

Abstract
Previous liver regeneration studies demonstrated that the mouse forkhead box M1B (FoxM1B) transcription factor regulates hepatocyte proliferation through expression of cell cycle genes that stimulate cyclin-dependent kinase 2 (Cdk2) and Cdk1 activity. In this study, we demonstrated that disruption of the FoxM1B Cdk1/2 phosphorylation site at Thr residue 596 significantly reduced both FoxM1B transcriptional activity and Cdk phosphorylation of the FoxM1B T596A mutant protein in vivo. Retention of this FoxM1B 596 Cdk phosphorylation site was found to be essential for recruiting the histone acetyltransferase CREB binding protein (CBP) to the FoxM1B transcriptional activation domain. Consistent with these findings, dominant negative Cdk1 protein significantly reduced FoxM1B transcriptional activity and inhibited FoxM1B recruitment of the CBP coactivator protein. Likewise, Cdc25B-mediated stimulation of Cdk activity together with elevated levels of the CBP coactivator protein provided a 6.2-fold synergistic increase in FoxM1B transcriptional activity. Furthermore, mutation of the FoxM1B Leu 641 residue within an LXL motif (residues 639 to 641) inhibited recruitment of Cdk-cyclin complexes and caused significant reduction in both FoxM1B transcriptional activity and in vivo Cdk phosphorylation of the FoxM1B Thr 596 residue. We demonstrated that FoxM1B transcriptional activity requires binding of either S-phase or M-phase Cdk-cyclin complexes to mediate efficient Cdk phosphorylation of the FoxM1B Thr 596 residue, which is essential for recruitment of p300/CBP coactivator proteins.
Cellular proliferation involves stimulation of the mitogen-activated protein kinase (MAPK) pathway, consisting of the Ras/Raf1/MEK/MAPK cascade (19, 47), and activation of the phosphoinositol 3-kinase (PI3K) pathway, consisting of the PI3K/phosphoinositide-dependent kinase 1 (PDK1)/Akt cascade (5). However, cell division is tightly regulated at the G1/S (DNA replication) and G2/M (mitosis) transitions of the cell cycle by temporal activation of multiple cyclin-dependent kinases (Cdks) complexed with their corresponding cyclin regulatory subunits. Cdk2-cyclin E or A and Cdk1-cyclin B kinase activity is essential for progression through the G1/S and G2/M transitions of the cell cycle, respectively. Furthermore, Cdk activity is negatively regulated by phosphorylation of Thr 14 and Tyr 15 by the dual specific Myt1 kinase (6, 30, 31, 37). Both MAPK and PDK1 phosphorylate and activate the downstream p90 ribosomal S6 kinase (Rsk) (4, 21, 51), which provides inhibitory phosphorylation to the dual Myt1 kinase (44, 45). Simulation of Cdk1 or Cdk2 activity involves removal of inhibitory phosphates at Thr 14 and Tyr 15 by either Cdc25A phosphatase (G1/S phase), Cdc25B (late S phase), or Cdc25C phosphatase (G2/M phase) (7, 41, 62). In addition, Cdk activity requires phosphorylation of Thr 160 by the Cdk-activating kinase (CAK) protein, which is also a direct target for MAPK phosphorylation (14, 28, 55).
Cdk1 and Cdk2 proteins are proline-directed kinases that phosphorylate either Ser or Thr residues within a defined consensus sequence, X-S/T-P-X-R/K. It is well established that Cdk2 activity in complex with either cyclin E or cyclin A cooperates with cyclin D-Cdk4 or Cdk6 to phosphorylate the retinoblastoma (RB) protein, which results in release of bound E2F transcription factor, allowing stimulation of genes required for S phase (16, 20). The Cdk1-cyclin B complexes drive M-phase progression through phosphorylation of protein substrates that are essential for chromosome segregation, breakdown of the nuclear envelope, and cytokinesis (3, 23, 40, 42, 43, 56).
The forkhead box (Fox) transcription factors are an extensive family of transcription factors, consisting of more than 50 mammalian proteins (22) that share homology in the winged helix DNA binding domain (12, 34). The FoxM1B protein (previously known as HFH-11B, Trident, and WIN) is a proliferation-specific transcriptional activator that is expressed in all replicating cells so far examined, but its expression is extinguished in terminally differentiated cells (26, 66, 68). During liver regeneration, FoxM1B expression is markedly induced at the G1/S transition and continues throughout hepatic cell proliferation (68). Premature hepatic expression of FoxM1B (HFH-11B) in regenerating liver of transgenic mice accelerated the onset of hepatocyte DNA replication and mitosis by stimulating earlier expression of cell cycle-regulatory genes (13, 58, 67). Furthermore, maintaining hepatocyte expression of FoxM1B in 12-month-old (old-aged) transgenic mice is sufficient to increase regenerating hepatocyte proliferation to levels found in young regenerating liver (61). In addition to reestablishing expression of cell cycle-regulatory genes, p27kip1 protein levels were also diminished when FoxM1B expression was restored in regenerating old-aged mouse liver (13, 27, 60). These results suggest the hypothesis that FoxM1b controls the transcriptional network of genes essential for progression through the cell cycle.
Consistent with this hypothesis, we used the albumin enhancer- and promoter-driven Cre recombinase transgene (Alb-Cre) to mediate hepatocyte-specific deletion of the Foxm1b floxed/floxed (fl/fl) allele and found that FoxM1b deficiency resulted in significant reduction in hepatocyte DNA replication and mitosis following partial hepatectomy (59). Reduced hepatocyte DNA replication was associated with increased protein levels of Cdk inhibitor p21Cip1 and reduced protein expression of Cdc25A phosphatase, leading to decreased Cdk2 activation and progression into S phase (59). Diminished hepatocyte mitosis was associated with undetectable expression of the Cdc25B phosphatase and delayed accumulation of cyclin B1, which is required for cyclin B-Cdk1 kinase activation and entry into mitosis. Cotransfection assays with the cytomegalovirus (CMV)-FoxM1B expression vector and a luciferase plasmid driven by the Cdc25B promoter, containing 200 bp of upstream sequence demonstrated that FoxM1B is capable of directly activating transcription of this Cdc25B minimal promoter region (59). Taken together, these liver regeneration studies indicated that Foxm1b deficiency caused diminished hepatocyte proliferation due to altered expression of proteins that limit Cdk1 and Cdk2 activity required for normal cell cycle progression into DNA replication and mitosis.
In this study, we demonstrated that the FoxM1B transcriptional activation domain recruits Cdk-cyclin complexes through an LXL motif (residues 639 to 641), which mediates efficient Cdk phosphorylation of FoxM1B Thr residue 596. FoxM1B transcriptional activity requires Cdk-cyclin-dependent phosphorylation of FoxM1B Thr residue 596, which results in recruitment of the p300/CREB-binding protein (CBP) histone acetyltransferase proteins. Consistent with these findings, inhibition of Cdk-cyclin activity caused diminished FoxM1B transcriptional activity and association with the CBP coactivator protein. Conversely, increasing Cdk1 activity by overexpression of Cdc25B in addition to elevated levels of CBP augmented FoxM1B-dependent transcription. We demonstrated that FoxM1B transcriptional activity requires binding of Cdk-cyclin complexes to mediate efficient phosphorylation of FoxM1b Thr residue 596, which is essential for recruiting the CBP/p300 coactivator proteins.
MATERIALS AND METHODS
Generation of FoxM1B expression plasmids and luciferase reporter plasmid.
The CMV-FoxM1B expression plasmid was generated by PCR amplification of the CMV human FoxM1B expression plasmid (68) with the 5′ EcoRI T7 epitope-tagged FoxM1B primer 5′-gcggaattcaccatggctagcatgactggtggacagcaaatgggtTGGCAGAACTCTGTGTCTGAG and a 3′ antisense primer that hybridized to the CMV expression vector simian virus 40 poly(A) region, 5′-gtttgtccaattatgtca (lowercase letters indicate the EcoRI site and T7 tag; uppercase letters indicate the FoxM1B sequence). The resulting 3.3-kb FoxM1B PCR product was digested with EcoRI and HindIII, generating the 2.5-kb EcoRI-HindIII T7-tagged FoxM1B cDNA fragment and removing 800 nucleotides from the 3′ untranslated region. This FoxM1B cDNA fragment was subsequently cloned in the EcoRI and HindIII sites in the CMV expression vector (46). We generated the CMV pEGFP-T7-FoxM1B expression plasmid by liberating a 2.5-kb EcoRI-HindIII fragment from the CMV T7-FoxM1B expression vector. The HindIII site was made blunt by the T4 polymerase fill-in reaction, and then the FoxM1B cDNA fragment was cloned into the EcoRI and SmaI sites of the pEGFP-C2 expression plasmid (Clontech).
We generated the CMV tetracycline operator (CMV-TO) FoxM1B expression plasmid by excising an EcoRI-BamHI fragment from pEGFP-T7-FoxM1B expression plasmid. The BamHI site was made blunt by a T4 polymerase reaction, and then the FoxM1B cDNA fragment was cloned into EcoRI and EcoRV sites of the pCDNA4-TO expression plasmid (T-Rex System; Invitrogen). The 6× FoxM1B/FoxA TATA-luciferase utilized six copies of the FoxM1B/FoxA binding site (TTTGTTTGTTTG) from the cdx-2 promoter region adjacent to the TATA box sequence (from the CMV promoter) driving expression of the luciferase reporter gene as described previously (49, 54, 68). We generated FoxM1B amino acid point mutations with the Altered Sites II mammalian mutagenesis system (Promega). Briefly, the 2.5-kb human FoxM1B cDNA fragment was cloned into EcoRI and NotI sites of the pAlter-Max vector by liberating the EcoRI/NotI fragment from the CMV-TO-T7-FoxM1B expression plasmid. Mutagenesis was performed as described by the manufacturer, and the FoxM1B point mutations were confirmed by sequencing (University of Chicago DNA Sequencing Facility). The following 5′-phosphorylated (5′P) antisense primers were used to introduce site-directed mutations in the FoxM1B coding region: T585A, 5′P-TTCCTTAATGGGTGCCTTAAAAGGTCC; T596A, 5′P-AGATTTGCTCGGGGCGGAGGAGATGGG; L641A, 5′P-GCTGAGATCCATCGCCCCCAGGGGGTC; and S657A, 5′P-GAGCCTTTGCGGTGCTTCAAGGGGGGG.
Cell culture and transient cotransfections.
Human osteosarcoma U2OS cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 1× penicillin-streptomycin, and 1× l-glutamine (Gibco). For transient transfection, U2OS cells were plated in six-well plates and transfected with Fugene 6 reagent (Roche) according to the manufacturer's protocol. Cells were transfected with 500 ng of either CMV FoxM1B or CMV FoxM1B site-directed mutants with 1.5 μg of a 6× FoxM1B TATA-luciferase reporter with 10 ng of CMV-Renilla luciferase internal control. Twenty-four hours posttransfection, cells were harvested and protein extracts were prepared for dual luciferase assays (Promega). Luciferase activity was determined as a percentage of wild-type FoxM1B activity following normalization to Renilla luciferase activity. Experiments were performed at least four times in triplicate, and statistical analysis was performed with Microsoft Excel tools.
