

Abstract
Despite recent progress in “shotgun” peptide separation by integrated liquid chromatography and mass spectrometry (LC/MS), proteome coverage and reproducibility are still limited with this approach and obtaining enough replicate runs for biomarker discovery is a challenge. For these reasons, recent research demonstrates that there is a continuing need for protein separation by two-dimensional gel electrophoresis (2-DE). However, with traditional 2-DE informatics, the digitized images are reduced to symbolic data through spot detection and quantification before proteins are compared for differential expression by spot matching. Recently, a more robust and automated paradigm has emerged where gels are directly aligned in the image domain before spots are detected across the whole image set as a whole. In this chapter, we describe the methodology for both approaches and discuss the pitfalls present when reasoning statistically about the differential protein expression discovered.
Keywords: 2-D gel electrophoresis, Image alignment, Spot detection, Spot matching, Differential expression analysis, Clustering, DIGE
1. Introduction
Since its beginnings in 1975 (1, 2), two-dimensional gel electrophoresis (2-DE) has established itself as the principal approach for separating proteins from cell and tissue samples (3). While recent progress in “shotgun” peptide separation with liquid chromatography and mass spectrometry (LC/MS) (4, 5) has brought some significant analytical benefits, recent bench comparisons have shown that proteome coverage is complementary to 2-DE rather than encompassing (6). Furthermore, currently there are issues with the reproducibility of LC/MS that are difficult to correct retrospectively by alignment, plus there are practical issues limiting the number of replicate runs that can be made and therefore experimental power for biomarker discovery. For these reasons, protein modelling, quantification, and differential expression analysis with 2-DE continues to be an important workhorse method for proteomics research.
The first step in proteomic informatics analysis is image acquisition, either of gels or mass spectra (in LC/MS). The traditional 2-DE informatics pipeline then attempts to identify spot boundaries and quantify individual spots on each gel before proteins are compared for differential expression by matching cognate spots between gels. With existing commercial software, errors in each step contribute to a highly subjective and labour-intensive correction. For example, it has been noted that increasing the number of gels in an experiment dramatically reduces the percentage of correct automated spot matches (7). Recently, a more robust and automated concept has emerged (8), where gels are directly aligned in the image domain (9, 10) so that subsequent spot detection can be based on the integration of the spot appearances in every gel. It has been shown that through preservation of the raw image information contained in each spot and its statistical “fusion” over the gel set, increased power and reliability in quantification is possible which further improves as the sample size is increased (11). Statistical rather than deterministic treatment is key to this new paradigm (12).
In this chapter, we describe the methodology for both approaches and discuss the pitfalls present when reasoning statistically about differential protein expression, with particular emphasis given to the need to perform power analyses and control the false discovery rate. In the remainder of this section, an overview of the established proteome informatics methods will be provided so that the choice of software detailed in Subheading 2 can be better understood. Subheading 3 then details step-by-step instructions for performing the analyses.
1.1. Proteome Informatics
There are a number of challenges in 2-DE proteome informatics (13). Despite the high resolution, diversity of cellular proteins often leads to spot co-migration. Some spots also tend to have severe tails in either dimension, confounding spot modelling. Contrast variations due to stain exposure, sample loading errors, and protein losses during processing inhibit the reliability of volume quantification. Furthermore, geometric distortions due to casting, polymerization, and the running procedure make the deduction of corresponding spots between gels demanding and therefore differential analysis challenging. The DIGE protocol (14) allows up to three samples to be run on the same gel with consequently little geometric discrepancy between them. However, typical experiments require a considerably greater sample size than a single DIGE gel to attain adequate power, and so inter-gel alignment is still a problematic issue. The typical steps in proteome informatics for 2-DE are (13):
Image acquisition: This prepares each raw acquisition for subsequent comparative analysis. After scanning, the images are pre-processed by cropping (manual delineation), noise suppression, and background subtraction (e.g., with mathematical morphology or smooth polynomial surface fitting).
Conventional analysis (Spot Detection » Spot Matching): Each protein spot is delineated and its volume quantified. Typically, the spots are segmented first by the watershed transform (15), where spots are treated as depressions in a landscape which is slowly immersed in water. Spot boundaries (watersheds) are constructed where the pools start to meet. Co-migrating spots with separate peaks are then separated by parametric spot mixture modelling e.g., optimizing the parameters of one or more 2-D Gaussians to minimize the squared residuals. Point pattern matching is then employed to match the spots between gels, which finds the closest spot correspondence between a point pattern (source spot list) and a target point set (reference spot list).
