan troglodytes (XP_518861.2). To tag PHF10L with HA and FLAG epitopes, its ORF was PCR amplified using appropriate primers (see fig. 1, black arrows) and cloned in frame into pTRE-Tight · (HA)BamH1(FLAG) · PGK-Puro vector at the BamH1 site. Each cDNA was sequenced to confirm its integrity. To clone (HA)PHF10L(FLAG) and PHF10b(HA) cDNAs into a retroviral vector pLPCX (Clontech), these cDNAs were released from pTRE-Tight · (HA)PHF10L (FLAG) · PGK-Puro and pTRE-Tight · PHF10b(HA) · PGK-Puro plasmids, respectively, by digesting with EcoRI and ClaI, and then each HA- and/or FLAG-tagged cDNA was cloned into pLPCX under the control of CMV promoter. cDNA of MLLT4 was provided by R. Dalla-Favera (New York).
Fig. 1.

PHF10 gene, its mRNA species and respective protein isoforms. APHF10 is comprised of 12 exons with a coding potential of 498 aa. Exons and introns are not drawn to scale. B This mRNA lacks exons 1 and 2, and codes for a 410-aa protein (PHF10a) with 2 zinc finger domains (blue rectangles). C This mRNA lacks exons 1 and 2, and codes for a 408-aa protein (PHF10b) due to alternative splicing (represented by a line within the mRNA and by a small blank rectangle in the protein). D Analysis of the published sequence of PHF10 by us revealed an ORF that included exon 2. This resulted in the addition of 41 aa at the 5′ end to the published isoforms shown in B or C, and thus increased their coding capacity to 451 or 449 aa. A cDNA corresponding to a 451-aa protein (PHF10L) was cloned by RT-PCR using primers originating at 5′ (99 bp) and 3′ (1,454 bp). Black arrows indicate the position of primers used to amplify the coding regions of respective cDNAs by PCR or RT-PCR.
Western Blot Analysis
Total protein was isolated from different normal and SV40-transformed cell lines in RIPA buffer (10 mM Tris, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS), 1 mM sodium orthovanadate, 1 mM phenylmethyl-sulfonyl fluoride, 40 μg/ml aprotinin). After quantitation, 30 μg protein of the cell extracts were electrophoresed in a 7.5% denaturing polyacrylamide gel and blotted onto a membrane. The membrane was then probed with anti-MLLT4 monoclonal antibody (BD Biosciences). After washing, the blot was exposed to horseradish peroxidase-conjugated secondary antibody and protein visualized using the chemiluminescence detection system (PerkinElmer™ Life Sciences). Anti-α-actinin antibody (Sigma-Aldrich) was used as a control for sample loading.
Northern Blot Analysis
Total RNA from different cell lines was isolated using TRIzol (Invitrogen). PolyA+ RNA was isolated using Oligotex mRNA Midi kit (Qiagen). 3 μg of PolyA+ RNA was fractionated on 0.8% agarose-RNA borate formaldehyde gels. After blotting, membranes were hybridized with 32P-labeled probes (whole cDNA or fragments derived from respective genes) overnight using PerfectHyb Plus hybridization buffer (Sigma). After further washing, membranes were exposed to X-ray films. An RNA ladder was used as marker (Invitrogen).
Retroviral Production
10 μg of different retroviral vectors were transfected by the calcium phosphate transfection method in Phoenix packaging cells (gift from G. Nolan) plated 20–24 h before in 100-mm dishes. Briefly, after 12 h of transfection, cells were fed with fresh medium, and different retroviruses were harvested after 36 h. After filtration with 0.45-μm filter, aliquots of 1.5 ml were made and stored frozen at −80°C. A frozen-thawed aliquot of each retrovirus (supplemented with 8 μg/ml polybrene, Sigma) was titered on NIH3T3 cells to determine virus infectious units. Viruses that gave reproducibly similar numbers of puromycin-resistant colonies on NIH3T3 cells were used in growth studies on the normal, telomerase-immortalized and SV40-transformed human cells.
Growth Assays
Different normal, telomerase- and SV40-immortalized fibroblast cells were plated at 2 × 105 cells per 60 mm dish. After 12 h, aliquots corresponding to different retroviruses containing PHF10b(HA) and (HA)PHF10L(FLAG) cDNAs or empty vector were thawed, supplemented with 8 μg/ml of polybrene, and then used for transduction. After 48 h, cells were selected with fresh medium containing 0.3–0.7 μg/ml puromycin. Cells were fed every 4 days with culture medium containing puromycin until day 12. Cells were washed with PBS, fixed with methanol and then stained with 0.1% crystal violet for 15–20 min. After rinsing with water, plates were dried and scanned. Each experiment was performed in triplicate and each experiment was repeated twice.
Efficiency of colony formation was determined by plating 600 cells of different cell lines in 100-mm dishes in 3 replicates. Cells of a stable clone (cl39/Tet-On · PHF10b(HA)) carrying PHF10b (HA) under the control of pTRE-Tight promoter were either treated with DOX (1 μg/ml) to induce PHF10b(HA) expression or kept in culture medium containing Tet-free serum as a control. Cl39T/Tet-On cells were also used as control. Cells were fed every 3 days with medium with or without DOX. After 21 days, cells were fixed, stained with crystal violet and colonies counted. Each experiment was repeated 3 times.
RNAi
To down-regulate the expression of PHF10 in normal cells, different shRNAs (shRNA#1: 5′ CAGCATTGCGCAGTGATGA 3′; shRNA#2: 5′CAGCCAAACATGCTGAGTA 3′; shRNA#3: 5′ CAGAAAGTTGAAGCCAGTA 3′, and shRNA#4: 5′ AGGTCAGTTCTTACCCAGT 3′) were synthesized along with their complementary strands, annealed and then cloned into a retroviral vector, pSIREN-RetroQ (Clontech). The control shRNA (5′CATCATGGCGTAGTGGTGA 3′) was synthesized by making 4 nucleotide substitutions in the shRNA#1 as shown by bold letters. Only RNAi sequences used to generate shRNAs are shown. Retroviruses encoding these shRNAs were generated as described above.
Results and Discussion
Generation and Identification of Mouse/HAL Somatic Cell Hybrids Containing Chromosome 6 or a Fragment of Chromosome 6
To separate human chromosome 6q* from the 6qt fragment present in HALneo [see fig. 2, Banga et al., 1997], we generated mouse/HAL cell hybrids as described in Materials and Methods. Using this strategy, we isolated 130 hybrid clones. To identify mouse/HAL hybrids containing either chromosome 6q* or fragment 6qt, these clones were screened by PCR using a polymorphic marker D6S264 (derived from 6q26) which identifies 2 different alleles in HS74 and HALneo DNAs (see fig. 3A). Twelve of the hybrids amplified only 1 allele; 9 showed the upper allele while 3 amplified the lower allele (fig. 3A), indicating physical separation of both chromosome 6q* and a fragment 6qt in mouse cells. The concordance of the D6S264 (lower) allele with that amplified from AR5 confirmed previous data.
