

Abstract
Cohesin is a ring-form multifunctional protein complex, which was discovered during a search for molecules that keep sister chromatids together during segregation of chromosomes during cell division. In the past decade, a large number of results have also demonstrated a need for the cohesin complex in other crucial events in the life cycle of the cell, including DNA duplication, heterochromatin formation, DNA double-strand break repair, and control of gene expression. The dynamics of the cohesin ring are modulated by a number of accessory and regulatory proteins, known as cohesin cofactors. Loss of function of the cohesin complex is incompatible with life; however, mutations in the genes encoding for cohesin subunits and/or cohesin cofactors, which have very little or a null effect on chromosome segregation, represent a newly recognized class of human genetic disorders known as cohesinopathies. A number of genetic, biochemical, and clinical approaches, and importantly, animal models, can help us to determine the underlying mechanisms for these human diseases.
Keywords: cohesin, cohesinopathies, Cornelia de Lange syndrome, Roberts syndrome, control, gene expression, insulators
Introduction
Cells have precise mechanisms for controlling cohesion of sister chromatids during cell division to ensure appropriate distribution of genetic material to daughter cells. The main molecular entity in this process is the evolutionarily conserved cohesin complex. The canonical cohesin complex consists of four subunits known as SMC1 and SMC3 (from the structural maintenance of chromosomes family) and SCC1 and SCC3 (sister chromatid cohesion). The latter two subunits are also known as RAD21 (SCC1, kleisin) and STAG (SCC3) in mammals (Table 1). The cohesin complex adopts a ring-like structure, which traps the replicated DNA and mediates cohesion between sister chromatids (Figure 1). The dynamics of the cohesin ring are regulated by an undetermined number of proteins that interact with cohesins (Table 1). These cohesin regulators were first characterized and studied in relation to the cohesive function of cohesin complexes, most of which are involved in the dynamics of the association and/or dissociation of the cohesin complex with chromatin. Loading of cohesins into chromosomes depends on the adherin complex, which is composed of two proteins, SCC2 (also named NIPBL/Nipped B/delangin) and SCC4 (MAU2, Figure 1). However, this loading is not sufficient for correct cohesive function and requires the participation of ESCO1/2 acetyltransferases for effective cohesion. The substrate of this acetylation is the SMC3 subunit of the cohesin complex (Figure 1). Others cohesin cofactors are PDS5 (precocious dissociation of sisters), WAPL (wings apart-like) and Sororin, which are implicated in the maintenance and/or removal of cohesins from chromatin (Figure 1). Although it is necessary to mention the role of these cohesin regulators, this is not the principal focus of this review, and there are already at least two excellent published reviews offering a detailed description of the function and molecular mechanisms of cohesin regulators.1,2
Table 1.
Subunits of cohesin complexes and cohesin regulators implicated in cohesinopathies
| Saccharomyces cerevisiae | Schizosaccharomyces pombe | Drosophila | Xenopus | Mammals | |
|---|---|---|---|---|---|
| Structural maintenance of chromosomes (SMC) | Smc1 | Psm1 | Smc1 | XSMC1 | SMC1α |
| Smc3 | Psm3 | Cap/Smc3 | XSMC3 | SMC3 | |
| Meiosis-specific | SMC1β | ||||
| α-kleisin (from Greek for “closure”) | Mcd1/Scc1 | Rad21 | DRad21 | XRAD21 | RAD21 |
| Meiosis-specific | Rec8 | Rec8 | XREC8 | REC8, RAD21L | |
| Stromalin/HEAT repeat domain | Scc3/IRR1 | Psc3 | DSA1 | XSA1, XSA2 | STAG1, STAG2 |
| Adherin (loading) | Scc2 | Mis4 | Nipped-B | Nipbl/Scc2/delangin | NIPBL/SCC2/DELANGIN |
| Scc4 | Ssl3 | Mau2/Scc4 | MAU2/SCC4 | ||
| Regulator (maintenance) | Pds5 | Pds5 | Pds5 | PDS5A | PDS5A |
| PDS5B/AS3/Aprin | PDS5B/AS3/APRIN | ||||
| Cohesin acetyltransferase (establishment) | Eco1/Ctf7 | Eso1 | Eco/Deco San |
Esco1 Esco2 |
ESCO1 ESCO2 |
| Cohesin deacetylase | Hos1 | ? | ? | Hdac8 | HDAC8 |
Figure 1.
