

Abstract
Branched sugar nucleosides have attracted much attention due to their biological activities. We have demonstrated that epoxy-sugar nucleosides serve as versatile precursor for the stereo-defined synthesis of these nucleoside derivatives on the basis of its ring opening with organoaluminum or organosilicon reagents. In this review article, novel methods for the synthesis of nucleoside analogues branched at the 1′ and 4′-position will be described. During this study, we could discover an anti-HIV agent, 4′-ethynylstavudine (Festinavir).
Festinavir showed more potent anti-HIV activity than the parent compound stavudine (d4T). Other significant properties of Festinavir are as follows: 1) much less toxic to various cells and also to mitochondorial DNA synthesis than d4T, 2) better substrate for human thymidine kinase than d4T, 3) resistant not only to chemical glycosidic bond cleavage but also to catabolism by thymidine phosphorylase, 4) the activity improves in the presence of a major mutation, K103N, associated with resistance to non-nucleoside reverse transcriptase inhibitors. Detailed profile of the antiviral activities, biology and pharmacology of Festinavir are also described.
Keywords: Epoxide, sugar, nucleoside, organoaluminum reagent, NRTIs, stavudine, anti-HIV-1 agent
(I) CHEMISTRY OF EPOXYSUGAR NUCLEOSIDES AND SYNTHESIS OF 2′,3′-DIDEHYDRO-3′-DEOXY-4′-ETHYNYLTHYMIDINE (4′-ETHYNYLSTAVUDINE)
(1) Background
Human Immunodeficiency virus (HIV) is causative agent of acquired immunodificiency syndrome (AIDS) and it is estimated that more than 33.3 million individuals worldwide are infected with this pathogen [1]. HIV is RNA virus and belongs to retrovirus family. Retrovirus has characteristic key enzyme, reverse transcriptase (RT), which catalyzes reverse transcription of viral RNA into provirus DNA. The enzyme is essential for lifecycle of HIV. Because the activity of RT in host-cell is quite low, it is rationale that RT is a target enzyme for developing anti-HIV agent.
In 1987, AZT was approved as the first anti-HIV drug, which is nucleoside reverse transcriptase inhibitor (NRTI) (Fig. 1). The discovery of AZT as antiretroviral agent stimulated the synthesis of sugar-modified nucleosides and the evaluation of their anti-HIV activity. These studies provided other clinically used anti-HIV derivatives such as zalcitabine (ddC), didanosine (ddI) and stavudine (d4T). However, the long-term usage of these anti-viral drugs lead to delayed toxicity in patients and/or emergence of drug resistant HIV variants. These circumstances necessitate novel anti-HIV agent with lower toxicity and wider anti-viral spectrum [2], and extensive synthetic research for developing novel anti-viral sugar-modified nucleosides has continued [3–6].
Fig. 1.
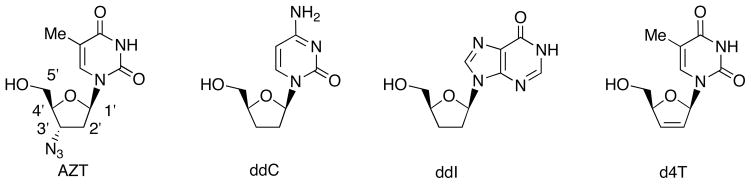
Anti- HIV Drugs: Nucleoside Reverse Transcriptase Inhibitors (NRTIs).
Among sugar-modified nucleosides, compounds substituted with carbon-substituent at the 1′-, 2′-, 3′-, 4′- or 5′-position of the pentose moiety are called branched-sugar nucleosides. Biologically active branched-sugar analogues are shown in Fig. 2. Anti-tumor nucleoside antibiotic angustmycin C, which is structurally-unique in being branched at the anomeric position of adenosine [6]. 2′-Methyladenosine has been reported to possess anti-HCV activity [5]. On the other hand, 3′-ethynylcytidine exhibits anti-tumor activity [8]. Furthermore, 4′-cyanothymidine shows significant inhibitory activity to HIV [9].
Fig. 2.
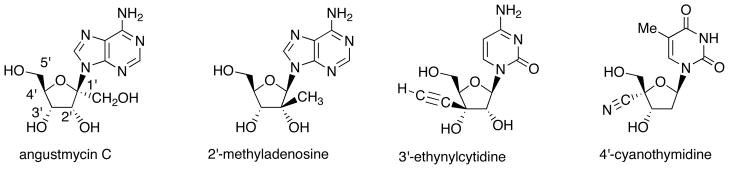
Biologically-active Branched-Sugar Nucleosides.
2′-Branched ribonucleosides such as 2′-methyladenosine have been synthesized by means of addition of carbon nucleophile to 2′-keto derivative available from oxidation of 2′-hydroxyl group of naturally occurring ribonucleosides (Fig. 3). Likewise, 3′-keto-nucleosides have been utilized for the synthesis of 3′-branched nucleosides exemplified by 3′-ethynylcytidine.
Fig. 3.
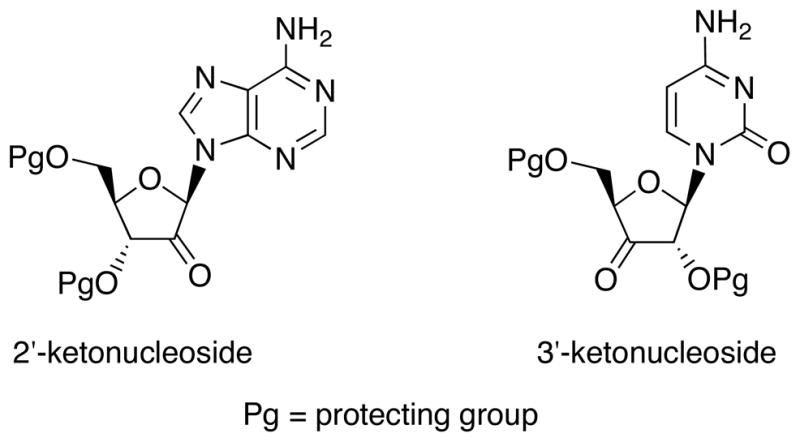
2′-Keto and 3′-Ketonucleosides Utilizing for the Synthesis of 2′-and 3′-Branched Derivatives.
On the contrary, the 1′ or 4′-position of nucleosides are inert for introducing the carbon-substituent because no functional groups are present at the position. Therefore, synthesis of 1′- or 4′-branched nucleosides such as angustmycin C and 4′-cyanothymidine has been difficult. Therefore, development of efficient method leading to the synthesis of these nucleosides has been challenging and arduous task in the nucleoside chemistry.
The first synthesis of 1′-branched derivatives starting from naturally occurring nucleoside has been carried out on the basis of nucleophilic substitution of 1, which was prepared from bromo-pivaloyloxylation of 1′,2′-unsaturated nucleosides, with organosilicon or organoaluminum reagent (Scheme 1) [10]. In this transformation, the desired 1′-α-carbon-substituted nucleoside 2 was obtained via neighboring group participation exerted by 2′-β-bromine atom of nucleoside anomeric carbenium ion I. Although 2 was converted into 2′-deoxy- and arabinofuranosyl nucleosides 3, the corresponding ribofuranosyl counterpart could not be synthesized.
Scheme 1.
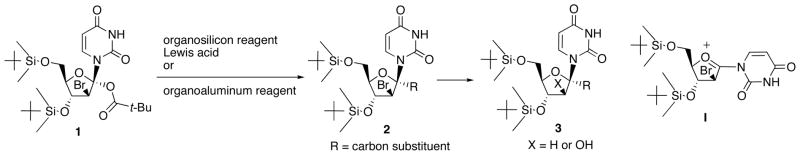
Synthesis of 1′-Branched Nucleosides 3 via 2 obtained on thde basis of Nucleophilc Substitution of 1.
To overcome this problem, reaction of samarium enolate II, generated by SmI2-mediated reduction of α-ketophenylselenide 4, with carbon electrophile has been reported (Scheme 2) [11]. Thus, reaction of 4 with aldehyde gave aldol 5, which was subjected to β-face-selective hydride reduction to provide 1′-carbon-subjected ribonucleoside 6.
Scheme 2.
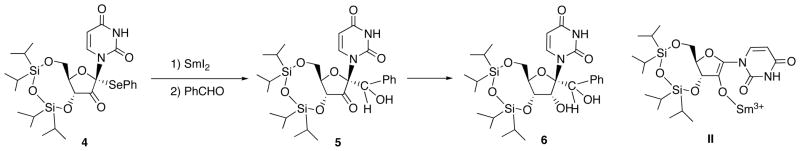
Synthesis of 1′-Branched Nucleosides on the basis of Electrophilic Substitution.
In the meantime, nucleoside anomeric radical has also been utilized as intermediate for the synthesis of 1′-branched nucleoside. The first report on the basis of this strategy is shown in Scheme 3 [12]. Thus, when bromo-pivaloyloxy derivative 7 was treated with tributyltin radical, the incipient 2′-carbon radical III was rearranged to anomeric radical IV through 1,2-acyloxy migration and subsequent reaction of IV with allyltributyltin gave 1′-allyl-arabinofuranosyluracil 8. In this case, sp2 hybridized anomeric radical reacted at α-face to give β-anomer as a sole product.
Scheme 3.
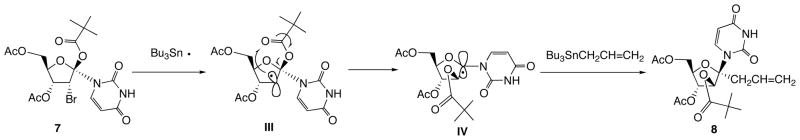
Nucleoside Anomeric Radical based Synthesis of 1′-Branched Nucleosides.
The other anomeric radical-based methods are shown in Schemes 4 and 5. The method shown in Scheme 4 is intermolecular radical reaction of 1′-phenylsulfanyldeoxyuridine 9 with allyltributyltin to give 1′-allyl-2′-deoxynucleoside (10) [13]. The other method depicted in Scheme 5 is intramolecular radical cyclization of 11 (Scheme 5) [14]. The cyclized product 12 could be transformed into 1′-vinyluridine (13) by means of fluoride ion-mediated E2-elimination. These above-mentioned novel methods for the synthesis of 1′-branched nucleosides have some drawbacks such as limited substituent to be introduced and difficulty of selective synthesis of the β-anomers.
Scheme 4.
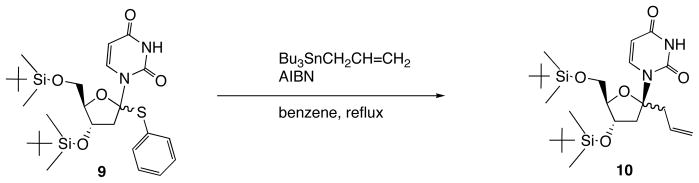
Radical Reaction of 1′-Phenylsulfanyl Nucleoside 9 under Radical Reaction leading to1′-Allyldeoxyuridine 10.