Fig. 2.

A metaphase preparation of HALneo cells showing 6q* and 6qt chromosomes (see arrows) using chromosome 6-specific painting probe.
Fig. 3.

Identification of mouse/HAL somatic cell hybrids that contain 6q* or 6qt chromosomes by PCR. A Separation of 6q* from 6qt. Genomic DNAs of different cell lines or mouse/HAL hybrids were amplified with the D6S264 polymorphic marker (from the 6q26 region) that amplifies 2 alleles in HS74 and HALneo. Nine hybrids retain the upper allele, whereas 3 hybrids retain the lower allele that is preserved in AR5. B Identification of hybrids. Genomic DNAs from a subset of these hybrids were amplified with D6S300 that amplifies a HALneo specific allele.
To specifically identify hybrids containing chromosome 6q* and which contain a 6qt, we performed PCR with another polymorphic marker, D6S300 (derived from 6q14), that amplifies only 1 (upper) allele in HALneo and thus distinguishes chromosome 6q* from the 6qt (see fig. 3B). Analysis of the mouse/human hybrids with this marker revealed that 9 of the hybrids (data for 5 of those hybrids are shown in fig. 3B) amplified the same upper allele that is present in the HALneo. The remaining 3 hybrids did not show any amplification with this marker indicating these contain chromosome 6qt; data for 2 of those hybrids are presented in figure 3B. Taken together, these results indicated that hybrids that amplified upper alleles with D6S264 and D6S300 markers carry chromosome 6q* (hereon referred to as Type I hybrids) and the hybrids which amplified the lower allele (D6S264 marker but not with D6S300 marker amplified) have chromosome 6qt (hereon referred to as Type II hybrids). These results also revealed that chromosome 6qt is derived from the chromosome that is retained in AR5, since both amplified the same allele.
Characterization of the Type I and Type II Somatic Cell Hybrids
Different polymorphic markers, non-polymorphic markers, expressed sequence tags, sequence tag sites or genes derived from the 6q25–q27 regions were used to characterize 6q* and 6qt chromosomes by PCR with the intent to identify homozygous deletions or other chromosomal rearrangements that may define the location of SEN6. Summary of these results is presented in figure 4. Analysis with the polymorphic markers confirmed that the 6qt chromosome is derived from the chromosome retained in AR5 whereas the 6q* chromosome has undergone a terminal deletion, consistent with our earlier published work [Banga et al., 1997]. Overall, our data are consistent with published mapping and sequencing data, suggesting no further large rearrangements or deletions have occurred in 6q* and 6qt chromosomes in the 6q25–q27 region.
Fig. 4.

Summary of characterization data of Type I and Type II somatic cell hybrids. Different genes (bold type), ests (underlined), sts (italicized) and markers (normal type) derived from the 6q25–q27 region were amplified by PCR using DNA from either Type I or Type II mouse/HAL hybrids.
Identification of a Chromosomal Break in MLLT4 in the 6q* Chromosome
Our FISH experiments involving 4,839 kb MLLT4 cDNA (NM_005936.2) and a 2.3-kb genomic fragment from the 5′ end (gift from R. Hauptschein and R. Dalla-Favera, New York) as probes on the HALneo cells showed a strong hybridization signal on the 6qt chromosome as compared to the 6q* chromosome (data not shown). This observation suggested that 6q* might have undergone a chromosomal break in MLLT4. To further investigate this observation, we amplified Type I and Type II hybrid DNAs with 2 primer pairs from the 5′ end within the first exon (from 1 to 90 nt and from 1 to 105 nt) as well as with 2 primer pairs from the 3′ end that amplify exon 28 (from 3,834 to 4,203 nt and from 4,077 to 4,839 nt) of the MLLT4 cDNA (NM_005936.2). Data for one primer pair from the 5′ end and one from the 3′ end is shown in figure 5. Indeed, both types of hybrids amplified the 105-bp fragment (from 1 to 105 nt) with the 5′ end primer pair, indicating no loss of these sequences occurred from the 5′ end of MLLT4. On the other hand, amplification of DNAs of both types of hybrids using primers from the 3′ end of MLLT4 resulted in the amplification of the expected 763-bp band (from 4,077–4,839 nt) only in a Type II hybrid whereas no amplification was observed in 3 Type I hybrids (see fig. 5) examined. These observations clearly demonstrate that MLLT4 has undergone a chromosomal break in the 6q* chromosome retained in HALneo immortal cell line. Further localization of the break point was not undertaken because of the large size of MLLT4 (∼140 kb). The data presented here also suggest that SEN6 is located at or distal to MLLT4 between MLLT4–TBP on 6q27. Alternatively, MLLT4 itself could be a candidate gene for SEN6.
Fig. 5.

Identification of a chromosomal break in MLLT4. PCR amplification of different DNAs with either primer from the 5′ end (5′ATGTCGGCGGGCGGCCGTG3′ and 5′CCAAATCCTCGGTCGGCTGG3′) or with primer from the 3′ end (5′TGCCGGTGATTTCGATGGAATG3′ and 5′CTATCCCCATGGGAAACACGC3′) which amplified 112- and 763-bp fragments, respectively. Italicized nucleotides in one of the first primer pairs do not match with the first exon and the underlined nucleotide is next to the end of the first exon with the result that this primer pair amplifies the entire first exon (105 bp) but the length of the amplified fragment is 112 bp. HSF43 and HS74 are 2 normal human diploid fibroblast cell lines. HALneo and AR5 are immortal cells lines. Type I and Type II mouse/HAL hybrids carry 6q* and 6qt chromosomes, respectively.
Expression Analysis of Genes at 6q27 within the Deleted Region, MLLT4–TBP
Sequencing of the human genome has identified a number of genes within the deleted region (http://genome.ucsc.edu). To gain insight whether these genes present within the candidate region are expressed in our immortal or normal cell lines, we performed RT-PCR using total RNAs. Data presented in table 1 show the expression pattern of different genes. Interestingly, we could not detect the expression of 5 genes (C6orf54, FRMD1, DACT2, SMOC2 and TCTE3) in the parental cell line HS74 or in a preimmortal SV40-transformed cell line by RT-PCR. Our repeated efforts to amplify the above mentioned genes failed to reveal any gene-specific expression, indicating that either these genes are not expressed at all or are expressed at levels below the detection threshold. These observations suggest that these genes are not likely candidates for SEN6, because they are not expressed in the parental cell lines. On the other hand, we are able to detect the expression of MLLT4, KIF25, PHF10, DLL1, THBS2, FAM120B, and WDR27, and they are, therefore, more likely candidate genes for SEN6.
