

Abstract
Large-scale cancer genomics, proteomics and RNA-sequencing efforts are currently mapping in fine detail the genetic and biochemical alterations that occur in cancer. However, it is becoming clear that it is difficult to integrate and interpret these data and to translate them into treatments. This difficulty is compounded by the recognition that cancer cells evolve, and that initiation, progression and metastasis are influenced by a wide variety of factors. To help tackle this challenge, the US National Cancer Institute Physical Sciences-Oncology Centers initiative is bringing together physicists, cancer biologists, chemists, mathematicians and engineers. How are we beginning to address cancer from the perspective of the physical sciences?
There is a rich history of the physical sciences contributing to cancer research and treatment. Max Delbrück, a physicist, was one of the pioneers of molecular genetics. In collaboration with the biologist Salvador Luria, he showed that phage resistance in a population of bacteria is caused by random mutations. The equations they developed to model this process are still used to predict how cancers gradually become resistant to chemotherapy. Francis Crick and Maurice Wilkins, two physicists, the biologist James Watson and the chemist Rosalind Franklin discovered the structure of DNA, and thus laid the foundation for cancer genomics and much of contemporary biology. Indeed, concepts from other fields as wide-ranging as agriculture (the seed-and-soil hypothesis1), developmental biology (Folkman’s antiangiogenesis strategy2) and mathematics (Nowell’s clonal evolution model3 and the multistep theory of tumorigenesis4,5) are at the core of cancer biology.
Treatments have also been influenced by the physical sciences. Chemotherapy began in chemistry laboratories, in which chemists sought to develop new dye molecules. Radiation oncology, which is a cornerstone of cancer therapy, originated from basic physical chemistry research. Physics and mathematics are central to designing the accelerators that are used to generate radiation and the algorithms that are used to determine where the radiation should be delivered and how much radiation should be used. Most recently, the availability of fairly inexpensive high-throughput sequencing is making it possible to contemplate highly personalized cancer therapies, in which patients are treated with drug regimens that are specifically tailored to their disease. In addition to laying the foundation for new, personalized treatments, these large-scale sequencing efforts have also helped scientists to delineate the enormous complexity of the disease and the degree to which signalling, drug resistance and genomic alterations vary from patient to patient and even within one patient.
This new vista of cancer in all its heterogeneity and complexity suggests additional ways in which the physical sciences can assist cancer researchers and clinicians. For decades, physical scientists have been grappling with systems that are composed of many interacting parts and that exhibit considerable local variation, much like tumours in individual patients. Entire scientific fields, such as the study of superconductivity and the fractional quantum hall effect, are devoted to understanding the unexpected things that can happen when large numbers of simple pieces interact. It is very difficult, or it may even be impossible, to predict the aggregate behaviour of these systems even if all the laws that are relevant to each constituent are known. Ultimately, physical scientists were forced to invent a completely new set of theoretical and computational tools, such as the Monte-Carlo method, to explore and to simulate systems with many coupled degrees of freedom.
Cancer is perhaps such a system. It has now become clear that cancer is not a strictly deterministic disease that progresses through a simple, fixed succession of specific mutations in two or three genes. Rather, there are many molecularly distinct routes to clinically identical cancers, and the final development of malignancy is influenced by a multitude of factors, encompassing the immune system, ageing, nutrition and microenvironmental details within particular tissues. Like other emergent phenomena, cancer cannot be readily understood by merely characterizing all its components. Developing a fundamental understanding of cancer that recognizes and embraces the great heterogeneity of tumours and their emergent properties may benefit from integrated teams of physicists, cancer biologists, mathematicians and engineers.
In this Review, we provide examples from four broad areas to illustrate the idea of the physical sciences contributing to cancer biology. These four areas have well-established clinical relevance, and in each of these areas there is also evidence, owing to decades of preceding research, that physics, mathematics, chemistry and engineering are able to contribute, sometimes decisively, to breakthroughs in cancer research. These areas are cancer mechanics, cancer evolution, information coding and decoding, and transport and delivery in cancer.
A physicist’s view of cancer mechanobiology
Since Egyptian times6, physicians have noted that tumours are typically harder than the tissue that surrounds them. This observation gave rise to the word oncology (from the Ancient Greek ‘onkos’, which means ‘a mass’) and continues to be widely used to detect cancers. However, the connections between tissue mechanics, cancer progression and patient outcomes are only now being established7–9. Much of what we know about the role of mechanics in biological function (and dysfunction) comes from studies of organ development10 and the investigation of clinical specimens with new tools such as magnetic resonance elastography11 and highly sensitive tissue indenters12. Tensile forces within developing organs are master regulators of cell sorting and packing, thereby specifying overall tissue architecture. For example, differential cell cortex tension is a key factor in progenitor cell sorting and thus germ-layer organization13. Tensile forces arise from cell–cell and cell–matrix adhesion, surface tension, and intracellular molecular machines and cytoskeletal elements. From the interplay of cell mechanics and geometrical constraints, constructed by the gene expression of cytoskeletal elements and adhesion complexes, emerge the approximately 250 distinct cell shapes and sizes found in the human body.
Once a tissue has formed, it remains sensitive to alterations to the shape and mechanics of all its constituents. Cells change their shape when the subtle balance of forces that define their shape is modified — this is analogous to how a small stumble can immediately alter, and can quickly end, a game of tug-of-war. When changes of cell shape and mechanics spread in a tissue, as is the case in cancer, the organization and shape of the entire tissue is necessarily altered. Communications between and among cells are mediated through cell surface receptors and a network of signal transduction reactions. Mechanical forces actively alter large-scale spatial organization of signalling molecules14, providing a mechanism for physical forces to directly regulate chemical signal transduction processes. These, in turn, can activate or repress genes, modifying cell and extracellular matrix mechanics, and so on.
The physics of soap bubbles is a simple starting point for thinking about how cells may change their shape during tumour progression. Single soap bubbles are spherical, the one shape that minimizes their surface area and thus their elastic energy. For bubbles, the principle of energy minimization is equivalent to surface area minimization15. This makes it possible to precisely calculate the shapes of collections of soap bubbles. Developmental biologists have recently discovered intriguing similarities between soap bubble configurations and growing and migrating cells. For example, surface mechanics seem to mediate pattern formation in the developing Drosophila melanogaster retina16 (FIG. 1). Despite the enormous obvious differences between soap bubbles and living cells, simple calculations and simulations invoking only cell–cell adhesion, cell contractility and energy minimization reproduce the intricate six-cell ommatidium cell clusters that are found in the D. melanogaster retina17,18. The key equation used in this approach (FIG. 1a) is worthy of discussion. The first term comes straight out of a physics textbook — from elasticity theory. The second and third terms relate to the levels and effective stickiness of N-cadherins and E-cadherins. These two terms blend at least five broad areas of science: genomics, cancer biology, thermodynamics, soft condensed matter physics, and structural and membrane biology. Taken together, this then yields the characteristic six-cell clusters of the D. melanogaster retina17,18. This example illustrates the power of bringing together the physical sciences and biology. It is tempting to speculate, but by no means proved, that similar integrated approaches will help to reveal why particular changes to cell and tissue architecture are so useful for detecting, identifying and staging cancer (FIG. 1b).
Figure 1. Similarities among soap bubbles, cells in the Drosophila Nature Reviews eye and loss of Cancer tissue organization in cancer.

The normal ommatidium structure within the Drosophila melanogaster retina is shown (part a). Note the beautifully regular pattern of six cells, four in the middle and two around, which constitutes the basic organizational unit. The geometry of the ommatidium mirrors that of four soap bubbles (part b), suggesting that surface tension has an important role in defining the shape of the ommatidium. Cells misexpressing N-cadherin are marked with green fluorescent protein (GFP; false-coloured purple) (part c). Cell outlines are visualized with fluorescent E-cadherin (green). A simplified form of an equation is shown, which was used by Hilgenfeldt et al.18 to model the interplay of physics (for example, membrane mechanics) and biology (for example, N- and E-cadherin levels). Kronecker delta function terms representing homophilic interactions are not shown for clarity. Equation terms are explained in the text. Alterations in cell and tissue architecture in colorectal cancer are shown (part d). The arrows represent normal and dysplastic crypts (with diameters of 75 microns and 150 microns, respectively). Parts a–c are reproduced, with permission, from REF. 16 (2004) Macmillan Publishers Ltd. All rights reserved. Part d is courtesy of B. Vogelstein, John’s Hopkins University, USA.
Moving beyond the fairly simple D. melanogaster retina to more complicated tissues with vasculature and dozens of different cell types will require qualitatively different approaches, some of which still need to be invented. The major issues are that cell–cell and cell–matrix interfaces are neither uniform nor static, and that there might be multiscale mechanochemical feedback within tissues, potentially yielding extremely complicated dynamics. For example, unlike soap bubbles with their homogeneous interfaces, cells and tissues have complex cell–cell and cell–matrix interfaces that change with time. Each cell inside a tissue monitors its surrounding tensile forces and chemicals. Different mechanochemical inputs to the cell change the genes that it expresses, altering its cytoskeleton and changing its stiffness. Thus, the global mechanochemical state of a tissue may modulate the mechanical properties of each of its constituent cells, and these in turn can initiate mechanochemical state changes that can ripple through the tissue (FIG. 2). This is perhaps the most difficult challenge in understanding the interplay of cell mechanics, signalling, genetics and tissue function: how do cause and effect interact in a tissue, when small changes may be synergistically amplified?