Scheme 5.
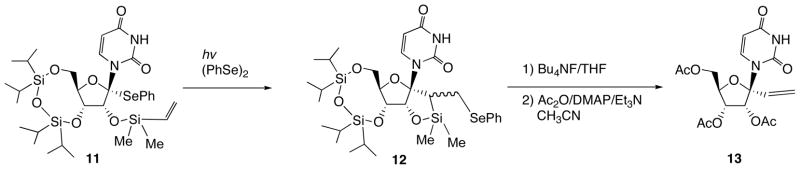
Intramolecular Radical Cyclization of 11 and Subsequent Transformation of 12 to 1′-Vinyl Uridine 13.
On the other hand, 4′-branched nucleosides such as 4′-cyanothymidine have been synthesized from common key intermediate 4′-hydroxymethylnucleoside 16 (Scheme 6) [15]. Thus, aldol-Cannizzaro reaction of thymidine 5′-aldehyde 14 gave 15. Selective tritylation of α-hydroxyl group, silylation of β-hydroxyl group of 15 and subsequent removal of the trityl group provide 4′-α-hydroxymethyl nucleoside 16. Oxidation of the primary hydroxyl group of 16 gave the corresponding aldehyde. Finally, reaction of the aldehyde with hydroxylamine and E2 elimination of the mesylate furnished 17 after deprotection. Although the method has been utilized most frequently, it required tedious steps for the synthesis of hydroxymethyl derivative 16 and the introduction of 4′-substituents has been limited.
Scheme 6.
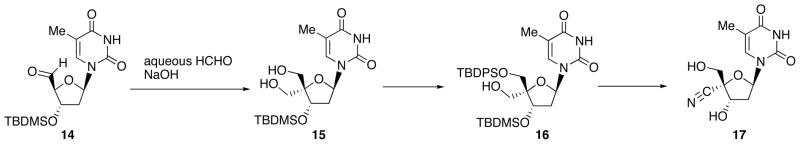
Synthetic Sequence for 4′-Branched Nucleosides by means of Aldol-Cannizzaro Reaction.
(2) Synthetic use of epoxy-sugar nucleoside
Ring opening of epoxides with carbon nucleophile have been recognized as one of the important synthetic operation for construction of carbon-carbon bond [16]. Epoxy-sugar nucleosides consist of four possible derivatives; 1′,2′-, 2′,3′-, 3′,4′-, and 4′,5′-epoxides (Fig. 4). Among these epoxy-sugar nucleosides, ring opening of 2′,3′-epoxy derivative with carbon nucleophile has been only precedent. Thus, Walker et al. has reported that 2′,3′-lyxo-epoxyuracil nucleoside 18 underwent regioselective ring opening by the reaction with LiC≡CH to give 3′-ethynylnucleoside 19 (Scheme 7) [17].
Fig. 4.
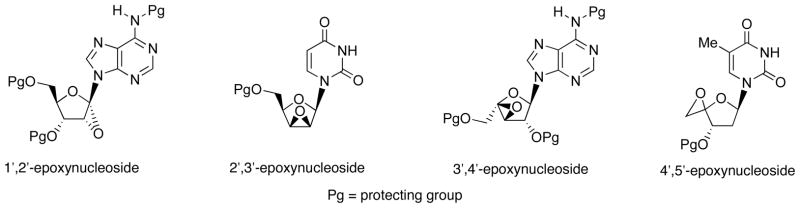
Structures of Epoxy-Sugar Nucleosides.
Scheme 7.
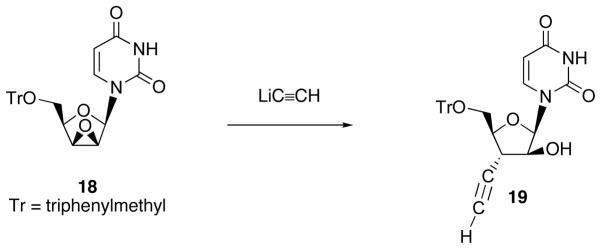
Synthesis of 3′-Branched Uracil Nucleoside 19 by Means of Ring Opening of 2′,3′-Lyxo-epoxide 18 with LiC≡CH.
The other three epoxy-sugar nucleosides are structurally unique in that the carbon bonded to the furanose ring oxygen is directly attached to the epoxide ring. These epoxides, therefore, are expected to readily undergo regioselective nucleophilic ring opening because of concomitant formation of oxonium ion. By the same reason, the preparation of these epoxides from the corresponding unsaturated-sugar nucleosides has to be carried out under non-nucleophilic conditions. In fact, 1′,2′-epoxynucleoside 20 exists as an equilibrium mixture with oxonium ion 21. Under aqueous conditions, 20 is readily converted into uracil and ribonolactone via hemiacetal 22 (Scheme 8).
Scheme 8.
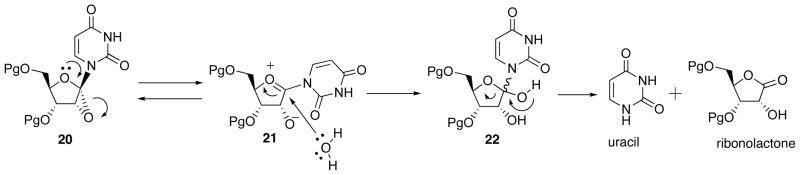
Characteristic Behaviour of 1′,2′-Epoxynucleoside 20.
We have envisioned that dimethyldioxirane (DMDO) would be suitable oxidizing reagent for the preparation of the above unstable 1′,2′-, 3′,4′-, and 4′,5′-epoxides because DMDO is able to transform an alkene into the respective epoxide in aprotic solvent under neutral conditions. DMDO is prepared by oxidation of acetone by potassium peroxomonosulfate (Oxone) (Scheme 9) [18–20].
Scheme 9.
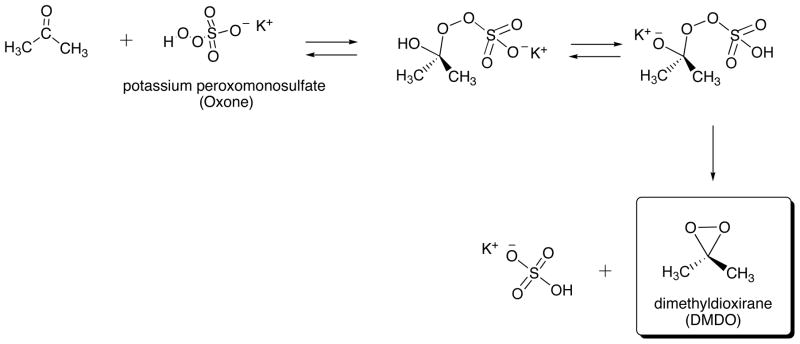
Preparation of Dimethyldioxirane (DMDO).
As expected, 1′,2′-, 3′,4′- and 4′,5′-epoxynucleosides could be prepared by DMDO-meditade oxidation of the corresponding unsaturated nucleosides (Scheme 10). Reaction of 1′,2′-epoxy-nucleoside with organoaluminum reagent gave “syn-opened” product stereoselectively. On the contrary, “anti-opened” product was obtained as a major product from the reaction of 3′,4′-epoxy-nucleoside with trimethylaluminum reagent. Interestingly, stereoselective 4′-α-carbon-carbon bond formation occurred in the SnCl4-initiated ring opening of 4′,5′-epoxynucleoside with organosilicon reagent.
Scheme 10.

Preparation and Ring-Openig of Epoxy-Sugar Nucleosides (Pg = protecting group).
In this review article, we describe stereo-defined synthesis of 1′-α- and 4′-α-branched nucleosides on the basis of ring opening of epoxy-sugar nucleosides. During this study, we have discovered an anti-HIV agent, Festinavir (4′-ethynylstavudine or 4′-Ed4T). Structure–activity relationships, detailed profile anti-HIV activity, biology and pharmacology of Festinavir are dicussed.
(3) Ring opening of nucleoside 1′,2′-epoxides with organoaluminum reagents: stereoselective entry to ribonucleosides branched at the anomeric position [21]
When TBDMS-protected 1′,2′-unsaturated uridine (23) was epoxidized with DMDO in CH2Cl2 at −30 °C, the reaction was completed for 30 min (Scheme 11). Because of its instability, the resulting epoxide could not be characterized by 1H NMR. Therefore, the reaction mixture was subsequently treated with Me3Al to give 1′-methyluracil nucleoside (25α) as a sole product. The depicted structure of 25α was determined on the basis of NOE correlation between H-6/H-4′, CH3-1′/H-3 and H-2/H-4′. The configuration of 2′-OH of 25α revealed that the epoxidation of 23 gave 1′,2′-“up”-epoxide 24 selectively. Similarly, Me3Al-mediated ring opening of the epoxide formed from the epoxidation of 1,1,3,3-(tetraisopropyldisiloxane-1,3-diyl) (TIPDS)-protected 26 provided 27α as a major isomer. These stereochemical outcome indicated that the epoxidation of TBDMS- and TIPDS-protected 1′,2′-unsaturated uracil nucleosides proceeded at the β-face predominantly.
Scheme 11.
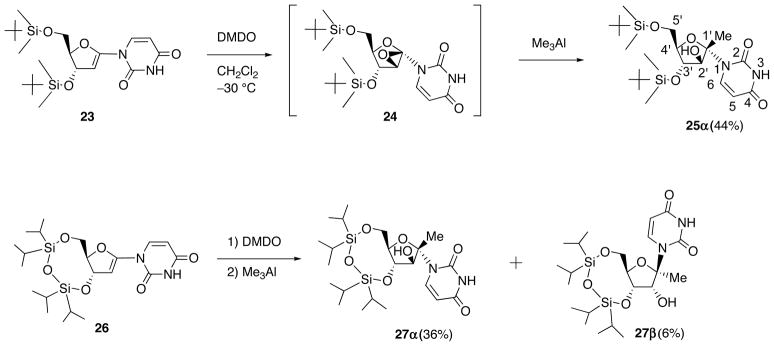
Epoxidation of 1′,2′-Unsaturated Uracil Nucleosides 23 and 26: Ring Opening of 1′,2′-Epoxynucleosides with Me3Al.
