

Abstract
Unidirectional fluid flow plays an essential role in the breaking of left-right (L-R) symmetry in mouse embryos, but it has remained unclear how the flow is sensed by the embryo. We report that the Ca2+ channel Pkd2 is required specifically in the peri-nodal crown cells for sensing the nodal flow. Examination of mutant forms of Pkd2 shows that the ciliary localization of Pkd2 is essential for correct L-R patterning. Whereas Kif3a mutant embryos, which lack all cilia, failed to respond to an artificial flow, restoration of primary cilia in crown cells rescued the response to the flow. Our results thus suggest that nodal flow is sensed in a manner dependent on Pkd2 by the cilia of crown cells located at the edge of the node.
(Please start the introduction with a more general sentence about LR asymmetry to draw in the broad reader —which organs are asymmetric, etc.— and define the ventral node for the nonspecialist.) Most of the visceral organs in vertebrates exhibit left-right (L-R) asymmetry in their shape or position. The breaking of L-R symmetry in the embryos of many vertebrates is mediated by a unidirectional fluid flow in the ventral node (an embryonic cavity at the midline filled with extra-embryonic fluid) or its equivalent structure (1, 2). The flow may transport a determinant molecule or provide mechanical force. However, it has remained unclear how the flow is sensed. We have now addressed this issue with the use of mice lacking Polycystin-2 (Pkd2, also known as TRPP2), a gene encoding a Ca2+-permeable cation channel implicated in polycystic kidney disease in humans(3, 4).
Pkd2 is required only in peri-nodal crown cells
The Pkd2−/− mouse was previously shown to lose Nodal expression in the lateral plate mesoderm (LPM) and to exhibit typical L-R patterning defects(5). Pkd2 is expressed ubiquitously at the early somite stage of mouse embryos(5) (fig. S1); however, it has been unclear whether the function of Pkd2 in L-R patterning is executed in the node, LPM, or other region of the embryo. We first examined if the LPM of the Pkd2−/− embryo is competent for Nodal signaling. When an expression vector for Nodal was introduced to a small region of the right LPM of the Pkd2−/− embryo, expression of endogenous Nodal gene was induced in the entire region of the right LPM (fig. S2), suggesting that the LPM of the Pkd2−/− embryo remains competent to express Nodal. To further localize the site of Pkd2 action, we established three types of transgenic mice that express Pkd2 specifically in the node and examined whether such transgenes were able to prevent the L-R defects of the Pkd2−/− mouse (Fig. 1; fig. S3). In one transgene, Foxa2NNE-hsp-Pkd2-IRES-LacZ (Fig. 1A), Pkd2 expression is driven by a node/notochord-specific enhancer of the mouse Foxa2 gene. This transgene is expressed in the midline including the node (Fig. 1A), and it restored left-sided expression of Nodal and Pitx2 in the LPM of Pkd2−/− embryos (rescued/tested embryos: 6/6 for Nodal and 6/6 for Pitx2) (Fig. 1C).
Figure 1. Specific requirement for Pkd2 in crown cells of the node for correct L-R patterning.

(A, B) Schematic representation of two types of node-specific Pkd2 transgeneare shown on the top. Their expression patterns were examined by staining of transgenic embryos at embryonic day (E) 8.0 with X-gal. Schematic (left panels) and actual (middle panels) views of whole embryos, and transverse sections of the node (right panels) are shown. Inset of the middle panel in B shows node from ventral view at higher magnification. Note that expression of the NDE-driven transgene is highly specific to crown cells of the node (B). A, anterior; P, posterior; L, left; R, right. Scale bars, 50μm. (C) Whole-mount in situ hybridization analysis of the expression of Nodal and Pitx2 in E8.0 embryos of the indicated genotypes. Left-sided expression of Nodal and Pitx2 in LPM is lost in Pkd2−/− embryos but is restored (red arrowheads) in Pkd2−/−;Foxa2-Pkd2 and Pkd2−/−;NDE-Pkd2 embryos. Scale bar, 500μm.
The ventral node contains two different types of ciliated cells: pit cells located in the central region of the node and crown cells located at the edge (fig. S3A)(1). The second Pkd2 transgene studied, NDE-hsp-Pkd2-IRES-LacZ (Fig. 1B), is driven by the node-specific enhancer (NDE) of Nodal, which is active specifically in the crown cells. This transgene was able to rescue left-sided expression of Nodal and Pitx2 in the LPM of Pkd2−/− embryos (6/6 and 3/3, respectively) (Fig. 1C). Finally, expression of Pkd2 specifically in the pit cells with dgFoxa2-hsp-Pkd2-IRES-LacZ (fig. S3B) was unable to rescue the left-sided expression of Nodal and Pitx2 (0/1 and 0/3 embryos, respectively) (fig. S3C). These results thus suggested that Pkd2 is required exclusively in crown cells of the node for correct L-R determination.