Image-based analysis (Gel alignment » Consensus Spot Modelling): With current techniques, a “reference” gel is chosen and the other “source” gels are aligned to it in pair-wise fashion. For the new image-based paradigm, “direct image registration” is applied which defines a transformation that warps (deforms) the source gel and a similarity measure which quantifies the quality of alignment between the warped source gel and the reference gel. The aim is to automatically find the optimal transformation that maximizes the similarity measure. Spot detection is then performed on an image or “spot mask” created from the set, which is then propagated to each individual gel for spot quantification.
Differential analysis : At this stage, we have a list of spots, and for each spot, a quantified abundance in each gel. The abundances are first normalized to remove systemic biases between gels and between channels in DIGE gels. Variance stabilization can then be employed to remove the dependence between the mean abundance of a protein and its variance e.g., a simple logarithmic transformation to fold-change values. Significance tests are then performed to obtain p-values for rejecting the null hypothesis that the mean spot abundance between groups is unregulated.
Advanced techniques : Since multiple hypothesis testing leads to a large number of false positives, it is essential to control the False Discovery Rate (FDR). The FDR is the estimated percentage of false positives within the detected differential expression rather than within the set of tests as a whole. Power analysis is also essential, which estimates the false negative rate that determines the optimal sample size needed to detect a specific fold change to a particular confidence level. Typical software packages do not contain these important methods.
Diagnostics: It is useful to look at various diagnostics to assess quality control of the gels in a given experiment, and to search for evidence of any artifacts that may indicate some problems in the gels. Hierarchical clustering can be used to assess which gels are most similar to each other, which can reveal experimental design or other quality control issues in the data. Further, one should visually assess any spots detected as differentially expressed to ensure that outliers unrelated to the biological groupings do not drive the result.
2. Materials
2.1. Image Acquisition
An image capture device is required, for which there are three main categories:
Flatbed scanner: This mechanically sweeps a standard charge-coupled device (CCD) under the gel and can be used to obtain 12–16 bits of greyscale or colour densitometry from visible light stains. Noise can be an issue due to size and cooling restrictions on the moving sensor and the need for reconstruction through image “stitching.” Calibration is often required to provide linearity. Based on high-end document scanners but fully sealed, flatbed scanners are typically the least expensive offerings. Examples: ImageScanner (GE Healthcare, Chalfont St. Giles, UK), ProteomeScan (Syngene, Cambridge, UK) and GS-800 (Biorad, Hercules, CA).
CCD camera: Since the sensor is fixed, its greater size and cooling provides a dramatic improvement in noise and therefore dynamic range (up to 104). Different filters and transillumination options allow a wide range of stains to be imaged, including visible light, fluorescent, reverse, chemiluminescent, and radioactive signals. However, the fixed sensor limits image resolution, while vignetting (reduction of brightness at the periphery) and barrel distortion requires dark frame and flat frame correction respectively, affecting quantification. Examples: LAS (Fuji Photo Film, Tokyo, Japan), ImageQuant (GE Healthcare), Dyversity (Syngene), BioSpectrum2D (UVP, Upland, CA, USA) and VersaDoc (Biorad).
Laser scanner: Photomultiplier detectors are combined with laser light and optical or mechanical scanning to pass an excitation beam over each target pixel. While slower than CCD cameras, spatial resolution is excellent and logarithmic response leads to a dynamic range of up to 105. However, acquisition is limited to dyes whose excitation spectra match that of the installed laser sources, which are costly. With some products, visible light stains can be negatively imaged by using a fluorescent back board. Examples: FLA (Fuji Photo Film), Typhoon (GE Healthcare) and PharosFX (Biorad).
Please see (16) for further details. Most specialized acquisition devices come with software to crop the resulting scans, but if this is unsuitable, the packages described in the next two sections have this facility, as does ProteomeGRID (http://www.proteomegrid.org/). See Note 1.
2.2. Conventional Analysis Software
A commercial software package is required, such as:
ImageMaster 2D or DeCyder (GE Healthcare, Chalfont St. Giles, UK)
Dymension (Syngene, Cambridge, UK)
Melanie (GeneBio, Geneva, Switzerland)
PDQuest (BioRad, Hercules, CA)
ProteinMine (BioImagene, Cupertino, CA)
These products are all quite expensive, so comparative personal evaluation is essential. As a guide, comparative assessments appear in the literature (7).
