

Abstract
Objective
Mitochondrial ATP-sensitive potassium channels have been proposed to be myoprotective. The relevance and specificity of this mechanism in cardiac surgery was unknown. The purpose of this study was to examine the effects of the mitochondrial potassium ATP-sensitive channel opener diazoxide on regional and global myocardial protection using a model of acute myocardial infarction.
Methods
Pigs (n = 19) were placed on total cardiopulmonary bypass and then subjected to 30 min normothermic regional ischemia by snaring the left anterior descending coronary artery (LAD). The aorta was then crossclamped and cold blood Deaconess Surgical Associates cardioplegia (DSA; n = 6) or DSA containing 50 μM diazoxide (DZX; n = 6) was delivered via the aortic root and the hearts subjected to 30 min hypothermic global ischemia. The crossclamp and snare were removed and the hearts reperfused for 120 min.
Results
No significant differences in preload recruitable stroke work relationship, Tau, proximal, distal or proximal/distal coronary flow, regional or global segmental shortening, systolic bulging or post-systolic shortening were observed within or between DSA and DZX hearts during reperfusion. Infarct was present only in the region of LAD occlusion in both DSA and DZX hearts. Infarct size (% of area at risk) was 33.6 ± 2.9% in DSA and was 16.8 ± 2.4% in DZX hearts (P < 0.01 versus DSA). Apoptosis as estimated by TUNEL positive nuclei was 120.3 ± 48.8 in DSA and was significantly decreased to 21.4 ± 5.3 in DZX hearts. Myocardial infarct was located centrally within the area at risk in both DSA and DZX hearts but was significantly increased at borderline zones within the area at risk in DSA hearts.
Conclusions
The addition of diazoxide to cardioplegia significantly decreases regional myocardial cell necrosis and apoptosis in a model of acute myocardial infarction and represents an additional modality for achieving myocardial protection.
Keywords: Mitochondrial ATP-sensitive potassium (mitoKATP) channels, Myocardial cell necrosis, Apoptosis, Cardioplegia, Surgery
1. Introduction
Previously we have shown that the mitochondrion plays a central role in the molecular events leading to tissue damage occurring under conditions of ischemia [1–3]. Myocardial tissue is primarily aerobic and its metabolism is closely dependent upon oxygen, as confirmed by the abundance of mitochondria. The high energy requirement of the myocardium is almost exclusively met by mitochondrial oxidative phosphorylation [4]. This leads to a high sensitivity of the myocardial cell to oxygen deficiency [5].
We and others have suggested that mitochondrial ATP-sensitive potassium (mitoKATP) channels play an important role in modulating infarct size [2,6,7]. Recently we have shown that the cardioprotection afforded by magnesium supplemented potassium (K/Mg) cardioplegia is modulated by mitoKATP channels both during ischemia and reperfusion, with infarct modulation occurring primarily during ischemia in the isolated buffer-perfused rabbit heart [2]. We have also shown that the selective mitoKATP channel opener diazoxide, when added to K/Mg cardioplegia, significantly decreased infarct size in the isolated buffer-perfused rabbit heart, suggesting that opening of mitoKATP channels in concert with cardioplegia would allow for additive cardio-protection significantly enhancing the infarct size reduction afforded by cardioplegia alone [3]. The relevance and specificity of this mechanism in cardiac surgery in a blood perfused model was unknown.
To determine if the addition of the selective mitoKATP channel opener, diazoxide, to Deaconess Surgical Associates (DSA) cardioplegia afforded enhanced cardioprotection as compared to DSA cardioplegia alone, a model of acute myocardial infarction was used in the in situ blood perfused pig heart which has little or no collateral circulation to allow for amelioration of infarct size [8]. Our results indicate that the addition of the selective mitoKATP channel opener, diazoxide, to DSA cardioplegia significantly decreased myocar-dial cell necrosis providing an additional modality for achieving myocardial protection.