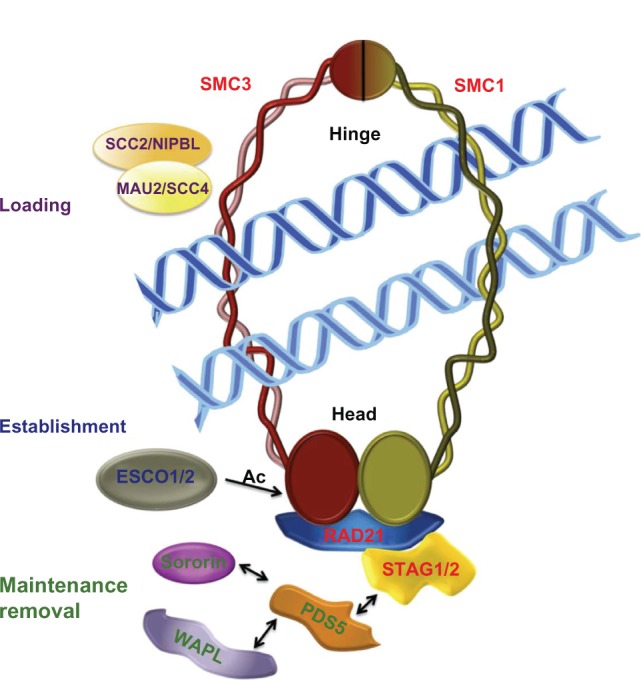
Cohesin complex and cohesin regulators.
Notes: Ring-embraced model of cohesion. The cohesin complex is formed by four proteins, ie, SMC1, SMC3, SCC1/RAD21, and SCC3/STAG. The SMC1, SMC3, and RAD21/SCC1 subunits form a ring-like structure. RAD21/SCC1 is the subunit that closes the ring formed by SMC (structural maintenance of chromosomes) subunits and has been named α-kleisin (closure). The SCC3/STAG protein interacts with RAD21/SCC1 to complete the cohesin complex. The adherin loading complex consists of two proteins, ie, SCC2/NIPBL and MAU2/SCC4. The acetyltransferases (ESCO1/2) acetylate the SMC3 cohesin subunit to establish cohesion of the chromatids. The cofactors, PDS5, wAPL, and Sororin, are involved in association and/or dissociation with chromatin.
In the last few years, new functions of the cohesin complex have emerged in terms of genome structure and dynamics. The requirement for cohesin subunits and regulators in repair of DNA damage in different models has been extensively reported.3–6 Alteration of these two essential cell life processes by defects in the cohesin network may underlie chromosomal instability and aneuploidy, which are two principal hallmarks of tumor formation and progression.7 One of the most surprising findings was the discovery in 2004 of a link between cohesin gene mutations and human developmental abnormalities.8–10 Four years later, three research groups reported results indicating the molecular mechanisms by which cohesin metabolism could be related to development control. Taking into account the fact that cohesins are also expressed in post-mitotic cells, different groups have investigated these newly discovered cohesin/chromatin interactions. The search for cohesin binding sites in the human genome showed overlap with CCCTC binding factor (CTCF) sites.11–13 CTCF is an insulator protein that participates in blocking enhancer-promoter interaction. These works pioneered the study of the function of this newly identified cohesin, suggesting involvement of cohesin in regulation of gene expression. The present review focuses on the links between cohesin networks and the genetic and molecular basis of human diseases now known as cohesinopathies.
Gene mutations in the cohesin network and human developmental disorders
Mutations in the cohesin complex subunits, or cofactors, which participate in cohesin ring dynamics, were described some years ago as the main genetic factors triggering a variety of rare human diseases, known as cohesinopathies. The two best described cohesinopathies are Cornelia de Lange syndrome (CdLS, OMIM 122470, 300590, 610759, 614701, 300882) and Roberts syndrome (and its variant SC phocomelia, OMIM 268300). CdLS is an autosomal dominant multiple neurodevelopmental disorder, with an estimated occurrence of one in every 10,000. It is characterized by mental retardation, facial dysmorphism, upper limb abnormalities, and growth delay. Roberts syndrome is an autosomal recessive disorder related phenotypically to CdLS, with affected patients having craniofacial abnormalities, limb reduction, and growth retardation.
Cornelia de Lange syndrome
Patients with CdLS have mutations in the gene encoding SCC2/NIPBL, a protein in the adherin complex required for loading of the cohesin complex to chromatin (Figure 2).8,9 In situ analyses of NIPBL expression in mouse embryos showed accumulation in the limb buds, branchial arch, and craniofacial mesenchyme.8 Similar experiments in human embryonic tissue sections revealed NIPBL expression in the primordial cartilage of the ulna and various hand bones.9 The investigators performing these studies also observed expression of this gene in craniofacial tissue, the spinal column, and the heart, with defects clearly related to the CdLS phenotype. In 2009, Kawauchi et al reported generation of mice heterozygous for a gene-trap mutation of the first coding exon of the NIPBL gene.14 This mouse model showed many features of CdLS, including growth retardation, cardiac defects, delayed bone development, and hearing deficits, providing the first experimental evidence that NIPBL mutations are the cause of CdLS. SCC2/NIPBL/delangin is a protein with two major isoforms, ie, delangin A (long) and delangin B (short). Both have the first 2683 amino acid residues in common, but have different carboxy-terminal tails containing 121 amino acids (long) and 14 amino acids (short).9 NIPBL contains a predicted nuclear localization sequence (residues 1108–1124) and five HEAT (protein interaction) repeat regions approximately spanning residues 1750–2350. NIPBL is able to mediate local chromatin modifications by recruitment of histone deacetylases and a large number of the CdLS mutations found in this protein that dramatically affect its function. These results suggest a contribution to the syndrome distinct from its canonical role in loading cohesin complexes to chromatin.15
Figure 2.