When choosing an acquisition device, it is important to ensure that the output format is compatible with the input format of the analysis software. While typically this involves standardized interchange with the TIFF format, few vendors adhere fully to the standard and therefore incompatibilities do occur. TIFF is also limited to 16 bits of linear dynamic range so some packages implement formats such as Fuji “IMG”, which supports logarithmic image capture, and GE Healthcare “GEL”, which supports square-root image capture. See Note 2.
2.3. SEA Image-Based Analysis Pipeline
Two commercial packages exist that adopt elements of the image-based analysis paradigm:
Delta2D (Decodon, Greifswald, Germany)
Progenesis SameSpots (Nonlinear Dynamics, Newcastle, UK)
Both packages perform image alignment before consensus spot detection. However, the alignment performed is only semi-automated with considerable user interaction, and the quantification is based on heuristic delineation of spot boundaries rather than more reliable peak detection (11). To utilize automated image-based alignment and fully harness strength borrowed from the whole gel set in the spot modelling phase, the following techniques can be combined:
RAIN (9, 10) (http://www.proteomegrid.org/) for automatic gel alignment.
Pinnacle (11) for automated spot detection and quantification that borrows strength between gels in determining what is a real spot.
2.4. Differential Analysis
The commercial packages described in Subheading 2.2 and 2.3 contain the standard tools for determining the statistical significance for regulation of an isolated protein between treatment groups. However, at the time of writing only Delta2D (Decodon) and the add-on Progensis Stats module (Nonlinear Dynamics) have facilities to correct for multiple hypothesis testing with FDR. If your software does not include FDR estimation or you wish to use freely available tools, one of the following microarray analysis suites can be used:
The R language and BioConductor repository (http://www.bioconductor.org/)
TM4 (http://www.tm4.org/)
The above suites also contain the more advanced normalization and power analyses described herein.
For data quality assessment and to investigate hidden factors in the data, the majority of commercial packages described in Subheading 2.2 and 2.3 contain basic data mining techniques. If a required technique is not available in your software, a range of advanced classification and data summarization methods can be found by using the microarray analysis suites above or with commercial solutions including:
Progenesis Stats (Nonlinear Dynamics)
Decyder EDA (GE Healthcare)
Genedata Expressionist (Genedata, Basel, Switzerland)
3. Methods
Today, typical experimental design should include enough biological replicate gels in each treatment group to confidently detect differential expression, though the optimal number is highly dependent on the tissue, sample preparation, and running protocols. It is therefore necessary to perform a few test experiments to optimize power as detailed in subheading 3.4 step 4. A good example of such a study is by Hunt et al. (17), where they determined that a sample size of 7–8 biological replicates would permit detection of a 50% change in protein expression in plasma samples. Since proteomics studies are challenging and time consuming, thorough planning of the experimental design is needed to protect against systematic bias. Therefore, standard design principles such as blocking and randomization of sample runs should be applied (18). Also, technical replicates should never be run at the expense of biological replicates. The study by Hunt et al. makes this point in dramatic fashion, showing much greater improvements in statistical power by increasing sample size rather than numbers of technical replicates. Nevertheless, if both must be mixed in the same experiment, both sources of error should be handled, see subheading 3.4 step 5.
A number of suggestions to bear in mind during 2-D gel running if informatics is to be facilitated are:
In general, the second dimension running should be consistent between samples. The spot matching and gel alignment algorithms will be confounded if some spots are visible in some gels and not in others. In any case, these spots will lack full statistical support for ascertaining differential expression.
Similarly, if one gel looks markedly different than other gels in the same treatment group, it should be discarded rather than incorporated into the analysis, since this will likely add significant outliers and therefore violate the assumption that the biological variation is normally distributed.
Saturation of abundant spots must be avoided as this will introduce increased error into the quantification (19). Moreover, the splitting of saturated complex spots is inaccurate regardless of the approach used.
Background and noise should be minimized otherwise dynamic range will be compromised, resulting in impaired sensitivity and specificity in spot detection. Danger areas include inadequate sample preparation and destaining, contaminated gels and too high laser scanner photo multiplier tube (PMT) voltages.