Table 1.
Expression analysis of different genes from the 6q27 region (between MLLT4 and TBP) in normal (HS74) and preimmortal (SV.RNS/HF) cell lines
| Genes/ESTs | Fragment size | Starting nucleotide position of the forward primer | Forward primer sequence (5'→3') | Reverse primer sequence (5'→ 3') | HS74 RNA | Preimmortal (SV.RNS/HF) RNA |
|---|---|---|---|---|---|---|
| MLLT4 | 723 | 168,095,029 | tgccggtgatttcgatggaatg | ctatccccatgggaaacacgc | + | + |
| C6orf54 | 171 | 168,137,343 | cagagtgccgagaccctgctag | gagacgtgggtggaaggtggtt | – | – |
| KIF25 | 240 | 168,182,158 | gaccttctggccaaagacag | gcaggaggctgtggttagag | + | + |
| FRMD1 | 286 | 168,207,245 | gactacatcctccggcacat | tctccagcttctttcccaga | – | – |
| DACT2 | 109 | 168,451,285 | ctcagagccctccaagcactcg | tcgctgctgctggactcacg | – | – |
| SMOC2 | 180 | 168,585,308 | tgcagaccccggacaagtggga | ccaaagcggtcgggagaacacg | – | – |
| THBS2 | 252 | 169,367,375 | cgtggacaatgaccttgttg | caaatatcaccccgtccatc | + | + |
| WDR27 | 279 | 169,804,358 | ctgagacttgcaccctgtga | gatctccaacacggcaatct | + | + |
| PHF10 | 244 | 169,852,305 | aagctgccactccaagaaaa | cactgccatgggtaggtctt | + | + |
| TCTE3 | 254 | 169,893,470 | acagagcgagatggagaagc | ttaattttgccagggcactc | – | – |
| OLLI | 232 | 170,436,652 | tccgctatccaggctgtctc | Gcagctccctccgttcttac | + | + |
| FAM120B | 199 | 170,474,109 | cgtccaagcagaaagctacc | ccactccttgacagctaggc | + | + |
Expression of the above genes/ESTs was detected by RT-PCR using total RNA. Each RT-PCR experiment was repeated at least twice. In some cases another set of primers were also used to confirm the absence of expression. One tenth of the RT-PCR products were electrophoresed in 4% agarose gels containing ethidium bromide to visualize amplified products.
− indicates no amplification of the expected DNA fragment, whereas + indicates amplification of the expected gene fragment.
Identification of Growth Suppressor at 6q27
We analyzed 4 genes, MLLT4, DLL1, FAM120B and PHF10, as candidate genes for SEN6.
1. Analysis of MLLT4 in SV40-immortalized cells. The identification of a chromosomal break in MLLT4 in HALneo makes this gene a strong candidate for SEN6. Interestingly, this gene has been disrupted in acute myeloid leukemia with t(6;11)(q27;q23) [Prasad et al., 1993; Tanabe et al., 1996]. MLLT4 encodes a multidomain protein with 2 RAS-binding domains, a forkhead-associated domain, a dilute domain, 2 proline-rich domains, one PSD-95/Dlg-1/ZO-1 domain (PDZ) and an F-actin-binding domain [Kuriyama et al., 1996; Saito et al., 1998; Schneider et al., 1999]. MLLT4 expressess 3 transcripts of 11.2, 7.3 and 5.5 kb which encode 3 proteins (MLLT4 type 2 − 1,816 aa, MLLT4 type 3 − 1,743 aa and MLLT4 type 1 − 1,611 aa) that differ at their 3′ terminal regions [Saito et al., 1998; Lorger and Moelling, 2006]. Afadin, a mouse homolog of the human MLLT4 protein, shows 2 splice variants: a short variant (s-afadin) and a long variant (l-afadin) which correspond to MLLT4 type 1 and MLLT4 type 3 proteins. The shortest form of MLLT4 protein (MLLT4 type 1) lacks the actin-binding domain [Saito et al., 1998; Lorger and Moelling, 2006] and is localized at the cell membrane as well as in the nucleus [Mandai et al., 1997; Buchert et al., 1999, Buchert et al., 2007], whereas the long isoform of MLLT4 protein (MLLT4 type 3) has an actin-binding domain and is primarily localized to the adherens junctions in epithelial cells [Buchert et al., 1999, 2007]. MLLT4 type 2 protein is not well studied. In addition to its interaction with many proteins including Ras, MLLT4 also interacts with Bcr, a fusion partner of Abl in Philadelphia chromosome, t(9;22), which is found in 95% of patients with chronic myelogenous leukemia [Faderl et al., 1999]. These observations suggest that MLLT4 plays an important role in cell signaling at sites of cell-cell contact to maintain cells in a nonproliferating state [Radziwill et al., 2003]. Loss of expression of MLLT4 protein has recently been reported in breast cancer, further suggesting its role as a tumor suppressor [Letessier et al., 2007]. These features of MLLT4/afadin make it likely to be a growth suppressor, and therefore we investigated this gene in more detail.
a. Expression analysis of MLLT4 in normal, preimmortal and SV40-immortalized cell lines. Since the MLLT4 gene produces multiple alternatively spliced transcripts [Saito et al., 1998], we did QRT-PCR using primers and TaqMan probes (Applied Biosystems) that specifically identifies each of the 3 transcripts. All are expressed in the fibroblast cell lines. Data for MLLT4 type 2 is not shown because it does not vary among different cell lines. Data shown in figure 6 for MLLT4 type 1 and MLLT4 type 3 indicate that expression of the transcripts is quite varied among normal (HS74), preimmortal (SVtsA/HF-A, SV.RNS/HF) and immortal (AR5, HALneo, SV.RNS/HF-1, cl39T, SVtsA/HF-CH2) cell lines. However, expression of the 2 transcripts within each cell line is similar. Notably, the MLLT4 type 1 and MLLT4 type 3 transcripts in HALneo are reduced as compared to other cell lines, including the normal (HS74) and its preimmortal (SVtsA/HF-A) parental cell line. Reduced expression of the corresponding MLLT4 type 1 and MLLT4 type 3 proteins in HALneo is further observed by Western blot analysis (fig. 7). Taken together, reduced expression of MLLT4 in HALneo indicated that the gene might be mutated in other SV40-immortalized cell lines.
Fig. 6.