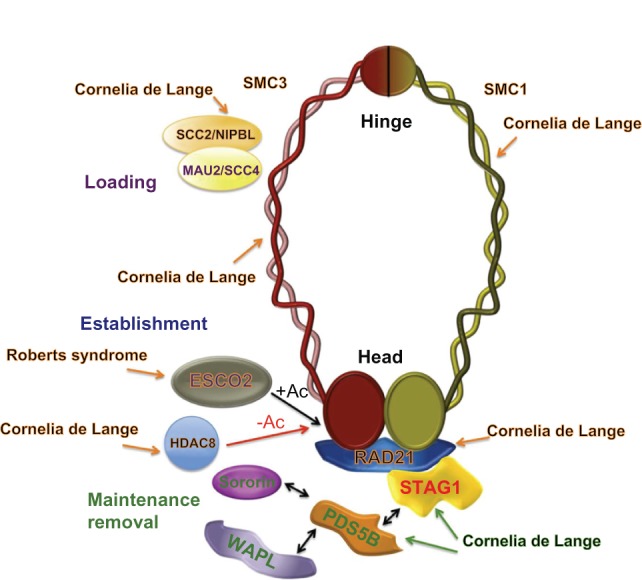
Cohesin and cohesin regulators in human cohesinopathies.
Notes: Gene mutation occurs in human CdLS and RBS (orange arrows), and results in mouse models suggest involvement of cohesin and cohesin regulator genes in these human disorders (green arrows). Mutations in the SCC2/NIPBL gene cause the most severe phenotype in patients with Cornelia de Lange syndrome. Mutations in genes encoding for the cohesin subunits SMC1α, SMC3, and RAD21, and for the histone deacetylase, HDAC8, cause a mild variant of this syndrome. Although the phenotype of mice lacking PDS5A and/or PDS5B function show developmental abnormalities like those in patients with Cornelia de Lange syndrome, currently there are no data supporting mutations of these genes as a cause of this cohesinopathy in humans. Similarly, to the author’s knowledge, no data have been reported on STAG1 mutations in patients with Cornelia de Lange syndrome. A human ortholog of yeast Eco1 named ESCO2 is mutated in Roberts syndrome, which has a phenotype closely related to that seen in Cornelia de Lange syndrome.
Abbreviation: Ac, Acetylation.
After these early findings, numerous studies have been done in patients to identify other mutations in proteins related to the dynamics of cohesin. Reports of several cases of this syndrome show that mutations in the SMC1α and SMC3 cohesin subunits cause a mild variant of CdLS.16,17 The disorder triggered by mutations in the human gene encoding the SMC1α cohesin subunit is frequently denominated as X-linked CdLS because the human homolog of yeast Smc1, known as SMC1L1, is located in the human X chromosome.16 Later, a single mutation was identified in human SMC3 in one of 96 probands tested.17 In CdLS patients with mutated SMC1L1 and SMC3, a milder phenotype characterized by less significant psychomotor and growth retardation, a lower incidence of major malformations, and milder limb anomalies has been reported. Because SMC1 and SMC3 are the cohesin subunits mostly involved in formation of the ring-like structure through their hinge and head domains (Figure 1), Revenkova et al studied the affinity of mutated SMC1α and SMC3 proteins for DNA, focusing on dimeric hinge domains where several CdLS mutations clustered. They found that SMC1α and SMC3 mutations mapping to the hinge domains produced higher binding affinity between the cohesin complex and DNA than wild-type proteins in vitro, suggesting modified dynamics of chromatin association-dissociation for mutated cohesin complexes.18
To identify additional potential causal loci for CdLS, Deardorff et al performed a genome-wide array-based copy number analysis for 101 individuals with typical CdLS and 189 with overlapping features who were negative for mutations in previously reported NIPBL, SMC1L1, and SMC3 genes associated with CdLS.19 They identified one boy with a 8q24.1 interstitial microdeletion that included RAD21, which encodes the kleisin subunit that interacts with the other three subunits, SMC1α, SMC3, and STAG/SCC3, as well as with other regulatory proteins to maintain the ring-like structure of the cohesin complex (Figures 1 and 2). In addition, these authors identified two additional probands with atypical clinical features which demonstrated heterozygous de novo RAD21 missense mutations (c.1127C > G [p.Pro376 Arg] and c.1753T > C [p.Cys585 Arg], respectively).