Cerl2 is the major target of Pkd2-mediated signaling
Initial breaking of L-R symmetry by leftward nodal flow is followed by asymmetric (R>L) expression of Cerl2 in crown cells(6, 7). Given that Pkd2 is required specifically in crown cells, we examined crown cell–specific markers in Pkd2−/− embryos. Gdf1, which is bilaterally coexpressed in crown cells and is required for subsequent Nodal expression in left LPM(8, 9), was normally expressed in Pkd2−/− embryos (fig. S4A). However, L-R asymmetric gene expression in crown cells was impaired in the mutant embryos. Expression of Cerl2 in crown cells is greater on the right side in wild-type embryos (fig. S4A)(6), but it was expressed equally on the two sides in Pkd2−/− embryos (fig. S4A). Nodal expression, which shows a subtle asymmetry in the wild-type embryo (right expression lower than left) (10), was bilaterally equal in Pkd2−/− embryos (fig. S4A). These results suggested that crown cells are correctly specified but lose asymmetric gene expression in the absence of Pkd2.
The higher expression of Cerl2 on the right in crown cells is required for subsequent asymmetric expression of Nodal in LPM(6). To clarify the relation between Pkd2 and Cerl2, we examined possible interaction between the two genes by analyzing Pkd2−/−;Cerl2−/− double-mutant embryos (fig. S4B). Nodal expression in LPM was always absent in Pkd2−/− embryos (5/5), whereas it was either left-sided (2/11), right-sided (3/11), bilateral (2/11), or absent (4/11) in Pkd2−/−;Cerl2−/− embryos (fig. S4B, C). The Nodal expression pattern in the double mutant was thus similar to that in Cerl2−/− embryos (fig. S4B, C), indicating that Cerl2 is the major target of Pkd2-mediated signaling for L-R asymmetric patterning (fig. S4D).
Pkd2 in crown cells is required for sensing nodal flow
Particle image velocimetry (PIV) revealed that nodal flow is maintained in the Pkd2−/− mutant (Fig. 2A), suggesting that Pkd2 might serve to sense a flow-derived signal(s) in crown cells. We tested this possibility with the use of a transcriptional enhancer that responds to nodal flow. This enhancer (ANE) is derived from the human Lefty1 gene and exhibits asymmetric (L>R) activity in crown cells (Fig. 2B)(11). Furthermore, ANE was found to respond to artificial flow. The relative L-R activity of ANE was thus reversed when a rightward flow was imposed on the wild-type embryo (Fig. 2C, D). In Pkd2−/− embryos, ANE activity was R = L (Fig. 2B), despite the fact that the mutant possesses morphologically normal cilia (fig. S5) and functional flow (Fig. 2A), suggesting that crown cells of Pkd2−/− embryos fail to sense the flow.
Figure 2. Pkd2 is necessary for sensing of nodal flow.