Expression analysis of MLLT4 mRNAs by QRT-PCR: For quantitative TaqMan RT-PCR of MLLT4 (3 variants), total RNA was isolated from cells using TRIzol Reagent (Invitrogen) and QRT-PCR was performed as described in the Materials and Methods section using reagents from Applied Biosystems. The blue bar represents probe Hs0984497_m1 that detects the shortest type 1 transcript (NM_005936.2) and the red bar represents probe Hs00988622 that detects type 3 transcript (NM_001040000.1). All samples were tested in triplicate, and average values were used for quantification (see Materials and Methods).
Fig. 7.
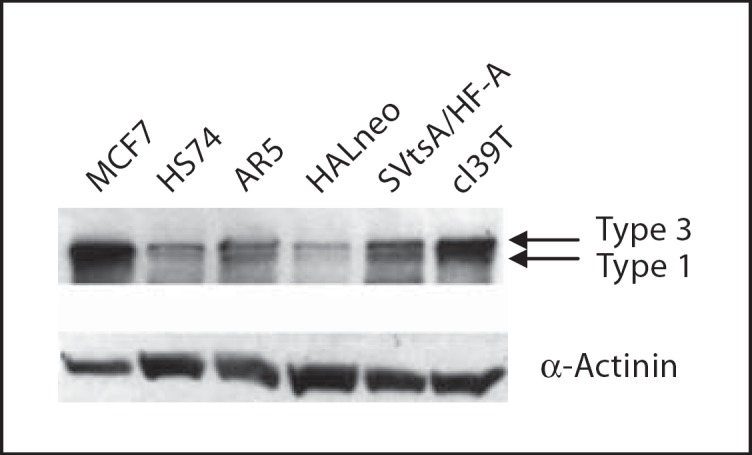
Expression of MLLT4 in SV40-immortalized human cells. 30 μg of the protein of each cell line was loaded and electrophoresed in 7.5% denaturing SDS polyacrylamide gel. After protein transfer the blot was probed with MLLT4-specific antibody and with the secondary antibody to visualize MLLT4 proteins. The same blot was probed with anti-α-actinin antibody for loading control. See text for different cell line designations. It is difficult to distinguish the larger transcripts of MLLT4 from each other. MCF7, a breast epithelial cancer cell line, was used as a positive control.
b. Mutation analysis of MLLT4 in SV40-transformed cell lines. To determine whether MLLT4 is mutated in SV40-immortalized cells (AR5, HALneo, SV.RNS/HF-1, SVtsA/HF-CH2 and cl39T), we sequenced the entire cDNAs of the 3 transcripts [Saito et al., 1998] by a combination of RT-PCR and genomic sequencing. Although sequence analysis confirmed the presence of published polymorphisms, it did not reveal any inactivating mutations in the coding region of any of the 5 immortal cell lines analyzed (data not shown). Most important, there were no inactivating mutations in AR5 and HALneo, which would be expected to share a mutation since they are both derived from the parental preimmortal SVtsA/HF-A and retain the same intact allelic copy of the MLLT4-coding region. This result suggested that inactivation of MLLT4 is not responsible for the immortalization of SV40-transformed human fibroblasts. This observation is consistent with results reported by Minaguchi et al. [1999], who did not find any mutations in the MLLT4 gene in various ovarian carcinomas showing LOH in 6q27. It is recognized that epigenetic changes or alterations other than mutation in the coding region could be responsible in HALneo, but the absence of reduced expression in other immortal cell lines makes it less likely to be the shared SEN6 mechanism.
2. Analysis of DLL1 and FAM120B (BC012177, KIAA1838) as growth suppressors. We then investigated the role of DLL1 a ligand to NOTCH receptor and BC012177 (a putative transmembrane encoding protein of unknown function) [Holden and Raymond, 2003] as growth suppressors by cloning their respective cDNAs in pTRE-Tight·PGK.Puro vector and by stably expressing them in cl39T-Tet-On cells (stably expressing rtTA, reverse tetracycline transactivator). Clones were selected by puromycin selection in the absence of doxycyline and were then subjected to growth inhibition studies in the presence as well as absence of doxycycline. Both genes did not show any effect on overall growth of the cells (data not shown). Moreover, Northern blot and RT-PCR analyses also did not show any change in gene expression for both these genes in a number of SV40-immortalized cell lines tested (data not shown). These observations eliminated DLL1 and FAM120B as putative candidate genes for SEN6.
3. Analysis of PHF10 as a growth suppressor. PHF10 belongs to a family of plant homeodomain (PHD) containing proteins which play a role in transcription regulation via chromatin remodeling [Aasland et al., 1995]. The NCBI database initially described that PHF10 had 2 known splice variants which encode 410-aa (PHF10a, NP_060758.1) or 408-aa (PHF10b, NP_579866.1) proteins based upon the model of 2 untranslated exons 1 and 2 (see fig. 1B and C). During the course of this study, the NCBI database has revised its interpretation to include exons 1 and 2 as part of the ORF. PHF10 has a SAY (supporter of activation of yellow) domain and 2 PHD finger motifs. Although PHF10 is evolutionarily conserved among species, its function in humans is unknown. However, the Drosophila homolog of PHF10, SAYP, regulates transcription both in euchromatin as well as in heterochromatin, and is an essential gene [Shidlovskii et al., 2005]. Recently SAYP has been shown to be a novel signature subunit of the PBAP chromatin remodeling complex [Chalkley et al., 2008].
a. Expression of PHF10 in different SV40-immortalized cell lines. To determine whether PHF10 gene expression is affected in different preimmortal and immortal cell lines, we performed Northern blot analysis using a 500-bp fragment from the 5′ end of PHF10bcDNA as a probe. This analysis identified 4 different PHF10 transcripts (1.9, 2.5, 4.3 and 5.0 kb) that were expressed in all cell lines (fig. 8A). Although expression of these transcripts is quite variable among different immortal cell lines as compared to preimmortal SV.RNS/HF and normal HS74 cells, expression of all transcripts was reduced in AR5 and especially SV.RNS/HF-1 cell lines. Despite the fact that PolyA+ RNA of immortal SV.RNS/HF-1 is underloaded in figure 8A as compared to its preimmortal parent SV.RNS/HF, the results of QRT-PCR (data not shown) and Northern blot analysis (see fig. 8B) using additional RNA preparations confirmed that overall expression of PHF10 transcripts is reduced in SV.RNS/HF-1. In cl39T, larger transcripts (4.3 and 5.0 kb) were more abundantly expressed as compared to normal (HS74) or preimmortal (SV.RNS/HF) cells (fig. 8A). In contrast, expression of all 4 transcripts was higher in HALneo. Reduced expression of all of the PHF10 transcripts in AR5 and SV.RNS/HF-1 immortal cell lines might be due to mutation(s) in PHF10 or due to regulation by epigenetic mechanisms.
Fig. 8.