FoxM1B transient-transfection assays with dominant negative kinases and pharmacological kinase inhibitors.
Dominant negative (DN) kinase mammalian expression vectors were obtained from the following laboratories: DN-RasN17, Ravi Misra, Medical College of Wisconsin; DN-p90Rsk, John Blenis, Harvard Medical School; DN-Akt, Nissim Hay, University of Illinois—Chicago; WT-Myt1, Helen Piwnica-Worms, Washington University; and DN-Cdk1, David Ucker, University of Illinois—Chicago. For dominant negative kinase cotransfection experiments and pharmacological kinase inhibitors, we used a tetracycline-inducible expression system with U2OS cells containing an episomal tetracycline repressor expression plasmid (Invitrogen, catalogue number R712-07). The episome was maintained by growth in medium containing 50 μg of hygromycin B per ml as described by Yao et al. (65). U2OS-Tetr cells were transiently cotransfected with reporter plasmid 6×-FoxM1B-TATA-luciferase and CMV-TO-FoxM1B in the presence or absence of either DN-RasN17, DN-p90Rsk, DN-Akt, DN-Cdk1, WT-Myt1, Cdc25B, or Cdc25C. Twenty-four hours posttransfection, tetracycline was added to the cell medium at 1 μg/ml to induce expression of the FoxM1B protein. Twenty-four hours following FoxM1B induction, cells were harvested and analyzed for dual luciferase activity.
For pharmacological inhibitor cotransfection experiments, U2OS-Tetr cells were transiently cotransfected with reporter plasmid 6×-FoxM1B-TATA-luciferase and expression plasmid CMV-TO-FoxM1B. Twenty hours posttransfection and 5 h prior to the induction of FoxM1B expression with 1 μg of tetracycline per ml, the medium was supplemented with the following pharmacological kinase inhibitors: U0126 (50 μM; Promega), Ly294002 (50 μM), alsterpaullone (100 nM, 1 μM, or 10 μM; Calbiochem), or Akt inhibitor (25 μM; Calbiochem); 20 h following induction of FoxM1B expression, cells were harvested and analyzed for dual luciferase activity. Luciferase activity was determined as a percentage of wild-type FoxM1B activity following normalization to Renilla luciferase activity. Experiments were performed at least four times in triplicate unless otherwise stated. Statistical analysis was performed with Microsoft Excel tools. U2OS cells were transiently transfected in two-well chamber slides (Nunc) with CMV pEGFP-T7-FoxM1B expression constructs in the presence or absence of either dominant negative kinases or pharmacological inhibitors. At 48 h posttransfection, cells were fixed in 4% paraformaldehyde for 20 min at room temperature. Green fluorescent protein (GFP) florescence was visualized with either a Nikon or Zeiss microscope.
Immunoprecipitation kinase assays, coimmunoprecipitation-Western blot assays, and electrophoretic mobility shift assays.
U2OS cells were plated on a 100-mm plate and transiently transfected with 5 μg of CMV pEGFP-T7-epitope-tagged (T7)-FoxM1B or CMV T7-FoxM1B expression plasmid, and cells were harvested at 48 h following transfection in phosphate-buffered saline and then pelleted by centrifugation. The cell pellet was resuspended in 500 μl of 1× NP-40 lysis buffer containing both protease and phosphatase inhibitors. Cells were vortexed and incubated on ice for 45 min with repetitive vortexing to ensure complete cellular lysis. Following cell lysis, cellular debris was removed from the supernatant by centrifugation at 10,000 rpm for 5 min at 4°C. Protein concentrations were determined by the Bradford method with the Bio-Rad protein assay reagent (Bio-Rad). The NP-40 lysis buffer consisted of 50 mM Tris (pH 7.5), 100 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1% NP-40, 5% glycerol and freshly added 1× Complete Mini protease inhibitor cocktail (Roche), 2 mM phenylmethylsulfonyl fluoride, and 20 mM beta-glycerolphosphate, 2 mM NaF, and 2 mM NaVO4 as phosphatase inhibitors.
For coimmunoprecipitation assays, we used antibodies (3 μg/coimmunoprecipitation) specific to either Cdk1 (NeoMarkers), Cdk2 (Santa Cruz Biotech), cyclin E (Santa Cruz Biotech), cyclin B1 (Santa Cruz Biotech), Cdc25B (Santa Cruz Biotech), or CBP (C-1 monoclonal antibody; Santa Cruz Biotech), retinoblastoma protein (RB; Monoclonal Pharmingen), and P-selectin antibodies (Pharmingen). Protein lysates prepared from U2OS cells transfected with either CMV GFP-T7-FoxM1B or CMV T7-FoxM1B expression vectors were immunoprecipitated with the antibodies listed above and then coimmunoprecipitated proteins were detected by Western blot analysis with monoclonal antibodies against either the GFP protein (Clontech) or the T7 epitope tag (Novagen). Untransfected U2OS protein lysates were immunoprecipitated with the above cell cycle antibodies and CBP antibodies, followed by Western blot analysis with affinity-purified rabbit polyclonal antibody specific to the FoxM1B protein (67, 68). The signals from the primary antibody were amplified by horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Bio-Rad, Hercules, Calif.) and detected with Enhanced Chemiluminescence Plus (ECL-Plus; Amersham Pharmacia Biotech, Piscataway, N.J.).
For the coimmunoprecipitation Cdk kinase reaction, protein lysates from U2OS cells transfected with the CMV GFP-T7-FoxM1B expression vector was coimmunoprecipitated overnight with either anti-Cdk1 (NeoMarkers; 3 μg) or anti-Cdk2 (M2; Santa Cruz) antibody with continuous rocking at 4°C as described previously (59). Lysates were then incubated with 20% protein A-Sepharose CL-4B (Amersham) for 4 h with rocking at 4°C. The beads were pelleted by centrifugation, washed three times with NP-40 lysis buffer, equilibrated in kinase reaction buffer by washing three times, and then used for kinase assays with [γ-32P]ATP isotope (59, 60). Phosphorylated proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10% polyacrylamide gel) and detected by autoradiography.
Electrophoretic mobility shift assay experiments were performed with the HeLa cell nuclear extract provided in the Gel Shift Assay System (Promega), following the manufacturer's instructions. We used 3.5 pmol of the double-stranded FoxM1B binding site oligonucleotide (from the 6× FoxM1B TATA-luciferase reporter gene) for radioactive labeling with [γ-32P]ATP isotope (ICN, Irvine, Calif.) and T4 polynucleotide kinase (Promega). Formation of the FoxM1B protein-DNA complex was performed by incubating approximately 0.2 pmol of 32P-labeled FoxM1B binding site oligonucleotide, 2 μg of HeLa cell nuclear protein extract, and 1× gel shift buffer (Promega) for 20 min at room temperature. We then added antibodies specific to either Cdk1, cyclin B, Cdk2, cyclin B1, Cdc25B, RB, CBP, FoxM1B, or P-selectin (control) proteins to the binding reaction mixture for an additional 45 min of incubation. The FoxM1B protein-DNA complexes were resolved by electrophoresis on a nondenaturing 9% polyacrylamide gel run at 4°C and detected by autoradiography as described previously (50).
We also performed DNA competitions that included in the binding reaction mixture a 100-fold molar excess of either unlabeled FoxM1B binding site oligonucleotide or an unrelated SP1 transcription factor binding site oligonucleotide (control). The human FoxM1B C-terminal region (amino acids 365 to 748) was cloned into the p28A+ expression vector (Novagen) in frame with the histidine (His)-tagged epitope. The His-tagged FoxM1B 365-748 protein was expressed in Escherichia coli BL21(DE3) bacteria, protein extracts were made, and the His-tagged FoxM1B 365-748 protein was affinity purified on a nickel column (Novagen) with the manufacturer's protocol. Mice were immunized with the His-tagged human FoxM1B 365-748 protein, and mouse serum was isolated by the University of Illinois at Urbana-Champaign Immunological Resource Center.
Phosphorylation time course and coimmunoprecipitation assays following serum stimulation of U2OS cells.
Asynchronously growing U2OS cells were transiently transfected with 500 ng of CMV T7 epitope-tagged (T7) FoxM1B expression plasmid with the Fugene 6 transfection reagent (Roche). Cells were rendered quiescent at 16 h posttransfection by washing plates three times in 1× phosphate-buffered saline followed by the addition of DMEM containing 0.1% serum for 48 h. Cells were stimulated to proliferate by the addition of DMEM supplemented with 20% fetal calf serum and harvested in phosphate-buffered saline at 4, 12, 18, 22, and 28 h following serum addition. Cell protein extracts were prepared by the NP-40 detergent lysis method as described above. We used 500 μg of total cell protein lysate for immunoprecipitation with a monoclonal antibody recognizing the T7 epitope tag (Novagen) overnight at 4°C. Lysates were then incubated with 20% protein A-Sepharose CL-4B beads (Amersham), and incubation was continued for an additional 3 h with rocking at 4°C. Following precipitation, the beads were washed three times with NP-40 detergent lysis buffer. The immunoprecipitated proteins were released by heating for 5 min at 100°C in SDS-PAGE sample buffer and resolved by SDS-PAGE (12% polyacrylamide gel). After electrophoretic transfer onto polyvinylidene difluoride, we detected either Cdk1 or Cdk2 phosphorylation of FoxM1B in vivo by Western blot analysis with mouse monoclonal antibody MPM2 (Upstate; 1:1,000 dilution) against the phosphorylated protein sequence phosphoserine/phosphothreonine-proline.
Furthermore, coimmunoprecipitation experiments were performed with protein extracts prepared from synchronized CMV T7-FoxM1B-transfected U2OS cells at 4 h (G1 phase), 12 h (S phase), or 18 h (G2 phase) following serum addition. These synchronized protein extracts were used for coimmunoprecipitation experiments with antibodies specific to either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, retinoblastoma protein, CBP, or P-selectin (control), followed by Western blot analysis with a monoclonal antibody against the T7 epitope tag. To examine Cdk phosphorylation in vivo, protein extracts prepared from unstimulated or serum-stimulated U2OS cells (12 or 18 h after serum addition) transfected with either the CMV T7-FoxM1B wild-type, CMV T7-FoxM1B T596A, or CMV FoxM1B L641A expression construct. These protein extracts were immunoprecipitated with T7 (Novagen), and then Western blot analysis with the MPM2 monoclonal antibody (Upstate; 1:1,000 dilution) was used to determine Cdk phosphorylation in vivo.