Figure 2. Tissues are complex dynamic systems that feature multiscale mechanochemical coupling.

Progress has been made particularly in delineating the molecular mechanisms of force generation by the cytoskeleton, the details of cell–cell adhesion and force sensing by proteins such as talin 1 and vinculin. However, mechanical effects in biology are inherently multiscale, in the sense that single cells can generate stresses and strains that contribute to the mechanics and the organization of entire tissues, and in turn, millimetre-scale tension fields within tissues can provide signals that are sensed by potentially millions of cells within that tissue. Understanding this interplay will require new types of experiments that can interrogate all relevant scales simultaneously, and broad conceptual and theoretical advances. ECM, extracellular matrix; FAK, focal adhesion kinase; P, phosphorylation; ROCK, Rho-associated, coiled-coil containing protein kinase; SFK, SRC family kinase.
One insight from the physical sciences and neurobiology is that complicated chemical, physical or biological systems are best approached by tightly integrating manipulation (where possible), measurement and simulation. Simulations allow powerful experiments to be designed, and precision manipulation and measurement allow models to be decisively tested and gradually refined. Implicit in such an approach is that experiments are carried out in a way that allows error estimates to be assigned to each measurement. For example, in particle physics, new particles are announced in terms of their sigmas: the probability of scientists being wrong. Overall, the triad of manipulation, measurement and simulation allows physical scientists and engineers to simulate the Earth’s global climate with increasing reliability, investigate the time evolution of the universe, and design and flight-test aircraft purely in silico. This approach is also showing impressive results in systems and synthetic biology19.
Another insight that is relevant to cancer mechanics is the utility of measuring system dynamics, which stems from the fundamental link between the forces that drive a system from one state to another and the dynamics with which that change takes place. A good example is an electron moving through space. A single picture of this process, however detailed, contains little information beyond revealing the presence of the electron. By contrast, a movie of this process allows the forces that control the motion of the electron to be accurately inferred; this is how electrodynamics, the theory of charge, light, radio waves and electricity, was developed. Essentially, all known physical laws were discovered by watching how things move, whether they are planets or atoms. In a cancer mechanics context, the ideal experiment would be to watch the cell boundaries within a living tumour and its surrounding tissue move with great accuracy and over long time periods. Such a movie could then be inverted to reveal the alterations of the normal cellular ‘tug-of-war’ that take place in a tissue during carcinogenesis and subsequent metastasis. However, this inversion will require the generation of new and complex mathematical models.
A final consideration is the value of simultaneously determining correlations among many parameters. For example, perturbing a cell and then quantifying the degree to which the fluctuations in the concentration of, or the location of, two or more proteins are correlated can reveal important information about signalling network topology, feedback loops and the role of noise in gene expression19,20. The cancer research literature still features reports of single genes or proteins that are asserted to cause cancer. The flood of results from the cancer genomics and cancer systems biology efforts makes one wonder, however, about the actual utility of relating any one isolated gene or protein to the disease. A new generation of measurement technologies that are able to simultaneously measure many cellular and tissue parameters, thereby relating them and thus allowing cause and effect to be distinguished, would be of great use. Imagine, for example, being able to mechanically manipulate cells within a tissue and then following the activity of all 50-plus proteins of the RAS effector pathways within single cells and thus seeing how the network is influenced by local mechanics and how it might work around a mechanically induced or a drug-induced reduction of kinase activity.
So far, we have been emphasizing the role of mechanics in development and disease, and we have also tried to provide a glimpse of related ideas that may be useful for cancer research. This is only one small part of a much larger puzzle. An equally important way to approach cancer is by studying how it evolves, as this reveals the physiological, mechanical, genetic and biochemical forces that guide the disease and its progression.
Evolution and evolutionary theory of cancer
Tumours result from evolutionary processes within tissues3,21–25. From an evolutionary standpoint, tumours can be regarded as collections of cells that accumulate genetic and epigenetic alterations, which are then subjected to the selection pressures operating on the cells that harbour them. These alterations have heritable effects on the fitness of cells and may thus lead to rapid increases or decreases of mutant clones within the tumour26,27. Beneficial alterations can generate adaptations, such as an increased growth rate, motility and ability to invade into surrounding tissue, as well as the induction of angiogenesis and evasion of the immune system. The fitness of a tumour cell thus results both from the accumulation of alterations and from the interaction with cells and other components of its microenvironment. Changes that are beneficial to the cell are normally detrimental to the organism and thus neoplastic processes are an example of conflicting selection acting on different hierarchical levels28: evolution and natural selection generally lead to increased proliferation, survival and evolvability on the cellular level and results in progression, invasion and resistance. Selection at the level of organisms and genes has led to the evolution of oncogenes and tumour suppressors in the genome23,29.
Viewing neoplasms as a result of evolutionary forces operating on tissues within multicellular organisms provides physicists, mathematicians and population geneticists with an opportunity to use their tools to describe the evolution and ecology of cancer cells with mathematical constructs. Such theoretical modelling, together with the principles of evolutionary biology, has been successfully used to study the mechanisms and dynamics of tumour initiation4,5,30,31 and progression32,33, as well as the response to treatment and the emergence of resistance34–36. For example, an interest in understanding and preventing the evolution of resistance against anticancer therapy has inspired the development of several mathematical approaches. Coldman and co-authors pioneered the field by introducing stochastic models of resistance to chemotherapy to guide the selection of treatment schedules35,37. The thought process introduced by these investigators was later applied to study the risk of pre-existing resistance38–41, resistance emerging during treatment39,40,42 and the optimal scheduling of treatment administration under various circumstances43–47. Related efforts have led to such seminal results as the discovery of tumour suppressor genes30,48 and the multistage theory of carcinogenesis4,49.
The recognition of cancer as a disease that is caused by the accumulation of several somatic alterations has motivated recent large-scale efforts to annotate the cancer genome and epigenome for many human cancers50–52. When combined with computational approaches that can distinguish significant, recurrent events from the background noise in high-resolution data sets, these cancer genome and epigenome surveys yield molecular portraits that are specific for each cancer type and consistent across multiple sample sets in that they uncover a subset of events in many samples of the same cancer type53,54. These emerging, large cross-sectional data sets are the basis for investigations by computational biologists, physicists, mathematicians and evolutionary biologists to address a multitude of questions about the generation and persistence of genetic and epigenetic alterations in cancer. Here, we summarize two recent advances in a mechanistic understanding of these alterations — one addressing the propensity of genetic alterations to arise at particular loci in the genomes of evolving cancer cells, and the other concerning the deduction of the temporal order in which genomic alterations arise during tumorigenesis using evolutionary mathematical approaches.
Which factors determine the mutagenic potential of the genome?
An unstable genome is a hallmark of many cancers55. The mechanisms of the generation of genomic variation, however, have not yet been entirely elucidated. It is unclear, for example, whether some mutagenic features that drive somatic alterations in cancer are encoded in the genome sequence or whether they can operate in a tissue-specific manner. Therefore, a genome-wide analysis of the properties associated with DNA breakpoints that are related to somatic alterations in cancer is of fundamental interest for many areas in biology, including cancer genomics, genome informatics and evolution.
Many exogenous and endogenous factors, as well as molecular mechanisms, can cause double-strand breaks and erroneous DNA repair, leading to genomic alterations in cancer genomes56–58. Under certain circumstances, DNA can adopt non-B conformations, which can similarly contribute to DNA damage59–61. Guanine-rich sequences (G3+N1–7G3+N1–7G3+N1–7G3+) can adopt four-stranded structures called G-quadruplexes (G4s)62–64. As these sequences occur frequently in the human genome, they could potentially contribute to DNA damage in multiple areas of the genome. Indeed, G4 structures have the potential to obstruct the movement of DNA polymerase65, thereby increasing the risk of DNA breakage or of non-allelic homologous recombination. A recent genome-wide analysis of DNA breakpoints that are associated with somatic copy number alterations (SCNAs) from 2,792 cancer samples classified into 26 cancer subtypes led to the identification of SCNA hotspots66. Despite a subset of these hotspots being present in the genomes of apparently healthy individuals, this investigation uncovered that G4 structures could be causally implicated in genomic instability and the generation of DNA breakpoints in cancer. The genomic alterations that were associated with DNA breaks had a strand-specific pattern that was consistent with a causal role of G4 structures in their generation. An analysis of methylation data from several different tissue and cancer types subsequently led to the finding that abnormal hypomethylation in genomic regions that are enriched in G4 sequences is likely to be a key mutagenic factor that is associated with tumorigenesis. These findings are consistent with observations that G4 structures are implicated in germline deletion67–68 and recombination69 events. These studies suggested a mechanistic model for the generation of tissue-specific mutational landscapes in cancer, showcasing the ability of computational approaches, together with modern cancer data sets, to provide mechanistic insights into the evolution of cancer genomes.