Expression analysis of PHF10 in normal, preimmortal and SV40-immortalized cell lines. A 3 μg of polyA+ RNA from the indicated cell lines was fractionated in 0.8% sodium borate-formaldehyde agarose gels. After blotting, the membrane was hybridized with 32P-labeled 500-bp fragment of PHF10b cDNA from the 5′ end using PerfectHyb Plus hybridization buffer (Sigma). After washing, the membrane was exposed to X-ray film for 3 h. B Northern blot of the indicated cell lines probed as in panel A except that polyA+ RNA was fractionated in 1% sodium borate-formaldehyde agarose gels. The polyA+ RNAs were prepared independently in panel A and B.
The 4 transcripts of PHF10 in normal and SV40 immortalized cell lines could arise due to alternate promoter usage, alternative splicing or site of polyadenylation. Molecular structure and identity of the 2.5-, 4.3- and 5.0-kb transcripts are unknown, although our repeated efforts to clone larger transcripts were unsuccessful by 5′RACE. We believe that the 1.9-kb transcript most likely represents NM_018828.2 and NM_013325.1, 2 known isoforms of PHF10 (fig. 1). It is likely that the 4.3-kb transcript represents EST AJ420510 which has a long 3′UTR. This EST lacks exons 7 and 10–12 present in other PHF10 cDNAs.
b. Analysis of mutations in the PHF10 gene. To determine whether reduced expression of PHF10 transcripts is due to mutations, we have amplified the presumptive coding region (222–1,454 nt) of PHF10 in 4 different SV40-immortalized cell lines (AR5, cl39T, SV.RNS/HF-1 and HALneo). Sequence analysis of at least 2 clones from each immortal cell line revealed no mutations within this region. Interestingly, both alternatively spliced forms of cDNA (PHF10a and PHF10b) were expressed in all these cell lines. Absence of mutations in multiple SV40-immortalized cell lines in PHF10 sequence suggests that epigenetic pathways could be playing a role in the regulation of PHF10 expression. It should be noted that we were unable to amplify 5′ exon 1 and promoter region of PHF10 because of its high GC-rich region, and therefore might have missed mutation(s) in that region.
c. Ectopic expression of PHF10b and PHF10L results in growth suppression of normal, telomerase-immortalized and SV40-immortalized cells. In initial experiments, we observed that doxycycline-regulated stable overexpression of PHF10b(HA) cDNA resulted in the partial suppression of growth in cl39T-Tet-On immortal cells (fig. 9). These data suggested that PHF10 may have growth-suppressive properties. To eliminate the possibility that the modest effect we observed is due to clonal variation, and to extend this observation to other immortalized cell lines, we cloned PHF10b(HA) and (HA)PHF10L(FLAG) (see fig. 1C and D) in a retroviral vector pLPCX. The cloned cDNAs expressed corresponding proteins by Western blot analysis after transient transfection assays in 293T cells (data not shown). Four independently derived SV40-immortalized cell lines (cl39T, SV.RNS/HF-1, AR5, CT10–2A) were transduced with retroviruses carrying (HA)PHF10L(FLAG), PHF10(HA) or empty vector. Overexpression of PHF10b(HA) and (HA)PHF10L(FLAG) clearly exhibited reduced growth of all 4 cell lines used irrespective of their origin as compared to empty vector (fig. 10, compare columns 3 and 4 with column 2). To further investigate whether this inhibition of cell proliferation is specifically restricted to SV40-immortalized cells, we transduced 2 independent normal fibroblast (HS74, HSF43) and 3 independently derived telomerase-immortalized human fibroblast cell lines (HS74-hTERT; HSF43-hTERT; IMR90-hTERT, lung fibroblast cell line immortalized with hTERT) with retroviruses carrying (HA)PHF10L(FLAG), PHF10b(HA) or empty vector. Interestingly, both cDNAs also affected growth of normal and telomerase-immortalized cell lines as compared to empty vector control indicating that growth inhibition by these PHF10 cDNAs is not limited to SV40-immortalized cells but instead is global in nature. Similar, inhibition of growth has been observed by the overexpression of PHD finger containing proteins such as members of the ING family of proteins (ING1–5) [Gunduz et al., 2008].
Fig. 9.

Effect of overexpression of PHF10b(HA) in cl39T/Tet-On cells as monitored by colony formation assay. 600 cells of different cell lines were plated on 100-mm dishes in 3 replicates. Cells from a stable clone carrying PHF10b(HA) under the control of pTRE-tight promoter were either treated with DOX (1 μg/ml) to induce PHF10b(HA) expression or kept in culture medium containing Tet-free serum as a control. Cells were fed every 3 days with medium containing DOX or without DOX. After 21 days, colonies were fixed, stained with crystal violet and counted. Data presented are the mean of 3 independent experiments. Column 1 – cl39T Tet-on cells, column 2 – cl39T/Tet-On·PHF10b(HA) cells (DOX–), and column 3 – cl39T/Tet-On·PHF10b(HA) cells (DOX+).
Fig. 10.

Growth inhibition of normal, telomerase-immortalized and different SV40-immortalized fibroblasts. Each cell line was transduced with retroviruses containing PHF10b(HA) (column 3), (HA)PHF10L(FLAG) (column 4) cDNAs or empty vector (column 2) as described in Materials and Methods. Untransduced cells are shown in column 1. After selection for puromycin resistance (0.3–0.7 μg/ml for different SV40-immortalized cell lines and 0.6 μg/ml for normal and telomerase-immortalized cell lines) for 12 days, dishes were stained with crystal violet to visualize growth. The AR5 cell line was cultured at 35°C.