2. Materials and methods
2.1. Animals
Animals were housed individually and provided with laboratory chow and water ab libitum. All experiments were approved by the Beth Israel Deaconess Medical Center Animal Care and Use Committee and the Harvard Medical Area Standing Committee on Animals (institutional animal care and use committee) and conformed to the U.S. National Institutes of Health guidelines regulating the care and use of laboratory animals (NIH publication 5377-83, 1996).
2.2. Surgical preparation
Yorkshire pigs of either sex (32–42 kg, n = 19) were sedated with ketamine hydrochloride (20 mg/kg, intramuscularly, Abbott Laboratories, North Chicago, IL), and anesthetized with a bolus infusion of thiopental sodium (Baxter Healthcare Corporation, Inc, Deerfield, IL; 5.0–7.0 mg/kg iv, through an ear vein). A tracheotomy was performed through a midline cervical incision (36 Fr. Argyle, Mallinckrodt, St. Louis, MO), and ventilation begun with a volume-cycled ventilator (North American Drager, model Narkomed II-A, Telford; oxygen, 40%; Tidal volume, 1000 ml; Ventilation rate, 12 breaths/min; Positive end-expiratory pressure, 3 cm H2O; Inspiratory to expiratory time ratio, 1/2). The left internal jugular vein was cannulated for intravenous access and the left common carotid artery was cannulated for arterial blood sampling and intraarterial blood pressure monitoring (Millar Instruments, Houston, TX). General anesthesia was maintained with 3.0% of sevoflurane (Ultane; Abbott Laboratories, Inc, North Chicago, IL) at the beginning of the surgical preparation, the concentration was then reduced to 1.0% and maintained at the same concentration throughout the experiment. During cardiopulmonary bypass (CPB), propofol (Baxter Healthcare Corporation, Inc, Deerfield, IL; 0.5–0.7 mg/kg per min iv) was continuously infused via jugular vein. Heparin sodium (Elkins-Sinn, Inc, NJ; 5000 IU iv) and 1% lidocaine (Elkins-Sinn, Inc, NJ; 5 ml iv) were given before sternotomy. Heparin was administered at the same dose every 30 min to the end of the experiment. The pericardial sac was exposed through a median sternotomy and was opened to form a pericardial cradle. A catheter-tipped manometer (Millar Instruments, Houston, TX) was introduced through the apex into the left ventricle (LV) to record LV pressure. The proximal third portion of the left anterior descending coronary artery (LAD) was dissected to continuously monitor the coronary flow by a transit time ultrasonic flow probe (3 mm RS-Series, Transonic Systems Inc.; Ithaca, NY) with a T206 flowmeter (Transonic Systems Inc.; Ithaca, NY). A silk thread (0 silk, K834H, Ethicon, Inc., Somerville, NJ) was passed around the distal third of the LAD, and both ends of the silk tie were threaded through a small vinyl tube to form a snare, which was then secured by clamping the tube with a mosquito clamp. At the same site, the distal LAD flow was measured continuously. Time course changes in the relationship between proximal and distal coronary arterial flow were expressed using distal/ proximal flow ratio to minimize anatomical variability among individual animals.
2.3. Experimental protocol for acute myocardial infarction
The experimental protocol is shown in Fig. 1. In brief pigs were divided randomly into two groups. The animals were then placed on CPB, which was initiated at a flow rate of 75 ml/kg per min, with a two-staged cannula for venous return and the right subclavian artery for arterial inflow. To maintain hematocrit levels, CPB pumps were primed with autologous blood drawn from a donor pig. All animals were allowed to stabilize for 30 min following cannulation for CPB. Following 30 min stabilization, initial myocardial functional acquisition was performed (Fig. 1, 30–40 min) by switching to modified right heart bypass, as previously described [9]. LV contractility was assessed by the preload recruitable stroke work relationship (PRSW), which uses the slope of the relationship between preload (end-diastolic volume) and external work as an estimate of contractility. PRSW was evaluated during incremental volume loading from 75 to 150 ml/kg per min in increments of 25 ml/kg per min, during modified right heart bypass. Diastolic function was assessed by Tau, the time constant of isovolemic pressure decay (LV global function) [9]. Tau, was assessed with inflow to LV maintained at 75 ml/kg per min. LV volume was estimated by sonimicrometry using piezoelectronic digital probes placed parallel to the LV long axis, and to the short axis using an ellipsoid model.
