

Although there have been recent notable successes in the discovery of ligands that target stable, high affinity protein-protein interactions (PPIs), the transient and moderate affinity PPIs that underpin many fundamental cellular processes have proven to be far less tractable for ligand discovery.[1] Prime examples of this are the dynamic complexes formed between DNA-bound transcriptional activators and coactivators that are part of eukaryotic transcription initiation.[1b, 2] In this instance, complex formation is mediated through interactions that are transient and only of moderate affinity (KDs of 0.1-10 μM).[3] An additional complication common to transient/modest affinity PPIs is that one or both of the binding interfaces is often used for complex formation with a variety of partners (Figure 1a).[4] Specificity is often fine-tuned in these complexes by allosteric regulation, with the binding of one ligand impacting the affinity of another ligand (Figure 1b).[5] Small molecules that can take advantage of these dynamic binding interfaces could potentially modulate the binding of ligands at multiple different sites on a protein yet maintain specificity for the target protein.[6] Here we report the identification of two uniquely specific ligands for the coactivator CBP/p300 that are of the depside (sekikaic acid) and depsidone (lobaric acid) natural product family, a group first identified by Emil Fischer in the early 20th century as polypeptide-like small molecules consisting of a series of phenol carboxylic acids units.[7] Through interaction with a dynamic surface of the CBP/p300 GACKIX domain, these molecules effectively inhibit the ability of two distinct classes of activators to form a complex with the coactivator yet do not affect other related complexes. In addition, the IC50 values of the two natural products rank them among the most effective inhibitors of these dynamic binding surfaces, demonstrating the enormous potential of natural products for targeting difficult PPIs.
Figure 1.
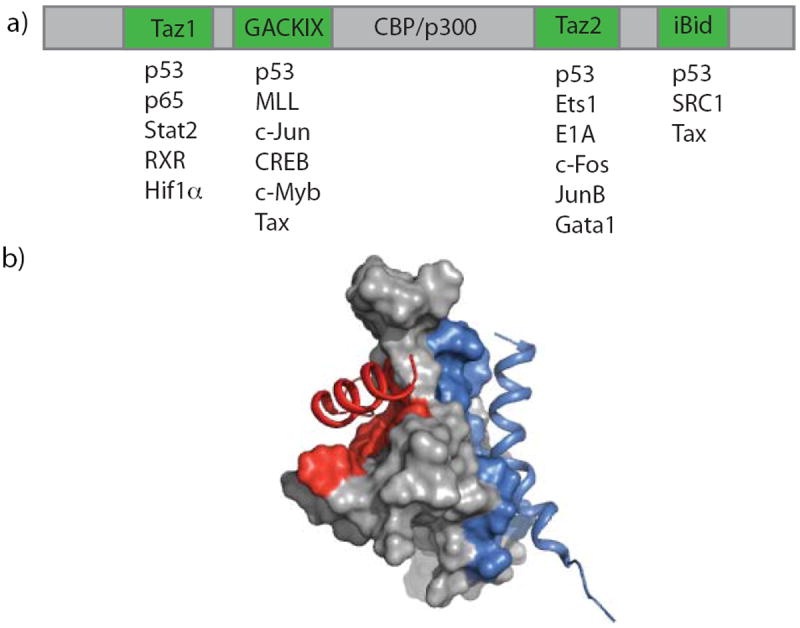
Activator-coactivator interactions. a) The coactivators CBP/p300 contain multiple distinct activator-interaction domains (green) that each are known to partner with several natural TADs b) The GACKIX domain of CBP/p300 interacts with activators using two allosterically regulated binding sites: a deeper binding cleft (red) for interaction with activators such as MLL and c-Jun and a shallower, broader binding site to interact with c-Myb and CREB (blue). (adapted from PDB ID: 2AGH)
The GACKIX domain of CBP/p300 is a prototypical activator binding motif in that it uses two distinct but allosterically connected binding surfaces to engage a variety of transcriptional activators and has been identified in several otherwise distinct coactivators.[5b, 8] The activators MLL, c-Jun, Myb and CREB each utilize this domain within the coactivator and histone acetyl transferase CBP/p300 as part of their transcription initiation function, with the first two examples interacting with a relatively deep binding cleft (red) while the latter two bind to a shallower, broader binding site (blue) (Figure 1b).[8a, 9] One of the three helices (α3) spans the two sites and enables communication between two bound activators, with, for example MLL and c-Myb binding cooperatively (2-fold) to the domain.[5b, 5c, 8a] These experimental[5b, 8a] and computational studies[5c] also reveal that large conformational changes within the flexible loop L12 and 310 helix G2 of GAXKIX are strongly coupled to the allosteric network of conformational changes in α3 upon MLL binding. Due to the role that GACKIX-targeting activators play in neurological disorders and in cancer,[10] there have been numerous efforts to identify modulators that would affect the binding of the activators to this domain.[11] With one exception,[11e, 12] efforts have focused on ligands binding the larger and shallower CREB/Myb site, which appears to be the more challenging of the two binding surfaces. Surprisingly, there is little functional or binding evidence suggesting allosteric modulation of the MLL/Jun binding surface in these cases.[11a-e] We hypothesized that by screening against an activator-GACKIX complex in which the activator was bound in the deeper and more flexible MLL/Jun binding surface, identification of inhibitors that affect the allosteric communication between the two sites would be more likely.