Therefore, until now, mutations in three cohesin subunits (SMC1α, SMC3, RAD21) and in one cohesin-interacting protein (NIPBL) have been found in patients with CdLS (Figure 2). However, there are results suggesting the possibility that mutations in other cohesins and cohesin regulators could also contribute to this syndrome in humans. In vertebrate somatic cells, there are two cohesin complexes that differentiate in the STAG subunit, consisting of SMC1α, SMC3, RAD21/SCC1, and either STAG1 or STAG2.20–22 Recently, Remeseiro et al studied the phenotypic characteristics of mouse embryos lacking STAG1 function.23 They found that loss of STAG1 function alters transcription of genes related to the CdLS phenotype, and hypothesized that impaired STAG1 function in gene expression underlies the molecular etiology of CdLS.
PDS5 is a regulatory component in the cohesin complex that interacts with the cohesin subunits and other cohesin cofactors (Figure 1).24 Vertebrates have two homologs of PDS5, ie, PDS5A and PDS5B, both of which contribute to the dynamics of cohesion. Mice that lack PDS5B have multiple developmental anomalies that resemble those found in humans with CdLS and die shortly after birth,25 whereas Pds5B−/− mouse cells show no defects in sister chromatid cohesion. In addition, Pds5A deficiency results in developmental abnormalities similar to those seen in Pds5B knockout mice.26 Although these results, reported in both PDS5 knockout models, suggest the involvement of these proteins in CdLS, a recent study looking for mutations in the PDS5A gene in 137 Italian patients with CdLS, who did not have mutations in NIPBL, SMC1L1, or SMC3, was negative,27 and mutations in PDS5A and/or PDS5B genes have not been reported in human patients with CdLS.
Roberts syndrome/SC phocomelia
Mutations in the gene encoding for human ESCO2, a homolog to yeast Eco1 acetyltransferase, were found to be responsible for the Roberts syndrome and its variant, SC phocomelia in humans (Figure 2).10 Cells from patients with Roberts syndrome show a lack of cohesion at the heterochromatic regions around centromeres and in the long arm of the Y chromosome.10 The sequence of human ESCO2 contains 601 amino acid residues, including two well conserved regions, a Zn finger-like motif and a conserved C-terminal acetyltransferase domain. Different mutations in the exons and introns of the human Esco2 gene have been reported in individuals with Roberts syndrome.28 Most, if not all, of these mutations cause loss of acetyltransferase activity of ESCO2, providing the first indication that loss of acetyltransferase function contributes to the pathogenesis of Roberts syndrome.29 Similarly to STAG and PDS5 proteins, ESCO acetyltransferases consist of two members, ie, ESCO1 and ESCO2, in reptiles, birds, fish, and mammals. Although ESCO1 mutations have not been reported until now in patients with Roberts syndrome, Slavin et al described a case with a phenotype of hemivertebral and rib anomalies. This individual showed a karyotype of 47 chromosomes and genes in the triplicated region, possibly contributing to her skeletal phenotype, including GATA6, MC2R, MC5R, RBBP8, ESCO1, and ROCK1 among others, and implicating the second acetyltransferase member, ESCO1, in her skeletal anomalies.30
The function of ESCO acetyltransferases is reversed by deacetylases. In humans, this deacetylase has been recently identified as HDAC8 (Figure 2), which is responsible for deacetylation of the SMC3 cohesin subunit to facilitate renewal of the cohesin complex after its dissociation from chromatin during prophase or anaphase.31 In this study, the authors screened this X-linked gene in 154 individuals with CdLS negative for mutations in NIPBL, SMC1a and SMC3, RAD21, STAG2, ESCO1, ESCO2, and SCC4. Four missense mutations and one nonsense mutation in HDAC8 were identified, indicating that loss of HDAC8 activity results in decreased cohesin at specific sites, causing clinical features of CdLS.
