

Abstract
Exchange of the glucocorticoid receptor (GR) at promoter target sites provides the only known system in which transcription factor cycling at a promoter is fast, occurring on a time scale of seconds. The mechanism and function of this rapid exchange are unknown. We provide evidence that proteasome activity is required for rapid GR exchange at a promoter. We also show that chaperones, specifically hsp90, stabilize the binding of GR to the promoter, complicating models in which the associated chaperone, p23, has been proposed to induce GR removal. Our results are the first to connect chaperone and proteasome functions in setting the residence time of a transcription factor at a target promoter. Moreover, our results reveal that longer GR residence times are consistently associated with greater transcriptional output, suggesting a new paradigm in which the rate of rapid exchange provides a means to tune transcriptional levels.
Based on fluorescence recovery after photobleaching (FRAP) analysis, most nuclear proteins are now known to be highly mobile (28). The list of rapidly moving proteins includes splicing factors (17), nucleolar proteins (9), histone H1 (19, 23), and several steroid receptor transcription factors (8, 22, 31, 34, 43). In all cases, transcription factor mobility is slower than that of GFP alone, demonstrating that all of these proteins transiently interact with nuclear binding sites of some sort. The majority of these sites cannot be specific promoters, given the numbers of expressed molecules (34). Rather, binding to chromatin or nuclear matrix is more likely (34, 43). Thus, these nuclear FRAP data provide insights about trafficking of proteins within the nucleus, but they do not directly address transcription factor binding to a promoter.
In a limited number of cases, binding of transcription factors to specific promoters has been studied. Here again, mobility has been detected, indicating that transcription factors do not remain permanently bound at a promoter but rather undergo cycles of binding and unbinding. The first evidence for this came from FRAP experiments using a tandem array of mouse mammary tumor virus (MMTV) promoter sites visualized with a (GFP)-tagged glucocorticoid receptor (GR) (22). Rapid exchange of this receptor was observed, with a total recovery time of less than a minute. Later studies using chromatin immunoprecipitation (ChIP) have shown that the estrogen receptor (ER) cycles at several different promoters but with a markedly longer periodicity, on the order of 1 h (31, 37). In addition to the transcription factors themselves, associated factors also exhibit exchange at promoters, but again with very different time scales depending on whether the experimental approach is FRAP, where rapid exchange is observed (3, 42), or ChIP, where in general slower cycling is detected (4, 31, 37). When the temporal resolution of ChIP was pushed to its limits, reciprocal cycling of two ER coactivator complexes (DRIP and p160) could be detected on a time scale as short as 2.5 min (4). These data suggest that much faster exchange exists in other systems but at or below the limits of ChIP temporal resolution.
Much remains to be learned about the mechanisms of transcription factor cycling. The slow cycling of ER requires proteasomal activity (31). In the case of rapid GR exchange, nothing is known except hints that chaperones and chromatin remodelers could be involved. Freeman and Yamamoto (12) showed that the molecular chaperone p23 can induce disassembly of thyroid receptor transcriptional regulatory complexes. They also found that in vivo targeting of a gal4-p23 fusion protein to a GR promoter could significantly reduce transcriptional activation there. Based on these and other experiments, they suggested that p23 could be involved in removing GR during rapid exchange. An in vitro chromatin-remodeling system has revealed that recruitment of Swi/Snf is accompanied by loss of GR, leading to the suggestion that chromatin remodelers may play a role in rapid GR exchange (11).
The function of transcription factor exchange is also not well understood. In the cases identified to date, it has been suggested that receptor cycling at a promoter is a mechanism to sense changes in hormone levels (12, 22, 31, 37). It has also been suggested that proteasomal removal of potent transcription factors from their promoters may be a means of limiting transcriptional output (24). However, beyond these hypotheses, no other functions have been proposed for transcription factor exchange.
Our aim in this study was to investigate the mechanism and function of rapid GR exchange observed in live cells at a tandem array of MMTV promoter sites. We found that both chaperones and proteasomes are present at the target sites. Disruption of either leads to opposite alterations in the exchange rate, indicating that the exchange rate is normally regulated in part by a balance between chaperone and proteasome activities. We also found a correlation between GR exchange and the amount of transcription, suggesting that longer GR residence times favor more transcription.
MATERIALS AND METHODS
Cell lines.
Cell line 3617 was used for most experiments. The cells contain 200 tandem repeats of a 9-kb element composed of the MMTV promoter followed by ras and BPV genes (16) and they stably express GFP-tagged GR under the control of a tetracycline-off system (44). Control experiments were done with the parental cell line 3134, which contains the tandem repeats but not GFP-tagged GR. To generate cells containing only GFP, 3134 cells were transfected with a GFP plasmid (pEGFPC1; Clontech). To generate GFP-HP1α-containing cells, 3134 cells were transfected with a plasmid for the construct (a kind gift of T. Cheutin and T. Misteli, Laboratory of Receptor Biology and Gene Expression, National Cancer Institute, Bethesda, Md.).
Cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL) supplemented with 2 mM glutamine (Gibco BRL), 10% fetal bovine serum (HyClone), and 5 μg of tetracycline/ml (to suppress GFP-GR expression). In preparation for microscopy experiments, cells were transferred to this medium and left there overnight, except that tetracycline was omitted, the fetal bovine serum was charcoal-dextran treated (Gemini Bio-Product) to remove the steroids that could activate GFP-GR, and phenol red-free medium was used to eliminate autofluorescence. Just prior to experiments, GFP-GR was activated by either the synthetic hormone dexamethasone (Sigma), the natural hormone corticosterone (Sigma), or the antagonist RU486 (a kind gift of Cathy Smith, Laboratory of Receptor Biology and Gene Expression), each at a concentration of 100 nM.
FRAP.
For FRAP experiments, cells were grown overnight in coverglass chambers (Lab-Tech) and then induced with hormone for 30 min. For hsp90 or proteasome inhibition, geldanamycin (2.5 μg/ml) or MG-132 (100 μM) (Calbiochem), respectively, was applied after hormone induction. In both cases, parallel control experiments with the vehicle (dimethyl sulfoxide) were conducted. After geldanamycin treatment, live-cell chambers were kept on the microscope stage for no more than 5 min with corticosterone or 30 min with dexamethasone to avoid stress leading to formation of GFP-GR aggregates. For the ATP depletion experiments, cells were treated with 10 mM sodium azide (Sigma) in glucose-minus DMEM supplemented with 6 mM 2-deoxyglucose (Sigma) or with glucose-minus DMEM without sodium azide and with 6 mM 2-deoxyglucose only. FRAP experiments were carried out on a Zeiss 510 confocal microscope with either a 40× 1.3-numerical-aperture (N.A.) or a 100× 1.3-N.A. oil immersion objective. The cells were kept at 37°C using an air stream stage incubator (Nevtek). Bleaching was performed with the 488- and 514-nm lines from a 45- mW argon laser operating at 75% laser power. A single iteration was used for the bleach pulse, which lasted 0.018 s for the 40× objective or 0.065 s for the 100× objective. Fluorescence recovery was monitored at low laser intensity (0.2% for a 45-mW laser) at 40- to 500-ms intervals, depending on the experiment.
FRAP analysis.
Approximately 10 separate FRAPs were performed and then averaged to generate a single FRAP curve. The temporal resolution was kept constant while recovery was measured, but this led to a very large number of closely spaced points in the second, slower phase of the recovery curve. To address this, we averaged 3 to 10 adjacent points in this slower part of the curve. This generated roughly equally spaced points along the recovery curve and therefore avoided overly weighting the slower phase of the curve during fitting.