d. Down-regulation of PHF10 expression causes loss of cell proliferation in normal HS74 fibroblast cells. Since overexpression of PHF10b(HA) or (HA)PHF10L(FLAG) causes reduced cell proliferation, we hypothesized that depletion of PHF10 function by RNAi should be growth stimulatory or should extend the life span of normal fibroblasts. With that rationale, we cloned different PHF10 shRNAs into a retroviral vector (pSIREN-RetroQ, Clontech) and then cotransfected these with PHF10b(HA) cDNA to identify shRNAs that are effective in down-regulating PHF10 protein. Western blot analysis shows that shRNA#1 and shRNA#4 both significantly reduced ectopically expressed PHF10b(HA) protein as compared to control shRNA, shRNA#2 and shRNA#3 (fig. 11). All classes of endogenous PHF10 mRNA are reduced in transfected SV40-immortalized cl39T cells treated with shRNA#1 (data not shown). To determine the effect of down-regulation of PHF10 function by shRNA#1 and shRNA#4 on the extension of life span of normal HS74 cells, we transduced these cells serially, first with retroviruses carrying shRNA#1 and then with shRNA#4 within 24 h. Surprisingly, depletion of PHF10 negatively affected growth of HS74 cells as well (fig. 12). This result is contrary to the growth inhibition data obtained with the expression of PHF10b(HA)and (HA)PHF10L(FLAG) cDNAs (see fig. 10). Since a similar effect was seen with shRNA#1 and shRNA#4 in a cancer as well as in a normal telomerase-immortalized cell line [Banga, unpubl. observations], we conclude that PHF10 function is required for cell growth. Accordingly, the growth suppression observed with different epitope tagged PHF10 cDNAs is most likely to be due to the following reasons. (1) At the time of cloning, the available PHF10 cDNAs or ests represented complete or full-length PHF10 cDNAs. Recently, NCBI has revised the full-length nature of PHF10 cDNA to include all 12 exons with a coding potential of 498 aa (see fig. 1). Therefore, it is now evident that we were examining the effect of overexpression of epitope-tagged truncated proteins in the cells, and therefore these proteins might be acting as dominant negative mutants. (2) This effect on cell proliferation is especially important for a protein whose function is essential for cell viability or survival. (3) This effect on growth suppression becomes more likely in the case of a protein that is a component of a multiprotein chromatin remodeling complex in humans [Banga, unpubl. observations] and in Drosophila [Chalkley et al., 2008]. It is also possible that growth inhibition could also be due to unregulated residual functioning of these overexpressed truncated PHF10 proteins during the cell cycle since they are driven by constitutively active CMV promoter. A similar effect on cell proliferation was previously observed when PW29, a nuclear protein involved in sister chromatid cohesion, was expressed as PW29–GFP fusion [Darwiche et al., 1999], whereas expression of H2B-GFP did not affect cell proliferation [Kanda et al., 1998]. Finally, it might be considered that marked alteration in the level of mutant PHF10 protein by overexpression or underexpression distorts a multiprotein complex dependent on PHF10 resulting in inhibition of cell proliferation or cell death.
Fig. 11.
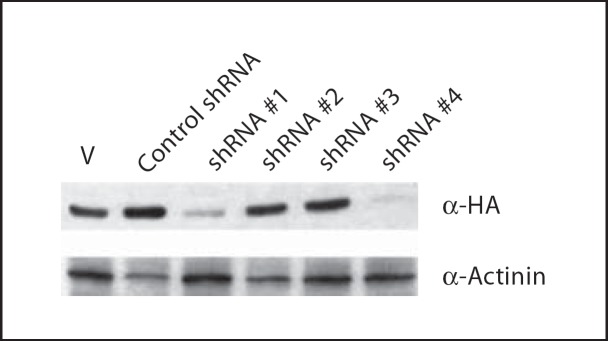
Identification of RNAi sequences effective in silencing PHF10 expression. One μg of PHF10b(HA) plasmid and 4 μg of pSIREN-RetroQ vector (V) or vector carrying respective hairpin shRNA sequences were transfected into 293T cells using Lipofectamine 2000 (Invitrogen). After 20–22 h of transfection, cell lysates were made using RIPA and analyzed by Western blotting (30 μg protein/lane). The same membrane was sequentially exposed to anti-α-HA high-affinity antibody (Roche), and to anti-α-actinin antibody (Sigma) as loading control.
Fig. 12.

Loss of cell proliferation in normal fibroblast cells by RNAi: HS74 cells were transduced with retroviruses containing shRNA#1, shRNA#4 or control shRNA as described in Materials and Methods. After selection for puromycin resistance (0.6 μg/ml) for 12 days, dishes were stained with crystal violet to visualize growth.
Our overexpression studies involving epitope-tagged PHF10 also revealed that PHF10-derived growth inhibition in 4 different SV40-immortalized and 3 telomerase-immortalized normal fibroblast cell lines is independent of the presence of the catalytic subunit of telomerase, hTERT. For example, cl39T immortal cells have long telomeres but do not express telomerase whereas AR5 (long telomeres) and SV.RNS/HF-1 (short telomeres) both express high levels of telomerase [Macera-Bloch, 2002]. In addition, all 4 cell lines used in this study were immortalized differently from each other. For example, CT10–2A was derived with wild-type SV40; cl39T was immortalized with origin defective SV40 early region DNA; AR5 was obtained with origin defective SV40 encoding temperature-sensitive T antigen tsA58 DNA; and SV.RNS/HF-1 was obtained with origin defective SV40 DNA encoding a wild-type T antigen. These observations suggest that some other mechanism(s) of growth suppression independent of T antigen (and thereby independent of pRb and p53) is involved with PHF10.
In summary, we have further refined the location of the senescence-inducing gene defective in SV40-immortalized cell lines, the putative growth suppressor SEN6, to the 6q27 region between MLLT4 and TBP by characterizing Type I (6q*) and Type II (6qt) mouse/human hybrids with polymorphic and non-polymorphic markers. Generation of Type I and Type II hybrids made the 6q27 region more amenable to molecular analysis with different classes of DNA markers using agarose gels, and allowed us to identify the chromosomal break in MLLT4. This strategy should be applicable to analyze any chromosome or chromosomal fragment from human cells of interest at the physical and expression level. Analysis of MLLT4, DLL1, FAM120B and PHF10 genes located in the deleted region and expressed in immortal, preimmortal and normal cells did not show features consistent with expectations for SEN6. In the case of epitope-tagged PHF10 cDNAs, growth suppression observed in normal, telomerase-immortalized and SV40-immortalized fibroblast cells could be due to a dominant negative effect or unregulated overexpression of PHF10b or PHF10L cDNAs, and hence PHF10 is also not a candidate gene for SEN6. This conclusion is consistent with the observation that PHF10 function is required for cell proliferation based upon RNAi studies.
Acknowledgements
We thank Robert Hauptschein and Riccardo Dalla-Favera (New York) for providing MLLT4 cDNA, the 5′ genomic fragment of MLLT4 and many primers. We thank Alexandra Soggiu for assistance in tissue culture and Sutapa Sahay for help with Western blots. We also thank the Molecular Resource Facility of New Jersey Medical School for help in synthesis of primers and sequencing. This work was supported by research grants from NIH National Institute on Aging (AG04821 to H.L.O.), foundation of UMDNJ (to S.S.B.) and NJMS-UH Cancer Research Program from the state of New Jersey.