Link between genotype and phenotype in cohesinopathies
Cohesin complex, insulator CTCF factor, and transcription regulation
The earliest results connecting the dynamics of the cohesin complex with control of gene expression date back to 1999, when a study by Rollins et al of the Nipped loci from Drosophila showed that Nipped-B (the Drosophila homolog of human NIPBL) mutations also magnify the effect of a gypsy transposable element insertion in the Ultrabithorax gene.32 Later, the same researchers found that, in contrast with the effect of Nipped-B, the Scc3 component of the cohesin complex inhibits long-range activation of the cut gene in Drosophila.33 Further, in human cells, STAG2 coactivates a multimeric NF-kB reporter construct and enhances activity of the p65/RelA transactivation domain.34 Three independent research groups have studied the localization of cohesin in mammalian chromosomes and found that numerous cohesin binding sites overlap with CTCF, an insulator protein that participates in blocking enhancer-promoter interactions (Figure 3A). Cohesins are also required for the CTCF insulation function and for control of H19/IGF2 locus imprinting in the G2 and G1 phases in mice. In G1 phase, there is no cohesion of sister chromatids, so the role of cohesins in the control of H19/IGF2 transcription is independent of their function in the cohesion of sister chromatids.11–13 These were the initial works suggesting a link between a new cohesin function and the molecular causes of cohesinopathies. Since then, a large body of research has confirmed this newly identified role of cohesin. The insulation-mediated CTCF/cohesin complex maintains formation of the chromatin loop and localization of transcriptional apparatus for promoters in the human apolipoprotein gene cluster.35
Figure 3.

Models for a chromatin architectural function of cohesin in control of gene expression. (A) Interaction of the cohesin complex with chromatin-bound CTCF maintains a chromatin structure in which the enhancer cannot interact with the promoter and behaves as an insulator, repressing gene expression. (B) Loop structure formed by the mediator/cohesin complex allows enhancer-promoter interactions promoting gene transcription.
Chromatin immunoprecipitation (ChIP) experiments have been very useful for identification of genes controlled by cohesin. The fact that RAD21 is recruited to CTCF sites in all Ig loci during development of B lymphocytes suggests that the cohesin/CTCF complex may promote formation of multiple loops and thus effective recombination of V(D)J.36 Liu et al, in a study of 16 mutant cell lines from severely affected CdLS probands, identified specific gene expression profiles for CdLS.37 The cohesin binding sites were significantly reduced in CdLS samples with mutant SCC2/NIPBL. However, of the altered control genes in the CdLS samples, about 60% were upregulated and 40% were downregulated, suggesting that SCC2/NIPBL and cohesin have both negative and positive effects on expression. In the last two years, a large number of results have been reported illustrating the role of the CTCF/cohesin insulation complex in the control of expression of distinct differentiation and developmental genes in the immune system, stem cells, and germ cells.38,39 Recently, in a transcriptomic analysis of marker genes for differences in intellectual disability found in patients with Down syndrome, Mégarbané et al showed that genes of major histocompatibility complex class II (HLA DQA1 and HLA DRB1) were significantly downregulated in patients with a low intelligence quotient.40 The intergenic region between these genes contains a consensus binding sequence for the CTCF/cohesin complex, indicating that regulation of gene expression by cohesin would extend beyond CdLS and Roberts syndrome.
Cohesin complex and CTCF-independent gene regulation
Initial results on this emergent cohesin function suggested a requirement for interaction between the cohesin complex and the CTCF homodimer (Figure 3A) to form a new cohesin/CTCF complex for control of cohesin-mediated gene expression, but later studies showed that the cohesin complex is also able to regulate the transcription of several genes by interacting with a specific transcription factor in a manner independent of CTCF.
Two laboratories have shown that the cohesin complex is required for morphogenesis of nondividing neurons in Drosophila. Using a modified piggyBac vector for insertional mutagenesis in a genetic screening41 and by generation of flies expressing a modified version of cohesin subunit RAD2142 (named RAD21TEV), they found that cohesin is essential for axon pruning. Looking for genes regulated by cohesin in this context, they found that expression of the steroid hormone receptor EcR gene, which encodes EcR-B1 protein, an essential regulator of axon pruning in the mushroom body, is reduced in SMC1α-depleted γ-neurons and that this pruning defect is reversed by overexpression of EcR-B1. In this case, the results indicated that cohesins facilitated EcR transcription. Further, zebrafish embryos lacking cohesin subunit RAD21 or SMC3 do not express runx3 and lose runx1 expression in early embryonic development.43 This positive regulation is the opposite of the effect previously described for cohesin function in insulators, but is similar to that previously reported for SA2/STAG2.34
Using (ChIP-seq) assays in human breast cancer (MCF-7) cells, Schmidt et al identified thousands of genomic sites that share cohesin and estrogen receptor-α yet lack CTCF binding.44 Further, estrogen-regulated genes are preferentially bound by both estrogen receptor-α and cohesin, and the silencing of cohesin causes aberrant re-entry of breast cancer cells into the cell cycle after hormone treatment. In a human hepatocellular carcinoma (HepG2) cell line, liver-specific transcription factors colocalized with cohesin independently of CTCF at liver-specific targets. These results show for the first time that the cohesin complex plays a role in estrogen-regulated gene expression independently of CTCF by interacting with specific transcription factors.45
Gene activation may involve a specific conformation of chromatin to facilitate interactions between enhancer-bound transcription factors/transcriptional coactivators and the transcription apparatus at the promoter. A multisubunit protein complex known as Mediator interacts with the RNA polymerase II enzyme and control of gene expression.45 ChIP-seq results showed that Mediator, cohesin, and the cohesin loading factor, SCC2/NIPBL, co-occupy thousands of sites in the embryonic stem cell genome. To confirm interactions between Mediator and the cohesin complex, Kagey et al, using antibodies against Mediator subunits (Med1, Med12) and cohesin (SMC1α, SMC3), coprecipitated both Mediator and cohesin subunits. Further, an antibody against NIPBL also coprecipitated both cohesin and Mediator subunits.46 These results confirm that Mediator, cohesin, and NIPBL interact, and also suggest that a Mediator/cohesin complex would function as a chromatin architect, maintaining the DNA loop at specific chromosome sites to activate gene expression (Figure 3B). Therefore, involvement of the cohesin complex in the regulation of positive and negative transcription points to the function of cohesin in gene expression being gene-dependent.