To screen for ligands that interact with GACKIX, we used a high-throughput fluorescence polarization assay with a fluorescein-labeled version of the MLL transcriptional activation domain. A 50,000 member compound collection consisting of a diverse set of molecules from commercial libraries of small drug-like molecules selected based on computed structural properties (LogP, polar surface area, number of rotatable bonds etc) was screened in this assay format. Although only moderately stringent conditions were used, no hits emerged from this exercise (see SI for details). In parallel, a diverse collection of ~15,000 natural product extracts isolated from marine sediment-derived microbes, cyanobacteria, lichens and sponges was screened. In contrast to the commercial compound collection, 64 of the natural product extracts inhibited the MLL-GACKIX interaction. Subsequently, follow-up assays using two protein-protein and one protein-DNA interaction counter screens resulted in two extracts that showed repeated and selective inhibition of MLL-GACKIX (Figure 2a). The active compounds were identified in HPLC fractionated extracts using NMR and mass spectrometry, yielding lichen-derived depsides sekikaic acid, microphyllinic acid and 4-O-demethylmicrophyllinic acid (Figure 2b).
Figure 2.
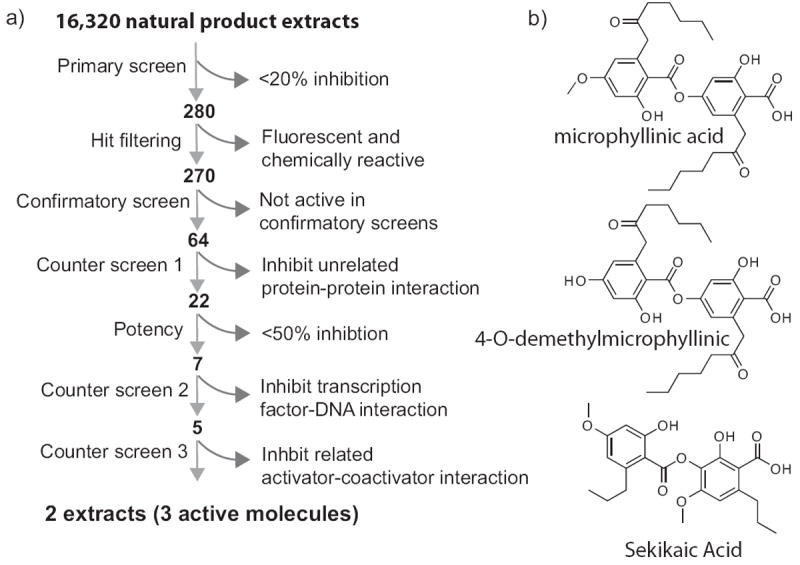
High-throughout screening for small molecules that target GACKIX. a) Flowchart for screening of natural product extracts for inhibiting the MLL-GACKIX interaction. 16,320 extracts were screened using a high-throughput FP assay and after a series of counter screens only 2 extracts were obtained that selectively inhibited the MLL-GACKIX interaction (see Supporting Information for experimental details). b) Structure of molecules found in active extracts: microphyllinic acid, 4-O-demethylmicrophyllinic acid and sekikaic acid.