References
- Aasland RT, Gibson TJ, Stewart AF. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci. 1995;20:56–59. doi: 10.1016/s0968-0004(00)88957-4. [DOI] [PubMed] [Google Scholar]
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banga SS, Hall KT, Sandhu Ak, Weaver DT, Athwal RS. Complementation of V(D)J recombination defect and X-ray sensitivity of scid mouse cells by human chromosome 8. Mutat Res. 1994;315:239–247. doi: 10.1016/0921-8777(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Banga S, Kim S, Hubbard K, Dasgupta T, Jha K, et al. SEN6, a locus for SV40-mediated immortalization of human cells, maps to 6q26–27. Oncogene. 1997;14:313–321. doi: 10.1038/sj.onc.1200842. [DOI] [PubMed] [Google Scholar]
- Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–56. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Reddel RR. SV40-induced immortalization of human cells. Crit Rev Oncog. 1994;5:331–357. doi: 10.1615/critrevoncog.v5.i4.10. [DOI] [PubMed] [Google Scholar]
- Buchert M, Schneider S, Meskenaite V, Adams MT, Canaani E, et al. The junction-associated protein AF-6 interacts and clusters with specific Eph receptor tyrosine kinases at specialized sites of cell-cell contact in the brain. J Cell Biol. 1999;144:361–371. doi: 10.1083/jcb.144.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchert M, Poon C, King JA, Baechi T, D’Abaco G, et al. AF6/s-afadin is a dual residency protein and localizes to a novel subnuclear compartment. J Cell Physiol. 2007;210:212–223. doi: 10.1002/jcp.20853. [DOI] [PubMed] [Google Scholar]
- Chalkley GE, Moshkin YM, Langenberg K, Bezstarosti K, Blastyak A, et al. The transcriptional coactivator SAYP is a trithorax group signature subunit of the PBAP chromatin remodeling complex. Mol Cell Biol. 2008;28:2920–2929. doi: 10.1128/MCB.02217-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwiche N, Freeman LA, Strunnikov A. Characterization of the components of the putative mammalian sister chromatid cohesion complex. Gene. 1999;233:39–47. doi: 10.1016/s0378-1119(99)00160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza AT, Hankins GR, Washington MK, Fine RL, Orton TC, Jirtle RL. Frequent loss of heterozygosity on 6q at the mannose 6-phosphate/insulin-like growth factor II receptor locus in human hepatocellular tumors. Oncogene. 1995;10:1725–1729. [PubMed] [Google Scholar]
- Devilee P, van Vliet M, van Sloun P, Kuipers Dijkshoorn N, Hermans J, et al. Allelotype of human breast carcinoma: a second major site for loss of heterozygosity is on chromosome 6q. Oncogene. 1991;6:1705–1711. [PubMed] [Google Scholar]
- Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology. 1999;13:169–180. [PubMed] [Google Scholar]
- Foulkes WD, Ragoussis J, Stamp GW, Allan GJ, Trowsdale J. Frequent loss of heterozygosity on chromosome 6 in human ovarian carcinoma. Br J Cancer. 1993;67:551–559. doi: 10.1038/bjc.1993.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidano G, Hauptschein RS, Parsa NZ, Offit K, Rao PH, et al. Deletions involving 2 distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood. 1992;80:1781–1787. [PubMed] [Google Scholar]
- Gunduz E, Gunduz M, Beder LB, Tamamura R, Nagatsuka H, Nagai N. Inhibitor of growth (ING) family: An emerging molecular target for cancer therapy. J Hard Tissue Biol. 2008;17:1–10. [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Holden S, Raymond FL. The human gene CXorf17 encodes a member of a novel family of putative transmembrane proteins: cDNA cloning and characterization of CXorf17 and its mouse ortholog orf34. Gene. 2003;318:149–161. doi: 10.1016/s0378-1119(03)00770-4. [DOI] [PubMed] [Google Scholar]
- Honchel R, McDonnell S, Schaid DJ, Thibodeau SN. Tumor necrosis factor-alpha allelic frequency and chromosome 6 allelic imbalance in patients with colorectal cancer. Cancer Res. 1996;56:145–149. [PubMed] [Google Scholar]
- Hubbard-Smith K, Patsalis P, Pardinas JR, Jha KK, Henderson AS, Ozer HL. Altered chromosome 6 in immortal human fibroblasts. Mol Cell Biol. 1992;12:2273–2281. doi: 10.1128/mcb.12.5.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha KK, Banga S, Palejwala V, Ozer HL. SV40-Mediated immortalization. Exp Cell Res. 1998;245:1–7. doi: 10.1006/excr.1998.4272. [DOI] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- Kuriyama M, Harada N, Kuroda S, Yamamoto T, Nakafuku M, et al. Identification of AF-6 and canoe as putative targets for Ras. J Biol Chem. 1996;271:607–610. doi: 10.1074/jbc.271.2.607. [DOI] [PubMed] [Google Scholar]
- Letessier A, Garrido-Urbani S, Ginestier C, Fournier G, Esterni B, et al. Correlated break at PARK2/FRA6E and loss of AF-6/Afadin protein expression are associated with poor outcome in breast cancer. Oncogene. 2007;26:298–307. doi: 10.1038/sj.onc.1209772. [DOI] [PubMed] [Google Scholar]
- Lorger M, Moelling K. Regulation of epithelial wound closure and intercellular adhesion by interaction of AF6 with actin cytoskeleton. J Cell Sci. 2006;119:3385–3398. doi: 10.1242/jcs.03027. [DOI] [PubMed] [Google Scholar]
- Macera-Bloch L.SV40-transformed human fibroblasts: An analysis of crisis and differential gene expression upon immortalization. Ph.D. Dissertation (University of Medicine and Dentistry of New Jersey, Newark 2002).