The temporal order in which genomic alterations arise during tumorigenesis
These emerging, cross-sectional cancer data sets have also recently been linked to a novel evolutionary approach for predicting the temporal order of somatic events that arise during tumorigenesis. Knowledge of this temporal order helps to guide the generation of the correct genomic context in animal models of human cancer, and aids in prioritizing the validation of potential drug targets, as changes that occur early in malignant transformation may result in the rewiring of the signalling circuitry or may confer a state of addiction to the new signal. A novel evolutionary approach, called retracing the evolutionary steps in cancer (RESIC)70, determines the sequence of genetic events using cross-sectional genomic data from a large number of tumours.
RESIC is based on the principles of population genetics71 (BOX 1). RESIC predicts the distribution of patients across possible mutational states that are defined by specific genotypes; this distribution is then compared with the numbers of clinical samples that contain the corresponding genotypes. This mapping is used to optimize the evolutionary parameters by minimizing the difference between the predicted and the observed frequencies in the data set. The output of RESIC is given as a percentage of the flux through the network through each particular evolutionary path, thus specifying the temporal sequence of somatic alterations in cancer samples.
Box 1. Retracing the evolutionary steps in cancer (RESIC).
Consider a population of N cells at risk of accumulating the genetic changes that lead to cancer. Cells proliferate according to a stochastic process: at each time step, a cell is chosen that is proportional to fitness to produce a possibly mutated daughter cell. Subsequently, another cell is chosen at random to die, and is replaced by the newly produced cell to maintain homeostasis. A mutated cell can take over the population (that is, reach fixation) or go extinct owing to stochastic fluctuations (see the figure). If the population size is smaller than the inverse of the mutation rate, then at any time, there are at most two types of cells in the population: type i and type j. Cells of type i differ from cells of type j by only one genetic alteration. Their respective fitness values (that is, growth rates) are denoted by ri and rj. The rate at which the population transitions from state i to state j is given by mi,j = Niui ρ(ri,rj), where ρ(ri,rj) = [1–1/(rj/ri)]/[1–1/(rj/ri)Ni], if ri ≠ rj, and ρ(ri,rj) = 1/Ni, if ri = rj. Depending on the order of appearance of alterations, the population follows different evolutionary paths towards the fully mutated state (part a).
Cancers are considered to originate from a single population of cells per person. Using this model, we study the evolutionary dynamics of individuals accumulating the mutations leading to cancer (part b). We consider the dynamics of patients in steady state: there is a constant influx into the unmutated state, representing diagnosis of disease, and a constant outflux from the fully mutated state, accounting for diagnosis and deaths of patients or their cure. The evolutionary dynamics of a population is described by X = XM + F, where the vector X(t) consists of the frequencies Xi(t), the matrix M contains the transition probabilities mi,j, and F = (f,0, …–f) represents the influx into the initial node and outflux from the fully mutated node. At steady state, the population is distributed across all possible states; this steady state distribution can be compared with the numbers of clinical samples that have the corresponding genotypes, where the total number of patients in a data set is equal to the sum of patients in all states (part b). This mapping is used to optimize a subset of parameters in the mathematical model (that is, the fitness values of cell types) by minimizing the difference between the prediction and the observed frequencies in the data set. Other parameters, such as cellular population size, mutation rate and influx rate, are estimated from experimental results and tested for robustness over several orders of magnitude. The output of RESIC is given as the percentage of flux through the network via each particular path, and can be used, together with cross-sectional cancer genome profiling studies, to identify the temporal sequence of events arising during tumorigenesis (part c).
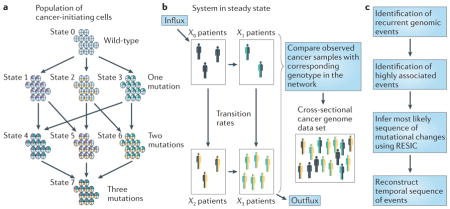
The established sequence of genetic events arising during the multistep process of colorectal carcinogenesis72, neurofibromin (NF1)-driven primary glioblastoma73 and secondary acute myeloid leukaemia74 provided an opportunity to validate the ability of RESIC to recover the orders of events from cross-sectional data sets70. This methodology was also applied to a large, integrated genomics data set of primary glioblastoma samples53. First, areas of significant gene copy number alterations were identified using Genomic Identification of Significant Targets in Cancer (GISTIC)75, and then alterations that were significantly positively correlated with each other were selected for further analysis; correlations between alterations are a prerequisite for the methodology, as the determination of an order of oncogenic events is only meaningful for those events that co-occur sufficiently often. This approach determined that homozygous deletions of the CDKN2A locus (which encodes INK4A and ARF) frequently co-occur with epidermal growth factor receptor (EGFR) and PTEN alterations (P value < 10–8) in primary glioblastoma. When studying this mutational network, RESIC predicts that the most common early alterations are EGFR low-level amplification and CDKN2A deletion, which have a similar likelihood of occurring. Although there is no single most frequent path through the network, the frequency of paths concluding with high-level amplification of EGFR is highest; the second most frequent final event is homozygous CDKN2A deletion70. These data suggest that glial progenitor cells can tolerate full EGFR activation only after the inactivation of CDKN2A or PTEN. This result agrees with the fact that EGFR overexpression is insufficient for tumorigenesis in mouse models of glioblastoma76,77, providing support for the temporal order of events predicted by RESIC.
Evolutionary methods of analysis such as the ones presented here will provide the research community with tools for the identification of tumourinitiating events using modern cancer genome and epigenome data sets; furthermore, such frameworks of tumorigenesis will help with the generation of hypotheses that can be tested using transgenic mouse models of human cancer. Many opportunities exist to use evolutionary approaches to further our knowledge of cancer initiation, cancer progression, the response to treatment and therapeutic resistance.
Information coding and transfer in cancer
Current thinking in information coding and decoding in biological systems generally implies a one-way information flow — from DNA to transcribed RNA to translation to protein. Recent studies in developmental biology and epigenetics, however, have demonstrated that this information can flow in both directions, and that this flow can be influenced by external physical forces, even if the underlying DNA sequence remains unaltered. It is becoming increasingly clear that this biological information system is not only made up of two-way communication, but that feedback loops, inter-connectivity and modulation by external environmental forces also introduce a previously unappreciated level of complexity. Moreover, studies are revealing that new kinds of biological information exist, ranging from genetic information that is encoded in the mechanical properties of DNA, to information in protein sequences that control the lifetime and post-translational processing of the protein. This complexity represents an opportunity for physicists, chemists and engineers to work together and has led to studies at the interface of physical science, genetics and oncology. These investigations are yielding rapid advances in our understanding of chromosome structure and function at multiple length scales and are shedding light on gene regulation in normal health and development, which in turn may help to explain, diagnose and treat gene misregulation in cancer. Here, we summarize two of these advances — one concerning fundamental physical chemistry in gene regulation that is related to the lowest levels of chromosome structural organization, and the other concerning a novel cancer diagnostic technique that seems to be measuring aspects of the highest levels of chromosome organization.
Sequence-dependent DNA mechanics, nucleosome positioning and gene regulation
An especially active research area concerns the rules governing the most fundamental level of chromosome architecture78, in which short stretches of DNA (147 bp) are wrapped locally in ~1.75 turns around octameric cores of his-tone proteins, creating nucleosomes79. Nucleosomes are separated from each other by ~10–50 bp stretches of unwrapped linker DNA; thus, only 75–90% of eukaryotic genomic DNA is wrapped in nucleosomes. DNA that is wrapped in nucleosomes is sterically occluded from most other DNA-binding proteins, and moreover is sharply distorted away from the DNA conformations that are favoured by most other proteins80. Consequently, the placement of nucleosomes along the DNA profoundly influences essential DNA interactions, such as gene regulation, transcription, replication, recombination, chromosome breakage, retroviral and transposon integration sites and DNA repair81–92.
It is perhaps not surprising, therefore, that recent studies have shown that nucleosome positioning is tightly regulated, and that there is an additional layer of genetic information, superimposed or multiplexed directly on top of other kinds of regulatory and coding information, which functions to bias where nucleosomes can be located along the DNA. The nature of this information lies in the sequence-dependent mechanics of DNA93. Different DNA sequences differ greatly regarding the ease with which they can bend around a nucleosome94,95, conferring differences of many thousand-fold or more on the affinity of nucleosomes for one DNA sequence versus another96. The concentration of nucleosomes in the cell is kept below 100% saturation of the genomic DNA, and thus different regions of DNA compete for nucleosome occupancy. DNA sequences can dictate which DNA regions will compete well for nucleosomes and have high intrinsic nucleosome occupancy, and which will not. Like transcription factor binding sites, natural genomic nucleosome positioning sequences are not determined purely through highest possible affinity96, allowing degeneracy in the choice of DNA sequence used. Thus, the degeneracy introduced by variations in the genetic code, transcription factor binding sites and the mechanical constraints of the nucleosome DNA sequence preferences97,98 allow the preferential locations of many nucleosomes to be specified alongside DNA coding and conformation changes that constitute genetic information and conventional gene regulatory information.