Fig. 1.
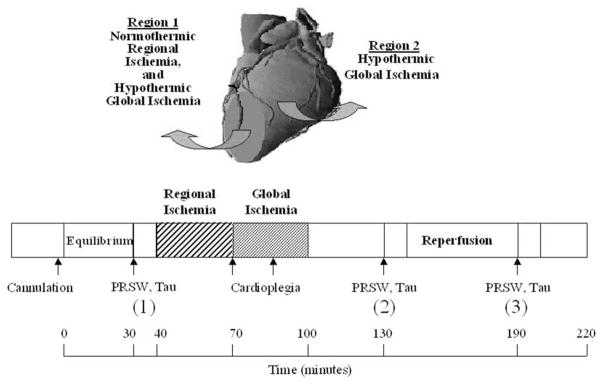
Experimental schema of acute myocardial infarct model in the in situ pig heart. Pigs were cannulated placed on cardiopulmonary bypass (CPB). Following 30 min stabilization (equilibrium), initial myocardial functional acquisition was performed (30–40 min) by switching to modified right heart bypass and PRSW, Tau and regional function (SS, SB, PSS) determined to allow for baseline comparison. The hearts were returned to CPB and then subjected to 30 min normothermic regional ischemia by snaring of the distal portion of LAD (Region 1). Immediately following 30 min normothermic regional ischemia, the aorta was crossclamped and cold blood DSA cardioplegia or DSA with the addition of 50 μM diazoxide (DZX) was administered via the aortic root. The hearts were then subjected to 30 min hypothermic global ischemia (Region 2). Following 30 min hypothermic global ischemia, the crossclamp and the snare were released and the hearts reperfused for 120 min. At 30 and 90 min reperfusion the hearts were again switched to modified right heart bypass to allow for a second (2) and third (3) evaluation (10 min each) of post-arrest mechanics.
Regional myocardial contractility of the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) was evaluated by segmental shortening (SS) using the equation; SS = 100 × (EDL − ESL)/EDL; with EDL representing end-diastolic segment length measured at the onset of positive dP/dt (the first derivative of pressure over time), and ESL representing end-systolic segment length (ESL) measured at peak negative dP/dt [11].
Wall motion abnormalities, systolic bulging (SB) and post-systolic shortening (PSS) were determined separately, by sonomicrometry (Sonomicrometrics Digital Ultrasonic Measurement System, Sonometrics Corp., ON, Canada) using digital piezoelectronic probes (2.0 mm) implanted in the subepicardial layer approximately 10 mm apart with probes being placed parallel to the minor and major axes of the heart in Region 1 and the LV area subjected to 30 min hypothermic global ischemia (Region 2; Fig. 1), and secured to the epicardium with polypropylene stitches (5-0 Prolene, 8580H, Ethicon, Inc., Somerville, NJ). Correct positioning of the probes was confirmed by cyanosis and re-confirmed by injection of monastryl blue pigment following the completion of the experiment. The probes were left in place until the end of the experiment. Digital data were inspected for correct identification of end-diastolic and end-systolic points using post-processing software (Sono-View, Sonometrics Corp., London, ON, Canada) [10]. Measurements were made over at least three cardiac cycles in normal sinus rhythm and then averaged. The ventilator was stopped during data acquisition to eliminate the effects of respiration. SB defined as the bulging of the myocardium after the end of diastole, was determined using the equation: SB = 100 × (Maximum segment length during systole − EDL)/EDL [12]. PSS defined as the shortening after the end of systolic ejection was determined using the equation: PSS = 100 × (ESL − Minimum segment length during diastole)/EDL [13].
2.3.1. Regional myocardial ischemia
The hearts were then subjected to 30 min normothermic regional ischemia by snaring of the distal portion of LAD. Myocardial ischemia was confirmed visually by regional cyanosis of the myocardial surface [10].