Between treatment groups, normalization can be greatly facilitated by employing DIGE and running a pooled sample on each gel as a paired control for gel normalization.
Within a given laboratory, studies should be performed to identify the key sources of experimental variability, and those factors should be accounted for using randomized block designs. These should be used to ensure that potentially important experimental factors are not confounded with factors of interest. See Note 3.
Another important design consideration is whether or not to pool samples. Pooling samples increases the protein load on each gel, which may reduce technical variability, but also results in loss of information about each sample. While sometimes necessary in order to obtain enough protein to reliably run the assay, pooling should be avoided since it results in a reduction of statistical power. When pooling samples, the key sample size factor is the number of pools, not the number of subjects. See Note 4.
3.1. Image Acquisition
The full dynamic range of the scanner should be utilized in order to maximize the number of weakly expressed spots visible above the noise floor. If your acquisition device does not provide automatic calibration, this can be done with a step tablet (e.g., UVP, Upland, CA) or a step wedge (e.g., Stouffer Industries, Mishawaka, IN). If unavailable, a generic IT8 scanner target can be substituted as the bare minimum.
Once scanned, accurate cropping of the gels is essential. All gel edges must be outside the cropped region otherwise the alignment algorithm will attempt to align the edges at the detriment of aligning the spot patterns and erroneous spots will often be detected along the edges. Special care should be taken to ensure that every spot is inside the cropping region in all gels. If your informatics package supports irregular inclusion and exclusion regions, any artifact that appears on only a subset of the gels should be cropped away, such as cracks, fingerprints, and smudges. Suitable cropping is illustrated in Fig. 1.
Fig. 1.
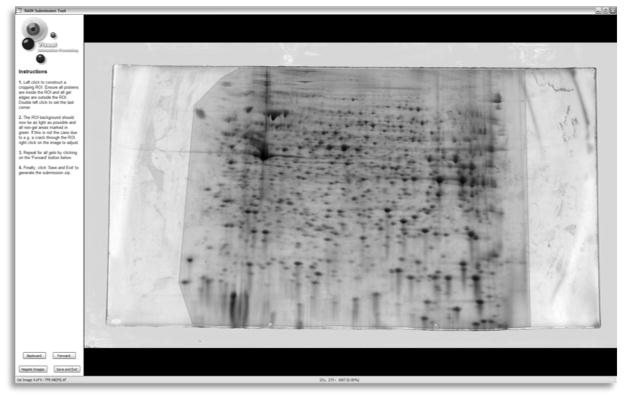
Gel image cropping with the RAIN submission tool. The shaded area shows an optimum polygonal crop that removes the gel edges and some artifacts while retaining the protein spots
3.2. Conventional Analysis Pipeline
Since spot detection is the first stage in conventional analysis, the gels must be first background subtracted to remove non-protein elements as well as all streaks and smears that do not adhere to the software’s protein spot model. However, the removal is subjective and can interfere with surrounding real spots. See Note 5.
The spot detection process is then initiated on each gel separately. Typically, spot detection is controlled by setting a handful of algorithm-specific parameters, which should be optimized for each experiment but fixed inside the same experiment. Unfortunately, optimization is a subjective process which requires a trade-off between false-positives (noise detected as spots, over-segmented spots) and false-negatives (spots failed to be detected, under-segmentation of merged spots).
Even with optimization, a significant amount of manual editing will be required post-hoc, which could introduce an element of subjective bias. In particular, calculation of spot boundaries is fraught with errors yet affects protein quantification significantly. See Note 6.
Once spots are quantified, a characteristic vector is extracted from each detected spot, which includes position, volume and perhaps shape and boundary information. These are combined to form a spot list for each gel. A reference gel is then manually chosen (or the software may suggest one), and in turn, each spot list is matched to the reference spot list. Since spots are matched between all the gels using the reference gel as an intermediary, any spots undetected on the reference will not be matched. See Note 7. Typically, the neighbourhood of each spot is used to facilitate the matching process, which is why outliers markedly affect the analysis.