Approaches that are more commonly used in the physical sciences are contributing to this work in two ways. Diverse new experimental studies99,100 and theoretical studies, ranging from atomic101 to multi-scale102 and mesoscopic97,98,103, seek to measure and explain the sequence-dependent mechanics of DNA, with the goal of being able to predict the influence of the genomic DNA sequence on nucleosome formation and on the stability or occupancy of other structures involving tightly bent DNA (such as, numerous sharply looped gene regulatory complexes) from first principles.
Additionally, physical science tools have already enabled advances in predicting important aspects of nucleo-some organization in vivo, using phenomenological definitions of the nucleosome DNA sequence preferences that have been obtained in direct binding experiments104. This prediction problem is complicated by the combination of the high concentration of nucleosomes along the DNA and the physical reality that nucleosomes cannot overlap along the DNA in any one cell at any one point in time. The problem is that where any one nucleosome resides along the DNA not only partly depends on the DNA sequence inside that nucleosome, but also on the positions of the neighbours of that nucleosome; however, where those neighbours are not only depends on the DNA sequences inside them, but also on where their neighbours reside — and so on, out to the ends of the chromosome. If nucleosomes occurred only rarely along the DNA, then one might be able to ignore the problem, as the chances of two favoured nucleosome locations overlapping on the DNA might be very low. This is the case in typical analyses of transcription factor binding sites, for example, as the recognition sites for any given transcription factor are typically sparsely distributed along the genome. But for nucleosomes this is definitely not the case; some sort of ‘holistic’ theoretical approach is required, which solves the nucleosome distribution problem for an entire chromosome all at once.
The assembly of nucleosomes in vitro from purified components104 has identified the sequences to which nucleosomes are more likely to bind. This information specifies an effective potential for a nucleosome to start at each basepair along the DNA. But calculating where the nucleosomes will actually be is complicated by competition between nucleosomes, which occupy space and cannot overlap. If one poses the hypothesis that nucle-osomes equilibrate their locations along the DNA — a conjecture that could at best be only approximately true — then this problem reduces to a famous problem in statistical mechanics, namely that of a one-dimensional solution of hard rods in an external potential, and thus can be approximately solved by Monte Carlo methods105 or exactly solved by numerical integration106, recursion107 or dynamic programming78,97 (BOX 2). The solution of these equations yields the probability of a nucleosome starting at each basepair, and the probability that each basepair is covered by any of the 147 different nucleosomes that could potentially cover it (as each nucleosome covers 147 bp). The solution bears a striking resemblance to the locations of nucleosomes genome-wide (and a highly significant genome-wide correlation) measured in vivo, suggesting that the assumptions made in the theoretical analysis could be reasonable.
Box 2. Nucleosome positioning.
Following the recursive approach, one defines a potential Vn for a nucleosome located at basepair n, Vn = −kBT0 ln Pn, where kB is Boltzmann’s constant, Pn is the likelihood of a nucleosome starting at basepair n in the absence of nucleosome–nucleosome interactions, given by a Markov model used to specify the intrinsic DNA sequence preferences of the nucleosome 78,104 and T0 is the reference temperature at which the Markov model is defined. Thus, the likelihoods given by the nucleosome–DNA interaction Markov model are treated as an apparent free energy landscape onto which nucleosomes will be placed at equilibrium subject to the rule that they cannot overlap in space and time.
One then defines a recursion relation (see equation 1):
| (1) |
where μ is the nucleosome chemical potential (related to the nucleosome concentration (c) by μ = μ0 +kBTln (c), where μ0 is a constant) and the Hn are given by equation 2:
| (2) |
for a nucleosome occupying (excluding another nucleosome for a length of) a basepairs. The Hn capture the different ways a given site can be blocked by neighbouring nucleosomes. One initiates the recursion by forbidding a nucleosome from occupying less than length a basepairs at the right-hand end of the DNA, setting the Hn values for those a–1 basepairs = 1. One then solves equations 1 and 2 iteratively from large to small n. The probability ρn of a nucleosome starting at each basepair n is then given by equation 3:
| (3) |
which is solved iteratively from small to large n. Finally, the occupancy σn of a given basepair by any of the a nucleosomes that potentially occlude it is given by equation 4:
| (4) |
Of course, the real problem in the cell nucleus is not simply one of nucleosome positioning; rather, there is an ongoing dynamic competition between nucleosomes and changing constellations of transcription factors and other DNA binding proteins, each seeking to bind to favoured sequences along the DNA. Transcription factors may have high sequence specificity but they are present at fairly low concentrations, whereas nucleosomes may have lower specificity but are present at high concentrations, and thus both make important contributions to the outcome. In a sense, therefore, the genome sequence is specifying how this dynamically evolving competition will play out.
Similarly, it can be assumed that the nucleosomes and changing sets of competing factors approximate a distribution equilibrium for any given window of time (that is, for any given set of competing factor concentrations), and the statistical mechanics model summarized in BOX 2 can be generalized to allow for competition not just between nucleosomes, but also between one or more different transcription factors with each other and with nucleosomes108. This problem can again be solved exactly by using dynamic programming97,109. An example of the competition of nucleosomes with themselves and with a single factor binding at two nearby sites is shown in FIG. 3. In this model, nucleosomes reconfigure in response to changing transcription factor concentrations simply because of the changing nature of the competition. The transcription factors influence the resulting distribution of nucleosome locations and occupancies, while the nucleosomes equally influence the distribution of bound transcription factors, as well as the occupancies of the transcription factors110.
Figure 3. Physical sciences shed light onto nucleosome and transcription factor competition and chromosome packaging.

Schematic illustration of the partial wave spectroscopy (PWS) experiment (part a). Cells from the tissue of interest are brushed from the tissue and studied ex vivo. Each cell is individually scanned in a two-dimensional lattice of diffraction-limited pixels (strictly voxels because of the thickness of the cell). The cells are illuminated with light of wavelength λ, and backscattered waves propagating along one-dimensional paths within each voxel are measured as functions of lateral position over the cell (x, y) and of wavelength λ. Wavelength-dependent variability in reflectivity R at each pixel arises from the interference of photons scattered from refractive index fluctuations within the cell at that location, providing information about internal cellular heterogeneity on subwavelength length scales. The magnitude of the spectral fluctuations at each pixel in the image are represented by a disorder strength, Ld = <Δn2> × lc, where Δn is the local fluctuations in intracellular refractive index and lc is the correlation length of these fluctuations. For many different cancer types, the measured disorder strength increases in cells that that are located some distance away from a cancerous lesion, even though the cells being analysed are themselves not cancerous. Parts b–g illustrate the free energy landscape from a typical section of genomic DNA97. The 10 bp oscillations in interaction energy arise from the ~10 bp helical symmetry of DNA, the approximately circular DNA wrapping in the nucleosome, and the mechanical nature of the nucleosome–DNA interaction (part b). Parts c–g illustrate nucleosome–transcription factor competition97. The purple curves in part d show the corresponding distribution of nucleosome start probabilities with no transcription factor present (from the solution of equation 3 in BOX 2), and the top two rows of orange ovals (part e) represent two of the many different nucleosome configurations, all of which have significant probability in the equilibrium distribution. The purple curves in part g show the corresponding distribution of nucleosome occupancy (summed over the full set of allowed configurations, from the solution of equation 4 in BOX 2). The free energy landscape of a transcription factor, shown in part c, highlights two specific binding sites with equal energies (affinities), as indicated by the two sharp valleys in energy (the two green lines). The transcription factor also has slightly varying affinities for nonspecific sequences (indicated here by the thickness of the green bar covering the remainder of the landscape). In this example, the model is solved for a transcription factor concentration that is sufficiently high so as to give high occupancy at one of the binding sites (probability approaching 1; green curve in part d). However, because this site overlaps a preferred nucleosome location, binding at this site requires redistribution of the local nucleosome organization, to a new distribution of nucleosome start probabilities (orange curves in part d). The bottom two rows of orange and green ovals in part f represent two of the many different configurations of nucleosomes and bound factors, respectively, which have significant probability in the resulting equilibrium distribution. The corresponding nucleosome and transcription factor occupancies are shown in orange and green, respectively, in part g. Even though the two transcription factor sites have identical intrinsic affinities for the transcription factor, the required nucleosome redistribution is energetically inexpensive for the right-hand transcription factor binding site, compared with the left-hand transcription factor site, which allows a higher occupancy at the right-hand site for a given transcription factor concentration. The image of the nucleus in part a is reproduced, with permission, from REF. 146 (1997) National Academy of Sciences. The graph in part a is reproduced, with permission, from REF. 112. Parts b–g are reproduced, with permission, from REF. 97. a.u., arbitrary units.