For in vivo 32P pulse labeling, CMV GFP-T7-FoxM1B-transfected U2OS cells were serum starved in phosphate-deficient DMEM (Gibco-Invitrogen) containing 0.1% dialyzed fetal calf serum for 48 h. The transfected cells were then serum stimulated in phosphate-deficient DMEM (Gibco-Invitrogen) containing 20% dialyzed fetal calf serum (4 ml per 10-cm plate) for 0, 4, 12, 18, 22, and 28 h. At 3 h prior to preparing the protein extracts, the cells were pulse labeled with 200 μCi of [32P]orthophosphate (ICN, Irvine, Calif.) as described previously (48). Cell extracts were prepared as described above and immunoprecipitated with T7 epitope-tagged antibody, the immunoprecipitated proteins were released by heating for 5 min at 100°C in SDS-PAGE sample buffer and resolved by SDS-PAGE (12% polyacrylamide gel), and phosphorylated GFP-T7-FoxM1B protein was detected by autoradiography.
RESULTS
FoxM1B protein is phosphorylated and transcriptionally active throughout the cell cycle.
Liver regeneration and cell culture studies demonstrated that FoxM1B regulates the expression of cell cycle genes, including cyclin D1, cyclin B1, and Cdc25B (29, 58-61, 67) and that FoxM1B protein mediates reduced expression of the Cdk inhibitor p21Cip1 protein (59). Furthermore, Western blot analysis of protein extracts from cycling cells demonstrated that the FoxM1B (MPP2) protein is immunoreactive with the MPM2 monoclonal antibody, which recognizes proteins phosphorylated by either Cdk1- or Cdk2-cyclin complexes in a phosphoserine/phosphothreonine-proline-dependent manner (29, 63).
To examine Cdk phosphorylation of the FoxM1B protein at different stages of the cell cycle, we used the MPM2 monoclonal antibody for Western blot analysis with protein extracts prepared from synchronized U2OS cells. Osteosarcoma U2OS cells were transiently transfected with the CMV T7 epitope-tagged FoxM1B expression plasmid, rendered quiescent by serum withdrawal for 48 h, and then stimulated to reenter the cell cycle by addition of medium containing 20% fetal calf serum. At either 0, 4, 12, 18, 22, or 28 h after serum addition, the U2OS cells were used to prepare protein extracts for immunoprecipitation with the T7 epitope tag monoclonal antibody, followed by Western blot analysis with the MPM2 monoclonal antibody (Fig. 1A). Replica synchronized U2OS cells were trypsinized, stained with propidium iodide, and used for flow cytometry to determine the stage of the cell cycle at each time point following serum stimulation (Fig. 1B).
FIG. 1.

Phosphorylation and transcriptional activity of FoxM1B requires serum stimulation and cell cycle progression. (A) FoxM1B protein is phosphorylated by Cdk1/2 protein during the cell cycle. U2OS cells were transiently transfected with GFP-T7-FoxM1B, followed by serum starvation (0.1%) for 48 h to render cells quiescent. The transfected cells were replenished with medium containing 20% fetal bovine serum, and cell lysates were prepared from these cells at the indicated time points following serum addition. Cell lysates were immunoprecipitated with a T7 antibody and then fractionated by SDS-PAGE. Western blot analysis was performed with monoclonal antibody MPM2, which is specific to phosphorylated Cdk1/2 sites (63), and demonstrates that FoxM1B is phosphorylated by Cdk1/2 between 4 and 28 h following serum addition. (B) Flow cytometry analysis of cells harvested at various times following replenishing of serum compared to those from an asynchronous cell population. This analysis indicates that S phase is maximal at 12 h following serum addition, while G2/M phase occurs at the 18-h time point. (C) FoxM1B protein is phosphorylated during the cell cycle. Serum-starved transfected cells were replenished with medium containing 20% fetal bovine serum, pulse labeled with [32P]orthophosphate for 3 h prior to preparation of cell lysates from these cells at the indicated time points after serum addition. Radioactively labeled cell lysates were immunoprecipitated with T7 antibody and fractionated by SDS-PAGE, and then labeled FoxM1B protein was detected by autoradiography. (D) FoxM1B tran-scriptional activity requires serum-dependent phosphorylation. U2OS cells were transiently cotransfected with the reporter 6×-FoxM1B/FoxA-TATA-luciferase (49) and wild-type (WT) FoxM1B. Transfected cells were serum starved for 48 h and either not stimulated or stimulated with serum for 4, 12, or 18 h, and cells were harvested and processed for dual luciferase assays. Results are expressed as the induction of transcriptional activity with respect to CMV-FoxM1B in serum-starved cells.
The MPM2 Western blot analysis demonstrated that Cdk-cyclin complexes initiated phosphorylation of FoxM1B protein as early as 4 h following serum addition and continued throughout proliferation of U2OS cells (Fig. 1A). In combination with flow cytometry analysis, these results indicated that FoxM1B phosphorylation initiated at the G1 phase (4 h) and continued through the S phase (12 h) and G2 phase (18 to 22 h) of the cell cycle (Fig. 1B). Furthermore, [32P]orthophosphate was used for in vivo labeling of synchronized U2OS cells transfected with CMV GFP-T7-FoxM1B, and protein extracts were prepared at the indicated times following serum addition, immunoprecipitated with T7 monoclonal antibody followed by SDS-PAGE and autoradiography. These in vivo labeling experiments confirmed that the FoxM1B protein is phosphorylated at all time points following serum stimulation of U2OS cells and that FoxM1B protein exhibited the highest 32P labeling at the G2 phase of the cell cycle (18 h; Fig. 1C).
In order to determine whether FoxM1B phosphorylation correlated with an increase in its transcriptional activity, we transiently transfected U2OS cells with the 6× FoxM1B/FoxA-TATA-luciferase reporter plasmid (49) with CMV, the FoxM1B wild-type expression vector. Transfected cells were serum starved for 48 h and either left without serum (0 h) or stimulated with serum for 4, 12, or 18 h, and the cells were harvested and processed for dual luciferase assays. These transcription assays demonstrated that FoxM1B transcriptional activity was significantly reduced in serum-starved cells but that FoxM1B transcriptional activity was markedly stimulated at all three time points following serum stimulation.
FoxM1B binds to distinct Cdk-cyclin complexes and RB at specific stages of the cell cycle.
Our studies with MPM2 antibody demonstrated that FoxM1B was phosphorylated by Cdk-cyclin complexes throughout the cell cycle (Fig. 1A), and putative Cdk phosphorylation sites were found in the FoxM1B carboxyl-terminal transcriptional activation domain (Fig. 2A). We therefore examined the association of the FoxM1B protein with the cell cycle-regulatory proteins Cdk2-cyclin E and Cdk1-cyclin B1 complexes and RB and CBP transcriptional coactivator. Coimmunoprecipitation assays were performed with protein extracts prepared from U2OS cells that were transiently transfected with the CMV T7-FoxM1B expression plasmid for 48 h. These protein extracts were coimmunoprecipitated with either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, CBP, or P-selectin (control) antibody, followed by Western blot analysis with a monoclonal antibody against the T7 epitope tag.
FIG. 2.

FoxM1B protein interacts with cell cycle-regulatory proteins and CREB binding protein. (A) Schematic representation of the Cdk1 phosphorylation sites within the transcriptional activation domain of FoxM1B. The PhosphoBase 2.0 program (http://www.cbs.dtu.dk/databases/PhosphoBase/) and Scansite (http://scansite.mit.edu/) were used to predict Cdk1 phosphorylation sites within the transcriptional activation domain of FoxM1B. Numbers indicate the FoxM1B amino acid residues of the potential Cdk phosphorylation sites. The 100-amino-acid winged helix DNA binding domain (WHD) is indicated by a black box. (B) Endogenous Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, and CBP proteins coimmunoprecipitate with transfected T7-FoxM1B protein. U2OS cells were transiently transfected with CMV T7-FoxM1B, and cell protein extracts were prepared 48 h posttransfection and used for coimmunoprecipitation (Co-IP) with antibodies specific to either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, CBP, or P-selectin (control). Coimmunoprecipitated proteins were detected by Western blot analysis with a monoclonal antibody against the T7 epitope tag. The P-selectin antibody was used as a negative control for coimmunoprecipitation experiments. Western blot analysis with T7 antibody demonstrated equal amounts of FoxM1B protein in all lanes (data not shown). (C) Endogenous FoxM1B protein coimmunoprecipitates with endogenous Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, and CBP proteins. Protein extracts were prepared from U2OS cells and used for coimmunoprecipitation with the same antibodies used above (B), followed by Western blot analysis with polyclonal antibody specific to FoxM1B protein (67, 68). (D) FoxM1B protein shows preferences for distinct Cdk-cyclin complexes and RB depending on the stage of the cell cycle. U2OS cells were transiently transfected with CMV T7-FoxM1B, followed by serum starvation (0.1%) for 48 h to render cells quiescent. The transfected cells were replenished with medium containing 20% fetal bovine serum, and cell lysates were prepared from these cells at 4 h (G1 phase), 12 h (S phase), or 18 h (G2 phase) following serum addition. These synchronized protein extracts were used for coimmunoprecipitation experiments with antibodies specific to either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, CBP, or P-selectin (control), followed by Western blot analysis with a monoclonal antibody against the T7 epitope tag. (E) FoxM1B protein-DNA complexes are associated with cell cycle proteins. Electrophoretic mobility shift assay experiments were performed with HeLa cell nuclear extracts, the FoxM1B binding site oligonucleotide, and antibodies specific to either cell cycle-regulatory or CBP proteins. Following formation of the FoxM1B protein-DNA complex, we added antibodies specific to either Cdk1, cyclin B, Cdk2, cyclin B1, Cdc25B, RB, CBP, FoxM1B, or P-selectin (control) proteins to the binding reaction mixture for an additional 45 min of incubation. The FoxM1B protein-DNA complexes were resolved by electrophoresis on a nondenaturing 9% polyacrylamide gel and detected by autoradiography as described previously (50). We also performed DNA competitions that included in the binding reaction mixture a 100-fold molar excess of either unlabeled FoxM1B binding site oligonucleotide (self-competition) or an unrelated SP1 transcription factor binding site oligonucleotide (control).
These coimmunoprecipitation experiments demonstrated that T7-FoxM1B protein efficiently associated with the endogenous S-phase Cdk2-cyclin E or G2-phase Cdk1-cyclin B1 complexes (Fig. 2B). Furthermore, FoxM1B associates with Cdc25B, which dephosphorylates and activates Cdk1 protein (7, 41, 62); with RB, which binds to and negatively regulates the E2F transcription factor (16, 17); and with the CBP histone acetyltransferase coactivator protein (Fig. 2B). We next examined whether these cell cycle-regulatory proteins would interact with endogenous levels of FoxM1B protein. Untransfected U2OS protein extracts were coimmunoprecipitated with either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, CBP, or P-selectin (control) antibody, followed by Western blot analysis with a FoxM1B N-terminal antibody (67, 68). These coimmunoprecipitation studies demonstrated that endogenous FoxM1B protein was also able to associate with Cdk2-cyclin E, Cdk1-cyclin B1, Cdc25B, and RB cell cycle-regulatory proteins as well as with the CBP coactivator protein (Fig. 2C).