It is expected that a significant number of weakly expressed spots will be detected only on a subset of the gels and therefore there will be a number of missing values in the resultant spot match list. These missing values reduce statistical power significantly and can introduce inadvertent bias. Thus, first of all, one should manually edit the spot detection and matching to maximize the number of successfully matched spots across gels. Invariably, there will still be some missing values in the spot match list, which must be dealt with in some way. Simply ignoring these spots for analysis introduces bias, since many of the gels with no matching spot likely had negligible or no expression of the corresponding protein. Substituting zeros or some other small value is a better option, but can still introduce bias, since it is expected that for some gels, there is evidence of some non-zero expression of the protein, but it simply fell below some arbitrarily specified detection threshold. Missing data is one of the major unsolved problems when using the conventional analysis pipeline.
Because of the nature of this traditional analysis pipeline, errors in automated spot matching increase as the experiment grows larger (7), meaning that the number of accurately matched spots decreases as increasing numbers of gels are run. This propagation of processing errors encourages researchers to run smaller studies that in turn are underpowered to statistically detect group differences when multiple testing is taken into account. The only current solution to this problem is to employ the image-based analysis paradigm instead.
3.3. Image-Based Analysis Pipeline
The first stage of the image-based pipeline is to manually select the gel with the most representative protein pattern and positions to be the reference image. The other gel images are then automatically warped so that their spot patterns are brought into alignment with that of the reference.
-
With Delta2D and Progenesis SameSpots, you must first manually identify a few spots that can be matched unambiguously in every gel in the set. The spots should be spread out evenly over the gel’s surface otherwise some regions will be aligned too poorly now to be corrected later. The software will then (or after every landmark) automatically generate a smoothly interpolated warp that aligns these landmarks and estimates the intermediary alignment between them. If available, a further automatic phase can be initiated that adjusts the intermediary alignment to better match the remaining spots. These matches can be iteratively accepted or modified by the user and the algorithm rerun. Finally, alignments must be completed by hand and a “spot mask” applied to the reference gel of each set.
With RAIN, fully automated image registration is performed by considering basic image gradients at several levels of detail and is therefore able to use extra image features such as global protein distribution, background, streaks, and smears in the alignment, as illustrated in Fig. 2. The set of gels is simply submitted to the ProteomeGRID web service (http://www.proteomegrid.org/), and after remote processing, the set of aligned images is available to download, together with visualisations to confirm the accuracy of the alignment.
The aligned images will then be automatically composed to create an image for subsequent spot detection. Pinnacle recommends computing an “average gel” that involves taking pixel-wise means of intensities across all gels. The key advantage of using the mean gel is that noise is reduced by √n for a set of n gels, while the signal for true spots is reinforced across gels, thus substantially improving the sensitivity of detection for weak expression whilst suppressing highly variable features expected to be artifacts. Spot detection on the average gel will tend to have increased sensitivity over individual spot detection on each gel for any proteins present in more than 1/√n of the gels (11). Furthermore, variability will decrease as the gel sample size increases. Delta2D’s fusion image is constructed by placing more emphasis on dark pixels likely to be protein matter (20). While the resulting image exhibits more spots than the average gel, statistically weak spots are artificially amplified so an increased number of false positives is possible.
Background subtraction and normalization may be applied to the average gel at this time. Background estimates can be global or local (11). Normalization adjusts for gel-specific effects such as protein load. One common global method for normalization is to divide by the total volume, or average intensity, on the background corrected gel. If performed after spot detection and quantification, the sum total of quantified spot protein abundance can be employed instead. In DIGE experiments with a common reference channel, dividing each spot volume by its corresponding reference channel spot volume provides for a more precise normalization.
The next step is to detect spots on the average gel while obtaining spot quantifications for each spot on each gel. Progenesis SameSpots and Delta2D compute spot volumes on individual gels after detection of spot boundaries on the fusion image. Spots that are too weak to be detected on some gels by conventional means are able to be quantified by the consensus approach. However, while the resultant spot match list has “no missing values”, the correctness of these values is exclusively dependent on their correct alignment. The spot detection results must be manually verified as described in Subheading 3.2 steps 2–3. See (7) for a comparison of Delta2D against conventional approaches. The spot boundaries are then copied onto each individual gel for quantification. See Note 8.
Fig. 2.