By complementing such an analysis of nucleosome and transcription factor binding configurations with information relating transcription factor binding and the eventual transcriptional output, it might be possible in the future to predict the transcriptional state of many genes in a cell given only the genomic DNA sequence, concentrations of key transcription factors, and the known sequence preferences of those transcription factors108. Such a predictive ability would in turn be valuable for understanding how a normal cell is transformed into a malignant one, and conversely, how a malignant cell might be transformed back into a non-malignant one. Currently, however, other outstanding issues remain, most notably, the extent to which these models will predict biological reality, as determined by experiments.
Higher order chromosome structure and novel cancer diagnostics
At the other, highly compacted, end of the structural hierarchy of the chromosome, little detailed structural information is currently available111, but there is great potential for important advances to be made from using approaches that are commonly used in the physical sciences. Indeed, methods from the physical sciences are already being used in cancer diagnostics.
Partial wave spectroscopy (PWS)112 takes advantage of the field effect, in which apparently normal cells, which are distant from a cancerous or potentially precancerous lesion, can develop anomalous properties that may be detectable even in the absence of knowledge regarding the exact location of the lesion itself (FIG. 3). The PWS experiment yields a signal, the magnitude of which can be a highly sensitive and specific predictor of the existence of a cancerous lesion some distance away in the body. This approach has great potential in the diagnosis of cancers, such as those of the colon, lung and pancreas, for which existing diagnostics are ineffective, unpleasant or have a substantial risk of complications. Although PWS was initially developed as a purely phenomenological indicator, its striking preliminary successes have heightened interest in better understanding the underlying physical and biological changes that it monitors. Quantitative studies on patient samples versus normal controls have shown that increased PWS disorder strength in cells that are distal to a cancerous lesion is a strong indicator of the existence of a lesion112,113. However, the PWS technology has not yet been applied to problems of distinguishing pre-malignant from malignant disease, which is an important goal of future studies.
PWS measures the refractive index, which is related to the polarization of molecules, and in turn is related to electron density. Of the main macromolecular constituents of a cell with amounts that might plausibly have significant variability on subwavelength length scales, nucleic acids and phospholipids stand out by virtue of having fairly high amounts of the electron-rich element phosphorous. Chromatin in particular is both highly phosphorous-enriched and highly heterogeneous in subcellular (and subnuclear) distribution, with notable regions of high local chromatin concentration observed cytologically as dense heterochromatic regions. Hence, one expects that dense nucleic acid-containing superstructures, such as large-scale regions of compact heterochromatin, might dominate the PWS disorder strength measurement.
Consistent with this expectation, direct tests show that both the nucleus and the cytoplasm contribute to the measured disorder strength, but that the nuclear contributions dominate114. Certainly within the nucleus, one expects chromatin structure to dominate refractive index inhomogeneities and so the PWS disorder strength115.
Therefore, for the case of PWS, physical science approaches have already made a valuable contribution to cancer diagnostics. This approach potentially provides diagnostic sensitivity and specificity in many cancer types that are comparable to or better than those that are presently available using much more invasive tests. PWS should also help in understanding the intracellular organization of chromatin, a problem that has resisted a definitive solution for many decades. Much anecdotal evidence suggests that there is a relationship between higher order chromatin compaction and transcriptional repression. Advances using PWS spectroscopy as a discovery tool, together with other approaches from physical sciences ranging from micromechanical studies on whole chromosomes, to novel imaging modalities such as super-resolution optical microscopy and electron microscopy using engineered nanoparticle markers, will probably shed much light on this longstanding problem in fundamental molecular biology. Furthermore, a better understanding of the chromatin organization and how it relates to the PWS disorder signal may in the future lead to further refinement of this promising diagnostic tool.
Deconvolving cancer complexity
Complex systems represent major areas of study for physical scientists. Fundamental insights from areas such as thermodynamics, fluid and classical mechanics, in combination with advanced computational visualization and simulation, could potentially aid in understanding cancer. Genomic instability is a fundamental characteristic of cancer, inherently variable between patients even with nominally the same disease and during cancer progression within the same patient116. The current clinical taxonomy of cancer technically defines more than 200 types, but closer inspection reveals that cancers are like ‘malignant snowflakes’, with no two cases identical at the cellular level117–119. Heterogeneity is higher in tumour types that originate later in life and increases during tumour progression120,121. As is particularly evident in pancreatic cancer122, tens or hundreds of different diseases coexist within the same person with metastatic dis- ease, with each metastasis having a distinct genetic profile and distinct signalling pathways, growth, interaction with the microenvironment and response to treatment.
Non-genetic and extrinsic factors greatly add to the complexity of cancer: the stochastic partitioning of proteins during cell division, which generates randomness in protein abundance123; epigenetic heterogeneity including DNA methylation, histone modifications, nucleosomal occupancy and remodelling, chromatin modifications and remodelling, non-coding RNAs124 and proteomic profiles125; microenvironmental heterogeneity between cancer types and metastases126, as well as within a single tumour; and the macroscopic heterogeneity of the patient, including age, gender, weight, immune status, lifestyle and mental health.
Faced with this overwhelming diversity, medicine has not substantially advanced in the treatment of metastatic cancer — and perhaps it never will, unless the root causes of this heterogeneity are clarified. Complexity theory in mathematics and physics is defined by the quest to understand and predict the emergence of order and structure in complex and apparently chaotic systems, such as turbulent flows. A major advance in breaking the overall problem into more manageable pieces lies in the answer to the question of what defines a cancer. Six fundamental, distinct hallmarks have been proposed55. It is possible that cancer can be understood in terms of these hallmarks, which is akin to resolving a six- dimensional vector into components along six coordinate axes. However, each ‘axis’ further comprises a limited number of mutated genes that define pathological pathways127. This is accompanied by the heterogeneity of genetic mutations even in two nominally identical tumours, owing to a large number of infrequently mutated genes, which may be crucial drivers of the development and the progression of tumours128. Systems biology129 offers a promising approach to the organization of genomic data into quanta of biological order, and information on molecular circuitry connecting cellular pathways and diseases states130. The quest for intelligible underlying structures in cancer to which complexity theory could be applied can be thought of as the search for ‘super-genes’ that are shared among cancer types, which form 3–5% of the mutated gene population, and which affect the key pathways in which the other mutations tend to cluster127,128. Even with this approach, the assessment of cancer complexity challenges our optimism about finding cures with the currently available molecularly targeted therapeutic approaches127,131,132.
The conventional approach to metastatic disease, which is what kills most cancer patients, involves the use of systemically administered agents, in the form of chemotherapy, radiotherapy or biomolecularly targeted therapeutics that have the capacity to reduce the selective fitness advantage of metastasizing and/or metastasized cells. Such chemical and biological agents are expected to simultaneously carry out a triad of functions: transport from the point of administration to the intended cancer target sites, preferential accumulation at these target sites and preferential cytotoxicity or cell signalling modulation. The failure to treat metastases stems from the failure to develop agents that are capable of reducing the reproductive success of metastatic cells, as well as the failure to address the three functions simultaneously, for all different presentations of the disease within a patient at any given time. One ‘blind spot’ has been the physics of mass and momentum transport. This is a starting point for a different perspective on cancer, termed Transport OncoPhysics133. This approach aims to reduce the complexity of apparently disparate biological hallmarks to the unifying notion that these hallmarks all reflect the deregulation of mass transport. In this framework, cancer is viewed as a disease of mass transport deregulation at multiple scales — bridging the molecular to the cellular, the microenvironmental, organ and organism levels (FIG. 4). An intriguing view emerges of a family of diseases characterized by pathological disruptions of mass transport, in hierarchically nested systems. For example, the defining aspect of malignancy, tissue invasion, is mass transport deregulation at the interface between the cell and the microenvironment. Metastasis defines the transition to lethality, and is a deregulation of local and distant cellular transport at the scale of the organism. Tumour-associated angiogenesis completely overhauls microenvironmental mass transport, and is another prime example of transport deregulation. The signalling pathologies that accompany the evasion of apoptosis, growth signal dependence and growth inhibitory messages from the immediate environment are also disruptions in molecular transport — for molecular signalling occurs by the transport of signalling molecules.
Figure 4. The application of physical science approaches for understandingNatur Reviews Cancer deregulated transport in cancer.

This is an illustration of the efforts to develop a broader understanding of the physical barriers and the biological factors that are involved in the progression of tumours and the efforts to design novel biocompatible delivery carriers that can overcome or take advantage of these barriers with favourable pharmacokinetics and tissue distribution profiles for highly efficient delivery of novel therapeutic and imaging agents. A physics- and biology-driven, and a mathematics-based, design of the engineered drug delivery vectors multiplies the probability of recognition of the novel targets, thus providing a synergistic solution for the imaging and therapy of tumours at the interface of physics, engineering, mathematics and cancer biology.