In order to determine whether FoxM1B protein exhibited differences in association with distinct Cdk-cyclin complexes, Cdc25B, or the RB protein depending on the stage of the cell cycle, U2OS cells were transiently transfected with CMV T7-FoxM1B followed by serum starvation (0.1%) for 48 h to render the cells quiescent. The transfected cells were replenished with medium containing 20% fetal bovine serum, and protein extracts were prepared from these cells at 4 h (G1 phase), 12 h (S phase), or 18 h (G2 phase) following serum addition. These synchronized protein extracts were used for coimmunoprecipitation experiments with antibodies specific to either Cdk1, cyclin B1, Cdk2, cyclin E, Cdc25B, RB, CBP, or P-selectin (control) antibody, followed by Western blot analysis with a monoclonal antibody against the T7 epitope tag (Fig. 2D). These synchronized coimmunoprecipitation experiments demonstrated that the FoxM1B protein displayed increased association with the Cdk2-cyclin E complex in G1- and S-phase protein extracts, whereas FoxM1B preferentially associated with Cdk1-cyclin B1 complex in G2-phase protein extracts (Fig. 2D). FoxM1B protein also displayed its strongest binding to Cdc25B in G1- and S-phase protein extracts (Fig. 2D). Furthermore, the FoxM1B protein only associated with RB in G1-phase protein extracts (Fig. 2D), suggesting that FoxM1B transcriptional activity may be negatively regulated by RB prior to S phase. These coimmunoprecipitation studies demonstrate that the FoxM1B protein interacts with distinct Cdk-cyclin complexes and RB, depending on the stage of the cell cycle.
In order to determine whether these cell cycle proteins are associated with the endogenous FoxM1B protein-DNA complex, electrophoretic mobility shift assay experiments were performed with untransfected HeLa cell nuclear extracts, 32P-labeled FoxM1B binding site oligonucleotide, and antibodies specific to either cell cycle-regulatory or CBP proteins (Fig. 2E). Following formation of the FoxM1B protein-DNA complex, we added antibodies specific to either Cdk1, cyclin B, Cdk2, cyclin B1, Cdc25B, RB, CBP, FoxM1B, or P-selectin (control) proteins to the binding reaction for an additional 45 min of incubation. The FoxM1B protein-DNA complexes were resolved by nondenaturing polyacrylamide gel electrophoresis and visualized by autoradiography.
Electrophoretic mobility shift assay experiments demonstrated that formation of the FoxM1B protein-DNA complex was specifically inhibited by including either unlabeled FoxM1B binding site oligonucleotide (100-fold molar excess) or the FoxM1B antibody to the binding reaction (Fig. 2E). Formation of the FoxM1B complex was specific because it was not disrupted by competition with either an unrelated SP1 transcription factor binding site oligonucleotide or the P-selectin (control) antibody (Fig. 2E). The electrophoretic mobility shift assay experiment demonstrated that formation of the specific FoxM1B protein-DNA complexes was significantly reduced with Cdk1, cyclin B1, or RB antibodies (Fig. 2E), suggesting that FoxM1B is able to associate with these cell cycle-regulatory proteins when it is bound to DNA. However, FoxM1B protein-DNA complex formation was only slightly decreased with antibodies against Cdk2, cyclin E, Cdc25B, or CBP (Fig. 2E). These results suggest that these regulatory proteins may have a reduced affinity for FoxM1B when it is bound to DNA. Alternatively, the protein epitopes for these antibodies may not be accessible in the FoxM1B protein-DNA complex in the electrophoretic mobility shift assay.
FoxM1B-dependent transcription requires Cdk phosphorylation site 596 and binding of Cdk1 and Cdk2 proteins through the FoxM1B LXL sequence.
Previous transfection studies demonstrated that the FoxM1B transcriptional activation domain was contained within the carboxyl-terminal 365 to 748 amino acid residues (68). Searching the FoxM1B C-terminal sequence for Cdk1/2 consensus phosphorylation sites, X-pS/T-P-X-R/K, revealed three potential Cdk1/2 sites at residues 585, 596, and 657 in the FoxM1B protein (Fig. 3A). In order to assess the transcriptional function of these potential FoxM1B Cdk1/2 sites, we used site-directed mutagenesis to alter either a Thr or a Ser residue to an Ala residue to prevent their Cdk phosphorylation in vivo. Transient-transfection assays with the 6× FoxM1B TATA-luciferase reporter and CMV vectors expressing either wild-type or Cdk1/2 mutant FoxM1B protein revealed that mutation of Cdk1/2 sites at either 585 or 657 resulted in only a marginal decrease (20% to 30%) in FoxM1B transcriptional activity (Fig. 3B). In contrast, mutation of the FoxM1B Thr 596 residue (FoxM1B T596A) caused an 80% decrease in transcriptional activity, suggesting that this particular Cdk1/2 phosphorylation site plays an important role in FoxM1B-dependent transcription (Fig. 3B). Moreover, FoxM1B was unable to activate expression of the TATA-luciferase control reporter in cotransfection assays, demonstrating that the multimerized FoxM1B binding sites were required for FoxM1B-dependent transcriptional activation (Fig. 3B).
FIG. 3.

FoxM1B transcriptional activity requires association with Cdk1 protein and phosphorylation by Cdk1. (A and B) Mutation of the Cdk1 phosphorylation site at position 596 and Leu residue at position 641 causes diminished FoxM1B transcriptional activity. All of the Thr or Ser residues were changed to Ala, and the second Leu residue at position 641 in the LXL motif was changed to Ala (A). U2OS cells were transiently cotransfected with the reporter 6×-FoxM1B/FoxA-TATA-luciferase (49) and either CMV-empty, wild-type (WT) FoxM1B, FoxM1B T585A, FoxM1B T596A, FoxM1B S657A, or FoxM1B L641A. Twenty-four hours posttransfection, cells were harvested and processed for dual luciferase assays. The results are expressed as a percentage of the activity with wild-type FoxM1B, where CMV-empty served as a control for basal expression levels of the FoxM1B reporter gene. We also performed cotransfection controls with CMV FoxM1B wild-type and TATA-luciferase (Luc.) constructs, demonstrating that FoxM1B transcriptional activation requires the FoxM1B recognition sequence. Four separate transfection experiments were performed in triplicate to calculate standard deviations. (C) Mutation of the Leu 641 residue in the FoxM1B LXL motif diminishes FoxM1B protein association with either Cdk1 or Cdk2. U2OS cells were transiently transfected with either CMV T7-FoxM1B (lane 1), CMV T7-FoxM1B T585A (lane 2), CMV T7-FoxM1B T596A (lane 3), CMV T7-FoxM1B L641A (lane 4), or CMV T7-FoxM1B S657A (lane 5). U2OS cell lysates were prepared 48 h after transfection and coimmunoprecipitated (Co-IP) with either Cdk1 or Cdk2 polyclonal antibody. The coimmunoprecipitated proteins were subjected to Western blot analysis with a T7 epitope antibody. (D) Mutation of the Cdk1 phosphorylation site at position 596 decreases FoxM1B phosphorylation by Cdk1. U2OS cells were transiently transfected with CMV GFP-T7-FoxM1B (lane 1), CMV GFP-T7-FoxM1B T585A (lane 2), CMV GFP-T7-FoxM1B S657A (lane 3), CMV GFP-T7-FoxM1B L641A (lane 4), or CMV GFP-T7-FoxM1B T596A (lane 5). Transfected U2OS cell lysates were coimmunoprecipitated with polyclonal Cdk1 antibody and subjected to radioactive Cdk1 in vitro kinase assay and SDS-PAGE and autoradiography to visualize radioactively labeled proteins. Note that FoxM1B L641A mutant is not phosphorylated by Cdk1 because it no longer coimmunoprecipitatedwith the Cdk1 protein (panel C). (E) The FoxM1B T596A Cdk mutant protein shows diminished in vivo phosphorylation by Cdk protein. U2OS cells were transiently transfected with either CMV T7-FoxM1B, CMV T7-FoxM1B T596A, or FoxM1B L641A, transfected cells were serum starved for 48 h and either not stimulated or stimulated with serum for 12 or 18 h, and cells were harvested and protein extracts were prepared. Protein extracts were immunoprecipitated (IP) with T7 epitope antibody and then subjected to Western blot analysis with the MPM2 monoclonal antibody, which recognizes phosphorylated Cdk sites. Western blot analysis with T7 antibody demonstrated equal amounts of FoxM1B protein in all lanes. The relative intensity of the MPM2 signal was determined, and the FoxM1B level in cells not stimulated with serum was set at 1.
To identify FoxM1B sequences involved in the interaction with Cdk proteins, we focused on the LXL (639 to 641) sequence (Fig. 3A), which has been shown to bind to Cdk-cyclin proteins as efficiently as the cyclin-binding Cy (RXL) motif (57, 64). We used site-directed mutagenesis to convert the Leu 641 residue to an Ala residue, thereby disrupting the FoxM1B LXL motif. Transient-transfection assays demonstrated that the FoxM1B L641A mutant protein displayed an 80% reduction in transcriptional activity (Fig. 3B). Furthermore, increasing amounts of the CMV FoxM1B L641A expression vector inhibited transcriptional activity of the wild-type FoxM1B protein in cotransfection assays (data not shown), suggesting that the CMV FoxM1B L641A mutant protein functioned as a dominant negative inhibitor. Moreover, both GFP-T7-FoxM1B L641A and GFP-T7-FoxM1B T596A mutant proteins were retained in the nucleus (Fig. 4A to C), indicating that their diminished transcriptional activity was not due to inhibition of nuclear localization.
FIG. 4.

Nuclear localization of GFP-T7-FoxM1B mutant protein and GFP-T7-FoxM1B wild-type protein following treatment with either pharmacological kinase inhibitors or dominant negative kinases. U2OS cells were transiently transfected with CMV GFP-T7-FoxM1B T596A (B), CMV GFP-T7-FoxM1B L641A (C), CMV GFP-T7-FoxM1B plus the indicated pharmacological kinase inhibitors (F and G), or dominant negative kinase expression vectors (D, E, H, and I). Cells were fixed 48 h after transfection, and GFP fluorescence was detected as described in Materials and Methods. Note that DN-p90Rsk causes perinuclear localization of the GFP-T7-FoxM1B fusion protein.
To determine whether the FoxM1B T596A or FoxM1B L641A mutant protein exhibited diminished protein association with either the Cdk1 or Cdk2 protein, we performed coimmunoprecipitation experiments with protein extracts prepared from U2OS cells transfected with either wild-type CMV T7-FoxM1B or mutant expression constructs (Fig. 3C). The transfected U2OS cell extracts were coimmunoprecipitated with either Cdk1 or Cdk2 antibody, and then FoxM1B protein was visualized by Western blot analysis with the T7 epitope tag monoclonal antibody. These studies demonstrated that CMV T7-FoxM1B L641A mutant protein was unable to interact with either Cdk1 or Cdk2 protein, whereas the FoxM1B mutant proteins disrupted in each of the Cdk1 phosphorylation sites could associate efficiently with the Cdk proteins (Fig. 3C). These results suggested that the second Leu residue within the LXL sequence is essential for interaction between FoxM1B and Cdk proteins and that FoxM1B binding of either Cdk1- or Cdk2-cyclin protein complexes is required for its transcriptional activity.