The effective diffusion fit was performed using the Soumpasis theory for a circular bleach spot (39) as implemented in Matlab with the nlinfit routine. In the Soumpasis theory, the FRAP rate is given by τ = w2/4D, where w is the bleach spot radius and D is the diffusion (or effective diffusion) constant. The FRAP data are given by frap(t) = e−2r/t[I0(2r/t) + I1(2r/t)], where t is time and I0 and I1 are modified Bessel functions. Bessel functions and their variants typically arise in differential equations with cylindrical symmetry (1), as occurs for a FRAP with a circular bleach spot (15). The Soumpasis theory presumes a normalized FRAP that ranges from 0 to 1. To accommodate this, we renormalized our FRAP data by setting the bleach depth to 0 and the final recovery level to 1. This factors out any differences between recoveries due to either the bleach depth or the final immobile fraction. Thus, the parameter τ directly measures the rate at which the recovery curve rises. For each FRAP, the estimate for τ yielded a standard error, which was then used in a t test to compute a P value in statistical comparisons of two recovery curves. These P values are shown in all comparison plots for GFP-GR recoveries.
The predicted diffusion constant can be used to estimate the mass of a molecule, assuming that there are no binding interactions. The Soumpasis fit for GFP-GR nuclear mobility leads to an estimate of DGFP-GR of 1.05 μm2/s. The Soumpasis fit for GFP only in nuclei of the same cell line leads to an estimate of DGFP of 15 μm2/s (data not shown), ∼15-fold slower than GFP-GR. Since D ∝ M−1/3, where M is mass, the FRAP data predict a 153 increase in mass of GFP-GR relative to GFP, or ∼95-MDa molecular mass for GFP-GR. This is much larger than any known molecular complex, and therefore the mobility of GFP-GR in nucleoplasm must be retarded by binding interactions.
When diffusion and binding interactions are present, the simplest scenario is effective diffusion (15). In effective diffusion, the FRAP mimics diffusion but at a lower rate given by the equation 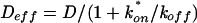 , where D is the cellular diffusion constant, koff is the off rate of binding, and
, where D is the cellular diffusion constant, koff is the off rate of binding, and 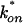 is the product of the on rate for binding times the equilibrium concentration of binding sites. The Soumpasis equation (39) then becomes
is the product of the on rate for binding times the equilibrium concentration of binding sites. The Soumpasis equation (39) then becomes 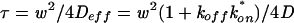 , where w is the bleach spot radius. To generate the family of curves in Fig. 1f, we used typical experimental parameters (τ = 0.5, w = 1.0 μm, and D = 15 μm2/s) to obtain a value for
, where w is the bleach spot radius. To generate the family of curves in Fig. 1f, we used typical experimental parameters (τ = 0.5, w = 1.0 μm, and D = 15 μm2/s) to obtain a value for 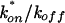 . Then, koff was varied as indicated in the figure legend to yield new values for τ, which were then used to produce new FRAP curves from the Soumpasis equation describing FRAP.
. Then, koff was varied as indicated in the figure legend to yield new values for τ, which were then used to produce new FRAP curves from the Soumpasis equation describing FRAP.
FIG. 1.

FRAPs at the MMTV array and elsewhere in the nucleus depend on bleach spot size and are well fit by an effective diffusion model. (a) 3617 cell after 30-min induction with 100 nM dexamethasone. The MMTV array is visible as a bright spot (circled) near one of the nucleoli. Scale bar, 10 μm. (b) For fast data collection during FRAP, images were collected only in the strip encompassed by the red rectangle in panel a. Selected time points (t) are shown. (c) Arrays were bleached with spot radii of either 0.9 or 1.8 μm. In all cases, the array was large enough to completely fill the bleach spot area. Larger bleach spots result in significantly slower recoveries. This indicates that diffusion contributes to the FRAP. For these and all succeeding FRAP curves, standard errors for all points were always <0.01 and therefore smaller than the dots used for plotting. (d) GFP-GR recovery elsewhere in the nucleus also shows dependence on bleach spot size, indicating a role for GFP-GR diffusion in nuclear mobility. (e) GFP-GR FRAP data both at the array and elsewhere in the nucleus were well fit by a single-parameter model for a diffusing molecule bleached by a circular spot with a recovery rate given by the fitting parameter, τ. Recoveries at the array are consistently slower than elsewhere in the nucleus. This difference is statistically significant, as indicated by a t test using the means for τ and their 95% confidence intervals. The computed P value (<0.0001) is highly significant. (f) Models for FRAPs that incorporate both diffusion and binding show that significant changes in binding parameters yield small changes in the FRAP curve. This is because diffusion remains unaffected after treatments that affect binding. Predicted curves are shown for a series of off rates.
Transcription assay.
Transcription levels were measured at the MMTV array by RNA fluorescent in situ hybridization (FISH) exactly as described by Müller et al. (25). The specific conditions for each transcriptional assay were as follows. When studying the effects of geldanamycin, cells were induced with hormone for 15 min to allow GFP-GR to translocate to the nucleus and were then treated for 45 min with the drug or with vehicle (dimethyl sulfoxide). To determine transcriptional levels following drug treatment, RNA FISH levels were corrected for transcription in the first 15 min when hormone was present but before any drug was added. This was done by measuring average transcriptional levels after 15 min of hormone treatment and then subtracting this value from the average RNA FISH intensity following the 45-min drug treatment.
For MG-132, cells were pretreated with the drug for 1.5 h and induced with hormone for 30 min before fixation. In all cases, average transcriptional levels were obtained from at least 35 cells. When transcriptional levels from condensed versus decondensed arrays were measured, 100 nM dexamethasone was added, and then the cells were prepared for RNA FISH after 3 h. A total of 113 cells were examined, and a 6-μm-diameter perimeter was chosen as the threshold value separating condensed from decondensed arrays.
Immunofluorescence.
Cells were grown overnight on 22-mm2 coverslips and then induced with 100 nM dexamethasone or corticosterone and fixed either with absolute methanol at −20oC for 15 min or for 5 min in 1 part 37% formaldehyde and 9 parts PEM buffer (100 mM PIPES, 5 mM EGTA, 2 mM MgCl2, pH 6.8, plus 0.2% Triton X-100). After either fixation, the cells were washed three times in phosphate-buffered saline (PBS) for 10 min each time. Similar immunofluorescence results were obtained with both fixation protocols, except that the methanol fix was required to detect any staining with the hsp90 antibody.
For detection of GFP-GR aggregates, corticosterone-induced cells were treated for 10 min with 2.5 μg of geldenamycin/ml followed by 20 min of either heat (45°C) or cold (on ice) shock and were fixed in 3.5% paraformaldehyde at room temperature for 15 min. Following formaldehyde fixation, the cells were permeabilized for 10 min with 0.5% Triton X-100 in PBS and washed three times in PBS for 10 min each time.
For all immunofluorescence experiments, cells were incubated overnight with the primary antibody diluted in PBS with 4% bovine serum albumin and 0.1% Tween 20. After incubation, the cells were washed three times in PBS for 10 min each time and then incubated for 1 to 2 h with the appropriate secondary antibody conjugated to either Texas red or rhodamine. The cells were then washed three more times in PBS before final mounting in PBS and examination on a Leica DMRA microscope with a Leica 100× 1.3-N.A. oil immersion objective. Images were acquired in green (GFP-GR) and red (antibody) fluorescence with a SenSys (Photometrics) camera with a KAF1400 chip configured to collect 0.067-μm-diameter pixels.