On the other hand, di-tert-butylsilylene (DTBS)-protected epoxide 29 derived from 28 was found to be stable for 1H NMR assignment of its structure (Scheme 12). The NOE experiment suggested that the epoxide 29 has 1′,2′-“down”-configuration. When the epoxide 29 was reacted with Me3Al, the desired syn-adduct 30α and its epimer anti-adduct 30α was obtained in a ratio of 5:1 in 86% isolated yield. On the basis of the molecular modeling study and the value of J3′,4′ (11.0 Hz) of 29, its Newman projection formulae of sugar portion was proposed (Fig. 5) [22]. As can be seen in the Figure, the hydrogen atom at the 3′-position occupies pseudo-axial position. When DMDO is approaching from the β-face of the enol ether, steric repulsion between the 3′-hydrogen and DMDO is seen. Therefore, the epoxidation of the double bond proceeded at the α-face to furnish 29 as a sole product. In the case of TBDMS-protected 23 and TIPDS-protected 26, these J3′,4′ values are 2.6 and 4.4 Hz, which suggest these 3′-hydrogen occupy the pseudo-equatorial position. In these cases, DMDO approaches from the α-face due to predominant steric hindrance of 3′-silyloxy-substituent leading to the formation of 1′,2′-“up”-epoxide 24 as a major product.
Scheme 12.
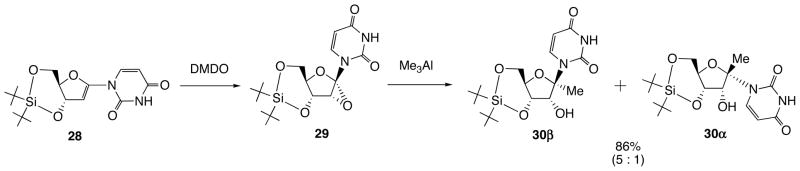
DMDO-mediated Epoxidation of DTBS-protected 1′,2′-Unsaturated Uracil Nucleoside 28 and Ring Opening of 1′,2′-“Down”-Epoxynucleoside 29 leading to 30.
Fig. 5.

Plausible Elucidation for α-Face-Selectivity of DMDO-Epoxidation of DTBS-protected 28.
To examine the scope and limitations, the ring-opening of 29 with other organoaluminum reagents was examined (Scheme 13) and these results are summarized in Table 1, which includes the results of the reaction with Me3Al in entry 1. Except for the result with triisobutylaluminum shown in entry 3, in which the isolated yield of 1′-isobutyl derivative (32) decreased to 32% due to concomitant hydride reduction, 1′-ethyl- (31), 1′-ethynyl- (33), 1′-vinyl- (34) and 1′-phenyluridine derivative (35) could be obtained in moderate to good yields (entries 2 and 4–6). As can be seen in the ratio of β- and α-anomers, the expected syn-ring-opened β-anomer was always accompanied with the anti-ring-opened α-uridine derivatives, except for the reaction of Ph3Al.
Scheme 13.
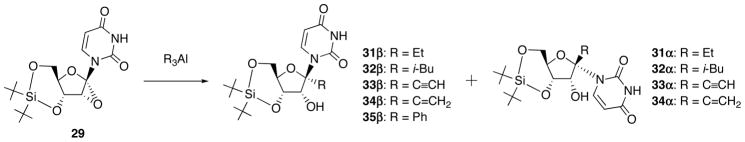
Reaction of 1′,2′-Epoxynucleoside 29 with Organoaluminum Reagents.
Table 1.
Reaction of 1′,2′-epoxynucleoside 28 with Organoaluminum Reagents
| entry | R3Al (equiv) | products | isolated yield (%) | ratio of β/α |
|---|---|---|---|---|
| 1 | Me3Al (3) | 30β/30α | 86 | 5/1 |
| 2 | Et3Al (3) | 31β/31α | 90 | 4/1 |
| 3 | i-Bu3Al (6) | 32β/32α | 35 | 9/1 |
| 4 | (HC≡C)3Al (6) | 33β/33α | 64 | 4/1 |
| 5 | (H2C=CH)3Al (6) | 34β/34α | 90 | 32/1 |
| 6 | Ph3Al (6) | 35β | 55 | – |
To explain these results, we have proposed a plausible mechanism for the reaction of 29 with R3Al (Scheme 14). Dissociation of an acidic N3-H of 29 with R3Al give V and subsequent coordination to the oxygen atom of the epoxide ring as well as to that of the C2-carbonyl of V would furnish VI, which in turn forms the oxonium intermediate VII. At this stage, if nucleophilic transfer of the aluminum ligand R takes place from the 2′-O-aluminate (path a), β-uridine derivatives 30β-35β should be formed, whereas such attack from the base moiety (path b) results in the formation of α-uridine derivatives 30α-34α or β-uridine derivatives depending upon the comformation about the N′-C′ pivot bond. The observed sole formation of 35β in the reaction of Ph3Al could be explicable in terms of inability of this bulky reagent to coordinate to the C2-carbonyl oxygen.
Scheme 14.
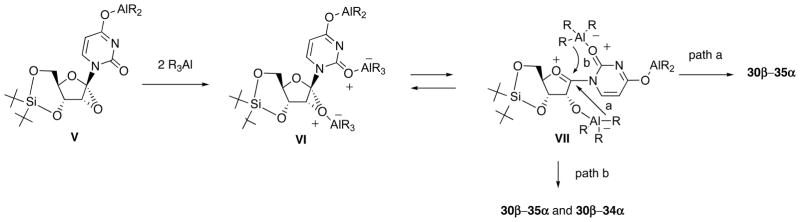
Plausible Mechanism for the Reaction of 1′,2′-Epoxynucleoside 29 with Organoaluminum Reagents.
Based on this mechanism, it would be reasonable to expect that the presence of a bulky protecting group at the N3-position will prevent coordination of R3Al to the C2-carbonyl oxygen. Reactions carried out along this line by employing the N3-protected substrates 36 and 37 uniformly gave the syn-ring-opened β-uridine derivatives 38-43 as a sole product (Scheme 15). Removal of N3-BOM group of 38-40 and N3-benzoyl group of 41-43 could be carried out by catalytic hydrogenolysis or treatment by methanolic ammonia, respectively.
Scheme 15.
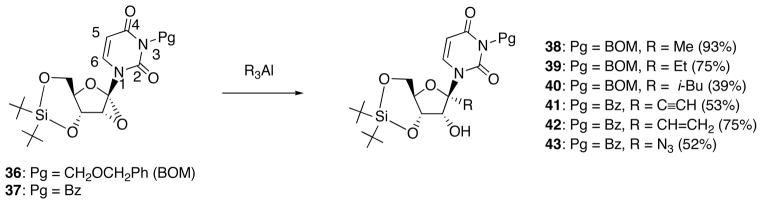
Reaction of N3-Protected 1′,2′-Epoxyuridine Derivatives 36 and 37 with Organoaluminum Reagents.
With the above successful results in hand, next, we have examined ring-opening of 1′,2′-epoxyadenosine derivative with organoaluminun reagents (Scheme 16). When 1′,2′-“down”-epoxide 44 was reacted with Me3Al, 1′-methyladenosine derivative 45 was obtained in 80% isolated yield. Likewise, 1′-ethyl- (46), 1′-isobutyl- (47), 1′-ethynyl- (48) and 1′-vinyl- (49) adenine nucleosides could be synthesized as a sole stereoisomer. 1′-Vinyl-adenosine 49 was transformed into protected angustmycin C 50 through OsO4-mediatde oxidative cleavage of vinyl group and subsequent hydride reduction of the resulting aldehyde. This is the first example that the nucleoside antibiotic was synthesized from adenosine.
Scheme 16.
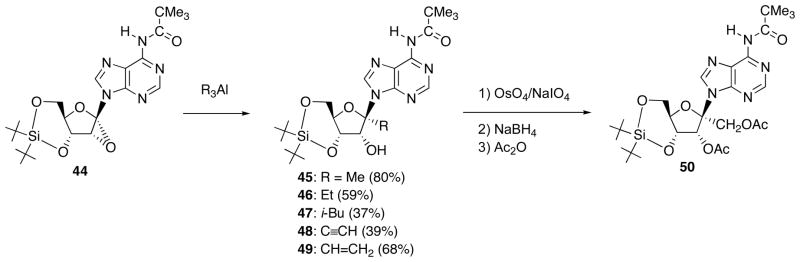
Reaction of 1′,2′-α-Epoxyadenosine Derivative 44 with Organoaluminum Reagents and Synthesis of Protected Angustmycin C.
As mentioned above, we have developed a novel method for the synthesis of 1′-branched uridine and adenosine derivatives by α-face-selective-epoxidation of 1′,2′-unsaturated nucleosides with DMDO and subsequent syn-ring-opening of the resulting 1′,2′-α-epoxides with organoaluminum reagents. These novel 1′-branched nucleosides did not show any anti-viral activities.
(4) Anti versus syn opening of epoxides derived from 9-(3-deoxy-β-D-glycero-pento-3-enofuranosyl)adenine with Me3Al: factors controlling the stereoselectivity [23]
Simple epoxides are known to react with Me3Al in a manner of anti-ring-opening, but no clear explanation is available for this stereochemical outcome. On the other hand, the epoxides derived from glycal, cyclic enol ethers, and 3,4-dihydro-2H-pyran give syn-ring-opened products. As shown in Scheme 17, by employing 3′,4′-β-epoxy nucleoside 52 derived from the DMDO-mediated epoxidation of 3′,4′-unsaturated adenine nucleoside 51, we investigated factors governing the stereoselectivity of its epoxy- ring-opening (anti- vs. syn-opening) with Me3Al.
Scheme 17.
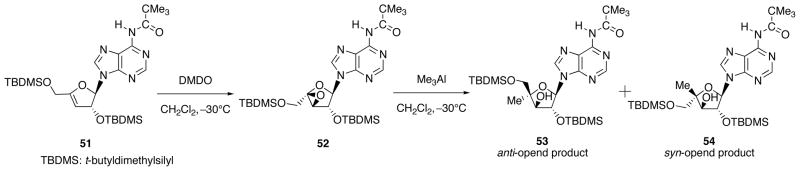
Epoxidation of 3′,4′-Unsaturated Adenine Nucleoside 51 and Ring-Opening of the Epoxy Nucleoside 52 with Me3Al.
Although 52 is a kind of glycal-derived epoxide, preferential formation of the anti-opened 53 was observed when the reaction was carried out in CH2Cl2. Also, it was found that the ratio of 53 (anti-opened)/54 (syn-opened) varied significantly (from 2/1 to 6/1) by increasing the amount of Me3Al (from 1.0 equiv. to 10 equiv.). In contrast to this, the same reaction carried out in THF, Et2O, or 1,4-dioxane by using 6.0 equiv. of Me3Al uniformly led to the exclusive formation of the syn-opened product 54. To see if the presence of the N6-pivaloyladenine base has any influence on the stereochemistry, the corresponding sugar epoxide 56 was prepared from 55 and reacted with Me3Al (6.0 equiv.). As shown in Scheme 18, although this reaction was carried out in CH2Cl2, the sole formation of the syn-opened product 57 was observed.