To examine whether the Cdk1-cyclin B complex phosphorylates the FoxM1B protein, we performed coimmunoprecipitation Cdk1 in vitro kinase assays with 32P-labeled ATP (see Materials and Methods). Protein extracts prepared from U2OS cells transfected with either CMV GFP-T7-FoxM1B wild-type or GFP-T7-FoxM1B Cdk mutant expression vectors were coimmunoprecipitated with Cdk-1 antibody and then used for the radioactive Cdk1 in vitro kinase assay. The proteins phosphorylated in the coimmunoprecipitation Cdk1 in vitro kinase reaction were resolved on SDS-PAGE and visualized by autoradiography. Consistent with reduced transcriptional activity, the Cdk1 coimmunoprecipitation kinase assay demonstrated that the GFP-T7-FoxM1B T596A mutation exhibited reduced phosphorylation by the Cdk1 protein, whereas Cdk1 phosphorylated the GFP-T7-FoxM1B T585A and GFP-T7-FoxM1B S657A proteins to levels found with the GFP-T7-FoxM1B wild-type protein (Fig. 3D). As expected, the GFP-T7-FoxM1B L641A mutant protein failed to interact efficiently with Cdk1 protein (Fig. 3C), and therefore only low levels of FoxM1B L641A mutant protein are available for Cdk1 phosphorylation in the coimmunoprecipitation Cdk1 kinase assay (Fig. 3D).
To examine Cdk phosphorylation in vivo, protein extracts were prepared from serum-stimulated U2OS cells transfected with either CMV T7-FoxM1B wild-type, CMV T7-FoxM1B T596A, or CMV FoxM1B L641A expression constructs. These protein extracts were immunoprecipitated with the T7 antibody, and then Western blot analysis with the MPM2 monoclonal antibody was used to determine Cdk phosphorylation in vivo. These results demonstrated that Cdk phosphorylation of T7-FoxM1B wild-type protein was increased following serum stimulation and that the FoxM1B Thr 596 residue was required for phosphorylation by the Cdk-cyclin complexes in vivo (Fig. 3E). Furthermore, in vivo Cdk phosphorylation of the T7-FoxM1B L641A mutant protein was significantly reduced (Fig. 3E), suggesting that recruitment of the Cdk-cyclin complex by the FoxM1B LXL sequence was critical for its efficient Cdk phosphorylation in vivo.
Increasing both Cdk activity and levels of CBP stimulates FoxM1B-dependent transcription.
In order to examine whether Cdk activity is necessary for FoxM1B transcriptional activity, we cotransfected CMV-FoxM1B and the 6× FoxM1B TATA-luciferase with increasing amounts of CMV-DN-Cdk1 or treated the cells with increasing concentrations of the pharmacological Cdk inhibitor alsterpaullone (Fig. 5A) as described in the Materials and Methods section. Inhibiting Cdk activity with either dominant negative Cdk1 or a pharmacologically active concentration of alsterpaullone (1 μM) caused an 80 to 90% reduction in FoxM1B transcriptional activity (Fig. 5C). Cotransfection studies demonstrated that DN-Cdk1 equally inhibited the transcriptional activity of wild-type FoxM1B protein, FoxM1B Cdk mutant proteins, and FoxM1B L641A mutant proteins (data not shown). Furthermore, neither DN-Cdk1 nor alsterpaullone (1 μM) altered the nuclear localization of transfected GFP-T7-FoxM1B protein (Fig. 4A, D, and E), suggesting that inhibiting Cdk activity was required for FoxM1B-dependent transcription. Furthermore, cotransfection of CMV WT-Myt1 kinase, which negatively regulates Cdk activity through phosphorylation (6, 30, 31, 37), resulted in a 64% reduction in FoxM1B transcriptional activity (Fig. 5C). Consistent with these findings, stimulation of Cdk activity by cotransfection of either the CMV Cdc25B or CMV Cdc25C expression vector (Fig. 5B) enhanced FoxM1B transcriptional activity by 3.4-fold and 1.7-fold, respectively (Fig. 5D). Because Cdc25A and Cdc25B phosphatases have the same substrate specificity and differ only in their expression pattern during the cell cycle (7, 41, 62), it is anticipated that increased Cdc25B levels will stimulate both Cdk1 and Cdk2 activity. Taken together, our results strongly support the hypothesis that Cdk activity is required to stimulate FoxM1B transcriptional activity.
FIG. 5.

Inhibition of Cdk1 activity (DN-Cdk1 and Myt1) diminishes FoxM1B transcriptional activity, while stimulation of Cdk1 activity (Cdc25B and Cdc25C phosphatases) potentiates FoxM1B function. (A) Cartoon depicting inhibition of Cdk1 kinase activity by either Myt1 phosphorylation, dominant negative (DN) Cdk1, or the Cdk1 inhibitor Alsterpaullone. (B) Cartoon depicting stimulation of Cdk1 activity by Cdc25B and Cdc25C dephosphorylation. (C) Inhibition of Cdk1 activity diminishes FoxM1B transcriptional activity in cotransfection assays. U2OS-Tetr cells were transiently cotransfected with the reporter 6×-FoxM1B-TATA-luciferase or CMV-TO-FoxM1B (500 ng) alone or with increasing amounts of either CMV-DN-Cdk1, the Cdk1 pharmacological inhibitor alsterpaullone, or CMV-Myt1. FoxM1B expression was induced by tetracycline at 24 h following transfection, and pharmacological kinase inhibitors were added 5 h prior to FoxM1B induction. Cellular protein extracts were isolated at 48 h after transfection and assayed for dual luciferase activity. Results are expressed as a percentage of the activity with wild-type FoxM1B in four separate transfection experiments performed in triplicate to calculate standard deviations. (D) Activation of Cdk1 activity by dephosphorylation with either Cdc25B or Cdc25C stimulates FoxM1B transcriptional activity, which is potentiated by increased CBP levels. FoxM1B transcription assays in U2OS-Tetr cells were performed as described in the legend to Fig. 3C with either CMV-Cdc25B phosphatase or CMV-CBP either alone or in combination, demonstrating that increased Cdc25B levels enhanced CBP-mediated stimulation of FoxM1B transcriptional activity. Cotransfection assays with CMV-CBP and CMV-E1A, which inhibits the histone acetyltransferase activity of the CBP protein (9), eliminated CBP-mediated stimulation of FoxM1B transcriptional activity. Also shown is cotransfection with CMV-Cdc25C alone.
Because FoxM1B protein coimmunoprecipitated with the CBP coactivator protein (Fig. 2), we examined whether FoxM1B transcriptional activity requires the CBP function. The adenovirus E1A protein is known to inhibit CBP histone acetyltransferase activity (1, 9), and therefore we performed cotransfections with CMV-CBP or CMV-adenovirus E1A alone and in combination. Cotransfection of CMV-CBP caused a slight increase in FoxM1B transcriptional activity, whereas inhibition of CBP function with E1A resulted in a 75% reduction in FoxM1B transcriptional activity (Fig. 5D). Furthermore, cotransfection of CMV Cdc25B and CMV CBP together significantly augmented CBP-mediated stimulation of FoxM1B transcriptional activity from a 1.4-fold increase to a 6.2-fold increase (Fig. 5D). These studies suggest that recruitment of the p300/CBP family of coactivator proteins is essential for FoxM1B transcriptional activation and that increased Cdk activity enhances CBP-mediated stimulation of FoxM1B transcriptional activity.
FoxM1B transcriptional activity involves recruitment of CBP through phosphorylation of the FoxM1B 596 Cdk site.
Because stimulation of Cdk activity potentiated CBP-mediated increase in FoxM1B-dependent transcription, we wanted to determine whether the critical Cdk1/2 phosphorylation site at FoxM1B residue 596 was required for recruitment of CBP protein. We transiently transfected U2OS cells with CMV-CBP and either the CMV GFP-T7-FoxM1B wild type, CMV GFP-T7-FoxM1B L641A (mutation of the LXL motif), or CMV GFP-T7-FoxM1B T596A (mutation of the Cdk1 site) expression vector. Protein extracts were prepared 48 h after transfection and used for coimmunoprecipitation experiments with CBP antibody, followed by Western blot analysis with GFP monoclonal antibody. These coimmunoprecipitation studies demonstrated that CBP protein efficiently interacted with either wild-type FoxM1B protein or FoxM1B L641A mutant protein (Fig. 6A), the latter of which exhibited reduced but detectable Cdk phosphorylation in vivo (Fig. 3E). In contrast, disruption of the FoxM1B Cdk1 phosphorylation site at Thr residue 596 significantly diminished FoxM1B's ability to associate with the CBP protein (Fig. 6A), suggesting that the FoxM1B Cdk phosphorylation site at position 596 is essential for recruitment of CBP. Consistent with this finding, inhibition of Cdk activity by cotransfection of U2OS cells with CMV-DN-Cdk1, CMV-CBP, and CMV GFP-T7-FoxM1B eliminated the interaction between FoxM1B and CBP proteins (Fig. 6B), demonstrating that Cdk phosphorylation is required for FoxM1B association with the CBP protein. Furthermore, increasing Cdk activity through CMV Cdc25B cotransfection stimulated the transcriptional activity of the FoxM1B T585A and FoxM1B S657A mutants to levels found with wild-type FoxM1B protein (Fig. 6C). In contrast, cotransfection of CMV Cdc25B resulted in reduced stimulation of transcriptional activity of the FoxM1B T596A and FoxM1B L641A mutant proteins compared to wild-type FoxM1B (Fig. 6C), a finding consistent with both FoxM1B mutant proteins' exhibiting diminished Cdk phosphorylation in vivo (Fig. 3E). Taken together, our results support the hypothesis that FoxM1B phosphorylation by Cdk-cyclin complexes is required for recruitment of the p300/CBP coactivator protein and thus functions as a mechanism for proliferation-specific stimulation of FoxM1B transcriptional activity.
FIG. 6.