2.3.2. Global myocardial ischemia
Immediately following 30 min regional ischemia, the ascending aorta was crossclamped and cold (4°C) cardioplegia (10 ml/kg) was administered antegradely through the aortic root using a cardioplegia needle (9F AR II aortic root cannula; Medtronic’s DLP, Inc, Grand Rapids, MI) inducing prompt diastolic arrest. DSA hearts (n = 6) received cold blood cardioplegia [DSA; K+, 60 mM; MgSO4, 8 mM; Dextrose, 2.5 mM; THAM (Tris (hydroxymethyl) amino-methane), 10 mM in normal saline, 0.9%)] mixed (1:4; v:v) with cold autologous blood (final concentration: Na+, 134 mEq/l; Cl−, 119 mEq/l; K+, 16 mEq/l; Ca2+, 2.2 mEq/l, Mg2+, 4.4 mEq/l). Dimethyl sulfoxide (DMSO, Fisher Scientific Co, Fair Lawn, NJ) was added as vehicle sham to DSA cardioplegia. DZX hearts (n = 6) initially received DSA cardioplegia (10 ml/kg) containing diazoxide (50 μM). Diaz-oxide was dissolved in DMSO before added into DSA solutions. The final concentration of DMSO was less than 0.1%. At 15 min global ischemia, 5 ml/kg of DSA cardioplegia, without diazoxide, was re-administered in both groups.
All hearts received topical hypothermia during global ischemia by packing ice around the myocardium to reduce myocardial temperature [9]. Following 30 min hypothermic global ischemia, the crossclamp and the LAD snare were released and the ice packing removed and the hearts kept on total CPB for 120 min reperfusion. Post-arrest myocardial function in Region 1 (Fig. 1) was assessed separately at 30–40 and 90–100 min reperfusion (2 and 3, respectively in Fig. 1; 130 and 190 min, Table 2). PRSW and Tau were also determined at 30–40 and 90–100 min reperfusion (2 and 3, respectively in Fig. 1; 130 and 190 min, Table 2). Hemodynamic variables were continuously acquired using Po-Ne-Mah digital data acquisition system (Gould, Valley View, OH), with an acquire plus processor board, and LV pressure analysis software, and a Gould ECG/biotach.
Table 2.
Myocardial function in the left ventricular area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1) and in the left ventricular area subjected to 30 min hypothermic global ischemia (Region 2)a
| Variables | Initial myocardial function (30–40 min) | Reperfusion
|
|||
|---|---|---|---|---|---|
| 130 min | 190 min | ||||
| Regional myocardial function (Region 1) | SS (%) | DSA | 10.6 ± 1.5 | 2.5 ± 1.5* | 4.3 ± 1.5* |
| DZX | 11.9 ± 2.6 | 4.4 ± 1.3* | 3.5 ± 1.9* | ||
| SB (%) | DSA | 0.2 ± 0.2 | 1.3 ± 0.7 | 1.1 ± 0.6 | |
| DZX | 0.5 ± 0.3 | 0.7 ± 0.3 | 1.2 ± 0.7 | ||
| PSS (%) | DSA | 1.7 ± 0.4 | 1.3 ± 0.4 | 1.7 ± 0.5 | |
| DZX | 1.2 ± 0.4 | 1.6 ± 0.3 | 2.8 ± 0.5 | ||
| Global myocardial function (Region 2) | Tau (msec) | DSA | 30.7 ± 2.5 | 57.0 ± 8.7* | 50.7 ± 11.5 |
| DZX | 32.3 ± 3.3 | 58.9 ± 12.5 | 45.8 ± 6.1 | ||
| PRSW (mJ/beat per ml) | DSA | 80.4 ± 8.0 | 42.0 ± 6.3* | 51.5 ± 7.2* | |
| DZX | 81.7 ± 6.6 | 46.4 ± 5.6* | 54.4 ± 5.8* | ||
| Percent recovery of EF (% of equilibrium) | DSA | – | 65.8 ± 9.3* | 75.9 ± 4.9* | |
| DZX | – | 66.5 ± 5.9* | 81.4 ± 8.3 | ||
All results are shown as mean ± SEM for each group.