Automatic image-based gel alignment by http://www.proteomegrid.org/. (a) Gels are cropped with the RAIN submission tool (see Fig. 1) and split into treatment groups. (b) The images are uploaded together with relevant metadata such as reference gel, stain/label used and DIGE channel. (c) Each gel is automatically aligned to the reference. The grid lines show the various levels of image warping needed. (d) Pixel-wise difference between reference and sample images before alignment. (e) After alignment, the differences should only be due to differential expression and artifacts, which will be differentiated by downstream spot modelling
After wavelet denoising the average gel image, Pinnacle focuses on peaks or “pinnacles” rather than spot boundaries and volumes in its detection and quantification algorithm. The idea is that non-saturated spots have well-defined pinnacles, and the intensity at this pinnacle is highly correlated with the spot volume but less affected by neighbouring spots. If saturation is avoided, peak detection on the average gel is sufficient to separate co-migrated spots, and furthermore, quantification using peak height only is more reliable and has greater validity than that derived through the spot boundary (11). Therefore, given a set of aligned images annotated by their treatment group, Pinnacle automatically outputs a list of peak intensities for each spot and each gel for downstream statistical significance testing as shown in Fig. 3.
Fig. 3.

Average gel computed after alignment using RAIN, with spots detected by Pinnacle marked with an “x”
3.4. Differential Analysis
After the previously described image processing, we are left with a matrix of spot quantifications for each spot across all gels. This matrix can be analyzed to discern which protein spots are differentially expressed across treatment groups.
Transformation: Frequently, the raw spot volumes or pinnacle intensities are highly skewed right, with many outliers, and the variance of a spot is related to its mean. These properties violate the assumptions underlying many statistical tests, such as t-tests or linear regression. To deal with this problem, it is possible to use some transformation of the spot quantifications before performing statistical analyses. Candidate transformations include the log, square root, and cube root transformations. Frequently, it helps to add a small constant (e.g., ½ or 1) before transforming the volumes in order to avoid artifacts near zero intensities. See Note 9. Choice of transformation can be assessed by QQ-plots and histograms of the residuals from the statistical test of interest.
Statistical tests for differential expression: In the past, one way to assess differential expression of spots is to simply flag spots with the largest fold-changes across groups. This approach is statistically flawed, since fold change does not take the variability in the data into account, and thus makes it impossible to gauge the level of false positives. Appropriate statistical tests which take into account experimental variability are Student’s t-test for two treatment groups, ANOVA for three or more treatment groups, and linear regression for quantitative correlative studies. If adequate normality cannot be obtained, the non-parametric Mann-Whitney and Kruskal-Wallis ANOVA tests can be substituted. After testing, each protein will be associated with a probability (the “p-value”) that the observed difference could occur by chance. A histogram of the range of p-values can be checked for a peak near 0, which is a promising sign for significant differential expression. A peak elsewhere suggests technical problems with the gels.
False-Discovery Rate-Based Thresholds: Since p-values are obtained for each of many spots (100’s or 1,000’s) in the experiment, a p-value threshold of 0.05 would typically lead to a great deal of false positives, since we expect 5% of all spots to have p-values less than 0.05 even if there are truly no proteomic differences between groups. In recent years, various methods to estimate and control the false discovery rate (FDR) have arisen, and can be used to find appropriate p-value thresholds for declaring statistical significance. Controlling the FDR at some level, say 0.05, means that of all spots we call differentially expressed, we expect only 5% of them to be false positives, and the other 95% true positives. See Note 10. Various other methods exist that are available for performing this analysis in Bioconductor/R, e.g. fdrtool. After using this method, a q-value or overall FDR threshold (typically 0.05, 0.10, or 0.20) is specified, and we obtain a list of differentially expressed spots.
Power Calculations: Must be performed since it will be necessary to redo the experiment with decreased variance or, usually more attainably, an increased sample size (number of replicates) if the statistical power is found to be too low. Software able to estimate the optimal sample size when the FDR is controlled is available as dictated in subheading 2.4. It is highly recommended that preliminary studies are performed so that the power calculations are based on the ranges of biological and technical variability for a particular experiment before a definitive protocol is laid down.
Mixed Effects: When multiple gels are obtained, one must take care in performing the statistical analysis since protein levels for replicate gels from the same individual are correlated with each other, violating the independence assumption underlying the test. One approach would be to average the spot quantifications across replicate gels to obtain one measurement per individual, and then analyze using a t-test or some other method assuming independence. Another alternative would be to use a method that takes this nested design into account. For example, a generalization of the t-test or ANOVA (21) or linear regression for correlated data would be a linear mixed model, including a fixed effect for treatment group, and random effect for the individual. Inference on the fixed effect from this model, then, yields a p-value that appropriately takes the correlation between gels from the same individual into account. Mixed models, e.g., PROC MIXED, Cary, NC, can be implemented in standard statistical software, including SAS and R. See Note 11.