Scheme 18.
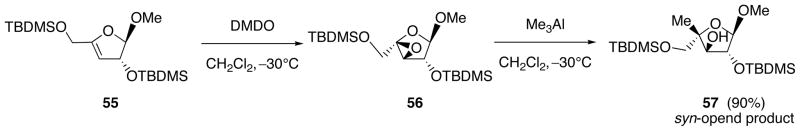
Epoxidation of 55 and Ring-Opening of the Sugar Epoxide 56 with Me3Al leading to 57.
These experimental results enable us to propose a possible reaction mechanism between 52 and Me3Al depicted in Scheme 19 (N6-pivaloyladenine moiety is omitted for simplicity). Highly oxygenophilic Me3Al would prefer coordination to the 3′,4′-epoxy structure of 52 to give VIII, which subsequently undergoes epoxide ring opening to form an oxonium ion that carries an alkoxyaluminate at the 3′-position. Two extreme conformers can be depicted for the oxonium ions as a result of rotation of the 3′-O-Al bond. In one conformer IX, Al is located above the furanose ring, and in the other conformer XI, it is outside the ring avoiding either steric or electronic repulsion with the adenine base.
Scheme 19.

Proposed Reaction Mechanism for the Ring Opening of Epoxide 52.
When the amount of the remaining Me3Al is limited or it is complexed with an ethereal solvent, intramolecular attack of the methyl ligand from IX would be inevitably take place to give XII (syn-opening), which is finally converted to 53. On the other hand, in the case of where non-coordinated Me3Al is sufficiently available in CH2Cl2, there is a good opportunity for X to transfer its methyl ligand to Me3Al, yielding tetramethylaluminate and XI. Under such circumstances, the presence of the adenine base as well as the 3′-alkoxyaluminum substituent in XI would render the stereochemical bias in favor of less hindered attack to lead to the dominant formation of XIII (anti-opening), which gives 54 after workup.
Such transfer of the methyl ligand from X to Me3Al would also be affected by the concentration of Me3Al in the reaction medium. In fact, when the reaction was carried out in a 50-fold diluted medium, the ratio of 53/54 was changed from 5/1 to 1.5/1.
One would imagine that the stereochemical outcome of this reaction also would be affected by the bulkiness of the 2′-O-silyl group. The ratio of 53/54 = 5/1 observed for 51 became 10/1 when the corresponding TES (triethylsilyl)-protected epoxide was employed (Scheme 20 and Table 2). It was beyond our expectation that the TBDPS (di-tert-butyldiphenylsilyl)-protected epoxide 61 gave the reverse stereoselectivity (62/63 = 1/7).
Scheme 20.
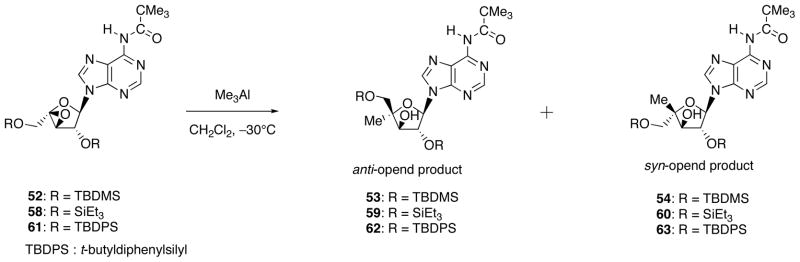
Effect of Bulkiness of the Protecting Group for the ratio of Anti-Opened/Syn-Opened Products.
Table 2.
Reaction of 52, 58 and 61 with Me3Al
| entry | epoxide | products | combined yield (%) of the two isomers | ratio of β-D-isomer/α-L-isomer |
|---|---|---|---|---|
| 1 | 52 | 53 and 54 | 90 | 53 and 54 = 5/1 |
| 2 | 58 | 59 and 60 | 89 | 59 and 60 = 10/1 |
| 3 | 61 | 62 and 63 | 94 | 62 and 63 = 1/7 |
A significant change of the ratio was also observed by varying the reaction temperature. At a higher temperature, almost equal amounts of 53 and 54 were formed (at 0 °C, 1.4/1; at room temperature, 0.7/1). At a lower temperature of −80 °C, the ratio was inverted and became 30/1. By combining the experimental results obtained thus far, the highest stereoselectivity (53/54 = 50/1, combined yield 90%) was attained by carrying out the reaction at −80 °C in CH2Cl2 employing the TES protected epoxide (Scheme 21).
Scheme 21.
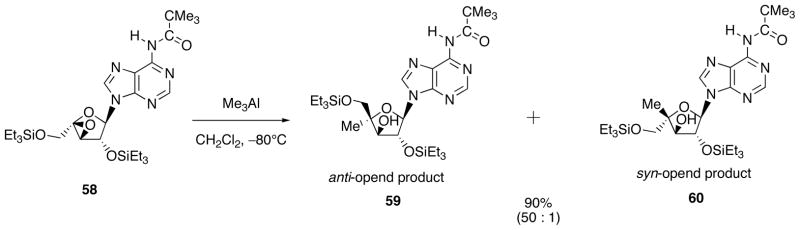
Optomized Reaction Conditions leading to Anti-Opened Product 59.
(5) Ring opening of 4′,5′-epoxy thymine nucleosides: finding of a promising anti-HIV-1 agent 4′-ethynylstavudine
This study was motivated by fairly recent reports that 4′-substituted nucleosides show significant inhibitory activity against HIV proliferation [24–29]. Since the most commonly utilized method for the synthesis of these compounds is manipulation of 4′-hydroxymethyl derivatives of nucleosides or sugars prepared via aldol-Cannizzaro reaction [30–31], we intended to develop a new and general method based on nucleophilic ring opening of a suitable 4′,5′-epoxy structure.
Ring opening of the epoxide 65 prepared from 4′,5′-unsaturated thymidine derivative 64 was first examined using Me3Al (Scheme 22) [32]. As a result, dominant formation of the 4′-methyl-α-L-isomer 67 was observed, the desired β-D-isomer 66 being formed only in 5% yield. This unsatisfactory outcome is assumed to be due to conformational preference of the oxonium intermediate depicted as XV, which can avoid the steric repulsion between the 5′-O-aluminate and the 3′-O-TBDMS group. In fact, the 4′,5′-epoxide 68 having the opposite 3′-configuration to 65 (Scheme 23), upon reacting with Me3Al, gave solely the 4′-methyl-β-D-isomer 69 (72%) through the aluminate XXVI. By applying this method, the 4′-vinyl 70 and 4′-ethynyl 71 derivatives also were prepared. This method was found to be applicable to the respective purine nucleosides.
Scheme 22.

Epoxidation of 4′,5′-Unsaturated thymidine 64 and Ring Opening of 4′,5′-Epoxide 65 with Me3Al.
Scheme 23.

Ring Opening of 4′,5′-Epoxythymine Nucleoside 68 with Organoaluminum Reagents.
To effect inversion at the 3′-position of these 4′-branched products by nucleophilic substitution, 4′-methyl derivative 69 was converted into chloromesyl nucleoside 73 following 2 steps (Scheme 24). When 73 was reacted with cesium acetate in the presence of 18-crown-6 in benzene under reflux conditions, the expected thymidine derivative 74 could not be obtained but the eliminated 4′-methyl d4T 75 was formed. The obtained nucleoside is 4′-methyl derivative of clinically used anti-HIV agent d4T (stavudine). Therefore, these 4′-branched products 69-71 were transformed into the respective d4T derivatives (Scheme 25).
Scheme 24.
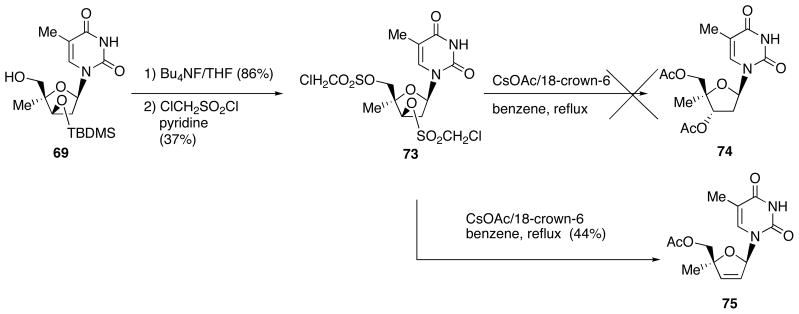
Attemtps to Effect Inversion at the 3′-Position of 69 and Formation of Elimination Products 75.
Scheme 25.
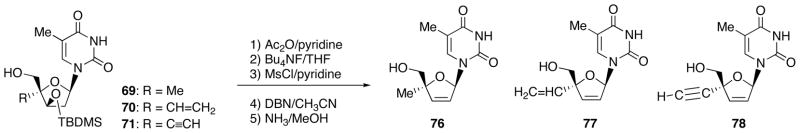
Synthesis of 4′-Substituted d4T 76-78.
Among these elimination products, 2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine (78, Ed4T) was found to be more inhibitory against HIV-1 than the parent compound d4T, and much less toxic to various cells and also to mitochondrial DNA synthesis (Table 3) [33]. This compound has several additional advantages as a promising anti-HIV-1 agent: 1) it is a better substrate for human thymidine kinase than stavudine [34], 2) it is very much more resistant to catabolism by thymidine phosphorylase [34], and 3) its anti-HIV activity is enhanced in the presence of a major mutation K103N [35], associated with resistance non-nucleoside reverse transcriptase inhibitors.
Table 3.
Anti-HIV-1IIIB activity of 4′-carbon-substituted stavudine 76-78a
| Compd | IC50 (μM)a | IC50 (μM)b |
|---|---|---|
| 76 | > 100 | > 100 |
| 77 | > 100 | > 100 |
| 78 (4′-ethynylstavudine) | 0.20 | > 100 |
| stavudine | 2.8 | 100 |
Data taken from ref. 33.
Inhibitory concentration required to achieve 50% protection of MT-2 cells against the cytopathic effect of HIV-1 IIIB.
Cytotoxic concentration required to reduce the viability of mock-infected MT-2 cells by 50%.
Some structure-activity relationship studies of Ed4T (78) also were carried out. For the analogues of 78 to be inhibitory against HIV-1, their 4′-carbon-substituent has to be sp-hybridized like ethynyl and cyano group [36]. Since methylethynyl nucleoside 79 decreased the activity [37], smaller size could be an additional requirement for the 4′-substituent. Although its carbocyclic analogue 80 and 81 resulted in total loss of the activity [38–39], the 4′-thio derivative 82 retains the activity (Fig. 6) [40].