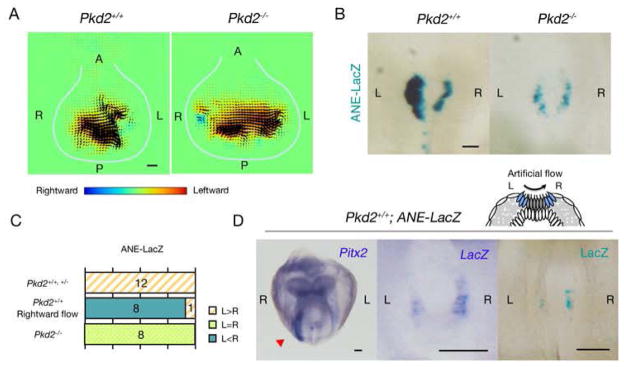
(A) Nodal flows in wild-type and Pkd2−/− embryos were examined by PIV analysis at E8.0. Flow is maintained in the Pkd2−/− embryo. Each arrowhead denotes the direction and speed of the flow at the indicated position. The color scale indicates the direction and magnitude of the flow velocity (leftward in yellow and red, rightward in blue). White lines indicate the outlines of the node. Scale bar, 10μm. (B) E8.0 Pkd2+/+ and Pkd2−/− embryos harboring the ANE-LacZ transgene were stained with X-gal. Scale bar, 50μm. (C) The L-R patterns of ANE activities in embryos of the indicated genotypes either in situ or under conditions of artificial rightward flow in vitro are summarized. The numbers of embryos showing each pattern are indicated. (Adjust the color/pattern for C. The small box definitions of colors will not show the pattern of top line after figure reduction. ---response: color/patterns have been changed and it is easy to see the difference in color/pattern) (D) Pkd2+/+ embryos with ANE-LacZ were cultured under the influence of a rightward artificial flow from early headfold to six-somite stages. Artificial rightward flow reversed the pattern of Pitx2 expression in LPM (left panel, red arrowhead) and that of ANE activity in crown cells, as detected by in situ hybridization of LacZ mRNA (middle) and X-gal staining (right). Scale bars, 100μm. Why does the “L” and “R” designation switch from left and right between the figure panels? Figure 1 shows “L” on the right side of figure; Figure 2 shows “L” and “R” in different panel sides in the figure. ---Response: embryos showing gene expression in the lateral plate such as ones in Fig. 1C are frontal views, so that “L ” is shown on the right side. Embryos showing gene expression in the node such as ones in Fig. 2B were back views, so that “L ” was shown on the left side. To avoid confusion, however, all embryo photos are shown in such a way that the right side of an embryo is always shown on the left side.) (Define N and C.---response: N & C are now deleted)
Given that Pkd2 encodes a Ca2+ permeable ion channel, we examined Ca2+ signaling in crown cells by generating a transgenic mouse that expresses Ca2+ indicator GCaMP2, which exhibits highly cooperative Ca2+ binding over the physiological range of cellular free Ca2+ (KD= 146 nM)(12), specifically in crown cells. Indeed Ca2+ signaling was detected in crown cells, but it was present bilaterally and was retained in Pkd2−/− embryos (fig. S6). It should be noted that Ca2+ asymmetry previously observed with exogenous dyes (13, 14) occurs in the endoderm, a different tissue around the node. To clarify the role of Ca2+ signaling, transgenic embryos harboring ANE-lacZ were incubated with various Ca2+ signaling blockers. GdCl3(an inhibitor of stretch-sensitive TRP channels), 2-ABP (an inhibitor of IP3 receptor) and Thapsigargin (an inhibitor of Ca2+-ATPase in ER) disrupted L>R asymmetry of ANE activity, whereas Ruthenium Red (a potent inhibitor of intracellular Ca2+release by ryanodine receptors) did not (Fig. 3). Treatment with Thapsigargin did not significantly reduce the level of Ca2+ signaling within crown cells (fig. S7), likely because a single inhibitor would only affect a portion of Ca2+ signaling within a cell. Rotational movement of pit cell cilia and ciliary localization of Pkd2 were maintained by treatment with GdCl3and Thapsigargin (fig. S8). These results suggest that Ca2+ signaling mediated by Pkd2 and IP3 receptor is essential for generating L-R asymmetry at the node (Fig. 3B).
Figure 3. Calcium signal is essential for L-R symmetry breaking in the node crown cells.

(A) Activity of ANE is disrupted when Ca2+ signal blockers are treated. Two pathways that lead to Ca2+ release from endoplasmic reticulum were examined (B). L>R asymmetric ANE activity is maintained in control and Ruthenium Red- treated embryos, whereas it is disrupted with Gd3+, 2-aminoethoxydiphenyl borate (2-APB) or Thapsigargin (TG). Scale bar, 50μm. (C) The effects of each reagent on L-R asymmetric ANE activity are summarized. The numbers of embryos showing each pattern are indicated.
Ciliary localization of Pkd2 is essential for correct L-R decision
Pkd2 protein resides in the primary cilia of renal epithelial cells(15). In crown cells as well as pit cells of the node, endogenous Pkd2 was also localized in the cilia (fig. S9A; (13)). We also examined the subcellular localization of Pkd2 with the use of a transgenic mouse that expresses a Pkd2::Venus fusion protein in crown cells (Fig. 4A). This fusion protein was shown to be functional by the observation that its expression in crown cells corrected the L-R defects of Pkd2−/− embryos, and was preferentially localized to the cilia of crown cells (Fig. 4A). Expression of Pkd2::Venus protein did not influence motility of cilia (movie S1). Notably, live imaging of Pkd2::Venus-labeled cilia at the presomite stage showed that most of the cilia were immotile (fig. S10; movie S2). The frequency of motile cilia gradually increased between the presomite stage and the three-somite stage (fig. S10). However, most (90%~) crown cell cilia were immotile while ~60% of pit cell cilia are already motile at the presomite stage (fig. S10), the stage at which Cerl2 expression in crown cells begins to exhibit L-R asymmetry (11).