Cdk1 phosphorylation of FoxM1B protein is required for efficient association with CBP. (A) Phosphorylation of the FoxM1B protein Cdk1 site at position 596 is required for efficient association with CBP in coimmunoprecipitation assays. U2OS cells were transiently transfected with CBP and either CMV GFP-T7-FoxM1B wild-type (lanes 1 and 2), CMV GFP-T7-FoxM1B L641A (lanes 3 and 4), or CMV GFP-T7-FoxM1B T596A (lanes 5 and 6) expression plasmid or mock transfected (lanes 7 and 8). Lysates were immunoprecipitated with a monoclonal antibody that recognizes CBP, and immunocomplexes were subjected to Western blot analysis with a GFP monoclonal antibody. The first lane of each set contains 1/10 of the input protein extract (50 μg), and the second lane contains the immunoprecipitated (IP) protein extracts. (B) Dominant negative Cdk1 inhibits FoxM1B-mediated recruitment of CBP. U2OS cells were transiently transfected with CBP and CMV wild-type GFP-T7-FoxM1B either alone or together with CMV dominant negative Cdk1 expression plasmids. Lysates were immunoprecipitated with a monoclonal antibody that recognizes CBP, and immunocomplexes were subjected to Western blot analysis with a GFP monoclonal antibody. These studies demonstrated that dominant negative Cdk1 inhibits coimmunoprecipitation between FoxM1B and CBP. (C) Increased Cdc25B levels displayed reduced stimulation of the FoxM1B T596A and FoxM1B L641A mutant proteins. U2OS cells were transiently cotransfected with the reporter 6×-FoxM1B/FoxA-TATA-luciferase and CMV-Cdc25B and either CMV FoxM1B wild-type, CMV FoxM1B S657A, CMV FoxM1B T585A, CMV FoxM1B L641A, or CMV FoxM1B T596A expression plasmid. Twenty-four hours posttransfection, cells were harvested and processed for dual luciferase assays. Results are expressed as a percentage of the activity with wild-type FoxM1B, where CMV-empty served as a control for basal expression levels of the FoxM1B reporter gene. Four separate transfection experiments were performed in triplicate to calculate standard deviations.
Blocking the Ras-MAPK and PI3K-PDK1 pathways diminished FoxM1B transcriptional activity, but inhibiting Akt did not influence FoxM1B-dependent transcription.
The MAPK pathway is required for cell cycle progression because it stimulates CAK-mediated phosphorylation, resulting in activation of Cdk proteins (28). Furthermore, activation of the MAPK and PI3K/PDK1 signaling pathways mediates phosphorylation and activation of the downstream p90 ribosomal S6 kinase (Rsk) (4, 21, 51), which inactivates the Myt1 kinase, which provides inhibitory phosphorylation of the Cdk1 and Cdk2 proteins (44, 45).
In order to determine the role of either the MAPK or PI3K pathway in regulating FoxM1B activity, FoxM1B transcription assays were performed in U2OS cells that were either treated with the pharmacological MEK1/2 inhibitor U0126 or PI3K inhibitor Ly294002 or cotransfected with CMV DN-RasN17 expression vector (Fig. 7A). These transfection studies demonstrated that inhibition of either MEK1/2, PI3K, or Ras caused a 70 to 80% reduction in FoxM1B-dependent transcription (Fig. 7C), a finding consistent with the important roles of the Ras/MAPK and PI3K/PDK1 pathways in Cdk1-cyclin B activation. In contrast, blocking the Akt pathway with either CMV DN-Akt or the Akt pharmacological kinase inhibitor did not significantly alter FoxM1B transcriptional activity (Fig. 7C). Furthermore, combining the MEK1/2 (U0126) and PI3K (Ly294002) inhibitors resulted in a 90% reduction in FoxM1B-dependent transcription, demonstrating the importance of the Ras/MAPK and PI3K/PDK1 pathways in regulating FoxM1B transcriptional activity (Fig. 7C). Cotransfection of CMV DN-p90Rsk (Fig. 7A) resulted in a 56% reduction in FoxM1B transcriptional activity (Fig. 7C), which is similar to the transcriptional reductions found with CMV WT-Myt1 (Fig. 5C), a kinase that is negatively regulated by p90Rsk. Addition of the Ras/MEK1/2 or PI3K/Akt pathway inhibitor neither diminished expression (Fig. 7B) nor altered the nuclear localization of GFP-T7-FoxM1B protein (Fig. 4A and F to I), suggesting that these inhibitors caused decreases in FoxM1B transcriptional activity. However, cotransfection of DN-p90Rsk resulted in redistribution of a portion of GFP-T7-FoxM1B fluorescence to the periphery of the nucleus (Fig. 4J), suggesting that p90Rsk signaling may influence the localization of FoxM1B within the nucleus. Taken together, our studies demonstrate that FoxM1B transcriptional activity requires Cdk-cyclin activation, which is mediated by growth factor stimulation of the Ras/MAPK and PI3K/PDK1 signaling cascades.
FIG. 7.

Inhibition of the Ras/MEK/MAPK/p90Rsk and PI3K/PDK1/p90Rsk pathways results in diminished FoxM1B transcriptional activity. (A) Cartoon depicting the Ras/MEK/MAPK/p90Rsk/Myt1 and PI3K/PDK1/p90Rsk/Myt1 pathways, which prevent Myt1 phosphorylation-mediated inhibition of Cdk1 and Cdk2 activity. Also shown is the action of DN-RasN17, the MEK1/2 inhibitor U0126, the PI3K inhibitor Ly294002, DN-Akt and the Akt pharmacological kinase inhibitor, and DN-p90Rsk. (B) Cotransfection of DN-p90Rsk or DN-Ras or treatment with the MEK inhibitor U0126, the PI3K inhibitor Ly294002, or the Akt pharmacological kinase inhibitor does not influence expression of the GFP-T7-FoxM1B protein. U2OS cells were transiently transfected with CMV GFP-T7-FoxM1B plasmid with either CMV DN-p90Rsk or CMV DN-RasN17 or 50 μM U0126, 50 μM PI3K inhibitor Ly294002, or 25 μM Akt inhibitor. Protein extracts were prepared 48 h following transfection and subjected to Western blot analysis with GFP antibody. (C) Inhibition of Ras/MEK/MAPK/p90Rsk and PI3K/PDK1/p90Rsk pathways results in diminished FoxM1B transcriptional activity. U2OS-Tetr cells were transiently cotransfected with the reporter 6×-FoxM1B-TATA-luciferase and CMV-TO-FoxM1B (500 ng) with CMV-DN-p90Rsk, CMV-DN-Ras, or DN-AKT or with 50 μM U0126 or Ly294002 either alone or together or with 25 μM Akt inhibitor. FoxM1B transcription assays in U2OS-Tetr cells with tetracycline-inducible FoxM1B protein were performed as described in the legend to Fig. 3C and in Materials and Methods. Four separate transfection experiments were performed in triplicate to calculate standard deviations.
DISCUSSION
Previous studies have demonstrated that the FoxM1B transcription factor is required for regenerating hepatocyte proliferation through transcriptional regulation of the Cdc25B phosphatase gene and controlling protein levels of Cdc25A phosphatase and Cdk inhibitor p21Cip1 (59). Activation of the Cdk2-cyclin E and A and Cdk1-cyclin B complexes is critical for cell cycle progression. These Cdk-cyclin complexes are activated by removal of inhibitory phosphates at Cdk residues Thr 14 and Tyr 15 by Cdc25A, Cdc25B, and Cdc25C phosphatases, which have identical substrate specificities but are expressed at different stages of the cell cycle (7, 41, 62). In this study, we demonstrated that Cdk-cyclin phosphorylation of FoxM1B protein at Thr residue 596 is required for FoxM1B transcriptional activation by mediating phosphorylation-dependent recruitment of the p300/CBP histone acetyltransferase protein (Fig. 8). The p300/CBP proteins activate transcription by acetylation of lysine residues of histone proteins, causing their local dissociation, and by interaction with the general transcriptional machinery (1, 9, 10, 38, 39, 53). Furthermore, we demonstrated that FoxM1B transcriptional activity required an LXL sequence (positions 639 to 641) for binding of either the Cdk2-cyclin E or Cdk1-cyclin B complex, which is required for efficient Cdk phosphorylation of FoxM1B Thr residue 596 that is essential for recruitment of the p300/CBP coactivators (Fig. 8).
FIG. 8.
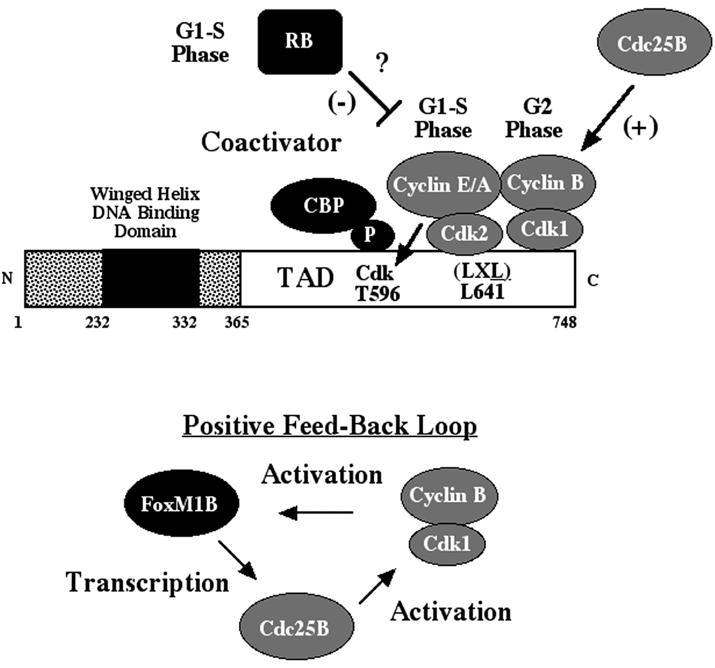
Model for Cdk-cyclin-mediated stimulation of FoxM1B-dependent transcription. FoxM1B transcriptional activity is restricted to proliferating cells through interaction with and phosphorylation by the Cdk-cyclin complexes. Activated Cdk2-cyclin E or A or Cdk1-cyclin B complexes phosphorylate FoxM1B Thr 596, which is required for the recruitment of CBP for FoxM1B transcriptional activation. FoxM1B transcriptional activity requires binding of Cdk1-cyclin B or Cdk2-cyclin E/A complexes through the FoxM1B LXL motif, which mediates efficient phosphorylation of the FoxM1B Cdk site at position 596. Furthermore, FoxM1B interacts with RB at the G1 phase of the cell cycle, and RB thus may negatively regulate FoxM1B transcriptional activity. We have previously demonstrated that FoxM1B is essential for transcription of the Cdc25B phosphatase gene (59), which is required for dephosphorylation and activation of Cdk1 protein (7, 41, 62). Taken together, these data establish a positive regulatory loop between FoxM1B, Cdc25B, and Cdk1.
We found that the Cdk phosphorylation of FoxM1B protein initiated as early as the G1 phase of the cell cycle in synchronized U2OS cells and that FoxM1B phosphorylation by Cdk-cyclin complexes continued throughout the proliferative stages of the cell cycle. Mutation of a FoxM1B Cdk1/2 phosphorylation site at Thr 596 to Ala caused an 80% reduction in FoxM1B transcriptional activity and resulted in diminished Cdk-dependent phosphorylation of the FoxM1B protein in vivo. We showed that either FoxM1B T596A mutation or cotransfection of wild-type-FoxM1B protein with dominant negative Cdk1 constructs prevented association with CBP histone acetyltransferase protein, suggesting that this Cdk site is essential for transcriptional activity because its phosphorylation recruits CBP coactivator function. Furthermore, inhibition of CBP histone acetyltransferase activity with the adenovirus E1A oncoprotein (1, 9) significantly decreased FoxM1B transcriptional activity. Consistent with these findings, Cdc25B-mediated stimulation of Cdk activity in combination with increased levels of the CBP coactivator provided a 6.2-fold synergistic increase in FoxM1B-dependent transcription, whereas inhibiting Cdk activity with DN-Cdk1 caused a 90% decrease in FoxM1B transcriptional activity.