Fig. 6.
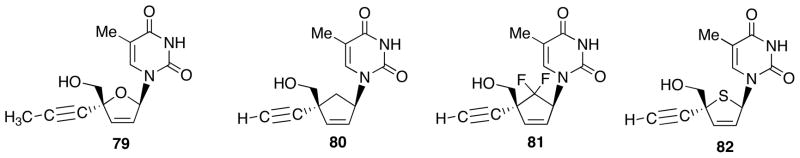
Structures of Ed4T analogs 79-82.
(II) ANTIVIRAL ACTIVITY OF 4′-ETHYNYLSTAVUDINE
The inhibitory effects of 4′-ethynylstavudine, stavudine, and lamivudine on HIV-1 (IIIB) replication have been evaluated in MT-2 and MT-4 cells. 4′-Ethynylstavudine exhibited higher activity than satavudine and lamivudine (Table 4). The 50% effective concentrations (EC50s) of 4′-ethynylstavudine were 0.25 and 0.07 μM in MT-2 cells and MT-4 cells, respectively. The activity of 4′-ethynylstavudine was 5.2-fold higher than that of d4T in MT-2 cells. The compound was 4.4- and 8.5-fold more potent inhibitor of HIV-1 replication in MT-4 cells than d4T and 3TC, respectively. Furthermore, 4′-ethynylstavudine was found to be active against both X4 (IIIB) and R5 (Ba-L) strains in peripheral blood mononuclear cells (PBMCs). On the other hand, cytotoxicity of 4′-ethynyl-stavudine appeared to be lower than that of d4T.
Table 4.
Anti-HIV-1 activity of 4′-ethynylstavudinea
The anti-HIV-1 activity of 4′-ethynylstavudine against various nucleoside reverse transcriptase inhibitor (NRTI)-resistant mutants has been evaluated (Tables 5 and 6). A012D contains four NRTI-associated mutations (NAMs) (D67K, K70R, T215F and K219Q), which confer a high level (210-fold) resistance to zidovudine [41]. The M184V mutation in reverse transcriptase (RT) also confers a high level resistance to lamivudine [42]. 4′-ethynylstavudine was 6.7-, 10-, and 2.9-fold less active against A012D and the M184V mutants of HXB-2 and NL4-3 compared to the wild-type, respectively (Table 5). The K65R mutation confers resistance to tenofovir and some NRTIs [43–45] and, the Q151M complex (A62V, V75I, F77L, F116Y, and Q151M) confers resistance to most of the clinically approved NRTIs [46]. While zidovudine, stavudine, didanosine, and lamivudine were 440-, 8.4-, 14- and 2.8-fold less active against the Q15M mutant of HXB-2, respectively, 4′-ethynyl-stavudine retained potent anti-HIV-1 activity against the mutants harboring the Q151M or K65R mutation (Table 5). The K103N mutation confers a high level resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs). In fact, nevirapine (NVP) was 55- and 70-fold less active against the K103N mutants of HXB-2 and NL4-3, respectively (Table 5). Interestingly, these mutants proved more susceptible to 4′-ethynylstavudine. Furthermore, 4′-ethynylstavudine exhibited its anti-HIV-1 activity against the KK strain, a clinical isolate from a treatment-naive patient, with an EC50 of 0.020 μM (Table 6). Although the activity of 4′-ethynylstavudine was 4.4 to 17.5-fold weaker against clinical isolates (HKW, HNK, HTN, and HTK) harboring multidrug-resistant mutations than that against the KK strain, the compound still retained considerable anti-HIV-1 activity against these mutants.
Table 5.
Anti-HIV-1 activity of selected RT inhibitors against various drug-resistant mutants in MAGI-CCR5 cellsa
| Virus | Mutation | EC50 (μM)b
|
|||||
|---|---|---|---|---|---|---|---|
| 4′-Ed4T | AZT | D4T | ddI | 3TC | NVP | ||
| A012B | Wild type | 0.49 ± 0.05 (1) | 0.037 ± 0.003 (1) | 0.33 ± 0.17 (1) | 1.1 ± 0.2 (1) | 0.27 ± 0.06 (1) | NDc |
| A012D | NAMsd | 3.3 ± 1.2 (6.7) | 7.7 ± 1.8 (210) | 0.59 ± 0.03 (1.8) | 3.6 ± 2.2 (3.3) | 1.1 ± 0.4 (3.3) | ND |
| HXB-2 | Wild type | 1.5 ± 0.2 (1) | 0.17 ± 0.06 (1) | 7.6 ± 3.2 (1) | 3.8 ± 0.8 (1) | 1 ± 0.3 (1) | 0.022 ± 0.012 |
| K65R | 1.3 ± 0.3 (0.87) | 0.085 ± 0.008 (0.50) | 7.8 ± 1.1 (1.1) | 10 ± 1 (2.6) | 4.7 ± 1.3 (2.6) | ND | |
| K103N | 0.46 ± 0.28 (0.31) | ND | 1.2 ± 0.5 (0.16) | ND | ND | 1.2 ± 0.5 (55) | |
| Y181C | 1.5 ± 0.5 (1) | ND | 5.2 ± 1.3 (0.68) | ND | ND | 3.8 ± 0.5 (170) | |
| M184V | 17 ± 2 (10) | 0.13 ± 0.02 (0.76) | 5.6 ± 0.4 (0.74) | 3.7 ± 0.5 (0.97) | > 100* (> 100) | ND | |
| MDRe | 1.1 ± 0.3 (0.73) | 74 ± 29 (440) | 64 ± 8 (8.4) | 52 ± 13 (14) | 2.8 ± 0.5 (2.8) | ND | |
| NL4-3 | Wild type | 0.27 ± 0.11 (1) | ND | 0.25 ± 0.05 (1) | ND | 0.21 ± 0.08 (1) | 0.061 ± 0.012 (1) |
| K103N | 0.16 ± 0.08 (0.59) | ND | 0.21 ± 0.04 (0.8) | ND | 0.23 ± 0.14 (1) | 4.3 ± 0.4 (70) | |
| M184V | 0.79 ± 0.15 (2.9) | ND | 0.22 ± 0.07 (0.88) | ND | > 100 (> 476.2) | 0.066 ± 0.025 (1) | |
| K103N + M184V | 0.31 ± 0.12 (1.1) | ND | 0.2 ± 0.05 (0.8) | ND | > 100 (> 476.2) | 2.3 ± 0.4 (37) | |
Data from reference 35.
All data represent means ± SD for three or four separate experiments. Values in parentheses represent fold increase (a ratio of EC50 for wild type to EC50 for mutant).
ND, not determined
Multidrug resistance: NRTI-associated mutations D67N, K70R, T215F, and K219Q
Multidrug resistance: Q151M complex (A62V, V75I, F77L, F116Y, and Q151M)
Table 6.
| Virus | Tropism | EC50 (μM) | Mutations associated with NRTI resistancec |
|---|---|---|---|
| KK | R5 | 0.020 ± 0.008 | Isolated from a treatment-naive patient |
| HKW | R5 | 0.35 ± 0.02 | M41L, V75L, D67N, M184I, T215Y, K219R |
| HNK | R5 | 0.089 ± 0.052 | D67N, T69D, K70R, M184V, T215F, K219D |
| HTN | R5 | 0.27 ± 0.15 | M41L, M184I, T215Y, K219R |
| HTK | X4 | 0.15 ± 0.08 | M41L, L74V, M184V, T215Y |
Data from reference 35.
Except for HKW, all data represent means ± SD for three or four separate experiments. For HKW, the EC50 represents the mean ± range for two separate experiments.
Mutations of HKW, HTK, HTN, and HNK were determined by Oka et al. (unpublished data).
To examine the emergence of resistance to 4′-ethynylstavudine, repeated passages of HIV-1 (IIIB)-infected MT-4 cells in the presence of escalating concentrations of the compound were conducted. Lamivudine induced a virus harboring the M184V mutation on day 29 (3TC29D), and the mutant was completely resistant to lamivudine (Table 7). However, 4′-ethynylstavudine was found to be only 2-fold less active against this mutant. 4′-ethynylstavudine also induced a virus harboring the M184V mutation on day 26 (4′-Ed4T26D), which was 1.7-fold less susceptible to the compound. 4′-Ed4T induced a resistant virus harboring three mutations (P119S, T165A, and M184V) on day 81 (4′-ethynylstavudine81D), yet 4′-ethynylstavudine still retained sufficient anti-HIV-1 activity against this mutant. We have previously reported that the EC50 of 4′-ethynylstavudine against 4′-ethynylstavudine81D was 13 μM, which was 130-fold resistant in comparison to the wild type IIIB [35]. Our recent experiment revealed that the EC50 against this strain was 2.3 μM (only 10-fold reduction in comparison to the wild type). The resistant virus 4′-ethynylstavudine81D had synonymous mutations at the codons correspond to the amino acid residues E40 and N136 (Table 8). It was reported that mutations in the connection domain of RT confers drug-resistance [47,48]. Among the mutations, N348I and A360V in the connection domain contribute to increasing resistance to zidovudine [49,50]. The N348I mutation was reported to be highly correlated with the M184V/I mutation [50]. The N348I and A360V mutations confer resistance to zidovudine by increasing excision of incorporated zidovudine through RNase H-dependent and independent mechanism [51]. However, the resistant virus 4′-ethynylstavudine81D did not contain any mutations at the thumb/connection/RNase H domain of RT (Fig. 7).
Table 7.
Anti-HIV-1 activity of 4′-Ed4T against wild-type and drug-resistant mutantsa
| Strain | EC50 (μM)b
|
|
|---|---|---|
| 4′-Ed4T | 3TC | |
| IIIB (wild type) | 0.22 ± 0.13 (1) | 2 ± 0.8 |
| IIIB (3TC29D) | 0.41 ± 0.08 (1.8) | > 20 |
| IIIB (4′-Ed4T26D) | 0.39 ± 0.13 (1.7) | > 20 |
| IIIB (4′-Ed4T81D) | 2.3 ± 1.4 (10) | > 20 |
| CC50(μM)c | > 20 | > 20 |
All data represent means ± standard deviations for three separate experiments
EC50: 50% effective concentration based on the inhibition of virus-induced cytopathicity in MT-4 cells
CC50: 50% cytotoxic concentration based on the reduction of viable cell number in mock-infected MT-4 cells
Table 8.