The following antibodies were used: anti-hsp70 (MA3-006; Affinity Bioreagents), anti-p23 (MA3-414; Affinity Bioreagents), antiproteasome (19S Subunit S10B; Affinity Bioreagents), and anti-hsp90 (SPA-835; Stressgen).
RESULTS
Characterization of FRAP curves.
To investigate the mechanism and function of rapid transcription factor exchange, we used the 3617 cell line in which GR exchange was previously characterized (22). These cells are mouse adenocarcinomas containing ∼200 tandem copies of the MMTV promoter with associated reporter genes (44). By quantifying GFP levels at the MMTV array, we have estimated that on average ∼700 GFP-GRs are present there, consistent with the expected number of GR binding sites on the array (10).
In cells treated with hormone, the MMTV array becomes visible as a bright spot (25) (Fig. 1a). FRAPs at this site reveal a very rapid recovery with a half time (time to 50% recovery) of ∼5 s (22). To achieve good temporal resolution of this recovery, FRAP data were collected at ≥40-ms intervals by collecting images only in a narrow strip encompassing the array (Fig. 1a and b). Approximately 10 cells were averaged to generate one FRAP curve for a single experiment, and all experiments reported here were performed on at least three different days to assure reproducibility.
FRAP measures mobility due to diffusion and, if present, transport or binding. Diffusion contributes to every recovery, but in some cases this contribution is small and/or rapid and can be ignored. To test the role of diffusion, we performed bleaches with different spot sizes at the array and observed differences in recovery (Fig. 1c). We also found differences in the recovery rate when other regions of the nucleus were bleached with different spot sizes, indicating that diffusion contributed to GFP-GR recoveries everywhere in the nucleus (Fig. 1d).
These recovery rates, however, were considerably lower than that of GFP alone, which recovered almost instantaneously with the spot sizes used in our experiments (data not shown). If GFP-GR recoveries were due to simple diffusion, then they would predict a GFP-GR mass of ∼95 MDa, significantly larger than any known complex (see Materials and Methods for details of this calculation). Therefore binding interactions must retard GFP-GR FRAPs both in the nucleus and specifically at the MMTV array.
The simplest scenario to explain combined diffusion and binding is effective diffusion (15). Here, the FRAP exhibits a slowed diffusion, with the “effective” diffusion constant retarded in proportion to the binding affinity. Consistent with effective diffusion, GFP-GR FRAP rates were well fit by a single-parameter model for a diffusing molecule bleached by a circular spot (Fig. 1e) (39).
The effective-diffusion fits are informative for several reasons. First, they provide a single-parameter fit to the FRAP rate (τ), allowing a statistical comparison of curves based on this parameter. For example, using the same bleach spot size, τ at the array is significantly larger than τ elsewhere in the nucleus (Fig. 1e). These τ values, with their 95% confidence intervals, can be used in a t test to compute a P value to assess whether the recoveries are significantly different. The P value for the nucleus recovery versus the array recovery is highly significant (P < 0.0001). This demonstrates that the array recovers significantly more slowly than other sites in the nucleus, which indicates tighter binding at the array, as expected for the locally high concentration of specific binding sites there.
Second, the effective-diffusion theory provides insight into how such FRAP curves change in response to changes in binding affinity. Doubling or halving the off rate leads to modest changes in recovery rate because diffusion, which does not change, also contributes (Fig. 1f). Thus, in the effective-diffusion scenario, the FRAP data must be collected and analyzed with precision in order to detect changes in binding affinity under different perturbations.
Finally, in its simplest form, the effective-diffusion theory predicts a pseudoequilibrium binding constant (15), but only for the case where the distribution of fluorescence is homogeneous. This should apply to generic locations in the nucleus, but at present such estimates cannot be obtained for the array because the theory must be extended to account for such a nonhomogeneous fluorescence distribution in which a bright region (the array) is surrounded by a dimmer region (the rest of the nucleus). Further theoretical work is needed to extract estimates of binding constants from these data.
In all subsequent experiments, we used the estimates of the recovery rate, τ, at the array only as a means to compare curves. For any pair of GFP-GR recoveries, we performed a t test on the τ values for the recovery rate. In each plot, we report a P value as a measure of whether there is a statistically significant difference between the curves.
Energy dependence of the GFP-GR recovery.
The simplest explanation for GFP-GR exchange at the array is that it reflects a passive process determined by the simple equilibrium of GFP-GR binding to the MMTV promoter. If so, then the exchange process should be energy independent. To test this, we depleted cellular ATP levels with sodium azide and deoxyglucose. FRAPs both at the array and elsewhere in the nucleus were radically altered (Fig. 2a and b). Both sites showed <100% recovery, with a more significant retardation at the MMTV array than anywhere else in the nucleus. This suggested that energy might be required for normal GFP-GR exchange at the array, but the large effect on recoveries elsewhere in the nucleus raised the concern of nonspecific effects. Therefore, we sought to identify conditions for ATP reduction that minimized the effects on FRAPs at other sites in the nucleus. This was achieved by incubation in deoxyglucose alone, which blocks only nonoxidative phosphorylation (36) and is therefore not as potent as azide and deoxyglucose together. FRAPs were slightly affected throughout the nucleus after deoxyglucose treatment, but recoveries at the array were now qualitatively different in that they still exhibited an immobile fraction (Fig. 2c and d). This immobile fraction disappeared after deoxyglucose washout, demonstrating that immobility was not a consequence of toxicity (Fig. 2e).
FIG. 2.

GFP-GR exchange at the array is an energy-dependent process. A 30-min treatment with 10 mM sodium azide and deoxyglucose leads to a marked reduction in the exchange rate at the array (a) and elsewhere in the nucleus (b). Transferring the cells to a glucose-free medium (deoxyglucose only) for 60 to 90 min induces an ∼5% immobile fraction at the array (c), but the exchange at other sites in the nucleus does not show an immobile fraction (d). (e) Deoxyglucose washout eliminates the immobile fraction at the array.
The complete recovery observed elsewhere in the nucleus suggests that reduced ATP levels first impact a subset of sites unique to the MMTV array before affecting generic GR-chromatin binding. This fraction of MMTV sites appears to require energy for GFP-GR release. Rapid exchange, however, still occurs at the remaining MMTV sites, thereby defining two classes of sites: a small fraction that is ATP sensitive and a larger fraction that is relatively ATP insensitive. These results are in marked contrast to many nuclear proteins that do not require energy for their mobility (19, 28).
Roles for chaperones in GFP-GR recovery.
A number of factors could contribute to the energy dependence at the ATP-sensitive fraction of MMTV sites. An ATP-dependent chaperone complex that includes hsp70, hsp90, p23, and several immunophilins associates with unliganded GR in the cytoplasm, allowing ligand binding (30). Once ligand is bound, the chaperone complex dissociates but later reassociates after the ligand dissociates from GR. This association and dissociation triggered by the loss of ligand is referred to as the chaperone cycle. In addition to its cytoplasmic role, the chaperone complex also plays a role in GR recycling within the nucleus (7, 20). Moreover, Freeman and Yamamoto (12) have proposed that chaperones disassemble transcriptional regulatory complexes at promoter sites. If this were the case for GR at the MMTV promoter, then disruption of chaperone function should lead to a slowdown in the FRAP at the MMTV array.