Although depsides have been reported to have anti-oxidant, antibiotic, anti-HIV activity and to be inhibitors of cellular biosynthetic processes, only recently have they been shown to affect protein-protein interactions.[13] We initially focused our attention on the most abundant depside observed in the extracts, sekikaic acid, and generated a dose-response inhibition curve of the MLL-GACKIX complex (IC50 34 μM)(Figure 3). This places sekikaic acid as the most potent small molecule inhibitor of the complex and among the most potent inhibitors of activator-coactivator complexes.[1b, 1d, 2c, 11b, 11e, 14] The GACKIX domain of CBP/p300 has two primary binding sites that make contacts with activators.[8a, 9a] The shallower and broader site interacts with the KID domain of the activator CREB.[15] To investigate if sekikaic acid can modulate binding of activators to both binding sites, a FP-based inhibition experiment was performed with KID. Sekikaic acid was also found to inhibit the complex of Fl-KID- GACKIX with an IC50 of 64 μM (Figure 3). Taken together, the inhibition experiments show that sekikaic acid is able to block activators at both binding sites on GACKIX, the first reported small molecule that can effectively perform this function.
Figure 3.
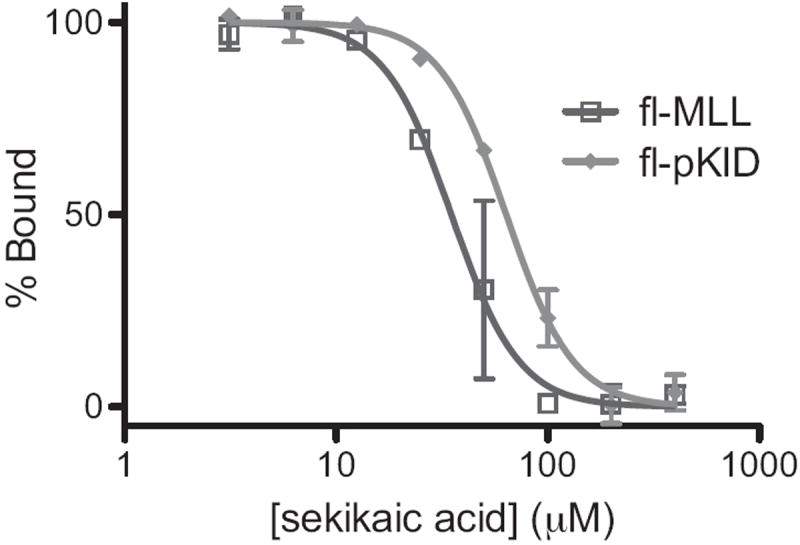
Inhibition of TAD-GACKIX complexes by sekikaic acid. Increasing concentrations of sekikaic acid were added to the GACKIX domain in complex with fluorescein-tagged MLL (fl-MLL: dark gray trace) or fluorescein-tagged pKID (fl-pKID light gray trace) and changes in fluorescence polarization monitored. Each curve represents at least three independent determinations with the indicated error (SDOM). See Supporting Information for additional experimental details.
The binding mode of sekikaic acid to GACKIX was further defined through small molecule- and protein-observed NMR experiments. In ligand-detected 1D- 1H-NMR studies, the addition of GACKIX to sekikaic acid leads to perturbation of the sekikaic acid chemical shifts of the aromatic proton resonances (Figure 4a). The simultaneous addition of MLL and KID peptide results in the sekikaic acid resonances reverting to their unbound state. These experiments show that binding of sekikaic acid is reversible and that the depside natural product is not inducing protein misfolding or aggregation. This was further supported by circular dichroism spectra taken of GACKIX in the presence and absence of sekikaic acid (see SI Figure 5). In addition, 1H,15N-HSQC NMR experiments with 15N-labeled GACKIX in the presence and absence of sekikaic acid revealed sizeable chemical shift perturbations surrounding the flexible loop that connects helices 1 and 2, proximal to the MLL/c-Jun binding site; Val 608, Leu 620, Lys 621, Arg 624, Met 625, Glu 626 and Phe 612 (an important contact for MLL and a key residue for allosteric signal transmission) showing significant changes in chemical shift (Figure 4b, SI Figure 1).[16] Smaller but still significant changes were also observed in the residues surrounding the broader shallower binding site, one of which is with Lys 662 that forms a key salt bridge with phosphoserine 133 of CREB (Figure 4b, SI Figure 1).[15b] Taken together with the FP experiments, these data are consistent with a binding model in which sekikaic acid targets a binding surface within GACKIX that overlaps with the MLL site, inducing conformational changes in the protein that further impact interaction at the allosterically-connected KID binding site.