Figure 4. Ciliary localization of Pkd2 is required for correct L-R determination.

(A) Schematic representation of NDE-driven Pkd2::Venus transgene is shown on the top. Crown cell-specific expression of the transgene in an E8.0 embryo was confirmed by whole-mount in situ hybridization with a Venus probe (upper right panel). Scale bar, 50μm. Left-sided expression of Nodal was restored in Pkd2−/−; NDE-Pkd2::Venus embryo (lower right panel), suggesting that Pkd2::Venus is functional. Scale bar, 500μm. Note that Pkd2::Venus protein is preferentially localized to cilia (left panel, red arrowheads). Scale bar, 10 μm. (B) Transgenic embryos expressing Pkd2(E442G)::Venus or Pkd2(D509)::Venus in crown cells. Note that both proteins are unable to localize to cilia. Scale bar, 10 μm. (Define N and C. ---response: N & C are removed) (C) Localization of Pkd2(E442G)::Venus was confirmed by immunofluorescence staining with antibodies to acetylated tubulin (red—difficult to see/read tubulin---response: we have changed the color brighter so that it is easier to see), to Venus; GFP (green) and phalloidin (cyan). Scale bar, 10μm. (D) Open probability of Pkd2(wt), Pkd2(E442G) and Pkd2(R6G-G819X) channels in the presence of increasing cytosolic Ca2+ concentration. Note that Pkd2(E442G) retains normal channel activity whereas Pkd2(R6G-G819X) loses it. Error bars represent ± SEM. (E) The relationship between ciliary localization, Ca2+ channel activity, ability of L-R rescue and interaction with Pkd1l1 is summarized for various Pkd2 mutants. (Please remove E from the figures section and make it a separate Table.---response: we have made E a separate Table).
Ciliary localization of Pkd2 may depend on its interaction with Pkd1l1, another ciliary protein whose deficiency in mice and zebrafish results in L-R defects similar to those of the Pkd2−/− mutant(16, 17). To test if Pkd2 may function in cilia of crown cells, various mutant forms of Pkd2 were examined for their abilities to localize to crown cell cilia and to correct the L-R defects of Pkd2−/− embryos (Fig. 4B-E; fig. S9; fig. S11). Although a missense mutation of Pkd2 (E442G) induces L-R defects identical to those of Pkd1l1 mutant mouse(18), a Pkd2(E442G)::Venus fusion protein was unable to localize to the cilia of crown cells (Fig. 4B, C) and cilia of cultured LLC-PK1 cells (fig. S12). Pkd2(E442G) can interact with Pkd1l1(fig. S13), suggesting that ciliary localization of Pkd2 requires Pkd1l1-independent mechanisms. Pkd2(R6G), a missense mutant unable to localize to primary cilia in cultured LLC-PK1 cells(19), was found to localize to the cilia of crown cells (fig. S9B) and rescued L-R patterning in the Pkd2−/− mutant (fig. S11). Pkd2( 5–73), which lacks the NH2-terminal region (residues 5 to 73) including the R6VxP motif(19), was also able to localize to the cilium when expressed in crown cells (fig. S9C). Pkd2(R6G-G819X), which harbors the R6G missense mutation and lacks the COOH-terminal region, was unable to localize to the crown cell cilium (fig. S9D) or to correct the L-R defects of the Pkd2−/− mutant (fig. S11). Similarly, Pkd2(D509V), a missense mutant associated with polycystic kidney disease in humans(20), failed both to localize to cilia of crown cells (Fig. 4B) and to normalize the Pkd2−/− phenotype (fig. S11). Two mutants that were unable to localize to crown cell cilia (E442G and R6G-G819X) were examined for their channel activity (Fig. 4D). Although Pkd2(R6G-G819X) lost channel activity, Pkd2(E442G) retained it (Fig. 4D). Our findings that Pkd2(E442G) retains channel activity yet is unable to localize to crown cell cilia (Fig. 4B, C) indicate that ciliary localization of Pkd2 in crown cell is essential for correct L-R decision.