Sequence analysis for the RT region of HIV-1 resistant to 4′-Ed4T
| Strain | Number of amino acid residue
|
|||||
|---|---|---|---|---|---|---|
| 40 | 119 | 136 | 165 | 184 | ||
| IIIB (wild type) | nucleotide | GAA | CCC | AAC | ACA | ATG |
| amino acid | E | P | N | T | M | |
| IIIB (4′-Ed4T81D) | nucleotide | GGG | TCC | AAT | GCA | GTG |
| amino acid | E | S | N | A | V | |
| IIIB (3TC29D) | nucleotide | GGA | CCC | AAC | ACA | GTG |
| amino acid | E | P | N | T | V | |
The mutated nucleotides or amino acids are indicated with underlines
Fig. 7.

Sequence Analysis for the Thumb/Connection/RNase H Domain of HIV-1 Resistant to 4′-Ed4T.
(III) BIOLOGY AND PHARMACOLOGY OF 4′-ETHYNYLSTAVUDINE
The finding of potent anti-HIV activity of 4′-ethynylstavudine without much impact on mitochondrial DNA (mt-DNA) concentration in drug treated cells indicated that this compound may have less toxicity than stavudine. Since this discovery, a lot of studies have been done to understand the biology and pharmacology of 4′-ethynylstavudine. In summary, 4′-ethynylstavudine promises to be an ideal nucleoside analog with higher potency and therapeutic index, longer half-life of active metabolites in cells, effective against multidrug resistance (MDR) HIV, and high genetic barrier to the development of resistance probably due to the interaction of the compound with HIV reverse transcriptase (RT) [33,34].
Among 4′-substituted compounds synthesized [34], 4′-ethynylstavudine had a higher anti-HIV activity than stavudine, the parent compound) and much less inhibitory activity against cell growth or mt-DNA than either stavudine or zidovidine [34]. The adverse effects of nucleoside analogs are mediated by their effects on host DNA polymerase activity. Mitochondrial DNA polymerase-γ, unlike nuclear DNA (n-DNA) polymerases, lacks the ability to effectively discriminate against some nucleoside analogs in favor of endogenous nucleic acids [52]. Nucleoside analog-induced inhibition of mt-DNA synthesis is proposed to induce depletion of cellular mt-DNA and is ultimately responsible for the delayed toxicity [53,54]. Both zidovudine and stavudine (stavudine≫zidovudine) cause delayed toxicity in HIV-1 patients due to their impact on mt-DNA and/or n-DNA of affected organs. Interestingly, the IC50 values for 4′-Ed4TTP to inhibit pol γ and pol β were both at least 100-fold greater than that for d4TTP (Table 9) [55]. This is consistent with the observation that 4′-ethynylstavudine caused much less cellular toxicity and mitochondrial DNA less than stavudine in cell culture studies [34]. The underlying molecular mechanism of less inhibition of 4′-Ed4TTP to pol γ and pol β than d4TTP could be due to subtle differences in the interaction of the 4′-position of these two compounds at the active site of at pol γ and pol β.
Table 9.
Action of 4′-Ed4TTP on major human DNA polymerases
| DNA Polymerase | IC50 (μM)a,b | |||
|---|---|---|---|---|
| 4′-Ed4TTP | D4TTP | ddTTP | Aphidicolin | |
| α | >100 | >100 | NDc | 5 |
| β | >100 | 1 | 0.3 | ND |
| γ | ~100 | 1 | 1 | ND |
| δ | 60 | 40 | >100 | ND |
| δ | >100 | >100 | ND | 5 |
The IC50 values represent means from at least three independent experiments with standard deviation less than 20%.
When 0.3μM dTTP was used in the assays.
ND, not determined.
The anti-HIV potency of 4′-ethynylstavudine has been explained by its binding to HIV RT. In steady-state enzymatic analyses, 4′-ethynylstavudine triphosphate (4′-Ed4TTP) inhibited the DNA polymerase activity of RT more efficiently than d4T triphosphate (d4TTP), and the inhibition was more effective on DNA replication with RNA template than with DNA template [55]. 4′-Ed4TTP had much lower Ki value in inhibition of RT compared with that for d4TTP also the efficiency of 4′-Ed4TMP incorporation by RT with DNA/RNA substrate was 3-fold lower than that of d4TMP incorporation. The difference between the inhibition and incorporation efficiencies of 4′-Ed4TTP implies that the binding of 4′-Ed4TTP to RT-P/T complex is a more important factor than actual 4′-Ed4TTP incorporation. Moreover, pre-steady-state kinetic studies and computer modeling have illustrated that 4′-Ed4TTP was a better RT inhibitor than d4TTP due to the additional binding of the 4′-ethynyl group at a presumed hydrophobic pocket in the RT active site, which is critical for HIV RT activity (Table 10) [56]. This hydrophobic pocket is formed by the side chains of A114, Y115, M184, F160 and D185 [52]. These residues are highly conserved, and mutations at this pocket (A114M, A114L, Y115Q, F160A, F160L, and M184F) except M184V and M184A caused complete loss of RT activity [56,57]. M184V could affect the binding of incoming dNTP due to a gap created between the polymerase and the DNA minor groove of the nascent base pair leading to 3 to 5-fold resistance to 4′-Ed4T.
Table 10.
Pre-steady-state kinetic parameters for dTMP, d4TMP, and 4′-Ed4TMP incorporation by wt RT and the M184V mutant with DNA/DNA and DNA/RNA P/Ts
| P/T | Enzyme | dTTP | d4TTP | 4′-Ed4TTP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kd(μM) | Kpol(s−1) | Kpol/Kd (μM−1s−1) | Kd(μM) | Kpol(s−1) | Kpol/Kd (μM−1s−1) | Selectivitya | Kd(μM) | Kpol(s−1) | Kpol/Kd (μM−1s−1) | Selectivitya | ||
| DNA/DNA | Wt RT | 15.4±2.9 | 22.6±1.3 | 1.47 | 48.0±4.8 | 16.0±0.5 | 0.33 | 4.5 | 15.8±2.4 | 12.1±0.5 | 0.77 | 1.9 |
| M184V RT | 73.2±8.0 | 22.4±0.9 | 0.31 | 605±285 | 29.8±10.4 | 0.05 | 6.2 | 168.1±25.6 | 18.9±1.1 | 0.11 | 2.8 | |
| DNA/RNA | Wt RT | 67.1±10.2 | 65.0±3.9 | 0.97 | 40.8±9.2 | 18.4±1.4 | 0.45 | 2.2 | 11.4±2.7 | 11.7±0.8 | 1.0 | 0.97 |
| M184V RT | 143.9±25.0 | 41.7±3.5 | 0.29 | 232.3±50.0 | 29.6±3.6 | 0.13 | 2.2 | 43.4±13.9 | 9.7±0.8 | 0.22 | 1.3 | |
All values are mean ± SD.
Selectivity is calculated by dividing the efficiency of dTTP (kpol/Kd) by the efficiency of d4TTP or 4′-Ed4TTP.
The sine qua non of HIV treatment since 1996 is combination of highly active antiretroviral drugs. When these drugs are used in combination, lower doses may achieve desired antiviral effect with less toxicity. In pre-clinical studies, 4′-ethynylstavudine had synergistic interactions with lamivudine and LFd4C against HIV and was additive in combination with didanosine or zidovudine [34]. Therefore, 4′-ethynylstavudine could be given in combination with several approved antiretroviral drugs in the clinic. 4′-ethynylstavudine is a thymidine analog in the same class as zidovudine and stavudine. However, the phosphorylation of 4′-ethynylstavudine by TK-1 is an essential step but may not be sufficient to explain its potent antiviral activity over stavudine. Since its antiviral effect could be neutralized by dThd but not by dCyd, 4′-ethynylstavudine (like stavudine) acts as a dThd analog; however, the antiviral mechanism of action of 4′-ethynylstavudine could still be quite different from that of stavudine. Stavudine could be more efficiently phosphorylated (using a CEM cellular extract supplemented with partially purified TK-1 and recombinant human dTMP kinase) to the triphosphate metabolite than 4′-ethynylstavudine. This raises the issue of whether the 4′-ethynyl d4TMP is one of the active metabolite instead of only 4′-ethynyl-d4TTP. Furthermore, 4′-ethynyl-stavudine is not a substrate for thymidylate phosphorlyase (TP) and may explain its pharmacokinetic advantages over stavudine [34].
In studies of intracellular metabolism of nucleoside analogs, the peak concentrations of the metabolites occurred at 2 h for zidovudine and at 12 h for both 4′-ethynylstavudine and stavudine. Interestingly, 4′-ethynylstavudine was phosphorylated to the triphosphate at a slower rate than that of stavudine, but faster than that of zidovudine. The amount of intracellular triphosphate metabolites of 4′-ethynylstavudine was higher than that of zudivudine at 24 h in culture [58]. The major metabolite of 4′-ethynylstavudine was the monophosphate, which contributed most to the linear increase in triphosphate over 12 hour. This was consistent with the behavior of 4′-ethynylstavudine toward TMP kinase, the rate limiting-step, observed previously [59]. Most importantly, 4′-Ed4TTP, the active metabolite, persisted significantly longer (t1/2 8.0 to 9.7 h) than diphosphate (t1/2, 2.4 to 5.1 h) and monophosphate (t1/2, 1.4 to 2.4 h) after removal of the drug from cell culture [57]. Further studies were done to find out whether the persistent intracellular 4′-Ed4TTP will translate to a more persistent anti-HIV-1 activity after removal of the drug from the culture medium. On the average, the ability of the inhibitors to protect cells from HIV infection after 48 h of removal of drug from cell culture was 4′-Ed4TTP > LFD4C > didanosine > stavudine > lamivudine > zidovudine > emtricitabine (FTC) > NVP [60]. That is, 4′-ethynylstavudine could protect uninfected cells against HIV replication longer than zidovudine, satavudine, and most of currently used HIV drugs. In a viral rebound studies, none of the inhibitors could completely prevent viral rebound after removal from culture. After 48 h of removal of inhibitor from cell culture, the fold-change in concentration of inhibitor required to keep viral rebound at 50% was in the order of didanosine < 4′-Ed4TTP < LFD4C < FTC < stavudine < lamivudine < NVP < zidovudine [61]. The persistence of antiviral activity of 4′-ethynylstavudine after removal of drug from culture may be due to; 1) the fact that the triphsphate once formed remains relatively stable and active in cells, and that the pool of the monophosphate (which is not effluxed out of the cells [60]) may continue to replenish the critical concentration of 4′-Ed4TTP, 2) less efficient removal of incorporated 4′-Ed4T by Exos from terminal viral DNA, and 3) inability of 4′-Ed4TTP metabolites to permeate the cell membrane [60] as compared to the metabolites of zidovudine [62].