To verify or disprove the Transport OncoPhysics approach, several concurrent, novel investigational modalities and tools are required, which are based on mathematics and the physical sciences: a multiscale mathematical theory of mass and momentum transport through the body; multiscale imaging that enables the tracking of mass transport in living organisms, with integrated resolution from subcellular to full body levels; and multiscale probes, in conjunction with imaging techniques, to determine the transport properties at various levels, as functions of the characteristics of the transported object. In the Center for Transport OncoPhysics, within the US National Cancer Institute’s Physical Sciences-Oncology Centers initiative134, we have focused on all three of these enabling aspects (the multiscale modelling of cancer growth and multiparameter response to therapy135,136; multiscale imaging137; and multiscale probes138), which also provide specific vectoring across biological barriers to target lesions139, yielding novel mechanisms of transport, accumulation and the release of the therapeutic payload. These have resulted in unprecedented therapeutic results140,141 through the use of systems of nested (multistage) particles, designed with explicit consideration of the physics and mathematics governing the convection, margination, adhesion and cellular uptake of the particles142–145. Once the physical laws of transport are determined, their parameters for individual lesions can be obtained from direct observation through suitable imaging techniques. Among these parameters are the blood flow velocity, shear stress at the vascular wall, vascular permeability and the density of the specific antigens of interest that are expressed on the vascular endothelium. Once these are observed, the mathematical routines for the rational design of particulates can be used to yield therapeutic systems that are individualized for the specific lesion — not only in the therapeutic payload but also in the therapeutic delivery vector itself. This opens new frontiers for the idea of personalized therapy.
Conclusion and outlook
The physical laws and principles that define the behaviour of matter are essential for developing an understanding of the initiation and progression of cancer at all length scales. The theoretical approaches that enable the definition of behaviour within complex systems offer opportunities for new insights into long-standing problems in cancer research. For example, the metastatic process, the generation and maintenance of heterogeneity within and among tumours, the emergence of drug resistance and the ecological behaviour of cell types with differing reproductive fitness and degrees of drug sensitivity, and the delivery of therapeutics to the core of a tumour, as well as its distant metastases, will all benefit from an application of physical science approaches to oncology — from mechanics to evolution, chemistry and nanotechnology. Thus, the successful integration of approaches from mathematics, physics and engineering with cancer biology may be our best hope to understand complex systems such as cancer and to develop effective strategies for a cure.
At a glance.
Approaches from the physical sciences can contribute to the rate at which powerful new diagnostic tools and therapies can be discovered and brought into the clinic. We provide examples from four areas to describe how teams of physical scientists, cancer biologists, clinicians and cancer advocates are tackling cancer from the perspective of the physical sciences.
The principles of evolutionary biology can be used to study the mechanisms and dynamics of tumour initiation, tumour progression, the response to treatment and the emergence of resistance. For example, large-scale cross-sectional genomic data sets can be combined with novel evolutionary approaches to predict the temporal order of somatic events that arise during tumorigenesis. Such knowledge helps to guide the generation of the correct genomic context in animal models of human cancer and helps to prioritize the validation of potential drug targets.
DNA in vivo is often sharply distorted away from the canonical Watson – Crick structure; different DNA sequences vary greatly in the ease with which such sharp distortions can be accommodated. Most of the eukaryotic genomic DNA is bent around histones to form nucleosomes. The capacity of the DNA sequence to undergo such distortion can influence the specific preferred locations for many of the nucleosomes.
The existence of a cancerous lesion can sometimes be detected through the analysis of the altered behaviour of cells that are located substantial distances away from the primary lesion, a phenomenon that is known as the ‘field effect’. Partial wave spectroscopy takes advantage of the field effect to allow for the sensitive and specific detection of cancers in tissues that are difficult to reach.
Cancer is an extraordinarily complex disease. Methods that are commonly used in physics can reduce the complexity of cancer to a manageable set of underlying principles and phenomena. In particular, Transport OncoPhysics views cancer as a disease of multiscale mass transport deregulation involving the biological barriers that separate different body compartments. Probes that can be used to investigate the mass transport properties of tissues can be used as directed vectors for the localized, preferential release of therapeutics into tumours.
Acknowledgments
The authors would like to acknowledge support from the US National Cancer Institute Physical Sciences-Oncology Center (PSOC) initiative to fund the Dana-Farber Cancer Institute PSOC (F.M.), Bay Area PSOC (J.L.), The Methodist Hospital Research Institute PSOC (M.F.) and the Northwestern University PSOC (J.W.). M.F.’s research for this article was furthermore supported by grants from DoD/BCRP (W81XWH-09-1-0212), as well as by the Ernest Cockrell Jr. Distinguished Endowed Chair. The authors would like to thank A. Sebeson for her invaluable help. This work is dedicated to Professor Jonathan Widom. With his passing, we have lost both a major intellectual force and a valued member of our community, as well as a trusted friend and colleague.
Glossary
- Superconductivity
A phenomenon of zero electrical resistance occurring in certain materials below a characteristic temperature
- Fractional quantum hall effect
A property of a collective state in which electrons bind magnetic flux lines to make new quasiparticles, and excitations have a fractional elementary charge
- Monte-Carlo method
A technique in which a large quantity of randomly generated numbers is studied using a probabilistic model to find an approximate solution to a numerical problem that would be difficult to solve by other methods
- Coupled degrees of freedom
The number of values in a study that are free to vary but that are constrained to vary together
- Emergent phenomena
Complex systems and patterns that arise from a multiplicity of relatively simple interactions
- Elastic energy
Energy stored in the configuration of a physical system as work is carried out to distort its volume or shape
- Population genetics
The mathematical study of the dynamics of genetic variation within populations
- Mesoscopic
A subdiscipline of condensed matter physics that deals with materials of an intermediate length scale, between the size of a quantity of atoms (such as, a molecule) and of materials measuring microns
- Probes
Nanoparticles or macromolecules that test the transport properties of tissues and biological barriers
- Delivery vector
A carrier nanoscale or microscale particle, for injection in the systemic circulation, that encapsulates anticancer therapy, and delivers it preferentially to target tissue
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Segal Lab of Computational Biology
http://genie.weizmann.ac.il/software/nucleoprediction.html
The Bay Area Physical Sciences-Oncology Center
The Dana-Farber Cancer Institute Physical Sciences-Oncology Center:
The Methodist Hospital Research Institute Physical Sciences-Oncology Center
http://www.methodisthealth.com/tmhri.cfm?id=39932
The Northwestern University Physical Sciences-Oncology Center
http://www.psoc.northwestern.edu/
The US National Cancer Institute’s Physical Sciences-Oncology Centers initiative
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 2.Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 3.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Armitage P, Doll R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 1957;11:161–169. doi: 10.1038/bjc.1957.22. One of the first mathematical approaches to explain age-specific cancer incidence curves. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fisher JC. Multiple-mutation theory of carcinogenesis. Nature. 1958;181:651–652. doi: 10.1038/181651b0. [DOI] [PubMed] [Google Scholar]
- 6.Vault RBNY, editor. The Edwin Smith Surgical Papyrus. Acad. Med. Rare Book Room; New York: [Author unknown.] (c1,600bce) [Google Scholar]
- 7.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nature Rev Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schedin P, Keely PJ. Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol. 2011;3:a003228. doi: 10.1101/cshperspect.a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montell DJ. Morphogenetic cell movements: diversity from modular mechanical properties. Science. 2008;322:1502–1505. doi: 10.1126/science.1164073. [DOI] [PubMed] [Google Scholar]
- 11.Mariappan YK, Glaser KJ, Ehman RL. Magnetic resonance elastography: a review. Clin Anat. 2010;23:497–511. doi: 10.1002/ca.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansma P, et al. The tissue diagnostic instrument. Rev Sci Instrum. 2009;80:054303. doi: 10.1063/1.3127602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krieg M, et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biol. 2008;10:429–436. doi: 10.1038/ncb1705. [DOI] [PubMed] [Google Scholar]
- 14.Salaita K, et al. Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor JE. Structure of singularities in soap-bubble-like and soap-film-like minimal surfaces. Ann Math. 1976;103:489–539. [Google Scholar]
- 16.Hayashi T, Carthew RW. Surface mechanics mediate pattern formation in the developing retina. Nature. 2004;431:647–652. doi: 10.1038/nature02952. [DOI] [PubMed] [Google Scholar]