Fish models of cohesinopathies
Two zebrafish (Danio rerio) models with deficiency in nipbl47 and esco248 have been generated to explore the molecular mechanisms causing the phenotypes observed in CdLS and Roberts syndrome. Using morpholino knockdown for the two nipbl genes in zebra fish, ie, nipbla and nipblb, it was found that a deficiency of these genes produces a spectrum of specific heart and gut/visceral organ defects with similarities to those found in CdLS. In addition, reduced nipbl function has quantitative effects on the expression of multiple genes also contributing to the appearance of developmental defects.47
Zebrafish embryos deficient for esco2 have phenotypes similar to those in humans with Roberts syndrome.48 Esco2-depleted fish have craniofacial abnormalities characterized by missing cartilaginous elements along with truncated pectoral fins, much like the phocomelia observed in patients with Roberts syndrome. Cells in esco2-mutant fish have mitotic defects characterized by disorganized chromosomes spread throughout the cell. This observation is consistent with the precocious separation of sister chromatids observed in cells of patients with Roberts syndrome.10 However, fish mutant for esco2 do not show other anomalies typical of Roberts syndrome. Ossification is normal, indicating that the developmental pathways for bone are intact in these embryos. High levels of cell death contribute to the morphology of esco2-depleted embryos without affecting specific developmental pathways, and it is suggested that cell proliferation defects and apoptosis could be the primary cause of the features seen in Roberts syndrome.
On the other hand, similar research in the medaka (Oryzias latipes) model of Roberts syndrome shows that mutation of esco2 affects expression of several genes.49 In this study, the investigators demonstrated that an esco2 mutation (R80S) actually suppresses expression of notch1a (a neural development marker) and its downstream genes, as well as notch3 (a vascular differentiation marker), suggesting an important effect of esco2 mutations on expression of developmental genes in Roberts syndrome.
These results show that mutations in different elements of the cohesion apparatus have distinct developmental outcomes, and suggest that different processes, dysregulation of gene expression, cell proliferation defects, and apoptosis could contribute to the anomalies seen in Roberts syndrome.
Concluding remarks
The cohesin complex, initially characterized as a ringlike protein complex that maintains cohesion between sister chromatids during segregation of chromosomes, is now considered an essential component of the family of modulators of chromatin structure during different dynamic processes in chromosomes. In many cases, these processes aim to safeguard the stability of genetic material and its proper distribution to daughter cells, and for those related to aneuploidy, genomic instability, and cancer (Figure 4). It is known that cohesins are also involved in control of gene expression, so it is not surprising that when there are errors or problems with the cohesin complex related to its role in transcription, one of the likely results is abnormal development of the organism.
Figure 4.
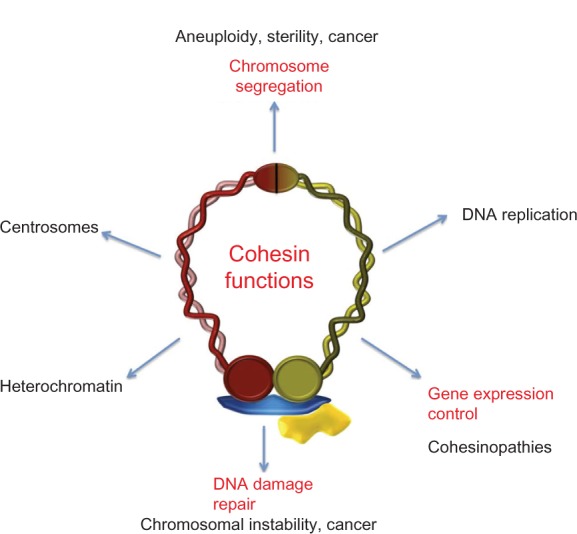
Cohesin metabolism and human disease.