As a first test for chaperone involvement, we used immunofluorescence to investigate the subcellular distribution of hsp90, hsp70, and p23 in the MMTV array cell line. We found, as expected, that these molecules were largely cytoplasmic, but some nuclear fluorescence was also detected. Significantly, we consistently found an association of hsp90, hsp70, and p23 with the MMTV array. Antibodies against these molecules colocalized with the GFP-GR signal at the array and also typically stained a region surrounding the array (Fig. 3). The size of the region stained by chaperones was always proportional to the size of the MMTV array, with larger arrays that contained more GFP-GR characterized by correspondingly larger regions of chaperone staining. These observations demonstrate that chaperones are recruited to the MMTV site and suggest that chaperones are poised to affect GFP-GR exchange at the array.
FIG. 3.

Immunofluorescence detection of chaperone proteins at the MMTV array (circled). Antibodies against hsp90 (a to c), hsp70 (d to f), or p23 (g to i) stain the cytoplasm as expected, but within nuclei, they also consistently colocalize with GFP-GR at the MMTV array. Insets contain higher-magnification views of colocalization at the array. Scale bar, 10 μm.
To disrupt chaperone activity, we first used the antibiotic geldanamycin (32), which specifically blocks hsp90 activity by binding to its N-terminal ATP site and preventing p23 binding (38). As expected, geldanamycin (2.5 μg/ml) blocked GFP-GR import in the array cell line (6), so to study nuclear events, we pretreated cells with dexamethasone and then added geldanamycin 30 min later, when GR nuclear import was complete. Although MMTV arrays began to disappear after 1 h of geldanamycin treatment (Fig. 4a), a normal number of arrays were still clearly visible after 30 to 60 min of geldanamycin treatment, thereby allowing measurement by FRAP of GFP-GR exchange at the array. In geldanamycin-treated cells, FRAPs at the array were faster than in control cells (Fig. 5a). Larger effects could be observed with higher concentrations of geldanamycin, but they also led to significant effects elsewhere in the nucleus (data not shown). Thus, the geldanamycin concentration used here minimizes the effects on the rest of the nucleus and therefore should reflect effects unique to GFP-GR binding to the MMTV sites. Further evidence for this comes from cells transfected with a different nuclear protein, GFP-HP1α, and treated with geldanamycin. No effect on GFP-HP1α recovery was found (Fig. 5b). Similar results were also obtained for GFP alone (data not shown). These findings indicate that geldanamycin did not generically alter protein mobilities in the nucleus.
FIG. 4.

Arrays (circled) disappear much more rapidly after geldanamycin treatment when corticosterone is the ligand than when dexamethasone is the ligand. In either case, geldanamycin induces arrays to disappear (red lines) faster than normal (black lines). An example of array disappearance for each case is shown in pseudocolor to accentuate the arrays. (a) When dexamethasone is the ligand, array disappearance is gradual and occurs over a 2-h time course. (b) When corticosterone is the ligand, the geldanamycin-induced disappearance of arrays is dramatically accelerated, occurring over a time course of 10 min. These results indicate that geldanamycin effects are exacerbated by corticosterone, presumably due to its more rapid exchange with GR. They also suggest that in the presence of geldanamycin, GR eventually becomes unliganded and incapable of binding to the MMTV sites. Scale bar, 5 μm.
FIG. 5.

Effects of hsp90 inhibition. FRAPs at MMTV arrays are faster after treatment with 2.5 μg of geldanamycin/ml (a) or 5 μg of radicicol/ml (c) than in control cells, but the speed-up is detectable immediately in corticosterone (e) while it appears only after 30 to 60 min in dexamethasone (a). (b) These changes are not caused by a generic effect on protein mobility, as cells transfected with GFP-HP1α show no effect on FRAPs after geldanamycin treatment. (Note that the GFP-HP1α recoveries are not fit by effective diffusion [data not shown], so a t test to compute a P value cannot be performed.) (d) Hormone withdrawal experiments demonstrate that, compared to dexamethasone, corticosterone exchanges much more rapidly with GR. Shown are the average transcriptional levels measured from 35 cells by RNA FISH. Cells were induced by 100 nM corticosterone (CORT) or dexamethasone (DEX) for 15 min and then washed three times over a 5-min period in hormone-free medium and left in that medium for 45 min. With corticosterone as a ligand, transcription is abolished after a 5-min wash, indicating complete exchange of the ligand with GR during the wash time. In the same wash period, a significant amount of dexamethasone remains bound, since transcription drops by only 50%. (f) Treatment with geldanamycin induces a progressive loss of GFP-GR from the array and a decrease in size, and this is accompanied by a loss of chaperones. Shown is the loss of p23 from the array following geldanamycin (GELD) treatment with dexamethasone as a ligand. Scale bar, 5 μm.
To corroborate the geldanamycin findings, we also treated cells with another drug, radicicol, which disrupts hsp90 activity by blocking its N-terminal ATP-binding site (35). Again, we found that FRAP at the array was faster (Fig. 5c).
Geldanamycin and radicicol are known to be specific for hsp90 (32), but this chaperone plays a role in many cellular processes (29). To test if geldanamycin directly affected GR, we substituted the natural hormone corticosterone for dexamethasone. Since the half time of corticosteroid binding to GR at 0°C is much shorter than that of dexamethasone (14, 26), we reasoned that corticosterone should exacerbate any effects of geldanamycin if they originate due to loss of ligand from GR and consequent passage through the chaperone cycle.
To confirm the difference in the binding affinities of these two steroids to GFP-GR in the MMTV array cell line at physiological temperatures, we used either corticosterone or dexamethasone as a ligand, washed the cells three times over a 5-min period in ligand-free medium, and compared the transcriptional outputs. After corticosterone washout, additional transcription was abolished, in contrast to dexamethasone washout, where transcription dropped by only 50% (Fig. 5d). This demonstrates that over the 5-min wash period, virtually all corticosterone became unbound from GR, whereas significant amounts of dexamethasone were still bound. These results demonstrate in vivo that dexamethasone remains much more tightly bound to GR than corticosterone.
As predicted, the effects of geldanamycin were accelerated in the presence of corticosterone. Disappearance of GFP-GR arrays proceeded much faster with corticosterone (Fig. 4b), and FRAPs became faster immediately after geldanamycin was added (Fig. 5e). This immediate response strongly suggests that the geldanamycin effect is specific for GR.
We next asked whether geldanamycin treatment affected the association of chaperones with the MMTV array. Using immunofluorescence, we found that p23 levels decreased in proportion to the duration of the geldanamycin treatment (Fig. 5f). Similar results were observed with hsp90 and hsp70. The gradual loss of chaperones was always accompanied by a decrease in the size of the array and the amount of associated GFP-GR. Thus, our data do not distinguish whether loss of the chaperones after geldanamycin treatment leads to loss of GFP-GR or vice versa.