- 17.Kafer J, Hayashi T, Maree AF, Carthew RW, Graner F. Cell adhesion and cortex contractility determine cell patterning in the Drosophila retina. Proc Natl Acad Sci USA. 2007;104:18549–18554. doi: 10.1073/pnas.0704235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilgenfeldt S, Erisken S, Carthew RW. Physical modeling of cell geometric order in an epithelial tissue. Proc Natl Acad Sci USA. 2008;105:907–911. doi: 10.1073/pnas.0711077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acar M, Pando BF, Arnold FH, Elowitz MB, van Oudenaarden A. A general mechanism for network-dosage compensation in gene circuits. Science. 2010;329:1656–1660. doi: 10.1126/science.1190544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sprinzak D, et al. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature. 2010;465:86–90. doi: 10.1038/nature08959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 22.Heppner GH, Miller FR. The cellular basis of tumor progression. Int Rev Cytol. 1998;177:1–56. doi: 10.1016/s0074-7696(08)62230-5. [DOI] [PubMed] [Google Scholar]
- 23.Crespi B, Summers K. Evolutionary biology of cancer. Trends Ecol Evol. 2005;20:545–552. doi: 10.1016/j.tree.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nature Rev Cancer. 2004;4:197–205. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- 25.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nature Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. Key reference elucidating evolutionary and ecological approaches to cancer. [DOI] [PubMed] [Google Scholar]
- 26.Brash DE, Zhang W, Grossman D, Takeuchi S. Colonization of adjacent stem cell compartments by mutant keratinocytes. Semin Cancer Biol. 2005;15:97–102. doi: 10.1016/j.semcancer.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Maley CC, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 28.Keller L. Levels of Selection in Evolution. Princeton Univ. Press; 1999. [Google Scholar]
- 29.Weinstein BS, Ciszek D. The reserve-capacity hypothesis: evolutionary origins and modern implications of the trade-off between tumor-suppression and tissue-repair. Exp Gerontol. 2002;37:615–627. doi: 10.1016/s0531-5565(02)00012-8. [DOI] [PubMed] [Google Scholar]
- 30.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haeno H, Levine RL, Gilliland DG, Michor F. A progenitor cell origin of myeloid malignancies. Proc Natl Acad Sci USA. 2009;106:16616–16621. doi: 10.1073/pnas.0908107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci USA. 1996;93:14800–14803. doi: 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desper R, et al. Inferring tree models for oncogenesis from comparative genome hybridization data. J Comput Biol. 1999;6:37–51. doi: 10.1089/cmb.1999.6.37. [DOI] [PubMed] [Google Scholar]
- 34.Goldie JH, Coldman AJ. Quantitative model for multiple levels of drug resistance in clinical tumors. Cancer Treat Rep. 1983;67:923–931. [PubMed] [Google Scholar]
- 35.Coldman AJ, Murray JM. Optimal control for a stochastic model of cancer chemotherapy. Math Biosci. 2000;168:187–200. doi: 10.1016/s0025-5564(00)00045-6. [DOI] [PubMed] [Google Scholar]
- 36.Michor F, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435:1267–1270. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 37.Coldman AJ, Goldie JH. A stochastic model for the origin and treatment of tumors containing drug-resistant cells. Bull Math Biol. 1986;48:279–292. doi: 10.1007/BF02459682. [DOI] [PubMed] [Google Scholar]
- 38.Skipper HE. The forty-year-old mutation theory of Luria and Delbruck and its pertinence to cancer chemotherapy. Adv Cancer Res. 1983;40:331–363. doi: 10.1016/s0065-230x(08)60683-1. [DOI] [PubMed] [Google Scholar]
- 39.Iwasa Y, Michor F, Nowak MA. Evolutionary dynamics of escape from biomedical intervention. Proc Biol Sci. 2003;270:2573–2578. doi: 10.1098/rspb.2003.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komarova NL, Wodarz D. Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci USA. 2005;102:9714–9719. doi: 10.1073/pnas.0501870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durrett R, Moseley S. Evolution of resistance and progression to disease during clonal expansion of cancer. Theor Popul Biol. 2010;77:42–48. doi: 10.1016/j.tpb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Harnevo LE, Agur Z. The dynamics of gene amplification described as a multitype compartmental model and as a branching process. Math Biosci. 1991;103:115–138. doi: 10.1016/0025-5564(91)90094-y. [DOI] [PubMed] [Google Scholar]
- 43.Goldie JH, Coldman AJ. The genetic origin of drug resistance in neoplasms: implications for systemic therapy. Cancer Res. 1984;44:3643–3653. [PubMed] [Google Scholar]
- 44.Day RS. Treatment sequencing, asymmetry, and uncertainty: protocol strategies for combination chemotherapy. Cancer Res. 1986;46:3876–3885. [PubMed] [Google Scholar]
- 45.Citron ML, et al. Randomized trial of dose-dense versus conventionally scheduled and sequential versus concurrent combination chemotherapy as postoperative adjuvant treatment of node-positive primary breast cancer: first report of Intergroup Trial C9741/Cancer and Leukemia Group B Trial 9741. J Clin Oncol. 2003;21:1431–1439. doi: 10.1200/JCO.2003.09.081. [DOI] [PubMed] [Google Scholar]
- 46.Komarova NL, Katouli AA, Wodarz D. Combination of two but not three current targeted drugs can improve therapy of chronic myeloid leukemia. PLoS ONE. 2009;4:e4423. doi: 10.1371/journal.pone.0004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foo J, Michor F. Evolution of resistance to anti-cancer therapy during general dosing schedules. J Theor Biol. 2010;263:179–188. doi: 10.1016/j.jtbi.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knudson AG. Two genetic hits (more or less) to cancer. Nature Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 49.Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varmus H. The new era in cancer research. Science. 2006;312:1162–1165. doi: 10.1126/science.1126758. [DOI] [PubMed] [Google Scholar]
- 51.Weir B, Zhao X, Meyerson M. Somatic alterations in the human cancer genome. Cancer Cell. 2004;6:433–438. doi: 10.1016/j.ccr.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 52.Futreal PA, et al. A census of human cancer genes. Nature Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 56.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nature Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 57.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nature Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang G, Christensen LA, Vasquez KMZ. DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci USA. 2006;103:2677–2682. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao J, Bacolla A, Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huppert JL. Structure, location and interactions of G.-quadruplexes. FEBS J. 2010;277:3452–3458. doi: 10.1111/j.1742-4658.2010.07758.x. [DOI] [PubMed] [Google Scholar]
- 63.Lipps HJ, Rhodes DG. quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009;19:414–422. doi: 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 64.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nature Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 65.Sun D, Hurley LH. Biochemical techniques for the characterization of G-quadruplex structures: EMSA, DMS footprinting, and DNA polymerase stop assay. Methods Mol Biol. 2010;608:65–79. doi: 10.1007/978-1-59745-363-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De S, Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nature Struct Mol Biol. 2011 Jul 3; doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kruisselbrink E, et al. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective. C elegans Curr Biol. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 68.Pontier DB, Kruisselbrink E, Guryev V, Tijsterman M. Isolation of deletion alleles by G4 DNA-induced mutagenesis. Nature Methods. 2009;6:655–657. doi: 10.1038/nmeth.1362. [DOI] [PubMed] [Google Scholar]
- 69.Boan F, Gomez-Marquez J. In vitro recombination mediated by G-quadruplexes. Chembiochem. 2010;11:331–334. doi: 10.1002/cbic.200900612. [DOI] [PubMed] [Google Scholar]
- 70.Attolini CS, et al. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc Natl Acad Sci USA. 2010;107:17604–17609. doi: 10.1073/pnas.1009117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hartl DL, Clark AG. Principles of Population Genetics. 4. Sinauer Associates; 2007. [Google Scholar]
- 72.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 73.Zhu Y, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abdel-Wahab O, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70:447–452. doi: 10.1158/0008-5472.CAN-09-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachoo RM, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 77.Zhu H, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci USA. 2009;106:2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. This paper showed that genomes encode intrinsically preferred locations for many of their nucleosomes, and showed that these positions seemed to facilitate diverse and specific aspects of chromosome function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 80.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 81.Field Y, et al. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nature Genet. 2009;41:438–445. doi: 10.1038/ng.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Field Y, et al. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010;24:748–753. doi: 10.1101/gad.1913210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Getun IV, Wu ZK, Khalil AM, Bois PRJ. Nucleosome occupancy landscape and dynamics at mouse recombination hotspots. EMBO Rep. 2010;11:555–560. doi: 10.1038/embor.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sasaki S, et al. Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Science. 2009;323:401–404. doi: 10.1126/science.1163183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanzer M, Wertheimer SP, de Bruin D, Ravetch JV. Chromatin structure determines the sites of chromosome breakages in Plasmodium falciparum. Nucleic Acids Res. 1994;22:3099–3103. doi: 10.1093/nar/22.15.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pryciak PM, Varmus HE. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell. 1992;69:769–780. doi: 10.1016/0092-8674(92)90289-o. [DOI] [PubMed] [Google Scholar]