We showed that both endogenous and transfected FoxM1B associated with either Cdk2-cyclin E or Cdk1-cyclin B complexes in coimmunoprecipitation experiments (Fig. 2). Mutation of the FoxM1B Leu at 641 to Ala within an LXL motif caused an 80% reduction in transcriptional activity and inhibited FoxM1B interaction with both Cdk1-cyclin B and Cdk2-cyclin B1 protein complexes. These studies show that the FoxM1B protein uses a LXL sequence to recruit the Cdk-cyclin complexes, which has been shown to bind to Cdk-cyclin proteins as efficiently as the cyclin-binding Cy (RXL) motif (57, 64). Furthermore, we found significant reduction in phosphorylation of the T7-FoxM1B L641A mutant protein in vivo (Fig. 3E), suggesting that recruitment of the Cdk-cyclin complex by the LXL FoxM1B sequence was critical for efficient Cdk phosphorylation. These results support the hypothesis that FoxM1B transcriptional activity requires binding of either S-phase or M-phase Cdk-cyclin complexes to mediate efficient Cdk phosphorylation of the FoxM1B Thr 596 residue, which is essential for recruitment of p300/CBP coactivator proteins (Fig. 8).
We propose a positive-feedback model in which FoxM1B transcriptional activity is stimulated during the G1/S phase of the cell cycle by Cdk2-cyclin E or A phosphorylation, allowing recruitment of the CBP coactivator and binding of the Cdk2-cyclin E or A complex (Fig. 8). Activation of the FoxM1B protein allows transcription of the Cdc25B phosphatase gene, which is a direct target for FoxM1B transcriptional activation (59). The increased levels of Cdc25B protein will activate the Cdk1-cyclin B complex through dephosphorylation, allowing Cdk1-cyclin B to maintain phosphorylation of FoxM1B protein and recruitment of the p300/CBP coactivator proteins during the G2 phase of the cell cycle (Fig. 8). The transcriptional activity of FoxM1B protein is also maintained by recruiting activated Cdk1-cyclin B complexes to maintain FoxM1B phosphorylation to recruit the p300/CBP transcriptional coactivators (Fig. 8). Activation of FoxM1B protein will allow transcriptional regulation of genes required to mediate the G2/M transition, such as Cdk1, cyclin B1, cyclin B2, and cyclin F (60, 61).
Stimulation of the cyclic AMP response element binding protein (CREB) by protein kinase A phosphorylation of the CREB transcriptional activation domain was the first transcription factor reported to display phosphorylation-dependent recruitment of the CBP (11). Recent studies have demonstrated that the Cdk2-cyclin E complex phosphorylates the activation domain of E2F-5 and mediates recruitment of p300/CBP coactivator proteins during S phase (36). Our FoxM1B study is the first reported demonstration that activation of a proliferation-specific transcription factor requires binding of either S-phase or M-phase Cdk-cyclin complexes for its efficient phosphorylation to mediate recruitment of p300/CBP coactivators (Fig. 8).
Similar to the E2F transcription factors (16, 20), we found that RB associates with FoxM1B in G1-phase protein extracts but not with S-phase and G2-phase protein extracts when RB is hyperphosphorylated (24, 32). These data suggest the hypothesis that RB negatively modulates FoxM1B transcriptional activity at G1 phases of the cell cycle but not during the S and G2 phases, when hyperphosphorylated RB (24, 32) fails to interact with the FoxM1B protein (Fig. 2D). In support of the hypothesis that RB negatively regulates FoxM1B transcriptional activity, a threefold increase in FoxM1B transcriptional activity was found in cotransfection assays with a FoxM1B-dependent reporter gene along with expression vectors for the FoxM1B (MPP2) protein and the human papillomavirus type 16 E7 protein (33). Because the human papillomavirus type 16 E7 protein is known to target the RB protein for degradation (18), these published cotransfection studies (33) support the hypothesis that diminished RB protein levels are sufficient to stimulate FoxM1B transcriptional activity.
We demonstrated that inhibition of the Ras/MAPK and PI3K/PDK1 pathways either separately or together resulted in significant decreases in FoxM1B-dependent transcriptional activity, indicating that these cascades are required for FoxM1B transcriptional activity (Fig. 7C). We propose the hypothesis that the Ras/MAPK and PI3K/PDK1 signaling cascades are required for FoxM1B-dependent transcription because they mediate activation of the Cdk-cyclin complexes (Fig. 8). Activation of the MAPK and PI3K/PDK1 signaling pathways mediates phosphorylation and activation of the downstream p90 ribosomal S6 kinase (Rsk) (4, 21, 51), which inactivates the Cdk1-inhibitory Myt1 kinase (44, 45). The MAPK signaling cascade also stimulates Cdk1 activity through CAK-mediated phosphorylation of Thr 160 (28) and by inducing expression of the Cdc25B (through FoxM1B) and Cdc25C phosphatases, which mediate the removal of inhibitory phosphates at Thr 14 and Tyr 15 (7, 41, 62). Furthermore, the PI3K/PDK1 signaling pathway is necessary for normal expression levels of cyclin B1 protein (52). However, we demonstrated that Akt phosphorylation is not necessary for FoxM1B transcriptional activity because inhibition of Akt activity did not significantly affect FoxM1B-dependent transcription (Fig. 7C). This is in striking contrast to the FoxO (Fkhr, Daf-16) transcription factors, in which Akt negatively regulates FoxO transcriptional activity by phosphorylation of the FoxO C terminus, resulting in its nuclear export and retention in the cytoplasm (2, 8, 15).
In proliferating mammalian cells, the PI3K/Akt signal transduction pathway is essential for G1-to-S-phase progression because it prevents transcriptional activity of the FoxO proteins, which stimulate transcription of the Cdk inhibitor p27 kip1 gene (35) and the retinoblastoma-like p130 gene (25). However, our studies clearly demonstrate that the FoxM1B protein is regulated by the activity of the Cdk-cyclin complexes and that this regulatory mechanism is distinct from Akt-mediated regulation of the related FoxO proteins. It is interesting that FoxM1B regulates the protein levels but not the transcription of the Cdk inhibitors p27kip1 and p21Cip1 and thus negatively regulates the activity of these Cdk inhibitors (59, 60). Our current studies also suggest that the ability of FoxM1B to diminish accumulation of the p27kip1 and p21Cip1 proteins will enhance FoxM1B transcriptional activity through increased Cdk activity.
In summary, we demonstrated that FoxM1B-dependent transcription requires binding of the Cdk-cyclin complexes through the FoxM1B LXL motif to allow efficient phosphorylation of the FoxM1B Thr 596 Cdk site in vivo. Phosphorylation of this Cdk site is essential for transcriptional activity through recruitment of p300/CBP coactivator proteins, thus providing FoxM1B protein with cell cycle-specific transcriptional activity.
Acknowledgments
We thank P. Raychaudhuri, X. Wang, Y. Tan, V. Kalinichenko, Y. Zhou, and F. Rausa for critically reviewing the manuscript, and we thank P. Raychaudhuri for helpful discussions. We also thank a number of investigators who provided us with dominant negative kinase expression vectors; these include Ravi Misra (Medical College of Wisconsin) for DN-RasN17, John Blenis (Harvard Medical School) for DN-p90Rsk, Nissim Hay (University of Illinois at Chicago) for DN-Akt, Helen Piwnica-Worms (Washington University) for WT-Myt1, and David Ucker (University of Illinois at Chicago) for DN-Cdk1.
This work was supported by NIH grant DK 54687-06 from the National Institute of Diabetes and Digestive and Kidney Diseases (R.H.C.).