Faster FRAPs in the presence of geldanamycin were also seen after more extended treatments (1 to 3 h with dexamethasone as a ligand), although arrays became harder to detect (Fig. 4a). After ∼30 min of imaging these cells treated with geldanamycin for 1 to 3 h, numerous GFP-GR enriched spots appeared throughout the nucleus (Fig. 6a). These spots were not arrays, as only one or two arrays are present in a nucleus. Spots could also be induced when geldanamycin treatment was combined with cold or heat shock. The spots colocalized with a proteasome component (Fig. 6b and c), suggesting that they could reflect abnormal accumulations of misfolded proteins (41). As has been observed for these other proteasome-associated inclusions, FRAPs at the GFP-GR spots revealed a slowdown, with an immobile fraction (Fig. 6d). This indicates that some fraction of GR is immobilized within each spot and forms a protein aggregate. Thus, we found that extended treatments with geldanamycin combined with additional stress (either heat, cold, or imaging) could induce nonspecific effects that resulted in a slower FRAP at GFP-GR aggregates. Once these aggregates had formed, arrays had disappeared, but until that point, FRAPs at arrays were always faster than in controls.
FIG. 6.

Nuclear GFP-GR aggregates. (a to c) Geldanamycin treatment combined with stress (heat, cold, or prolonged imaging) causes the disappearance of arrays and the formation of GFP-GR spots, which colocalize with a proteasome antibody. (d) In these spots, a fraction of GFP-GR is immobilized compared to other regions of the nucleus. During imaging, these aggregates appear more rapidly with corticosterone than with dexamethasone. Scale bar, 5 μm.
In sum, either geldanamycin or radicicol induced faster recovery at arrays. This speed-up occurred instantly in the presence of corticosterone, strongly suggesting a specific effect on GFP-GR. Therefore, the immobile fraction seen after ATP depletion does not arise from disruption of the chaperones.
Role for the proteasome in GFP-GR recovery.
The proteasome is an alternate candidate for regulating the ATP-sensitive fraction of MMTV sites. Both the 19S and 20S subunits of the proteasome require ATP (18). As noted in the introduction, increasing evidence points to a role for the proteasome in transcription factor regulation, and moreover, proteasomes often interact with chaperones.
As a first test for proteasome involvement, we stained cells with an antibody against the 19S component. This antibody consistently colocalized with the array and surrounding regions, suggesting that the proteasome was recruited to this site and could play a role in GFP-GR exchange there (Fig. 7a to c). As found for chaperone components, there was a direct correlation between the size of the array and the amount of proteasome staining (data not shown), suggesting that increasing amounts of GR recruit more proteasomes to the promoter.
FIG. 7.

Immunofluorescence detection of the proteasome at the MMTV array (circled) and FRAPs after perturbation of proteasome function. Considerable proteasome staining was found in the cytoplasm. (a to c) Within the nucleus, colocalization with the MMTV array was consistently observed. The inset contains a higher-magnification view of colocalization at the array. (d and e) Cells induced with dexamethasone or corticosterone and exposed to the proteasome inhibitor MG-132 exhibited slower FRAP at MMTV arrays. (f) This difference was not caused by a generic retardation of protein mobilities in the nucleus, as MG-132 treatment did not alter the recovery of GFP-HP1α. (Again, these GFP-HP1α recoveries are not fit by effective diffusion, so neither a recovery rate, τ, nor a P value for the comparison can be calculated.) Scale bar, 10 μm.
To test for a role for the proteasome in GFP-GR exchange at the MMTV array, we treated cells with the proteasome inhibitor MG-132 (18). This resulted in a slowdown of FRAPs with either dexamethasone or corticosterone as a ligand (Fig. 7d and e). Larger effects with MG-132 could be induced at higher concentrations or with longer treatments, but we selected a concentration and a treatment time so that effects of MG-132 were minimized elsewhere in the nucleus. This should help to ensure that the effects seen at the MMTV array were unique to GFP-GR binding to the MMTV sites. Additionally, MG-132 had no effect on the mobility of either GFP alone (data not shown) or GFP-HP1α (Fig. 7f), demonstrating that it did not nonspecifically disrupt protein mobility throughout the nucleus.
With evidence for both proteasome and chaperone activities at the MMTV sites, we then asked whether disruption of one system altered the other. We used immunofluorescence to examine levels of the 19S proteasome after geldanamycin treatment. As seen earlier for chaperone components, geldanamycin also led to a progressive loss over time of the 19S proteasome from the MMTV array (Fig. 8a to f). This was always correlated with a decrease in the size of the array itself and the amount of GFP-GR there, making it impossible to distinguish whether GFP-GR loss induced proteasomal loss or vice versa. Nevertheless, this gradual loss of the proteasome after geldanamycin treatment should by itself lead to slower FRAPs, thus counteracting the effect we detected, namely, faster FRAPs. This suggests that we might have observed even faster recoveries after geldanamycin had proteasome levels not been affected. In complementary experiments, we also used immunofluorescence to examine levels of p23 after MG-132 treatment and detected no change (Fig. 8g to l). This effect was seen with either dexamethasone or corticosterone, even though these different ligands affected transcription differently (see below). We conclude that the slowdown seen after MG-132 treatment is not due to loss of p23.
FIG. 8.

Effects of either geldanamycin treatment on proteasomes or MG-132 treatment on chaperones. (a to f) Progressive loss of the 19S proteasome is seen at the MMTV array (circled) with longer geldanamycin (GELD) treatment. Clear proteasomal staining is seen after 15 min of geldanamycin treatment (b), whereas much fainter staining at levels close to nuclear background is detected after 1 h of geldanamycin treatment (e). (g to l) MG-132 does not affect the levels of p23 with either dexamethasone (DEX) (g to i) or corticosterone (CORT) (j to l). Only one time point is shown for each ligand, since unlike geldanamycin treatment, there is no loss of GFP-GR from the array after proteasome inhibition. Scale bar, 5 μm.
In sum, our results suggest that the proteasome itself normally regulates GFP-GR exchange at the MMTV sites and is responsible for at least some of the ATP sensitivity at a fraction of these sites.
Transcriptional level and GFP-GR recovery.
The preceding experiments demonstrated that normal GFP-GR recovery is energy dependent and requires proteasome and chaperone functions. This shows that GFP-GR exchange is highly regulated. Since transcription is the primary function of GR binding to the MMTV promoter, we asked if there was any association between transcription and the exchange rate.
As a start, we investigated whether the exchange rate correlates with different endogenous transcription levels. Our previous studies have shown that the array size correlates with the transcription level as measured by RNA FISH. Condensed arrays are less transcriptionally active than decondensed arrays (25) (Fig. 9a and Table 1). Therefore, we performed FRAPs at condensed and decondensed arrays and found that recoveries at condensed arrays were slightly, but consistently, faster than at decondensed arrays (Fig. 9b). Although modest, this difference yields a highly significant P value (P < 0.001) because the data are fit so well by effective diffusion that the estimate of the recovery rate for each curve has a very small error.
FIG. 9.

Accelerated GFP-GR exchange is associated with less transcription. (a) Examples of a condensed array and a decondensed array are shown with the corresponding RNA FISH signals. (b) FRAPs of condensed arrays were consistently faster. (c and d) RU486 (100 nM) and corticosterone (100 nM) also yielded faster FRAPs than dexamethasone. Scale bar, 1 μm.
TABLE 1.