Hierarchical Clustering: This can be applied to the matrix of spot intensity values to see which samples cluster strongly together. In running this clustering, one can see how individuals within the same treatment group are similar. Also, these can be useful diagnostics to see whether there is some experimental factor that may have been strongly influential in the study. For example, if all samples run in the same IEF block cluster strongly together, that could indicate that something happened with that IEF block to make its gels different than the others. These can be valuable indicators to aid in the design of future studies.
Footnotes
Never manipulate the images in a generic image-editing package such as Photoshop (Adobe, San Jose, CA) before analysis. Even if the process appears risk-free such as cropping, a number of side-effects can occur silently. For example, calibration curves and metadata are likely to be lost, or the images may be quietly converted to 8 bit.
Never attempt quantification on images saved in a lossy compression format such as JPEG. Not only are these limited to 8 bits of dynamic range, but they also remove details that are essential for accurate protein quantification.
For example, if we have a case control study and the isoelectric focusing is performed in blocks of 8, ensure that for each run, 4 cases and 4 controls are run in the same block, with the positions determined based on a random number generator. This way, any variability in IEF runs will not mistakenly appear as a case/control effect, which can happen when IEF run and case/control are confounded.
It can be shown that maximizing the number of pools minimizes the total variance, yielding maximum power. A disastrous design would be to combine all cases into one pool and all controls into another pool, and then run replicate gels from each pool. If this design is used, it is impossible to assess biological variability, since the variation across gels would only capture technical variability. As a result, it would not be possible to do a valid statistical assessment of differential expression. Thus, if pooling is deemed necessary, one should maximize the number of pools, and make sure to have multiple pools per treatment group.
The background subtraction task is never perfectly discriminating, and therefore it is usually performed conservatively to ensure that the accuracy of protein quantification is not adversely affected.
In order to maximize the effectiveness of the spot-matching phase and minimize further manual verification, when using conventional analysis methods, it is important to make editing decisions consistently over the gel set. This is obviously a difficult task because of the migration variability between gels, and is a significant limitation of the conventional pipeline.
Some software may have an option to match every spot list to every other spot list to avoid this limitation. In this case, the reference gel is used only to define the fixed positional reference frame to which the other gel’s spots are migrated to.
Typical image-warping techniques do not preserve the amount of protein in each spot, leading to over-expression in areas of dilation and under-expression in areas of contraction. In order to avoid this issue, Progensis SameSpots and Delta2D unwarp each consensus spot boundary so that it can be applied to the original unaligned gels where quantification takes place. RAIN applies a volume-invariant warping procedure, wherein each pixel is weighted by its change in size, thereby allowing accurate quantification on warped gels.
One benefit of the log transformation is that a difference in the log scale corresponds to a multiplicative fold-change in the raw scale. For example, if a log2 transformation is used, a difference of 1 between groups corresponds to a two-fold multiplicative difference.
One simple method models the p-value histogram as a mixture of two distributions: the null distribution (true negatives and false positives) as the underlying uniform distribution; and the alternative distribution (true positives and false negatives) as a right skewed distribution with mode near zero. From this, it is possible to estimate the probability of a false positive for each p-value (this probability is called a q-value), and to estimate a cutpoint on p-values that controls the overall FDR at a prescribed level.
The mixed models can also be used in the design phase, using preliminary studies on the tissue of interest to estimate levels of technical and biological variability for spots in the study. These estimates can then be used to perform power calculations and make design determinations, e.g., if the technical variability is very large relative to biological variability, then it may be helpful to run several replicate arrays for each biological sample.