Figure 4.
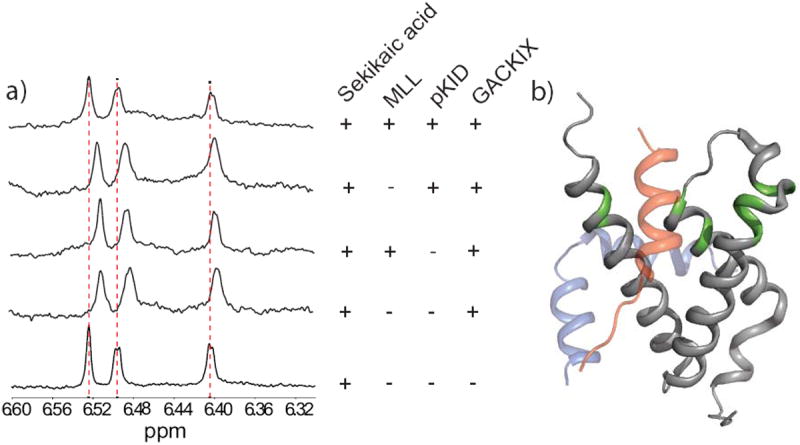
Binding mode of sekikiac acid. a) 1D- 1H-NMR studies of sekikaic acid binding to GACKIX. The aromatic 1H resonances of sekikaic acid (15 μM, bottom trace) are perturbed when GACKIX (5 μM) is added to the solution. The addition of MLL and pKID peptide (15 μM each) in combination (top trace) but not either peptide alone results in the resonances reverting back to the unbound state. b) Chemical shift perturbation data from a 1H-15N HSQC experiment of GACKIX complexed with sekikaic acid. GACKIX (gray) residues that display significant chemical shift upon sekikaic acid binding are colored in green (V608, F612, L620, K621, R624, M625, E626, K662 and L664) and are found in the region contacted by the MLL (red) and pKID (blue) TADs, consistent with the observed inhibition of both TADs. See Supporting Information for details of the NMR experiments. GACKIX structure adapted from PDB ID: 2AGH and 1KDX.
TAD-binding motifs in coactivators are typically hydrophobic, conformationally dynamic and adaptable for interaction with many distinct amphipathic transcriptional activation domains.[3b, 8a, 16-17] Further, each amphipathic TAD typically interacts with >3 distinct TAD-binding motifs.[4b-e, 18] For example, the well-characterized amphipathic TAD VP16 interacts in vitro with four distinct TAD binding motifs within the coactivator Med15.[4b] The implication for inhibitor discovery is that molecules selected or designed to bind to one TAD-binding motif in a coactivator will most often interact with multiple TAD-binding motifs.[12, 14, 19] This presents a significant barrier to inhibitor specificity. The counter screens leading to sekikaic acid discovery suggested that this natural product might have a unique specificity profile since it did not target at least two other activator-binding surfaces. To define this more rigorously, a competitive binding assay was performed with Med15(107-357), a sequence that contains both A and B box TAD interaction motifs, and a VP16-derived TAD, VP2.[4b] Notably, sekikaic acid did not significantly inhibit VP2-Med15(107-357) (Figure 5a), indicating that it does not interact with related TAD interacting domains but displays a remarkable degree of specificity for the GACKIX domain. This is likely related to its mixed direct/allosteric binding mechanism.
Figure 5.