Changes in GFP-GR recovery at the array are coupled to transcriptiona
| Condition studied | State 1 | State 2 | % Change in transcription (state 2 − state 1)/state 1 | Change in FRAP (state 2 vs. state 1) |
|---|---|---|---|---|
| Array state | Decondensed | Condensed | −70 | Faster (Fig. 9b) |
| Agonists and antagonists | Dexamethasone | RU486 | −90 | Faster (Fig. 9c) |
| Dexamethasone | Corticosterone | −50 | Faster (Fig. 9d) | |
| Chaperone perturbation | Dexamethasone | Geldanamycin + dexamethasone | −70 | Faster (Fig. 5a) |
| Dexamethasone | Radicicol + dexamethasone | −95 | Faster (Fig. 5c) | |
| Corticosterone | Geldanamycin + corticosterone | −70 | Faster (Fig. 5e) | |
| Proteasome perturbation | Dexamethasone | MG-132 + dexamethasone | +210 | Slower (Fig. 7d) |
| Corticosterone | MG-132 + corticosterone | −90 | Slower (Fig. 7e) |
The level of transcription was determined by measuring the relative RNA FISH signal intensities in the nuclei of 35 to 100 individual cells.
To investigate further a connection between the exchange rate and transcription, we measured FRAPs in the presence of the antagonist RU486. RU486 reduced transcription at the MMTV array to 10% of that with dexamethasone (Table 1). This reduced transcriptional level with RU486 was associated with faster FRAPs than with dexamethasone (Fig. 9c).
We also found for the MMTV promoter that the natural hormone corticosterone was a less efficient agonist than the synthetic hormone dexamethasone (Table 1). Reduced transcriptional levels with corticosterone were once again associated with faster GFP-GR exchange at the array than with dexamethasone (Fig. 9d).
Thus, for a purely endogenous difference (condensed versus decondensed arrays), or for differences induced by agonists or antagonists, reduction in transcriptional activity was accompanied by a higher GFP-GR exchange rate.
We then examined transcriptional levels after disruption of chaperone function. After hsp90 activity was blocked with either geldanamycin or radicicol, transcription levels dropped at the MMTV array (Table 1). This is consistent with previous studies (2), but in those studies it was not clear whether the reduced transcription arose from a failure of GR to bind to its target sites or even enter the nucleus. Under our conditions with geldanamycin, GR remains bound to the MMTV array for 10 min in corticosterone and for 1 h in dexamethasone. However, transcription still drops significantly in these time periods, demonstrating that some more subtle effect on transcription has occurred. One possibility is the higher exchange rate measured in all of these cases.
Finally, we measured transcriptional levels after proteasome inhibition. Here, we found a dependence on ligand. With dexamethasone, transcriptional levels rose, but with corticosterone, they dropped (Table 1). Deroo et al. (8) found that prolonged treatment (22 to 28 h) with MG-132 increased both GR levels and transcription in the presence of dexamethasone. However, the transcriptional changes that we detected were not due to changes in GFP-GR protein levels, since the mean fluorescence intensity of GFP-GR in nuclei treated with MG-132 for 1 h (97% ± 11%) was not significantly different from the intensity of the control nuclei (100% ± 8%). Despite the differences in transcription, FRAPs with either dexamethasone or corticosterone showed a slowdown and an immobile fraction, indicating that the ligand can have dramatically different effects on transcription when a fraction of receptors becomes immobilized.
In sum, in all cases so far we have seen that changes in the exchange rate are associated with changes in the transcriptional level, suggesting that the two are coupled.
DISCUSSION
We have investigated rapid GFP-GR exchange at a tandem array of MMTV promoter sites and have provided the first insights into its mechanism and function. Our results indicate that exchange is controlled at least in part by a balance between proteasome and chaperone functions. These two systems often act in concert, for example, in striking a balance between the refolding and degradation of cellular proteins (45). Our data suggest that a similar balancing act is performed at the MMTV promoter, with chaperones helping to stabilize GR binding and proteasomes helping to catalyze GR removal (Fig. 10). Exactly how this balance is struck is unknown, but factors such as CHIP and BAG-1, which are known to bridge the chaperone and proteasome systems, may well be involved (5, 21).
FIG. 10.

Model for proteasome-chaperone interaction at the MMTV template and association with transcription. Liganded GR binding to MMTV ultimately leads to initiation complex formation. Formation of the complex may be enhanced by longer GR residence at the MMTV template. The duration of GR occupancy at the template is determined in part by competition between proteasome and chaperone functions. Proteasome inhibition favors GR occupancy, leading to a slower FRAP with an immobile fraction. Chaperone inhibition by geldanamycin favors GR loss, leading to a faster FRAP. The equilibrium between these two components helps to set the transcriptional level and may be mediated in part by one or more factors that are known to couple chaperone and proteasome activities (5, 21).
We found that both chaperones and proteasomes were present at and around the MMTV array, demonstrating that both classes of molecules were specifically recruited to the site. These observations are consistent with ChIP studies demonstrating that either chaperones (12) or proteasomes (13, 31) are found at promoter sites, although until now not at the same promoter.
Proteasome function.
After disrupting proteasome activity, we found that GFP-GR recovered more slowly at the MMTV sites and attained only 95% of its starting intensity. The residual 5% corresponds to an immobile fraction of GFP-GR that remains stably bound to the promoter after proteasome inhibition. We conclude that removal of at least a fraction of GR molecules ordinarily requires the proteasome. Since both GR and various coactivators are ubiquitinated (5, 46), loss of GR at the promoter could reflect a direct effect of the proteasome on GR or an indirect consequence of the loss of a closely associated coactivator. Our data also do not distinguish whether GR loss normally involves degradation of this 5% fraction of GR at the promoter or simply ejection of the fraction from the promoter. We cannot rule out the possibility that proteasome treatment in some other way indirectly alters GR mobility by inducing, for example, a stress response (40). However, by using shorter MG-132 treatments that disrupt predominantly the FRAP behavior at the MMTV array, we reduce the possibility that the effects we observe are nonspecific. This issue can be addressed more directly in future studies by identifying and then perturbing specific molecules that couple proteasome activity with GR.
Proteasome inhibition is also known to affect the nuclear mobility of steroid receptors by inducing nuclear matrix binding (8, 34, 43). However, with the shorter incubations of the proteasome inhibitor that we used, the MMTV array remained visible and transcription continued, indicating that GR was still DNA bound at the MMTV sites. Our observation of an immobile fraction at specific sites after dexamethasone and proteasome inhibition is in contrast to the effect detected for GR elsewhere in the nucleus, where dexamethasone or other high-affinity ligands relieved the immobile fraction induced by proteasome inhibition (34). These differences indicate that the same treatment can yield different effects depending upon whether the binding site is a specific promoter or nuclear matrix.
A number of biochemical approaches have implied a role for proteasomes in transcription factor removal from promoter sites (27). Highly active transcriptional regulators, such as VP16 or myc, must be ubiquitinated to induce transcription and after further ubiquitination are degraded by the proteasome (33). This degradation may occur at the promoter, as it is enhanced by DNA binding (24). Recent studies have demonstrated that the slow cycling of ER is accompanied by and dependent upon proteasomal cycling (31).