REFERENCES
- 1.Arany, Z., W. R. Sellers, D. M. Livingston, and R. Eckner. 1994. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77:799-800. [DOI] [PubMed] [Google Scholar]
- 2.Biggs, W. H., III, J. Meisenhelder, T. Hunter, W. K. Cavenee, and K. C. Arden. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 96:7421-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blangy, A., H. A. Lane, P. d'Herin, M. Harper, M. Kress, and E. A. Nigg. 1995. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83:1159-1169. [DOI] [PubMed] [Google Scholar]
- 4.Blenis, J. 1993. Signal transduction via the MAP kinases: proceed at your own RSK. Proc. Natl. Acad. Sci. USA 90:5889-5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blume-Jensen, P., and T. Hunter. 2001. Oncogenic kinase signalling. Nature 411:355-365. [DOI] [PubMed] [Google Scholar]
- 6.Booher, R. N., P. S. Holman, and A. Fattaey. 1997. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 272:22300-22306. [DOI] [PubMed] [Google Scholar]
- 7.Borgne, A., and L. Meijer. 1996. Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 271:27847-27854. [DOI] [PubMed] [Google Scholar]
- 8.Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu, M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868. [DOI] [PubMed] [Google Scholar]
- 9.Chan, H. M., and N. B. La Thangue. 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114:2363-2373. [DOI] [PubMed] [Google Scholar]
- 10.Chen, H., M. Tini, and R. M. Evans. 2001. HATs on and beyond chromatin. Curr. Opin. Cell Biol. 13:218-224. [DOI] [PubMed] [Google Scholar]
- 11.Chrivia, J. C., R. P. Kwok, N. Lamb, M. Hagiwara, M. R. Montminy, and R. H. Goodman. 1993. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365:855-859. [DOI] [PubMed] [Google Scholar]
- 12.Clark, K. L., E. D. Halay, E. Lai, and S. K. Burley. 1993. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364:412-420. [DOI] [PubMed] [Google Scholar]
- 13.Costa, R. H., V. V. Kalinichenko, A. X. Holterman, and X. Wang. 2003. Transcription factors in liver development, differentiation, and regeneration. Hepatology 38:1331-1347. [DOI] [PubMed] [Google Scholar]
- 14.Desai, D., H. C. Wessling, R. P. Fisher, and D. O. Morgan. 1995. Effects of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol. Cell. Biol. 15:345-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo, S., G. Rena, S. Cichy, X. He, P. Cohen, and T. Unterman. 1999. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274:17184-17192. [DOI] [PubMed] [Google Scholar]
- 16.Harbour, J. W., and D. C. Dean. 2000. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14:2393-2409. [DOI] [PubMed] [Google Scholar]
- 17.Harbour, J. W., R. X. Luo, A. Dei Santi, A. A. Postigo, and D. C. Dean. 1999. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98:859-869. [DOI] [PubMed] [Google Scholar]
- 18.Helt, A. M., and D. A. Galloway. 2003. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 24:159-169. [DOI] [PubMed] [Google Scholar]
- 19.Hindley, A., and W. Kolch. 2002. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J. Cell Sci. 115:1575-1581. [DOI] [PubMed] [Google Scholar]
- 20.Ishida, S., E. Huang, H. Zuzan, R. Spang, G. Leone, M. West, and J. R. Nevins. 2001. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21:4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen, C. J., M. B. Buch, T. O. Krag, B. A. Hemmings, S. Gammeltoft, and M. Frodin. 1999. 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 274:27168-27176. [DOI] [PubMed] [Google Scholar]
- 22.Kaestner, K. H., W. Knochel, and D. E. Martinez. 2000. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 14:142-146. [PubMed] [Google Scholar]
- 23.Kimura, K., M. Hirano, R. Kobayashi, and T. Hirano. 1998. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science 282:487-490. [DOI] [PubMed] [Google Scholar]
- 24.Knudsen, E. S., and J. Y. Wang. 1997. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol. Cell. Biol. 17:5771-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kops, G. J., R. H. Medema, J. Glassford, M. A. Essers, P. F. Dijkers, P. J. Coffer, E. W. Lam, and B. M. Burgering. 2002. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol. Cell. Biol. 22:2025-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korver, W., J. Roose, and H. Clevers. 1997. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 25:1715-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krupczak-Hollis, K., X. Wang, M. B. Dennewitz, and R. H. Costa. 2003. Growth Hormone Stimulates Proliferation of Old-aged Regenerating Liver Through Forkhead Box m1b. Hepatology 38:1552-1562. [DOI] [PubMed] [Google Scholar]
- 28.Lents, N. H., S. M. Keenan, C. Bellone, and J. J. Baldassare. 2002. Stimulation of the Raf/MEK/ERK Cascade Is Necessary and Sufficient for Activation and Thr-160 Phosphorylation of a Nuclear-targeted CDK2. J. Biol. Chem. 277:47469-47475. [DOI] [PubMed] [Google Scholar]
- 29.Leung, T. W., S. S. Lin, A. C. Tsang, C. S. Tong, J. C. Ching, W. Y. Leung, R. Gimlich, G. G. Wong, and K. M. Yao. 2001. Over-expression of FoxM1 stimulates cyclin B1 expression. FEBS Lett. 507:59-66. [DOI] [PubMed] [Google Scholar]
- 30.Liu, F., C. Rothblum-Oviatt, C. E. Ryan, and H. Piwnica-Worms. 1999. Overproduction of human Myt1 kinase induces a G2 cell cycle delay by interfering with the intracellular trafficking of Cdc2-cyclin B1 complexes. Mol. Cell. Biol. 19:5113-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, F., J. J. Stanton, Z. Wu, and H. Piwnica-Worms. 1997. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol. 17:571-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundberg, A. S., and R. A. Weinberg. 1998. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18:753-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luscher-Firzlaff, J. M., J. M. Westendorf, J. Zwicker, H. Burkhardt, M. Henriksson, R. Muller, F. Pirollet, and B. Luscher. 1999. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene 18:5620-5630. [DOI] [PubMed] [Google Scholar]
- 34.Marsden, I., C. Jin, and X. Liao. 1998. Structural changes in the region directly adjacent to the DNA-binding helix highlight a possible mechanism to explain the observed changes in the sequence-specific binding of winged helix proteins. J. Mol. Biol. 278:293-299. [DOI] [PubMed] [Google Scholar]
- 35.Medema, R. H., G. J. Kops, J. L. Bos, and B. M. Burgering. 2000. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404:782-787. [DOI] [PubMed] [Google Scholar]
- 36.Morris, L., K. E. Allen, and N. B. La Thangue. 2000. Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat. Cell Biol. 2:232-239. [DOI] [PubMed] [Google Scholar]
- 37.Mueller, P. R., T. R. Coleman, A. Kumagai, and W. G. Dunphy. 1995. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 270:86-90. [DOI] [PubMed] [Google Scholar]
- 38.Naar, A. M., B. D. Lemon, and R. Tjian. 2001. Transcriptional coactivator complexes. Annu. Rev. Biochem. 70:475-501. [DOI] [PubMed] [Google Scholar]
- 39.Narlikar, G. J., H. Y. Fan, and R. E. Kingston. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108:475-487. [DOI] [PubMed] [Google Scholar]
- 40.Nigg, E. A. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell. Biol. 2:21-32. [DOI] [PubMed] [Google Scholar]
- 41.Nilsson, I., and I. Hoffmann. 2000. Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 4:107-114. [DOI] [PubMed] [Google Scholar]
- 42.Ohi, R., and K. L. Gould. 1999. Regulating the onset of mitosis. Curr. Opin. Cell Biol. 11:267-273. [DOI] [PubMed] [Google Scholar]
- 43.Ookata, K., S. Hisanaga, E. Okumura, and T. Kishimoto. 1993. Association of p34cdc2/cyclin B complex with microtubules in starfish oocytes. J. Cell Sci. 105:873-881. [DOI] [PubMed] [Google Scholar]
- 44.Palmer, A., A. C. Gavin, and A. R. Nebreda. 1998. A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 17:5037-5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer, A., and A. R. Nebreda. 2000. The activation of MAP kinase and p34cdc2/cyclin B during the meiotic maturation of Xenopus oocytes. Prog. Cell Cycle Res. 4:131-143. [DOI] [PubMed] [Google Scholar]
- 46.Pani, L., D. G. Overdier, A. Porcella, X. Qian, E. Lai, and R. H. Costa. 1992. Hepatocyte nuclear factor 3β contains two transcriptional activation domains, one of which is novel and conserved with the Drosophila forkhead protein. Mol. Cell. Biol. 12:3723-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pouyssegur, J., V. Volmat, and P. Lenormand. 2002. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 64:755-763. [DOI] [PubMed] [Google Scholar]
- 48.Qian, X., and R. H. Costa. 1995. Analysis of HNF-3β protein domains required for transcriptional activation and nuclear targeting. Nucleic Acids Res. 23:1184-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rausa, F., Y. Tan, and R. H. Costa. 2003. Association between HNF-6 and FoxA2 DNA binding domains stimulates FoxA2 transcriptional activity but inhibits HNF-6 DNA binding. Mol. Cell. Biol. 23:437-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rausa, F. M., Y. Tan, H. Zhou, K. Yoo, D. B. Stolz, S. Watkins, R. R. Franks, T. G. Unterman, and R. H. Costa. 2000. Elevated levels of HNF-3β in mouse hepatocytes influence expression of genes involved in bile acid and glucose homeostasis. Mol. Cell. Biol. 20:8264-8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richards, S. A., V. C. Dreisbach, L. O. Murphy, and J. Blenis. 2001. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol. Cell. Biol. 21:7470-7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts, E. C., P. S. Shapiro, T. S. Nahreini, G. Pages, J. Pouyssegur, and N. G. Ahn. 2002. Distinct cell cycle timing requirements for extracellular signal-regulated kinase and phosphoinositide 3-kinase signaling pathways in somatic cell mitosis. Mol. Cell. Biol. 22:7226-7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth, S. Y., J. M. Denu, and C. D. Allis. 2001. Histone acetyltransferases. Annu. Rev. Biochem. 70:81-120. [DOI] [PubMed] [Google Scholar]
- 54.Samadani, U., and R. H. Costa. 1996. The transcriptional activator hepatocyte nuclear factor six regulates liver gene expression. Mol. Cell. Biol. 16:6273-6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solomon, M. J. 1994. The function(s) of CAK, the p34cdc2-activating kinase. Trends Biochem. Sci. 19:496-500. [DOI] [PubMed] [Google Scholar]
- 56.Sutani, T., T. Yuasa, T. Tomonaga, N. Dohmae, K. Takio, and M. Yanagida. 1999. Fission yeast condensin complex: essential roles of non-SMC subunits for condensation and Cdc2 phosphorylation of Cut3/SMC4. Genes Dev. 13:2271-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeda, D. Y., J. A. Wohlschlegel, and A. Dutta. 2001. A bipartite substrate recognition motif for cyclin-dependent kinases. J. Biol. Chem. 276:1993-1997. [DOI] [PubMed] [Google Scholar]
- 58.Wang, X., N.-J. Hung, and R. H. Costa. 2001. Earlier expression of the transcription factor HFH 11B (FOXM1B) Diminishes Induction of p21CIP1/WAF1 levels and accelerates mouse hepatocyte entry into S-phase following carbon tetrachloride liver injury. Hepatology 33:1404-1414. [DOI] [PubMed] [Google Scholar]
- 59.Wang, X., H. Kiyokawa, M. B. Dennewitz, and R. H. Costa. 2002. The forkhead box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc. Natl. Acad. Sci. USA 99:16881-16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, X., K. Krupczak-Hollis, Y. Tan, M. B. Dennewitz, G. R. Adami, and R. H. Costa. 2002. Increased hepatic forkhead box M1B (FoxM1B) levels in old-aged mice stimulated liver regeneration through diminished p27Kip1 protein levels and increased Cdc25B expression. J. Biol. Chem. 277:44310-44316. [DOI] [PubMed] [Google Scholar]
- 61.Wang, X., E. Quail, N.-J. Hung, Y. Tan, H. Ye, and R. H. Costa. 2001. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevents age-related proliferation defects in regenerating liver. Proc. Natl. Acad. Sci. USA 98:11468-11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wells, N. J., N. Watanabe, T. Tokusumi, W. Jiang, M. A. Verdecia, and T. Hunter. 1999. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 112:3361-3371. [DOI] [PubMed] [Google Scholar]
- 63.Westendorf, J. M., P. N. Rao, and L. Gerace. 1994. Cloning of cDNAs for M-phase phosphoproteins recognized by the MPM2 monoclonal antibody and determination of the phosphorylated epitope. Proc. Natl. Acad. Sci. USA 91:714-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wohlschlegel, J. A., B. T. Dwyer, D. Y. Takeda, and A. Dutta. 2001. Mutational analysis of the Cy motif from p21 reveals sequence degeneracy and specificity for different cyclin-dependent kinases. Mol. Cell. Biol. 21:4868-4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yao, F., T. Svensjo, T. Winkler, M. Lu, C. Eriksson, and E. Eriksson. 1998. Tetracycline repressor, tetR, rather than the tetR-mammalian cell transcription factor fusion derivatives, regulates inducible gene expression in mammalian cells. Hum. Gene Ther. 9:1939-1950. [DOI] [PubMed] [Google Scholar]
- 66.Yao, K. M., M. Sha, Z. Lu, and G. G. Wong. 1997. Molecular analysis of a novel winged helix protein, WIN. Expression pattern, DNA binding property, and alternative splicing within the DNA binding domain. J. Biol. Chem. 272:19827-19836. [DOI] [PubMed] [Google Scholar]
- 67.Ye, H., A. Holterman, K. W. Yoo, R. R. Franks, and R. H. Costa. 1999. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol. Cell. Biol. 19:8570-8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye, H., T. F. Kelly, U. Samadani, L. Lim, S. Rubio, D. G. Overdier, K. A. Roebuck, and R. H. Costa. 1997. Hepatocyte nuclear factor 3/forkhead homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 17:1626-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]