In an initial selection for 4′-ethynylstavudine drug resistance study by Nitanda et al. [63] presented above, M184V mutation was observed on the 26th day and two additional mutations (P119S and T165A) of HIV RT were found on day 81. The M184V and triple (M184V/P119S/T165A) mutants were reported to confer 3–5-fold and 130-fold resistance to 4′-ethynylstavudine, respectively. This was puzzling as the P119S and/or T165A have not been observed previously in HIV-1-infected individuals. The clinical significance of these mutations was unknown. Several attempts at the Cheng lab to duplicate the initial selection for drug resistance failed (unpublished data). Therefore, the P119S, T165A, and M184V mutations were engineered into NL4-3 background to assess the contribution of each of these mutations to drug resistance, RT activity, and viral growth. Compared with wild type virus, variants with single RT mutations (P119S or T165A) did not show resistance to 4′-ethynylstavudine, however, the M184V and P119S/T165A/M184V strains conferred 3- and 5-fold resistance, respectively [56]. The P119S/M184V and T165A/M184V variants showed about 4-fold resistance to 4′-ethynylstavudine. The differences in the growth kinetics of the variants were less than 3-fold. The purified RT with P119S/M184V and T165A/M184V mutations were inhibited by 4′-Ed4TTP with 8 to13-fold less efficiency than wild type RT [55]. These findings led to reexamination of the viral strains from the original drug resistance selection. When the previous HIV resistant strain (P119S/T165A/M184V), with 130-fold resistance to 4′-ethynylstavudine, was recovered by Prof. Baba’s lab from storage and cultured in the absence of 4′-ethynylstavudine for reassessment of 4′-ethynylstavudine susceptibility, it was found to have only 3–5-fold resistance to 4′-Ed4T instead of the 130-fold resistance previously observed (Baba et. al., unpublished results). Could the previously observed 130-fold resistance of the P119S/T165A/M184V virus to 4′-ethynylstavudine be due to additional mutations in the RT and/or outside the RT sequence, or that this virus is either difficult to recover from storage due to a poor replication capacity? Based on recent studies and the structural modeling of interaction of HIV RT-primer complex and 4′-Ed4TTP, a virus with a high degree of resistance to 4′-ethynylstavudine (e.g., more that 50-fold resistance, highly resistant to 4′-ethynylstavudine) will be difficult to develop [57]. Mutations selected for during in vitro passage may not necessarily represent the mutation pathway that will evolve during clinical use, therefore, true resistance to 4′-ethynylstavudine is yet to be demonstrated in clinical studies. We surmise that clinical resistance to 4′-ethynylstavudine will be difficult to emerge in clinical studies with relevant dosage which will suppress M184V mutant virus, prerequisite for developing highly 4′-ethynylstavudine resistant virus if the initial resistance selection holds.
Interestingly, in pre-clinical studies by Oncolys BioPharma (Japan) 4′-ethynylstavudine was shown to be active against drug-resistant clinical isolates [35]. Moreover, the strains carrying the K65R or the Q151M complex were still susceptible to 4′-ethynylstavudine [34]. Furthermore, its efficacy against most isolates was at least equivalent to that of TDF. In a Phase Ia and Ib/IIa studies by Oncolys BioPharma (Japan), 4′-ethynylstavudine was well tolerated with no serious adverse events [63]. Early stage clinical studies suggest that 4′-ethynylstavudine is a promising candidate for HIV therapy. To date, the missing piece is whether resistance to 4′-ethynylstavudine will develop in the clinic. Ongoing and future clinical trials will inform on this.
Acknowledgments
Financial supports from the Japan Society for the Promotion of Science (KAKENHI No. 21590123 and 24590144 to K. Haraguchi) are gratefully acknowledged. Also, this work was supported by Public Health Service grant AI-38204 from NIAID to Y.C.C. Y.C.C. is a fellow of the National Foundation for Cancer Research. E.P was supported by grant K08AI074404 from NIAID.
The authors are also grateful to Miss Y. Odanaka and Mrs. M. Matsubayashi (Center for Instrumental Analysis, Showa University) for technical assistance with NMR, MS, and elemental analyses.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
References
- 1. [Accessed on 02/11/2012];Global report: UNAIDS report on the global AIDS endemic. 2010 http://www.unaids.org/globalreport/global_report.htm.
- 2.Tan JJ, Chong XJ, Hu LM, Wang CX, Jia L, Liang XL. Therapeutic strategies underpinning the development of novel techniques for the treatment of HIV infection. Drug Discov Today. 2010;15(5/6):186–97. doi: 10.1016/j.drudis.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuda A. Recent development of anti-HIV nucleosides. J Synth Org Chem Jpn. 1990;48(10):907–20. [Google Scholar]
- 4.Ichikawa E, Kato K. Sugar-modified nucleosides in past 10 years, a review. Current Medicinal Chemistry. 2001;8(4):385–423. doi: 10.2174/0929867013373471. [DOI] [PubMed] [Google Scholar]
- 5.Huryn DM, Okabe M. AIDS-driven nucleoside chemistry. Chem Rev. 1992;92(8):1745–68. [Google Scholar]
- 6.De Clercq E. The design of drugs for HIV and HCV. Nature Review. 2007;6:1001–18. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy JR, Jr, Robins RK, Robins MJ. Purine nucleosdes. XXII synthesis of angustmycin A (decoynine) and related unsaturated nucleosides. J Am Chem Soc. 1968;90(17):4993–9. doi: 10.1021/ja01020a038. [DOI] [PubMed] [Google Scholar]
- 8.Hattori H, Tanaka M, Fukushima M, Sasaki T, Matsuda A. Nucleosides and Nucleotides. 158. 1-(3-C-Ethynyl-β-D-ribo-pentofuranosyl)cytosine, 1-(3-C-ethynyl-β-D-ribo-pentofuranosyl)uracil and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J Med Chem. 1996;39(25):5005–11. doi: 10.1021/jm960537g. [DOI] [PubMed] [Google Scholar]
- 9.O-Yang C, Wu HY, Fraser-Smith EB, Walker KAM. Synthesis of 4′-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992;33(1):37–40. [Google Scholar]
- 10.Itoh Y, Haraguchi K, Tanaka H, Gen E, Miyasaka T. Divergent and stereocontrolled approach to the synthesis of uracil nucleosides branched at the anomeric position. J Org Chem. 1995;60(3):656–62. [Google Scholar]
- 11.Kodama T, Shuto S, Ichikawa S, Matsuda A. A highly stereoselective samarium diiodide-promoted aldol reaction with 1′-phenylseleno-2′-keto nucleosides. synthesis of 1′-α-branched uridine derivative. J Org Chem. 2002;67(22):7706–15. doi: 10.1021/jo020266j. [DOI] [PubMed] [Google Scholar]
- 12.Haraguchi K, Itoh Y, Matsumoto K, Nakamura KT, Tanaka H. Stereoselective synthesis of 1′-C-branched arabinofuranosyl nucleosides via anomeric radicals generated by 1,2-acyloxy migration. J Org Chem. 2003;68(5):2006–9. doi: 10.1021/jo020620d. [DOI] [PubMed] [Google Scholar]
- 13.Kumamoto H, Murasaki M, Haraguchi K, Tanaka H. Nucleophilic addition of benzenethiol to 1′,2′-unsaturated nucleosides: 1′-C-phenylthio-2′-deoxynucleosides as anomeric radical precursors. J Org Chem. 2002;67(17):6124–30. doi: 10.1021/jo0201934. [DOI] [PubMed] [Google Scholar]
- 14.Kodama T, Shuto S, Nomura M, Matsuda A. An efficient method for the preparation of 1′-branched-chain sugar pyrimidine ribonucleosides from uridine: the first conversion of a natural nucleoside into 1′-substituted ribonucleosides. Chem Eur J. 2001;7(12):2332–40. doi: 10.1002/1521-3765(20010601)7:11<2332::aid-chem23320>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 15.Sugimoto I, Shuto S, Mori S, Shigeta S, Matsuda A. Nucleosides and nucleotides. 183. synthesis of 4′α-branched thymidines as a new type of antiviral agent. Bioorg Med Chem Lett. 1999;9(3):385–8. doi: 10.1016/s0960-894x(99)00010-4. [DOI] [PubMed] [Google Scholar]
- 16.For a review, see: Smith JG. Synthetically useful reactions of epoxides. Synthesis. 1984:629–56.