Notes: Scheme of the main life processes of the cell in which cohesins have been functionally characterized and related diseases. The role of cohesin in control of gene expression (in red) is the principal focus of this review.
In recent years, an increasing body of scientific research has shown that the functions of cohesin are mediated by the actions of other proteins. These molecules, such as SCC2/SCC4, ESCO, HDAC8, PDS5, WAPL, and Sororin, may be necessary for the multiple functions of cohesin, or they may contribute to one specific role by spatial-temporal interaction with the cohesin complex, eg, CTCF and Mediator, in the control of gene expression. Specific post-translational modifications of cohesin subunits, such as acetylation, phosphorylation, and sumoylation, are also required for particular functions of cohesin, suggesting the existence of a cohesin code.
These findings point to the denomination of cohesin being currently partial and incomplete, and perhaps the term “chromosome structure architects” or “architectins” would be more appropriate. Therefore, interactions between the cohesin complex and specific regulator proteins model precise tridimensional structures at local regions of chromatin to perform different and specific functions depending on spatiotemporal requirements. This architectural function of cohesin would modulate the interaction of positive or negative regulatory elements in gene transcription during development, providing a molecular link between genotype and phenotype for the cohesinopathies discussed in this paper. Future research on the molecular mechanisms triggering alterations in the cohesin network in pathological conditions is crucial for determining the genetic and molecular causes of these neurodevelopmental disorders in humans, currently known as cohesinopathies.
Acknowledgments
This work was supported by the Spanish Ministerio de Ciencia e Innovación (BFU2009-08975/BMC) and Fundación Mutua Madrileña (AP98712012). Apologies are extended to all colleagues whose contributions could not be recognized for space reasons.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Haering CH, Jessberger R. Cohesin in determining chromosome architecture. Exp Cell Res. 2012;318:1386–1393. doi: 10.1016/j.yexcr.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 2.Xiong B, Gerton JL. Regulators of cohesin network. Annu Rev Biochem. 2010;79:131–153. doi: 10.1146/annurev-biochem-061708-092640. [DOI] [PubMed] [Google Scholar]
- 3.Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 4.Xu H, Balakrishnan K, Malaterre J, et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS One. 2010;5:e12112. doi: 10.1371/journal.pone.0012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heidinger-Pauli JM, Unal E, Koshland D. Distinct targets of the Eco1 acetyltransferase modulate cohesion in S phase and in response to DNA damage. Mol Cell. 2009;34:311–321. doi: 10.1016/j.molcel.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lightfoot J, Testori S, Barroso C, Martinez-Perez E. Loading of meiotic cohesin by SCC-2 is required for early processing of DSBs and for the DNA damage checkpoint. Curr Biol. 2011;21:1421–1430. doi: 10.1016/j.cub.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Barbero JL. Sister chromatid cohesion control and aneuploidy. Cytogenet Genome Res. 2011;133:223–233. doi: 10.1159/000323507. [DOI] [PubMed] [Google Scholar]
- 8.Krantz ID, McCallum J, DeScipio C, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 10.Vega H, Waisfsz Q, Gordillo M, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005;37:468–470. doi: 10.1038/ng1548. [DOI] [PubMed] [Google Scholar]
- 11.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wendt KS, Yoshida K, Itoh T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 13.Parelho V, Hadjur S, Spivakov M, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Kawauchi S, Calof AL, Santos R, et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahnke P, Xu W, Wülling M, et al. The cohesin loading factor NIPBL recruits histone deacetylases to mediate local chromatin modifications. Nucleic Acids Res. 2008;36:6450–6458. doi: 10.1093/nar/gkn688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musio A, Selicorni A, Focarelli ML, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 17.Deardorff MA, Kaur M, Yaeger D, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Revenkova E, Focarelli ML, Susani L, et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum Mol Genet. 2009;18:418–427. doi: 10.1093/hmg/ddn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deardorff MA, Wilde JJ, Albrecht M, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. 2012;90:1014–1027. doi: 10.1016/j.ajhg.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carramolino L, Lee BC, Zaballos A, et al. SA-1, a nuclear protein encoded by one member of a novel gene family: molecular cloning and detection in hemopoietic organs. Gene. 1997;195:151–159. doi: 10.1016/s0378-1119(97)00121-2. [DOI] [PubMed] [Google Scholar]