- Macera-Bloch L, Houghton J, Lenahan M, Jha KK, Ozer HL. Termination of lifespan of SV40-transformed human fibroblasts in crisis is due to apoptosis. J Cell Physiol. 2002;190:332–344. doi: 10.1002/jcp.10062. [DOI] [PubMed] [Google Scholar]
- Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, et al. Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. 1997;139:517–528. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millikin D, Meese E, Vogelstein B, Witkowski C, Trent J. Loss of heterozygosity for loci on the long arm of chromosome 6 in human malignant melanoma. Cancer Res. 1991;51:5449–5453. [PubMed] [Google Scholar]
- Minaguchi T, Matsushima M, Saito S, Kanamori Y, Shirahama S, et al. Complete DNA sequence and characterization of a 330-kb VNTR-rich region on chromosome 6q27 that is commonly deleted in ovarian cancer. DNA Res. 1999;6:131–136. doi: 10.1093/dnares/6.2.131. [DOI] [PubMed] [Google Scholar]
- Morita R, Saito S, Ishikawa J, Ogawa O, Yoshida O, et al. Common regions of deletion on chromosomes 5q, 6q, and 10q in renal cell carcinoma. Cancer Res. 1991;51:5817–5820. [PubMed] [Google Scholar]
- Neufeld DS, Ripley S, Henderson A, Ozer HL. Immortalization of human fibroblasts transformed by origin-defective simian virus 40. Mol Cell Biol. 1987;7:2794–2802. doi: 10.1128/mcb.7.8.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- Orphanos V, McGown G, Hey Y, Thorncroft M, Santibanez-Koref M, et al. Allelic imbalance of chromosome 6q in ovarian tumours. Br J Cancer. 1995;71:666–669. doi: 10.1038/bjc.1995.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Gu Y, Alder H, Nakamura T, Canaani O, et al. Cloning of the ALL-1 fusion partner, the AF-6 gene, involved in acute myeloid leukemias with the t(6;11) chromosome translocation. Cancer Res. 1993;53:5624–5628. [PubMed] [Google Scholar]
- Radna RL, Caton Y, Jha KK, Kaplan P, Li G, et al. Growth of immortal simian virus 40 tsA-transformed human fibroblasts is temperature dependent. Mol Cell Biol. 1989;9:3093–3096. doi: 10.1128/mcb.9.7.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziwill G, Erdmann RA, Margelisch U, Moelling K. The Bcr kinase downregulates Ras signaling by phosphorylating AF-6 and binding to its PDZ domain. Mol Cell Biol. 2003;23:4663–4672. doi: 10.1128/MCB.23.13.4663-4672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray FA, Kraemer PM. Frequent deletions in nine newly immortal human cell lines. Cancer Genet Cytogenet. 1992;59:39–44. doi: 10.1016/0165-4608(92)90155-2. [DOI] [PubMed] [Google Scholar]
- Rodier F, Kim SH, Nijjar T, Yaswen P, Campisi J. Cancer and aging: the importance of telomeres in genome maintenance. Int J Biochem Cell Biol. 2005;37:977–990. doi: 10.1016/j.biocel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Saha V, Lillington DM, Shelling AN, Chaplin T, Yaspo ML, et al. AF6 gene on chromosome band 6q27 maps distal to the minimal region of deletion in epithelial ovarian cancer. Genes Chromosomes Cancer. 1995;14:220–222. doi: 10.1002/gcc.2870140311. [DOI] [PubMed] [Google Scholar]
- Saito S, Saito H, Koi S, Sagae S, Kudo R, et al. Fine-scale deletion mapping of the distal long arm of chromosome 6 in 70 human ovarian cancers. Cancer Res. 1992;52:5815–5817. [PubMed] [Google Scholar]
- Saito S, Matsushima M, Shirahama S, Minaguchi T, Kanamori Y, et al. Complete genomic structure, DNA polymorphisms, and alternative splicing of the human AF-6 gene. DNA Res. 1998;5:115–120. doi: 10.1093/dnares/5.2.115. [DOI] [PubMed] [Google Scholar]
- Sandhu AK, Hubbard K, Kaur GP, Jha KK, Ozer HL, Athwal, RS. Senescence of immortal human fibroblasts by the introduction of normal human chromosome 6. Proc Natl Acad Sci USA. 1994;91:5498–5502. doi: 10.1073/pnas.91.12.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Buchert M, Georgiev O, Catimel B, Halford M, et al. Mutagenesis and selection of PDZ domains that bind new protein targets. Nat Biotechnol. 1999;17:170–175. doi: 10.1038/6172. [DOI] [PubMed] [Google Scholar]
- Schwartz D, Rotter V. p53-dependent cell cycle control: response to genotoxic stress. Semin Cancer Biol. 1998;8:325–336. doi: 10.1006/scbi.1998.0095. [DOI] [PubMed] [Google Scholar]
- Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- Shidlovskii YV, Krasnov AN, Nikolenko JV, Lebedeva LA, Kopantseva M, et al. A novel multidomain transcription coactivator SAYP can also repress transcription in heterochromatin. EMBO J. 2005;24:97–107. doi: 10.1038/sj.emboj.7600508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shridhar V, Staub J, Huntley B, Cliby W, Jenkins R, et al. A novel region of deletion on chromosome 6q23.3 spanning less than 500 Kb in high grade invasive epithelial ovarian cancer. Oncogene. 1999;18:3913–3918. doi: 10.1038/sj.onc.1202756. [DOI] [PubMed] [Google Scholar]
- Small MB, Gluzman Y, Ozer HL. Enhanced transformation of human fibroblasts by origin-defective simian virus 40. Nature. 1982;296:671–672. doi: 10.1038/296671a0. [DOI] [PubMed] [Google Scholar]
- Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996;273:63–67. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- Stein GH, Dulic V. Origins of G1 arrest in senescent human fibroblasts. Bioessays. 1995;17:537–543. doi: 10.1002/bies.950170610. [DOI] [PubMed] [Google Scholar]
- Steinemann D, Gesk S, Zhang Y, Harder L, Pilarsky C, et al. Identification of candidate tumor-suppressor genes in 6q27 by combined deletion mapping and electronic expression profiling in lymphoid neoplasms. Genes Chromosomes Cancer. 2003;37:421–426. doi: 10.1002/gcc.10231. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol. 2006;22:531–557. doi: 10.1146/annurev.cellbio.22.010305.104518. [DOI] [PubMed] [Google Scholar]
- Tanabe S, Zeleznik-Le NJ, Kobayashi H, Vignon C, Espinosa R, 3rd, et al. Analysis of the t(6;11)(q27;q23) in leukemia shows a consistent breakpoint in AF6 in three patients and in the ML-2 cell line. Genes Chromosomes Cancer. 1996;15:206–216. doi: 10.1002/(SICI)1098-2264(199604)15:4<206::AID-GCC2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Tibiletti MG, Bernasconi B, Furlan D, Riva C, Trubia M, et al. Early involvement of 6q in surface epithelial ovarian tumors. Cancer Res. 1996;56:4493–4498. [PubMed] [Google Scholar]
- Tibiletti MG, Trubia M, Ponti E, Sessa L, Acquati F, et al. Physical map of the D6S149-D6S193 region on chromosome 6q27 and its involvement in benign surface epithelial ovarian tumors. Oncogene. 1998;16:1639–1642. doi: 10.1038/sj.onc.1201654. [DOI] [PubMed] [Google Scholar]
- Tibiletti MG, Sessa F, Bernasconi B, Cerutti R, Broggi B, et al. A large 6q deletion is a common cytogenetic alteration in fibroadenomas, pre-malignant lesions, and carcinomas of the breast. Clin Cancer Res. 2000;6:1422–1431. [PubMed] [Google Scholar]
- Vaziri H, Benchimol S. Alternative pathways for the extension of cellular life span: Inactivation of p53/pRb and expression of telomerase. Oncogene. 1999;18:7676–7680. doi: 10.1038/sj.onc.1203016. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Wright WE, Pereira-Smith OM, Shay JW. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol. 1989;9:3088–3092. doi: 10.1128/mcb.9.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]