- 89.Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL. DNA transposon Hermes inserts into DNA in nucleosome-free regionsin vivo. Proc Natl Acad Sci USA. 2010;107:21966–21972. doi: 10.1073/pnas.1016382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palomera-Sanchez Z, Zurita M. Open, repair and close again: chromatin dynamics and the response to UV-induced DNA damage. DNA Repair. 2010;10:119–125. doi: 10.1016/j.dnarep.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 91.Bucceri A, Kapitza K, Thoma F. Rapid accessibility of nucleosomal DNA in yeast on a second time scale. EMBO J. 2006;25:3123–3132. doi: 10.1038/sj.emboj.7601196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prendergast JGD, et al. Chromatin structure and evolution in the human genome. BMC Evol Biol. 2007;7:72. doi: 10.1186/1471-2148-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q Rev Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 94.Cloutier TE, Widom J. Spontaneous sharp bending of double-stranded DNA. Mol Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- 95.Segal E, Widom J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol. 2009;19:65–71. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thåström A, Bingham LM, Widom J. Nucleosomal locations of dominant DNA sequence motifs for histone-DNA interactions and nucleosome positioning. J Mol Biol. 2004;338:695–709. doi: 10.1016/j.jmb.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 97.Morozov A, et al. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009;37:4707–4722. doi: 10.1093/nar/gkp475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tolstorukov MY, Colasanti AV, McCandlish DM, Olson WK, Zhurkin VB. A novel roll-and-slide mechanism of DNA folding in chromatin: implications for nucleosome positioning. J Mol Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Geggier S, Vologodskii A. Sequence dependence of DNA bending rigidity. Proc Natl Acad Sci USA. 2010;107:15421–15426. doi: 10.1073/pnas.1004809107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wiggins PA, et al. High flexibility of DNA on short length scales probed by atomic force microscopy. Nature Nanotech. 2006;1:137–141. doi: 10.1038/nnano.2006.63. [DOI] [PubMed] [Google Scholar]
- 101.Lavery R, et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010;38:299–313. doi: 10.1093/nar/gkp834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zakrzewska K, Bouvier B, Michon A, Blanchet C, Lavery R. Protein-DNA binding specificity: a grid-enabled computational approach applied to single and multiple protein assemblies. Phys Chem Chem Phys. 2009;11:10712–10721. doi: 10.1039/b910888m. [DOI] [PubMed] [Google Scholar]
- 103.Lankas F, et al. On the parameterization of rigid base and basepair models of DNA from molecular dynamics simulations. Phys Chem Chem Phys. 2009;11:10565–10588. doi: 10.1039/b919565n. [DOI] [PubMed] [Google Scholar]
- 104.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. This study measured intrinsic DNA sequence preferences of nucleosomes in a purely in vitro experiment involving purified yeast genomic DNA and purified histones only. A thermodynamic model of nucleosome–DNA interactions based on these data is highly predictive of the distribution of nucleosomes in vivo, proving that much of the in vivo nucleosome organization is explicitly encoded in the genomic DNA sequence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fraser RM, Allan J, Simmen MW. In silico approaches reveal the potential for DNA sequence-dependent histone octamer affinity to influence chromatin structure in vivo. J Mol Biol. 2006;364:582–598. doi: 10.1016/j.jmb.2006.08.092. [DOI] [PubMed] [Google Scholar]
- 106.Chevereau G, Palmeira L, Thermes C, Arneodo A, Vaillant C. Thermodynamics of intragenic nucleosome ordering. Phys Rev Lett. 2009;103:188103. doi: 10.1103/PhysRevLett.103.188103. [DOI] [PubMed] [Google Scholar]
- 107.Schwab DJ, Bruinsma RF, Rudnick J, Widom J. Nucleosome switches. Phys Rev Lett. 2008;100:228105. doi: 10.1103/PhysRevLett.100.228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Segal E, Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nature Rev Genet. 2009;10:443–456. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raveh-Sadka T, Levo M, Segal E. Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res. 2009;19:1480–1496. doi: 10.1101/gr.088260.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Segal E, Widom J. What controls nucleosome positions? Trends Genet. 2009;25:335–343. doi: 10.1016/j.tig.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Strukov YG, Belmont AS. Mitotic chromosome structure: reproducibility of folding and symmetry between sister chromatids. Biophys J. 2009;96:1617–1628. doi: 10.1016/j.bpj.2008.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Subramanian H, et al. Optical methodology for detecting histologically unapparent nanoscale consequences of genetic alterations in biological cells. Proc Natl Acad Sci USA. 2008;105:20118–20123. doi: 10.1073/pnas.0804723105. Key reference showing that partial wave spectroscopy can be a valuable tool for the diagnosis of cancerous lesions by imaging sites far removed from the lesion itself. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Subramanian H, et al. Nanoscale cellular changes in field carcinogenesis detected by partial wave spectroscopy. Cancer Res. 2009;69:5357–5363. doi: 10.1158/0008-5472.CAN-08-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Damania D, et al. Role of cytoskeleton in controlling the disorder strength of cellular nanoscale architecture. Biophys J. 2010;99:989–996. doi: 10.1016/j.bpj.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim JS, Pradhan P, Backman V, Szleifer I. The influence of chromosome density variations on the increase in nuclear disorder strength in carcinogenesis. Phys Biol. 2011;8:015004. doi: 10.1088/1478-3975/8/1/015004. [DOI] [PubMed] [Google Scholar]
- 116.Hudson TJ, et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 118.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Beerenwinkel N, et al. Genetic progression and the waiting time to cancer. PLoS Comput Biol. 2007;3:e225. doi: 10.1371/journal.pcbi.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yachida S, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Campbell PJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Huh D, Paulsson J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nature Genet. 2011;43:95–100. doi: 10.1038/ng.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. J Clin Oncol. 2011;29:1382–1391. doi: 10.1200/JCO.2010.28.2319. [DOI] [PubMed] [Google Scholar]
- 125.Hondermarck H. Breast cancer: when proteomics challenges biological complexity. Mol Cell Proteomics. 2003;2:281–291. doi: 10.1074/mcp.R300003-MCP200. [DOI] [PubMed] [Google Scholar]
- 126.Fidler IJ, Hart IR. Biological diversity in metastatic neoplasms: origins and implications. Science. 1982;217:998–1003. doi: 10.1126/science.7112116. [DOI] [PubMed] [Google Scholar]
- 127.Copeland NG, Jenkins NA. Deciphering the genetic landscape of cancer--from genes to pathways. Trends Genet. 2009;25:455–462. doi: 10.1016/j.tig.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 128.Wooster R, Bachman KE. Catalogue, cause, complexity and cure; the many uses of cancer genome sequence. Curr Opin Genet Dev. 2010;20:336–341. doi: 10.1016/j.gde.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 129.Auffray C, Imbeaud S, Roux-Rouquie M, Hood L. From functional genomics to systems biology: concepts and practices. C R Biol. 2003;326:879–892. doi: 10.1016/j.crvi.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 130.Liu ET, Kuznetsov VA, Miller LD. In the pursuit of complexity: systems medicine in cancer biology. Cancer Cell. 2006;9:245–247. doi: 10.1016/j.ccr.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 131.Check Hayden E. Cancer complexity slows quest for cure. Nature. 2008;455:148. doi: 10.1038/455148a. Fundamental reference for the understanding of the complexity of cancer. [DOI] [PubMed] [Google Scholar]
- 132.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 133.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28:181–188. doi: 10.1016/j.tibtech.2009.12.007. In this paper, cancer is presented as a disease of multiscale mass transport deregulation that requires multiscale physics for its investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moore NM, Kuhn NZ, Hanlon SE, Lee JS, Nagahara LA. De-convoluting cancer’s complexity: using a ‘physical sciences lens’ to provide a different (clearer) perspective of cancer. Phys Biol. 2011;8:010302. doi: 10.1088/1478-3975/8/1/010302. [DOI] [PubMed] [Google Scholar]
- 135.Bearer EL, et al. Multiparameter computational modeling of tumor invasion. Cancer Res. 2009;69:4493–4501. doi: 10.1158/0008-5472.CAN-08-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cristini V, Lowengrub J. Multiscale Modeling Of Cancer: An Integrated Experimental And Mathematical Modeling Approach. Cambridge Univ. Press; 2010. [Google Scholar]
- 137.Kim P, et al. In vivo wide-area cellular imaging by side-view endomicroscopy. Nature Methods. 2010;7:303–305. doi: 10.1038/nmeth.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ananta JS, et al. Geometrical confinement of gadolinium-based contrast agents in nanoporous particles enhances T1 contrast. Nature Nanotechnol. 2010;5:815–821. doi: 10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tasciotti E, et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nature Nanotechnol. 2008;3:151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 140.Tanaka T, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70:3687–3696. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ferrari M. Vectoring siRNA therapeutics into the clinic. Nature Rev Clin Oncol. 2010;7:485–486. doi: 10.1038/nrclinonc.2010.131. [DOI] [PubMed] [Google Scholar]
- 142.Decuzzi P, Ferrari M. Design maps for nanoparticles targeting the diseased microvasculature. Biomaterials. 2008;29:377–384. doi: 10.1016/j.biomaterials.2007.09.025. [DOI] [PubMed] [Google Scholar]
- 143.Decuzzi P, Ferrari M. The adhesive strength of non-spherical particles mediated by specific interactions. Biomaterials. 2006;27:5307–5314. doi: 10.1016/j.biomaterials.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 144.Gentile F, Ferrari M, Decuzzi P. The transport of nanoparticles in blood vessels: the effect of vessel permeability and blood rheology. Ann Biomed Eng. 2008;36:254–261. doi: 10.1007/s10439-007-9423-6. [DOI] [PubMed] [Google Scholar]
- 145.Serda RE, et al. Logic-embedded vectors for intracellular partitioning, endosomal escape, and exocytosis of nanoparticles. Small. 2010;6:2691–2700. doi: 10.1002/smll.201000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nickerson JA, Krockmalnic G, Wan KM, Penman S. The nuclear matrix revealed by eluting chromatin from a cross-linked nucleus. Proc Natl Acad Sci USA. 1997;94:4446–4450. doi: 10.1073/pnas.94.9.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]