- 21.Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3 subunits in the Xenopus and human cohesin complexes. J Cell Biol. 2000;150:405–416. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumara I, Vorlaufer E, Gieffers C, Peters BH, Peters JM. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol. 2000;151:749–762. doi: 10.1083/jcb.151.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Remeseiro S, Cuadrado A, Gómez-López G, Pisano DG, Losada A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 2012;31:2090–2102. doi: 10.1038/emboj.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losada A, Yokochi T, Hirano T. Functional contribution of Pds5 to cohesin-mediated cohesion in human cells and Xenopus egg extracts. J Cell Sci. 2005;118:2133–2141. doi: 10.1242/jcs.02355. [DOI] [PubMed] [Google Scholar]
- 25.Zhang B, Jain S, Song H, et al. Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development. 2007;134:3191–3201. doi: 10.1242/dev.005884. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B, Chang J, Fu M, et al. Dosage effects of cohesin regulatory factor PDS5 on mammalian development: implications for cohesinopathies. PLoS One. 2009;4:e5232. doi: 10.1371/journal.pone.0005232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliver C, Bedeschi MF, Blagowidow N, et al. Cornelia de Lange syndrome: extending the physical and psychological phenotype. Am J Med Genet. 2010;152A:1127–1135. doi: 10.1002/ajmg.a.33363. [DOI] [PubMed] [Google Scholar]
- 28.Van Den Berg DJ, Francke U. Roberts syndrome: a review of 100 cases and a new rating system for severity. Am J Med Genet. 1993;47:1104–1123. doi: 10.1002/ajmg.1320470735. [DOI] [PubMed] [Google Scholar]
- 29.Gordillo M, Vega H, Trainer AH, et al. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet. 2008;17:2172–2180. doi: 10.1093/hmg/ddn116. [DOI] [PubMed] [Google Scholar]
- 30.Slavin TP, Kuruvilla K, Curtis CA, Christ LA, Mitchell AL. Isolated skeletal malformations in a child with a small mosaic ring microduplication of 18 p11.21q11.2: genotype-phenotype correlations. Am J Med Genet. 2011;155A:618–621. doi: 10.1002/ajmg.a.33816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deardorff MA, Bando M, Nakato R, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rollins RA, Morcillo P, Dorsett D. Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics. 1999;152:577–593. doi: 10.1093/genetics/152.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rollins RA, Korom M, Aulner N, Martens A, Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol Cell Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lara-Pezzi E, Pezzi N, Prieto I, et al. Evidence of a transcriptional co-activator function of cohesin STAG/SA/Scc3. J Biol Chem. 2004;279:6553–6559. doi: 10.1074/jbc.M307663200. [DOI] [PubMed] [Google Scholar]
- 35.Mishiro T, Ishihara K, Hino S, et al. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28:1234–1245. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degner SC, Wong TP, Jankevicius G, Feeney AJ. Cutting edge: developmental stage-specific recruitment of cohesin to CTCF sites throughout immunoglobulin loci during B lymphocyte development. J Immunol. 2009;182:44–48. doi: 10.4049/jimmunol.182.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Zhang Z, Bando M, et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7(5):e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herold M, Bartkuhn M, Renkawitz R. CTCF: insights into insulator function during development. Development. 2012;139:1045–1057. doi: 10.1242/dev.065268. [DOI] [PubMed] [Google Scholar]
- 39.Holwerda S, de Laat W. Chromatin loops, gene positioning, and gene expression. Front Genet. 2012;3:217. doi: 10.3389/fgene.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mégarbané A, Noguier F, Stora S, et al. The intellectual disability of trisomy 21: differences in gene expression in a case series of patients with lower and higher IQ. Eur J Hum Genet. 2013 Feb 20; doi: 10.1038/ejhg.2013.24. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuldiner O, Berdnik D, Levy JM, et al. piggyBac-based mosaic screen identifies a postmitotic function for cohesin in regulating developmental axon pruning. Dev Cell. 2008;14:227–238. doi: 10.1016/j.devcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pauli A, Althoff F, Oliveira RA, et al. Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev Cell. 2008;14:239–251. doi: 10.1016/j.devcel.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horsfield JA, Anagnostou SH, Hu JK, et al. Cohesin-dependent regulation of Runx genes. Development. 2007;134:2639–2649. doi: 10.1242/dev.002485. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt D, Schwalie PC, Ross-Innes CS, et al. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20:578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taatjes DJ. The human mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci. 2010;35:315–322. doi: 10.1016/j.tibs.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2011;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muto A, Calof AL, Lander AD, Schilling TF. Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of Cornelia de Lange Syndrome. PLoS Biol. 2011;9:e1001181. doi: 10.1371/journal.pbio.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mönnich M, Kuriger Z, Print CG, Horsfield JA. A zebrafish model of Roberts syndrome reveals that Esco2 depletion interferes with development by disrupting the cell cycle. PLoS One. 2011;6:e20051. doi: 10.1371/journal.pone.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morita A, Nakahira K, Hasegawa T, et al. Establishment and characterization of Roberts syndrome and SC phocomelia model medaka (Oryzias latipes) Dev Growth Differ. 2012;54:588–604. doi: 10.1111/j.1440-169X.2012.01362.x. [DOI] [PubMed] [Google Scholar]