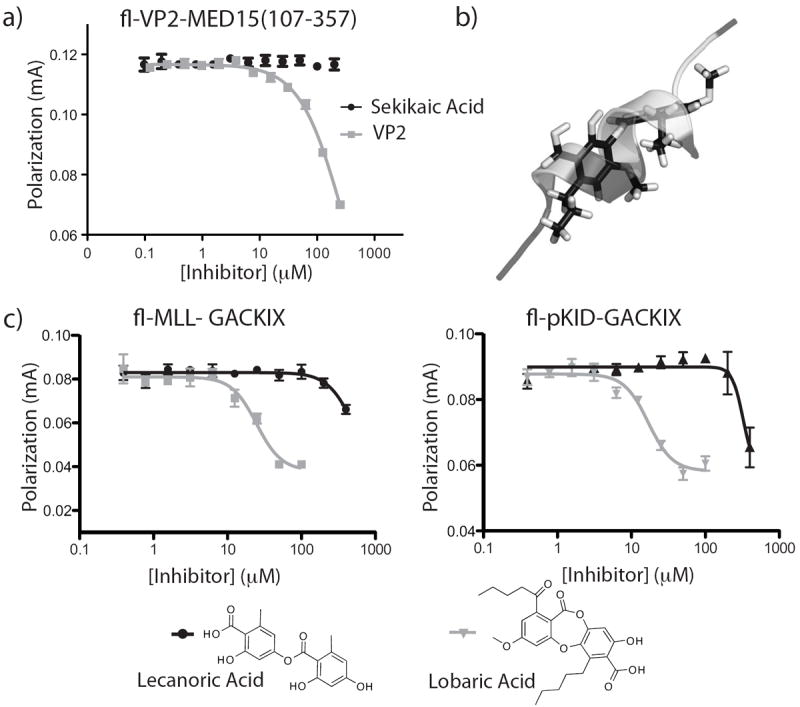
Specificity of sekikaic acid for GACKIX. a) FP assays with the coactivator Med15 that contains at least 3 TAD interaction motifs show that sekikaic is unable to significantly inhibit the interaction between the VP16-derived TAD VP2 and Med15(107-357). b) Overlay of sekikaic acid and an amphipathic helix. A prototypical amphipathic helical TAD p53 was overlayed with sekikaic acid using molecular dynamic simulations demonstrating that sekikaic acid adopts an amphipathic helix-like conformation. c) Impact of conformational rigidity on function. Lecanoric acid (black) computationally predicted to be more flexible than sekikaic acid is unable to significantly inhibit GACKIX-TAD interactions, while the conformationally more-constrained lobaric acid (gray) inhibits the interaction of MLL and pKID in FP assays. (see Supporting Information for experimental details).
To identify structural characteristics of sekikaic acid that contribute to its PPI function, we conducted molecular dynamics simulations of the small molecule. These indicated both that a significant barrier to rotation about the ester linkage exists and that the suite of lowest energy conformations produces an amphipathic helix mimic that overlays a classical helical conformation formed by several transcriptional activation domains (Figure 5b). We further investigated two structurally related molecules: the depside lecanoric acid that lacks the aliphatic side chains of sekikaic acid and the depsidone lobaric acid that has similar side chains but is structurally more rigid due to the central ring system (Figure 5c). MD simulations for both of these molecules suggested that lecanoric acid has significantly more conformational freedom than both lobaric acid and sekikaic acid (SI Figure 6). Competitive binding assays with Fl-MLL and Fl-KID revealed that lecanoric acid was unable to significantly inhibit binding of either activator while the conformationally more constrained lobaric acid did inhibit both activators with IC50s of 17 μM and 25 μM, respectively (Figure 5c). Additionally, consistent with the FP results, cell-based assays show sekikaic and lobaric acid causing a dose dependent down regulation of the c-Jun-driven gene Cyclin D1 (SI Figure 7). By contrast, lecanoric acid has no effect in cells, suggestive of a GACKIX-dependent transcriptional down-regulation mechanism for sekikaic and lobaric acid. Further cellular studies are underway to more rigorously characterize their mode of action and to develop these molecules as specific GACKIX-dependent activator probes.
In summary, we have identified a new class of natural product-based GACKIX inhibitors that demonstrate for the first time the ability of a small molecule to simultaneously modulate two distinct binding sites through interaction with a dynamic binding surface within the protein. Significantly, our findings demonstrate that depsides in general could be used as a new scaffold for the development of amphipathic TAD mimetics. Thus, this work opens new opportunities for the design of future generations of small molecules transcriptional modulators with significant utility as molecular probes and as potential drug leads.