References
- 1.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 2.Klose J. Protein mapping by combined iso-electric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik. 1975;26:231–43. doi: 10.1007/BF00281458. [DOI] [PubMed] [Google Scholar]
- 3.Rabilloud T. Two-dimensional gel electrophoresis in proteomics: old, old fashioned, but it still climbs up the mountains. Proteomics. 2002;2:3–10. [PubMed] [Google Scholar]
- 4.Wolters DA, Washburn MP, Yates JR. An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73:5683–90. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 5.Roe MR, Griffin TJ. Gel-free mass spectrometry-based high throughput proteomics: Tools for studying biological response of proteins and proteomes. Proteomics. 2006;6:4678–4687. doi: 10.1002/pmic.200500876. [DOI] [PubMed] [Google Scholar]
- 6.Reidegeld KA, Müller M, Stephan C, Blüggel M, Hamacher M, Martens L, Körting G, Chamrad DC, Parkinson D, Apweiler R, Meyer HE, Marcus K. The power of cooperative investigation. Proteomics. 2006;6:4997–14. doi: 10.1002/pmic.200600305. [DOI] [PubMed] [Google Scholar]
- 7.Clark BN, Gutstein HB. The myth of automated, high-throughput two-dimensional gel analysis. Proteomics. 2008;8:1197–1203. doi: 10.1002/pmic.200700709. [DOI] [PubMed] [Google Scholar]
- 8.Veeser S, Dunn MJ, Yang GZ. Multiresolution image registration for two-dimensional gel electrophoresis. Proteomics. 2001;1:856–870. doi: 10.1002/1615-9861(200107)1:7<856::AID-PROT856>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 9.Dowsey AW, Dunn MJ, Yang GZ. Automated image alignment for 2-D gel electrophoresis in a high-throughput proteomics pipeline. Bioinformatics. 2008;24:950–957. doi: 10.1093/bioinformatics/btn059. [DOI] [PubMed] [Google Scholar]
- 10.Dowsey AW, English J, Pennington K, Cotter D, Stuehler K, Marcus K, Meyer HE, Dunn MJ, Yang GZ. Examination of 2-DE in the human proteome organisation brain proteome project pilot studies with the new RAIN gel matching technique. Proteomics. 2006;6:5030–5047. doi: 10.1002/pmic.200600152. [DOI] [PubMed] [Google Scholar]
- 11.Morris JS, Walla BC, Gutstein HB. Pinnacle: A fast, automatic and accurate method for detecting and quantifying protein spots in 2-dimensional gel electrophoresis data. Bioinformatics. 2008;24:529–536. doi: 10.1093/bioinformatics/btm590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowsey AW, Yang GZ. The future of large-scale collaborative proteomics. Proceedings of the IEEE. 2008;96:1292–1309. [Google Scholar]
- 13.Dowsey AW, Dunn MJ, Yang GZ. The role of bioinformatics in two-dimensional gel electrophoresis. Proteomics. 2003;3:1567–1596. doi: 10.1002/pmic.200300459. [DOI] [PubMed] [Google Scholar]
- 14.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 15.Vincent L, Soille P. Watersheds in digital spaces: an efficient algorithm based on immersion simulations. IEEE Transactions on Pattern Analysis and Machine Intelligence. 1991;13:583–598. [Google Scholar]
- 16.Miura K. Imaging technologies for the detection of multiple stains in proteomics. Proteomics. 2003;3:1097–1108. doi: 10.1002/pmic.200300428. [DOI] [PubMed] [Google Scholar]
- 17.Hunt SMN, Thomas MR, Sebastian LT, Pedersen SK, Harcourt RL, Sloane AJ, Wilkins MR. Optimal replication and the importance of experimental design for gel-based quantitative proteomics. Journal of Proteome Research. 2005;4:809–819. doi: 10.1021/pr049758y. [DOI] [PubMed] [Google Scholar]
- 18.Box GEP, Hunter WG, Hunter JS. Statistics for experimenters: an introduction to design, data analysis, and model building. Wiley; New York: 1978. [Google Scholar]
- 19.Almeida JS, Stanislaus R, Krug E, Arthur JM. Normalization and analysis of residual variation in two-dimensional gel electrophoresis for quantitative differential proteomics. Proteomics. 2005 Apr;5:1242–1249. doi: 10.1002/pmic.200401003. [DOI] [PubMed] [Google Scholar]
- 20.Luhn S, Berth M, Hecker M, Bernhardt J. Using standard positions and image fusion to create proteome maps from collections of two-dimensional gel electrophoresis images. Proteomics. 2003;3:1117–1127. doi: 10.1002/pmic.200300433. [DOI] [PubMed] [Google Scholar]
- 21.Horgan GW. Sample size and replication in 2D gel electrophoresis studies. Journal of Proteome Research. 2007;6:2884–2887. doi: 10.1021/pr070114a. [DOI] [PubMed] [Google Scholar]