Although there is an intriguing similarity between these ChIP results for ER and our FRAP data for GR, they may reflect entirely different processes. This is because the time scales interrogated by the two techniques are radically different. The GFP-GR FRAP at the MMTV array is complete within 1 min. Thus, the average residency time of GFP-GR at the promoter can be no more than 1 min and could well be much shorter. Such rapid exchange cannot be detected by ChIP, since the formaldehyde fixation step in the procedure typically lasts much longer (∼10 min) than the GR cycling time. As a consequence, ChIP detects periodicities on time scales 1 to 2 orders of magnitude longer than those detected by FRAP. It is possible that a much slower cycling of GR is superimposed on the faster cycling that we measure by FRAP. The fraction of bound GR at the MMTV sites is determined by the binding affinity of GR for these sites. If this affinity slowly increases and then slowly decreases, then the amount of bound GR would slowly cycle, even though the individual GR molecules at any moment would exhibit the rapid exchange that we measure by FRAP. In principle, this could be detected in our live-cell system in two ways, neither of which has been carefully examined. If GR binding affinity to MMTV gradually increases, then the array intensity should gradually increase, and at the same time the FRAP rate should gradually decrease, since slower recoveries reflect higher-affinity binding. In sum, the same system can exhibit both fast and slow cycling. Fast cycling is best detected by FRAP at a promoter target array, and slow cycling is better observed by ChIP.
Chaperone function.
By disrupting chaperone activity with either geldanamycin or radicicol, we observed an accelerated GFP-GR exchange at the MMTV promoter sites. Faster GFP-GR exchange was detected instantly in the presence of geldanamycin and corticosterone, strongly supporting a specific effect on GFP-GR binding to MMTV.
Targeting one member of the chaperone complex, p23, to a series of GR promoter sites led to loss of GR binding there (12). Based on this and other observations, Freeman and Yamamoto proposed that p23 could repeatedly remove GR from a template and therefore give rise to the rapid GR exchange process observed in living cells. However, if this were the case, then in the simplest scenario we should have observed a slower FRAP after geldanamycin treatment, since the drug prevents p23 binding to the chaperone complex (38). This in turn should have prevented p23 from ejecting GR from the promoter. Instead, we detected a faster GFP-GR exchange, implying that the chaperones normally stabilize GR binding rather than catalyze its removal.
A number of explanations for this discrepancy are possible. A key difference between the two studies is that we have disrupted hsp90 function, whereas Freeman and Yamamoto altered p23 activity. Although p23 and hsp90 normally act in concert within the chaperone complex, they may not act together at a promoter. If so, then geldanamycin treatment would directly affect hsp90 but not p23. Then, our FRAP data in the presence of geldanamycin would imply that hsp90 is required to stabilize GR binding at the promoter, whereas Freeman and Yamamoto's ChIP data would suggest that p23 is required to destabilize it. Such antagonistic actions between hsp90 and p23 could play a role in setting GR residency times. More generally, a delicate balance must exist between the chaperone and proteasome functions at the MMTV promoter. Thus, even if geldanamycin treatment also disrupts p23 function at MMTV, it is possible that the specific method used to alter p23 activity may tilt the equilibrium between chaperones and proteasomes one way or the other, leading to opposite effects on GR binding. These discrepancies will probably be resolved by elucidating the molecular mechanism that establishes the interplay between chaperones and proteasomes at the MMTV promoter. At this time, we can say with certainty that both our results and those of Freeman and Yamamoto strongly support a role for chaperones in GR binding at a promoter.
Rapid-exchange function.
It has been suggested that transcription factor cycling at a promoter is a mechanism to sense environmental changes, for example, in ligand concentration (12, 22, 31, 37). After 5 min of corticosterone removal, we found that there was virtually no more transcription from the MMTV promoter. This is consistent with a sensing model for exchange, since the ∼1-min cycling time of GR is fast enough to show a response within 5 min after a drop in hormone concentration. Note, however, that such a rapid change in hormone levels could be sensed only by a rapid-exchange mechanism and not by the longer cycling times observed for ER by ChIP (31, 37).
In addition to a possible role in sensing altered hormone levels, we found a coupling between the transcriptional level and the exchange rate. In every case examined so far, when transcription levels changed, so did the exchange rate. At present, our data support a simple form of coupling, namely, that slower exchange is associated with more transcription. This could arise because a longer GR residence time could increase the chances of successful polymerase loading. We observed this correlation between the exchange rate and the transcription level in seven out of eight two-way comparisons (Table 1).
The one exception was with the proteasome inhibitor MG-132 and the ligand corticosterone. This combination yielded slower exchange and decreased transcription, whereas MG-132 and dexamethasone yielded slower exchange and increased transcription. One explanation for this discrepancy is the innate difference between the residence times for ligand binding to GR with dexamethasone and corticosterone. After proteasome inhibition by MG-132, a fraction of GR remains at the template. With corticosterone, this fraction will rapidly lose the ligand, whereas for dexamethasone it will not. Thus, longer residence of GR at the template may lead to increased transcription only if the ligand remains bound; otherwise, unliganded GR remains at the promoter, blocking access of liganded GR to those sites. Clearly, other interpretations are possible. Our simple model relating the exchange rate to the transcriptional level is no doubt complicated by a number of other factors that also contribute to the final transcriptional level. Nevertheless, our data suggest that GR residence time at a promoter is one factor in tuning the transcriptional output.
In sum, our results indicate that chaperones and proteasomes modulate rapid GR exchange at a promoter. If, as our data suggest, the exchange rate is intimately coupled with transcription, then it is likely that a number of other factors will impact this rate as a means of regulating transcriptional levels. Understanding how the exchange rate is tuned by contributions from different factors will be critical for understanding transcriptional regulation. Our data now underscore the importance of live-cell imaging for a complete understanding of transcriptional mechanisms.
REFERENCES
- 1.Abramowitz, M., and I. A. Stegun. 1970. Handbook of mathematical functions, p. 358-436. Dover Publications, Inc., New York, N.Y.