Experimental Section
See Supporting Information for detailed experimental procedures
Supplementary Material
Acknowledgments
A.K.M is grateful for financial support of this work through NIH RO1CA140667. D.H.S. and G.T.-C. are supported by NIH UO1 TW007404 (Costa Rica ICBG), C.L.B acknowledges GM037554. J.W.H. was supported by an ACS Division of Medicinal Chemistry graduate fellowship. W.C.P. acknowledges NIH F32 GM090550 and C.H.D. is a fellow of the UM Chemistry/Biology Interface Training Program (GM08597). We thank J.M.B. and C.A.F. for helpful discussions and Martha Larsen, Amy Danowitz and Jasmine Allen for technical assistance. We thank the National Conservation Areas of Costa Rica for issuing the collection
References
- 1.a) Arkin MR, Wells JA. Nature Reviews Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]; b) Berg T. Curr Opin Chem Biol. 2008;12:464–471. doi: 10.1016/j.cbpa.2008.07.023. [DOI] [PubMed] [Google Scholar]; c Majmudar CY, Mapp AK. Curr Opin Chem Biol. 2005;9:467–474. doi: 10.1016/j.cbpa.2005.08.012. [DOI] [PubMed] [Google Scholar]; d) Lee LW, Mapp AK. J Biol Chem. 2010;285:11033–11038. doi: 10.1074/jbc.R109.075044. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cummings CG, Hamilton AD. Curr Opin Chem Biol. 2010;14:341–346. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 2.a) Ptashne M, Gann A. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]; b) Ptashne M, Gann A. Genes & Signals. Cold Spring Harbor Laboratory; New York: 2001. [Google Scholar]; c) Arndt HD. Angew Chem Int Ed Engl. 2006;45:4552–4560. doi: 10.1002/anie.200600285. [DOI] [PubMed] [Google Scholar]
- 3.a) Wands AM, Wang N, Lum JK, Hsieh J, Fierke CA, Mapp AK. J Biol Chem. 2011;286:6238–16245. doi: 10.1074/jbc.M110.207589. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ferreira ME, Hermann S, Prochasson P, Workman JL, Berndt KD, Wright AP. J Biol Chem. 2005;280:21779–21784. doi: 10.1074/jbc.M502627200. [DOI] [PubMed] [Google Scholar]
- 4.a) Ansari AZ, Reece RJ, Ptashne M. Proc Natl Acad Sci U S A. 1998;95:13543–13548. doi: 10.1073/pnas.95.23.13543. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Majmudar CY, Wang B, Lum JK, Hakansson K, Mapp AK. Angew Chem Int Ed Engl. 2009;48:7021–7024. doi: 10.1002/anie.200902669. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Majmudar CY, Lee LW, Lancia JK, Nwokoye A, Wang Q, Wands AM, Wang L, Mapp AK. J Am Chem Soc. 2009;131:14240–14242. doi: 10.1021/ja904378z. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fishburn J, Mohibullah N, Hahn S. Mol Cell. 2005;18:369–378. doi: 10.1016/j.molcel.2005.03.029. [DOI] [PubMed] [Google Scholar]; e) Reeves WM, Hahn S. Mol Cell Biol. 2005;25:9092–9102. doi: 10.1128/MCB.25.20.9092-9102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lum JK, Majmudar CY, Ansari AZ, Mapp AK. ACS Chem Biol. 2006;1:639–643. doi: 10.1021/cb600363n. [DOI] [PubMed] [Google Scholar]
- 5.a) Swapna LS, Mahajan S, de Brevern A, Srinivasan N. BMC Struct Biol. 2012;12:6. doi: 10.1186/1472-6807-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bruschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M. J Am Chem Soc. 2009;131:3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]; c) Korkmaz EN, Nussinov R, Haliloglu T. PLoS Comput Biol. 2012;8:e1002420. doi: 10.1371/journal.pcbi.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Thompson AD, Dugan A, Gestwicki JE, Mapp AK. ACS Chem Biol. 2012 doi: 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Schon A, Lam SY, Freire E. Future Med Chem. 2011;3:1129–1137. doi: 10.4155/fmc.11.81. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shen A. Mol Biosyst. 2010;6:1431–1443. doi: 10.1039/c003913f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Berg T. Angew Chem Int Ed Engl. 2009;48:3218–3220. doi: 10.1002/anie.200806169. [DOI] [PubMed] [Google Scholar]; d) Thiel P, Kaiser M, Ottmann C. Angew Chem Int Ed Engl. 2012;51:2012–2018. doi: 10.1002/anie.201107616. [DOI] [PubMed] [Google Scholar]; e) Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Nat Chem Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fischer E. J Am Chem Soc. 1914;36:1170–1201. [Google Scholar]