- 17.Ashwell M, Jones AS, Walker RT. The synthesis of some branched-chain-sugar nucleoside analogues. Nucleic Acids Res. 1987;15(5):2157–66. doi: 10.1093/nar/15.5.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adam W, Balas J, Hadjarapoglou L. A convenient preparation of acetone solution of dimethyldioxirane. Chem Ber. 1991;124(10):2377. [Google Scholar]
- 19.Curci R, Fiorentiono M, Troisi L, Edwards JO, Pater RH. Epoxidation of alkenes by dioxirane intermediates generated in the reaction of potassium caroate with ketones. J Org Chem. 1980;45(23):4758–60. [Google Scholar]
- 20.Murray RW, Jeyaman R. Dioxiranes: synthesis and reaction of methyldioxiranes. J Org Chem. 1985;50(16):2847–53. [Google Scholar]
- 21.Haraguchi K, Kubota Y, Tanaka H. Ring opening of nucleoside 1′,2′-epoxides with organoaluminum reagents: stereoselective entry to ribonucleosides branched at the anomeric position. J Org Chem. 2004;69(6):1831–6. doi: 10.1021/jo030262u. [DOI] [PubMed] [Google Scholar]
- 22.Haraguchi K, Konno K, Yamada K, Kitagawa Y, Nakamura KT, Tanaka H. Electrophilic glycosidation employing 3,5-O-(di-tert-butylsilylene)-erythro-furanoid glycal leads to exclusive formation of the β-anomer: synthesis of 2′-deoxynucleosides and its 1′-branched analogues. Tetrahedron. 2010;66(25):4587–600. [Google Scholar]
- 23.Kubota Y, Haraguchi K, Kunikata M, Hayashi M, Ohkawa M, Tanaka H. Anti versus syn opening of epoxides derived from 9-(3-deoxy-β-D-glycero-pento-3-enofuranosyl)adenine with Me3Al: factors controlling the stereoselectivity. J Org Chem. 2006;71(3):1099–103. doi: 10.1021/jo052243l. [DOI] [PubMed] [Google Scholar]
- 24.Maag H, Rydzewski RM, McRoberts MJ, Crawford-Ruth D, Verheyden JPH, Prisbe EJ. Synthesis and anti-HIV activity of 4′-azido- and 4′-methoxynucleosides. J Med Chem. 1992;35(8):1440–51. doi: 10.1021/jm00086a013. [DOI] [PubMed] [Google Scholar]
- 25.O-Yang C, Wu HY, Fraser-Smith EB, Walker KAM. Synthesis of 4′-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992;33(1):37–40. [Google Scholar]
- 26.Sugimoto I, Shuto S, Mori S, Shigeta S, Matsuda A. Synthesis of 4′-α-branched thymidines as a new type of antiviral agent. Bioorg Med Chem Lett. 1999;9(3):385–8. doi: 10.1016/s0960-894x(99)00010-4. [DOI] [PubMed] [Google Scholar]
- 27.Nomura M, Shuto S, Tanaka M, et al. Synthesis and biological activities of 4′-α-branched-chain sugar pyrimidine nucleosides. J Med Chem. 1999;42(15):2901–8. doi: 10.1021/jm990050i. [DOI] [PubMed] [Google Scholar]
- 28.Ohrui H, Kohgo S, Kitano K, et al. Synthesis of 4′-C-ethynyl -D-arabino- and 4′-C-ethynyl-2′-deoxy-β-D-ribo-pentofuranosyl-pyrimidines and –purines and evaluation of their anti-HIV activity. J Med Chem. 2000;43(23):4516–25. doi: 10.1021/jm000209n. [DOI] [PubMed] [Google Scholar]
- 29.Kodama E, Kohgo S, Kitano K, et al. 4′-Ethynyl nucleoside analogs: potent inhibitors of multidrug-resistant human immunodeficiency virus varinats in vitro. Antimicrob Agents Chemother. 2001;45(5):1539–46. doi: 10.1128/AAC.45.5.1539-1546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nomura S, Shuto S, Tanaka M, et al. Synthesis and biological activity of 4′-alpha-C-branched-chain-sugar pyrimidine nucleosides. J Med Chem. 1999;42(15):2901–8. doi: 10.1021/jm990050i. [DOI] [PubMed] [Google Scholar]
- 31.Ohrui H, Kohgo S, Kitano K, et al. Synthesis of 4′-C-ethynyl beta-D-arabino and 4′-C-ethynyl-beta-D-ribo-pentofuranosylpyrimidine and -purines and evaluation of their anti-HIV activity. J Med Chem. 2000;43(23):4516–25. doi: 10.1021/jm000209n. [DOI] [PubMed] [Google Scholar]
- 32.Haraguchi K, Takeda S, Tanaka H. Ring opening of 4′,5′-epoxynucleosides: a novel stereoselective entry to 4′-C-branched nucleosides. Org Lett. 2003;5(9):1399–402. doi: 10.1021/ol020259h. [DOI] [PubMed] [Google Scholar]
- 33.Haraguchi K, Takeda S, Tanaka H, et al. Synthesis of a highly active new anti-HIV agent 2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine. Bioorg Med Chem Lett. 2003;13(19):3775–7. doi: 10.1016/j.bmcl.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Dutschman GE, Grill SP, Gullen EA, et al. Novel 4′-substituted stavudine analog with improved anti-human immunodeficiency virus activity and decreased cytotoxicity. Antimicrob Agents Chemother. 2004;48(5):1640–6. doi: 10.1128/AAC.48.5.1640-1646.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nitanda T, Wang X, Kumamoto H, et al. Anti-human immunodeficiency virus type 1 activity and resistance profile of 2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine in vitro. Antimicrob Agents Chemother. 2005;49(8):3355–60. doi: 10.1128/AAC.49.8.3355-3360.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haraguchi K, Itoh Y, Takeda S, et al. Synthesis and anti-HIV activity of 4′-cyano-2′,3′-didehydro-3′-deoxythymidine. Nucleosdes Nucletides Nucleic Acids. 2004;23(4):647–4. doi: 10.1081/NCN-120030721. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka H, Haraguchi K, Kumamoto H, Baba M, Cheng YC. 4′-ethynylstavudine (4′-Ed4T) has potent anti-HIV activity with reduced toxicity and shows a unique activity profile against drug-resistant mutants. Antiviral Chemistry & Chemotherapy. 2005;16(4):217–21. doi: 10.1177/095632020501600402. [DOI] [PubMed] [Google Scholar]
- 38.Kumamoto H, Haraguchi K, Tanaka H, et al. Synthesis of (±)-4′-ehtynyl- and 4′-cyano carbocyclic analogues of stavudine (d4T) Nucleosides Nucleotides Nucleic Acids. 2005;24(2):73–83. doi: 10.1081/NCN-51900. [DOI] [PubMed] [Google Scholar]
- 39.Kumamoto H, Haraguchi K, Ida M, et al. Synthesis of (±)-4′-ehtynyl-5′,5′-difluoro-2′,3′-dehydro-3′-deoxy-carbocyclic thymidine: a difluoromethylidene analogue of promising anti-HIV agent Ed4T. Tetrahedron. 2009;65(36):7630–6. [Google Scholar]
- 40.Kumamoto H, Nakai T, Haraguchi K, et al. Synthesis and anti-HIV-1 activity of 4′-branched (±)-4′-thiostavudines. J Med Chem. 2006;49(26):7861–67. doi: 10.1021/jm060980j. [DOI] [PubMed] [Google Scholar]
- 41.Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246(4934):1155–8. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- 42.Schinazi RF, Lloyd RM, Jr, Nguyen MH, et al. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob Agents Chemother. 1993;37(4):875–81. doi: 10.1128/aac.37.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Margot NA, Isaacson E, McGowan I, Cheng A, Miller MD. Extended treatment with tenofovir disoproxil fumarate in treatment-experienced HIV-1-infected patients: genotypic, phenotypic, and rebound analyses. J Acquir Immune Defic Syndr. 2003;33(1):15–21. doi: 10.1097/00126334-200305010-00003. [DOI] [PubMed] [Google Scholar]
- 44.Miller MD. K65R, TAMs and tenofovir. AIDS Rev. 2004;6(1):22–33. [PubMed] [Google Scholar]
- 45.White KL, Margot NA, Wrin T, Petropoulos CJ, Miller MD, Naeger LK. RT processivities in vitro, consistent with a potential fitness defect in vivo and the low prevalence of the K65R mutation among isolates from antiretroviral agent-experienced patients. Antimicrob Agents Chemother. 2002;46(10):3437–46. [Google Scholar]
- 46.Shirasaka T, Kavlick MF, Ueno T, et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc Natl Acad Sci U S A. 1995;92(6):2398–402. doi: 10.1073/pnas.92.6.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delviks-Frankenberry KA, Nikolenko GN, Barr R, Pathak VK. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3′-azido-3′-deoxythymidine resistance. J Virol. 2007;81(13):6837–45. doi: 10.1128/JVI.02820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nikolenko GN, Delviks-Frankenberry KA, Palmer S, et al. Mutations in the connection domain of HIV-1 reverse transcriptase increase 3′-azido-3′-deoxythymidine resistance. Proc Natl Acad Sci U S A. 2007;104(1):317–22. doi: 10.1073/pnas.0609642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hachiya A, Kodama EN, Sarafianos SG, et al. Amino acid mutation N348I in the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase confers multiclass resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J Virol. 2008;82(7):3261–70. doi: 10.1128/JVI.01154-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yap SH, Sheen CW, et al. N348I in the Connection Domain of HIV-1 Reverse Transcriptase Confers Zidovudine and Nevirapine Resistance. PLoS Med. 2007;4:e335. doi: 10.1371/journal.pmed.0040335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehteshami M, Beilhartz GL, Scarth BJ, et al. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3′-azido-3′-deoxythymidine through both RNase H-dependent and -independent mechanisms. J Biol Chem. 2008;283(32):22222–32. doi: 10.1074/jbc.M803521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37(29):10529–39. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 53.Chen CH, Cheng YC. Delayed cytotoxicity and selective loss of mitochondrial DNA in cells treated with the anti-human immunodeficiency virus compound 2′,3′-dideoxycytidine. J Biol Chem. 1989;264(20):11934–7. [PubMed] [Google Scholar]
- 54.Chen CH, Vazquez-Padua M, Cheng YC. Effect of anti-human immunodeficiency virus nucleoside analogs on mitochondrial DNA and its implication for delayed toxicity. Mol Pharmacol. 1991;39(5):625–8. [PubMed] [Google Scholar]
- 55.Yang G, Dutschman GE, Wang CJ, et al. Highly selective action of triphosphate metabolite of 4′-ethynyl D4T: a novel anti-HIV compound against HIV-1 RT. Antiviral Res. 2007;73(3):185–91. doi: 10.1016/j.antiviral.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Yang G, Wang J, Cheng Y, et al. Mechanism of inhibition of human immunodeficiency virus type 1 reverse transcriptase by a stavudine analogue, 4′-ethynyl stavudine triphosphate. Antimicrob Agents Chemother. 2008;52(6):2035–42. doi: 10.1128/AAC.00083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang G, Paintsil E, Dutschman GE, et al. Impact of Novel HIV-1 Reverse Transcriptase Mutations, P119S and T165A on 4′-ethynyl-thymidine Analog Resistance Profile. Antimicrob Agents and Chemother. 2009;53(11):4640–6. doi: 10.1128/AAC.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paintsil E, Dutschman GE, Hu R, et al. Intracellular metabolism and persistence of the anti-human immunodeficiency virus activity of 2′,3′-didehydro-3′-deoxy-4′-ethynylthymidine, a novel thymidine analog. Antimicrob Agents Chemother. 2007;51(11):3870–9. doi: 10.1128/AAC.00692-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu CH, Hu R, Dutschman GE, et al. Comparison of the phosphorylation of 4′-ethynyl 2′,3′-dihydro-3′-deoxythymidine with that of other anti-human immunodeficiency virus thymidine analogs. Antimicrob Agents Chemother. 2007;51(5):1687–93. doi: 10.1128/AAC.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Tanaka H, Baba M, Cheng YC. Study of the retention of metabolites of 4′-Ed4T, a novel anti-HIV-1 thymidine analog, in cells. Antimicrob Agents Chemother. 2009;53(8):3317–24. doi: 10.1128/AAC.00302-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paintsil E, Grill SP, Dutschman GE, Cheng YC. Comparative study of the persistence of anti-HIV activity of deoxynucleoside HIV reverse transcriptase inhibitors after removal from culture. AIDS Res Ther. 2009;6:5. doi: 10.1186/1742-6405-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimmerman TP, Mahony WB, Prus KL. 3′-azido-3′-deoxythy-midine. An unusual nucleoside analogue that permeates the membrane of human erythrocytes and lymphocytes by nonfacilitated diffusion. J Biol Chem. 1987;262(12):5748–54. [PubMed] [Google Scholar]
- 63.Mastuda Takuma, Paintsil Elijah, Ross Joel S, Schofield Jessica, Cheng Yung-Chi, Urata Yasuo. A Single Dose Escalation Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of OBP-601 (4′-Ed4T a Novel NRTI) in Healthy Subjects. 16th Conference on Retroviruses and Opportunistic Infections; Montreal, Canada. 2009. [Google Scholar]