Crown cell cilia function as sensors of nodal flow
Finally, we examined directly whether cilia of the crown cells function as sensors of nodal flow with the use of an IFT (intraflagellar transport) mouse mutant. Kif3a and Kif3b are expressed ubiquitously in mouse embryo and encode motor proteins that are required for formation of cilia(21–23). Kif3a−/− mutant embryos thus lack all node cilia, including those of both pit and crown cells and exhibit L-R defects(22, 23). A transgene that confers Kif3a expression specifically in crown cells (NDE-hsp-Kif3a-IRES-LacZ) restored cilia formation exclusively in these cells (Fig. 5A). Although Nodal expression in LPM was bilateral (L=R) in Kif3a−/− embryos (8/8), restoration of cilia formation in crown cells by the NDE-Kif3a transgene rescued left-sided Nodal expression in LPM in Kif3a−/− embryos (3/3) (Fig. 5B, C). PIV analysis revealed a weak leftward flow as well as vortical flow in the node of such embryos (fig. S14), likely because the number of motile cilia among crown cells is much smaller than that of immotile cilia (fig. S10). This result is consistent with our recent finding that as few as two rotating cilia are sufficient for symmetry breaking(24).
Figure 5. Nodal flow is sensed by the cilia of crown cells.

(A) An E8.0 transgenic embryo harboring NDE-Kif3a-IRES-LacZ was stained with X-gal, revealing crown cell specific expression of the transgene (left). Scale bar, 50μm. In scanning electron microscopy of the node of an E8.0 Kif3a−/− embryo with NDE-Kif3a, cilia are apparent at the edge (boxed by the red line), but not at the center (boxed by the blue line) of the node. Pale blue lines indicate the border between the endoderm and crown cells, with the dotted circle enclosing pit cells. The boxed regions on the left are shown at a higher magnification on the right. Scale bars, 5 μm. (B) Expression of L-R marker genes in embryos of the indicated genotypes that were examined at E8.0 (in vivo) or cultured under the influence of a rightward artificial flow before analysis. Note that Pitx2 expression pattern of the Kif3a−/−;NDE-Kif3a embryo responded to the flow and is right-sided (red arrowhead). Scale bar, 500 μm. (C) The numbers of embryos showing each pattern of gene expression are summarized.
We then examined whether the embryos with cilia only in the crown cells are able to respond to artificial fluid flow. The Kif3a−/−;NDE-Kif3a embryos, as well as control Kif3a−/− embryos, were thus subjected to rightward artificial flow (Fig. 5B), and L-R patterning as revealed by Pitx2 expression in LPM was examined. Whereas Kif3a−/− embryos (5/5) failed to respond to the rightward flow, the Kif3a−/−;NDE-Kif3a embryos (3/3) did respond by showing right-sided Pitx2 expression in LPM (Fig. 5B, C). These results thus demonstrated that nodal flow is indeed sensed by crown cell cilia located at the edge of the node.
Our results indicate that the fluid flow in the node is sensed by cilia of peri-nodal crown cells via ciliary-localized Pkd2. Many proteins are known to be localized to primary cilia, and they are implicated in cilium-mediated signaling. However, the role of their ciliary localization has not been rigorously established. Identification of Pkd2(E442G), a L-R defect-causing mutant form of Pkd2 that retains Ca2+ channel activity yet is unable to localize to the cilia, provides the direct evidence showing that ciliary localization of a protein is indeed essential for cilium-mediated signaling.
Several questions remain unanswered. It is still not clear what the cilia sense in the breaking of L-R symmetry: Do they sense flow-transported chemicals or flow-generated mechanical forces? Pkd2 is localized to the primary cilium of kidney cells and vascular endothelial cells and mediates mechanosensation(25, 26). Circumstantial evidence, including our recent observation that as few as two rotating cilia are sufficient for L-R symmetry breaking(24), favors the possibility that crown cell cilia sense mechanical force. Also unknown is how cilia signal to Cerl2, the major target of the signal transmitted by nodal flow and Pkd2. Finally, our results support the previous proposal (13, 27) that there are two populations of cilia in the node, motile and immotile cilia, and that the former generate nodal flow whereas the latter sense the flow. Given that most cilia of crown cells are immotile at the time when L-R symmetry is broken, it is likely that the flow is sensed by immotile cilia, although we are not able to rigorously exclude the possibility that motile cilia of crown cells also sense the flow. Further studies with diverse approaches are needed to address these issues.
Supplementary Material
Table 1.