- 8.a) De Guzman RN, Goto NK, Dyson HJ, Wright PE. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]; b) Novatchkova M, Eisenhaber F. Curr Biol. 2004;14:R54–55. doi: 10.1016/j.cub.2003.12.042. [DOI] [PubMed] [Google Scholar]; c) Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. J Biol Chem. 2002;227:43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]; d) Goodman RH, Smolik S. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 9.a) Campbell KM, Lumb KJ. Biochemistry. 2002;41:13956–13964. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]; b) Zor T, De Guzman RN, Dyson HJ, Wright PE. J Mol Biol. 2004;337:521–534. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 10.a) Saura CA, Valero J. Rev Neurosci. 2011;22:153–169. doi: 10.1515/RNS.2011.018. [DOI] [PubMed] [Google Scholar]; b) Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Oliveira AM, Abel T, Brindle PK, Wood MA. Behav Neurosci. 2006;120:724–729. doi: 10.1037/0735-7044.120.3.724. [DOI] [PubMed] [Google Scholar]; d) Kimbrel EA, Lemieux ME, Xia X, Davis TN, Rebel VI, Kung AL. Blood. 2009;114:4804–4812. doi: 10.1182/blood-2009-04-217794. [DOI] [PubMed] [Google Scholar]
- 11.a) Alluri P, Liu B, Yu P, Xiao X, Kodadek T. Mol Biosyst. 2006;2:568–579. doi: 10.1039/b608924k. [DOI] [PubMed] [Google Scholar]; b) Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Proc Natl Acad Sci U S A. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR. Nature Biotechnology. 2000;18:1080–1085. doi: 10.1038/80280. [DOI] [PubMed] [Google Scholar]; d) Rutledge SE, Volkman HM, Schepartz A. J Am Chem Soc. 2003;125:14336–14347. doi: 10.1021/ja034508o. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Bates CA, Pomerantz WC, Mapp AK. Biopolymers. 2011;95:17–23. doi: 10.1002/bip.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li BX, Xiao X. Chembiochem. 2009;10:2721–2724. doi: 10.1002/cbic.200900552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK. ACS Chem Biol. 2009;4:335–344. doi: 10.1021/cb900028j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Thadhani VM, Choudhary MI, Ali S, Omar I, Siddique H, Karunaratne V. Nat Prod Res. 2011;25:1827–1837. doi: 10.1080/14786419.2010.529546. [DOI] [PubMed] [Google Scholar]; b) Fujimoto A, Shingai Y, Nakamura M, Maekawa T, Sone Y, Masuda T. Bioorg Med Chem Lett. 2010;20:7393–7396. doi: 10.1016/j.bmcl.2010.10.040. [DOI] [PubMed] [Google Scholar]; c) Sperl B, Seifert MH, Berg T. Bioorg Med Chem Lett. 2009;19:3305–3309. doi: 10.1016/j.bmcl.2009.04.083. [DOI] [PubMed] [Google Scholar]; d) Muller K. Appl Microbiol Biotechnol. 2001;56:9–16. doi: 10.1007/s002530100684. [DOI] [PubMed] [Google Scholar]
- 14.Shimogawa H, Kwon Y, Mao Q, Kawazoe Y, Choi Y, Asada S, Kigoshi H, Uesugi M. J Am Chem Soc. 2004;126:3461–3471. doi: 10.1021/ja038855+. [DOI] [PubMed] [Google Scholar]
- 15.a) Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]; b) Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. J Mol Biol. 1999;287:859–865. doi: 10.1006/jmbi.1999.2658. [DOI] [PubMed] [Google Scholar]
- 16.Arai M, Dyson HJ, Wright PE. FEBS Lett. 2010;584:4500–4504. doi: 10.1016/j.febslet.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Dyson HJ, Wright PE. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]; b) Hermann S, Berndt KD, Wright AP. J Biol Chem. 2001;276:40127–40132. doi: 10.1074/jbc.M103793200. [DOI] [PubMed] [Google Scholar]
- 18.a) Teufel DP, Freund SM, Bycroft M, Fersht AR. Proc Natl Acad Sci U S A. 2007;104:7009–7014. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Krishnamurthy M, Dugan A, Nwokoye A, Fung YH, Lancia JK, Majmudar CY, Mapp AK. ACS Chem Biol. 2011;6:1321–1326. doi: 10.1021/cb200308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majmudar CY, Labut AE, Mapp AK. Bioorg Med Chem Lett. 2009;19:3733–3735. doi: 10.1016/j.bmcl.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.