- 2.Bamberger, C. M., M. Wald, A. M. Bamberger, and H. M. Schulte. 1997. Inhibition of mineralocorticoid and glucocorticoid receptor function by the heat shock protein 90-binding agent geldanamycin. Mol. Cell. Endocrinol. 131:233-240. [DOI] [PubMed] [Google Scholar]
- 3.Becker, M., C. Baumann, S. John, D. A. Walker, M. Vigneron, J. G. McNally, and G. L. Hager. 2002. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 3:1188-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burakov, D., L. A. Crofts, C. P. Chang, and L. P. Freedman. 2002. Reciprocal recruitment of DRIP/mediator and p160 coactivator complexes in vivo by estrogen receptor. J. Biol. Chem. 277:14359-14362. [DOI] [PubMed] [Google Scholar]
- 5.Connell, P., C. A. Ballinger, J. Jiang, Y. Wu, L. J. Thompson, J. Hohfeld, and C. Patterson. 2001. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3:93-96. [DOI] [PubMed] [Google Scholar]
- 6.Czar, M. J., M. D. Galigniana, A. M. Silverstein, and W. B. Pratt. 1997. Geldanamycin, a heat shock protein 90-binding benzoquinone ansamycin, inhibits steroid-dependent translocation of the glucocorticoid receptor from the cytoplasm to the nucleus. Biochemistry 36:7776-7785. [DOI] [PubMed] [Google Scholar]
- 7.DeFranco, D. B. 2002. Navigating steroid hormone receptors through the nuclear compartment. Mol. Endocrinol. 16:1449-1455. [DOI] [PubMed] [Google Scholar]
- 8.Deroo, B. J., C. Rentsch, S. Sampath, J. Young, D. B. DeFranco, and T. K. Archer. 2002. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol. Cell. Biol. 22:4113-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dundr, M., U. Hoffmann-Rohrer, Q. Hu, I. Grummt, L. I. Rothblum, R. D. Phair, and T. Misteli. 2002. A kinetic framework for a mammalian RNA polymerase in vivo. Science 298:1623-1626. [DOI] [PubMed] [Google Scholar]
- 10.Dundr, M., J. G. McNally, J. Cohen, and T. Misteli. 2002. Quantitation of GFP-fusion proteins in single living cells. J. Struct. Biol. 140:92-99. [DOI] [PubMed] [Google Scholar]
- 11.Fletcher, T. M., N. Xiao, G. Mautino, C. T. Baumann, R. Wolford, B. S. Warren, and G. L. Hager. 2002. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. Biol. 22:3255-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman, B. C., and K. R. Yamamoto. 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232-2235. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez, F., A. Delahodde, T. Kodadek, and S. A. Johnston. 2002. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296:548-550. [DOI] [PubMed] [Google Scholar]
- 14.Kaine, J. L., C. J. Nielsen, and W. B. Pratt. 1975. The kinetics of specific glucocorticoid binding in rat thymus cytosol: evidence for the existence of multiple binding states. Mol. Pharmacol. 578-587. [PubMed]
- 15.Kaufman, E. N., and R. K. Jain. 1990. Quantification of transport and binding parameters using fluorescence recovery after photobleaching. Potential for in vivo applications. Biophys. J. 58:873-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer, P. R., G. Fragoso, W. Pennie, H. Htun, G. L. Hager, and R. R. Sinden. 1999. Transcriptional state of the mouse mammary tumor virus promoter can affect topological domain size in vivo. J. Biol. Chem. 274:28590-28597. [DOI] [PubMed] [Google Scholar]
- 17.Kruhlak, M. J., M. A. Lever, W. Fischle, E. Verdin, D. P. Bazett-Jones, and M. J. Hendzel. 2000. Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J. Cell Biol. 150:41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, D. H., and A. L. Goldberg. 1998. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8:397-403. [DOI] [PubMed] [Google Scholar]
- 19.Lever, M. A., J. P. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 408:873-876. [DOI] [PubMed] [Google Scholar]
- 20.Liu, J., and D. B. DeFranco. 1999. Chromatin recycling of glucocorticoid receptors: implications for multiple roles of heat shock protein 90. Mol. Endocrinol. 13:355-365. [DOI] [PubMed] [Google Scholar]
- 21.Luders, J., J. Demand, and J. Hohfeld. 2000. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 275:4613-4617. [DOI] [PubMed] [Google Scholar]
- 22.McNally, J. G., W. G. Muller, D. Walker, R. Wolford, and G. L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287:1262-1265. [DOI] [PubMed] [Google Scholar]
- 23.Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature 408:877-881. [DOI] [PubMed] [Google Scholar]
- 24.Molinari, E., M. Gilman, and S. Natesan. 1999. Proteasome-mediated degradation of transcriptional activators correlates with activation domain potency in vivo. EMBO J. 18:6439-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müller, W. G., D. Walker, G. L. Hager, and J. G. McNally. 2001. Large-scale chromatin decondensation and recondensation regulated by transcription from a natural promoter. J. Cell Biol. 154:33-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munck, A., and N. J. Holbrook. 1984. Glucocorticoid-receptor complexes in rat thymus cells. Rapid kinetic behavior and a cyclic model. J. Biol. Chem. 259:820-831. [PubMed] [Google Scholar]
- 27.Muratani, M., and W. P. Tansey. 2003. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4:192-201. [DOI] [PubMed] [Google Scholar]
- 28.Phair, R. D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404:604-609. [DOI] [PubMed] [Google Scholar]
- 29.Picard, D. 2002. Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol. Life Sci. 59:1640-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pratt, W. B., and D. O. Toft. 1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 18:306-360. [DOI] [PubMed] [Google Scholar]
- 31.Reid, G., M. R. Hubner, R. Metivier, H. Brand, S. Denger, D. Manu, J. Beaudouin, J. Ellenberg, and F. Gannon. 2003. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 11:695-707. [DOI] [PubMed] [Google Scholar]
- 32.Roe, S. M., C. Prodromou, R. O'Brien, J. E. Ladbury, P. W. Piper, and L. H. Pearl. 1999. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 42:260-266. [DOI] [PubMed] [Google Scholar]
- 33.Salghetti, S. E., A. A. Caudy, J. G. Chenoweth, and W. P. Tansey. 2001. Regulation of transcriptional activation domain function by ubiquitin. Science 293:1651-1653. [DOI] [PubMed] [Google Scholar]
- 34.Schaaf, M. J., and J. A. Cidlowski. 2003. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol. Cell. Biol. 23:1922-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulte, T. W., S. Akinaga, T. Murakata, T. Agatsuma, S. Sugimoto, H. Nakano, Y. S. Lee, B. B. Simen, Y. Argon, S. Felts, D. O. Toft, L. M. Neckers, and S. V. Sharma. 1999. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol. Endocrinol. 13:1435-1448. [DOI] [PubMed] [Google Scholar]
- 36.Schwoebel, E. D., T. H. Ho, and M. S. Moore. 2002. The mechanism of inhibition of Ran-dependent nuclear transport by cellular ATP depletion. J. Cell Biol. 157:963-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shang, Y., X. Hu, J. DiRenzo, M. A. Lazar, and M. Brown. 2000. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843-852. [DOI] [PubMed] [Google Scholar]
- 38.Smith, D. F., L. Whitesell, S. C. Nair, S. Chen, V. Prapapanich, and R. A. Rimerman. 1995. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol. 15:6804-6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soumpasis, D. M. 1983. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 41:95-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stangl, K., C. Gunther, T. Frank, M. Lorenz, S. Meiners, T. Ropke, L. Stelter, M. Moobed, G. Baumann, P. M. Kloetzel, and V. Stangl. 2002. Inhibition of the ubiquitin-proteasome pathway induces differential heat-shock protein response in cardiomyocytes and renders early cardiac protection. Biochem. Biophys. Res. Commun. 291:542-549. [DOI] [PubMed] [Google Scholar]
- 41.Stenoien, D. L., M. Mielke, and M. A. Mancini. 2002. Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat. Cell Biol. 4:806-810. [DOI] [PubMed] [Google Scholar]
- 42.Stenoien, D. L., A. C. Nye, M. G. Mancini, K. Patel, M. Dutertre, B. W. O'Malley, C. L. Smith, A. S. Belmont, and M. A. Mancini. 2001. Ligand-mediated assembly and real-time cellular dynamics of estrogen receptor alpha-coactivator complexes in living cells. Mol. Cell. Biol. 21:4404-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stenoien, D. L., K. Patel, M. G. Mancini, M. Dutertre, C. L. Smith, B. W. O'Malley, and M. A. Mancini. 2001. FRAP reveals that mobility of oestrogen receptor-alpha is ligand- and proteasome-dependent. Nat. Cell Biol. 3:15-23. [DOI] [PubMed] [Google Scholar]
- 44.Walker, D., H. Htun, and G. L. Hager. 1999. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods 19:386-393. [DOI] [PubMed] [Google Scholar]
- 45.Wickner, S., M. R. Maurizi, and S. Gottesman. 1999. Posttranslational quality control: folding, refolding, and degrading proteins. Science 286:1888-1893. [DOI] [PubMed] [Google Scholar]
- 46.Yan, F., X. Gao, D. M. Lonard, and Z. Nawaz. 2003. Specific ubiquitin-conjugating enzymes promote degradation of specific nuclear receptor coactivators. Mol. Endocrinol. 17:1315-1331. [DOI] [PubMed] [Google Scholar]