Properties of various Pkd2 mutants
| Name | Mutation | cilia localization | LR rescue | Ca2+channel property | Interaction with Pkd1l1 | |
|---|---|---|---|---|---|---|
| node | LLC-PK1 | |||||
| R6G | R6VxP point mutation | yes | ND | yes | ND | ND |
| Δ (5–73) | R6VxP deletion | yes | ND | ND | ND | ND |
| R6G,G819X | Δ coiled-coil domain | no | ND | no | impaired | ND |
| E442G | Irm4* | no | no | no* (ND) | normal | yes |
| D509V | human D511V** | no | no | no | impaired** | ND |
Acknowledgments
We thank Y. Cai for YCB9 antibodies to Pkd2; S. Somlo for very helpful comments on Pkd2 function; D.P. Norris for Pkd1l1-CC-GFP plasmid and A. Fukumoto, Y. Ikawa, K. Miyama, K. Mochida, H. Nishimura, and S. Ohishi for technical assistance. This work was supported by a grant from CREST of the Japan Science and Technology Corporation to H.H., an NIH grant P30 DK090744 to B.E.E. and HFSP grant ST00246/2003C and the Deutsche Forschungsgemeinschaft grant PE 853/2 to P.P. S.Y. was supported by a fellowship from the Japan Society for the Promotion of Science for Japanese Junior Scientists. I.Y.K. was supported by the American Heart Association Postdoctoral Fellowship R10682.
References and Notes
- 1.Shiratori H, Hamada H. Development. 2006;133:2095. doi: 10.1242/dev.02384. [DOI] [PubMed] [Google Scholar]
- 2.Hirokawa N, Tanaka Y, Okada Y. Cold Spring Harb Perspect Biol. 2009;1:a000802. doi: 10.1101/cshperspect.a000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Perrett S, et al. Proc Natl Acad Sci U S A. 2001;98:1182. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koulen P, et al. Nat Cell Biol. 2002;4:191. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 5.Pennekamp P, et al. Curr Biol. 2002;12:938. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 6.Marques S, et al. Genes Dev. 2004;18:2342. doi: 10.1101/gad.306504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schweickert A, et al. Curr Biol. Apr 27;20:738. [Google Scholar]
- 8.Tanaka C, Sakuma R, Nakamura T, Hamada H, Saijoh Y. Genes Dev. 2007;21:3272. doi: 10.1101/gad.1623907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rankin CT, Bunton T, Lawler AM, Lee SJ. Nat Genet. 2000;24:262. doi: 10.1038/73472. [DOI] [PubMed] [Google Scholar]
- 10.Collignon J, Varlet I, Robertson EJ. Nature. 1996;381:155. doi: 10.1038/381155a0. [DOI] [PubMed] [Google Scholar]
- 11.Kawasumi A, et al. Dev Biol. 2012;353:321. doi: 10.1016/j.ydbio.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tallini YN, et al. Proc Natl Acad Sci U S A. 2006;103:4753. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Cell. 2003;114:61. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y, Okada Y, Hirokawa N. Nature. 2005;435:172. doi: 10.1038/nature03494. [DOI] [PubMed] [Google Scholar]
- 15.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Curr Biol. 2002;12:R378. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 16.Field S, et al. Development. 2011;138:1131. doi: 10.1242/dev.058149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamura K, et al. Development. 2011;138:1121. doi: 10.1242/dev.058271. [DOI] [PubMed] [Google Scholar]
- 18.Ermakov A, et al. Dev Dyn. 2009;238:581. doi: 10.1002/dvdy.21874. [DOI] [PubMed] [Google Scholar]
- 19.Geng L, et al. J Cell Sci. 2006;119:1383. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds DM, et al. J Am Soc Nephrol. 1999;10:2342. doi: 10.1681/ASN.V10112342. [DOI] [PubMed] [Google Scholar]
- 21.Nonaka S, et al. Cell. 1998;95:829. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 22.Takeda S, et al. J Cell Biol. 1999;145:825. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Proc Natl Acad Sci U S A. 1999;96:5043. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinohara K, et al. Nat Commun. 2012;3:622. doi: 10.1038/ncomms1624. [DOI] [PubMed] [Google Scholar]
- 25.Nauli SM, et al. Nat Genet. 2003;33:129. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 26.AbouAlaiwi WA, et al. Circ Res. 2009;104:860. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabin CJ, Vogan KJ. Genes Dev. 2003;17:1. doi: 10.1101/gad.1053803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.