Significant differences are shown as *P < 0.05 versus equilibrium hearts.
EF = ejection fraction; PRSW = preload recruitable stroke work; PSS = post-systolic shortening; SB = systolic bulging; and SS = segmental shortening.
Blood gases and hematocrits were monitored every 10–15 min using a Corning 238 pH/blood gas analyzer and a Corning 270 CO-oximeter (Chiron Diagnostics, MA.). Blood gases and acid-base parameters were maintained at PO2 > 100 mmHg. PaCO2 was maintained between 30 and 40 mmHg by adjustment of the ventilatory rate. pH was adjusted between 7.35 and 7.45 with intravenous sodium bicarbonate [9]. Core temperature was maintained at 37°C.
2.4. Measurement of infarct size
Area at risk (AAR) was delineated following 120 min reperfusion (220 min) by religation of the LAD and injection of monastryl blue pigment into the aorta. The heart was rapidly removed and sliced across the long axis of the LV, from apex to base, into 1 cm thick transverse sections and traced onto a clear acetate sheet over a glass plate, under room light. The sliced hearts were incubated in 1% triphenyl tetrazolium chloride (Sigma Chemical Co., St. Louis, MO) in phosphate buffer (pH 7.4) at 38°C for 20 min [9,10]. A copy of the stained heart slices were traced onto a clear acetate sheet over a glass plate under room light. AAR in the LV and the area of infarct size (IS) were measured by planimetry. The volumes of the infarcted zone and the AAR were calculated by multiplying the planimetered areas by the slice thickness. The ratio of ARR to LV weight was calculated. IS was expressed as a percentage of AAR for each heart (IS/AAR). Myocardial infarct size was determined in both the LV area subjected to 30 min normother-mic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) and in the LV area subjected to 30 min hypothermic global ischemia (Region 2; Fig. 1).
2.5. Terminal deoxyneuleotidyl transferase [TdT] mediated dUTP nick end labeling (TUNEL)
TUNEL was performed using the ApopTag detection system (Intergen, Gaithersburg, MD). Myocardial tissue samples (approximately 3 × 5 mm each) from Regions 1 and 2 consisting of epicardial, myocardial and endocardial tissue were removed at the end of each experiment and flash frozen and the samples sectioned (4–6 μm) and mounted on glass slides and used for in situ TUNEL. Five to six slides from each sample were assayed using both TUNEL and propidium iodide staining. Photomicrographs were taken in 10–15 random high powered (20 ×) fields using a Zeiss MC80DX camera and exposure meter. All cells were counted on each slide and TUNEL positive cells were expressed per 3000 myocardial cells [14]. Myocardial cell specificity was determined on opposite adjacent sequential serial slides (n = 5–6 for each sample) using the cardiac specific monoclonal antibody for troponin I (Spectral Diagnostics Inc., Toronto, Ont.) labeled with antimouse IgG conjugated to Alexa 350 (Molecular Probes, Inc. Eugene, OR). Troponin I was visualized using a DAPI filter (Chroma Technologies, Battleboro, VT). Evaluation of TUNEL and myocyte morphology was performed by a blinded independent examiner. TUNEL positive cells were expressed per 3000 myocardial cells [14].
2.6. Effects of anesthetic on the cardioprotection afforded by DSA and DSA cardioplegia with diazoxide
A separate study was performed using isoflurane as the anesthetic agent (n = 3 each for DSA and DZX heart). All protocols and methods were as described in Sections 2.2–2.5.
2.7. Statistical analysis
Statistical analysis was performed using SAS (version 6.12) software package (SAS Institute, Cary, NC). The mean ± SEM for all data was calculated for all variables. Statistical significance was assessed using repeated measures analysis of variance with group as a between subjects factor and time as a within subjects factor. Post hoc comparisons between groups for both the average effect and at individual time points were made using a Bonferroni correction to adjust for the multiplicity of tests. A one-way analysis of variance was used for area of infarction. Significant values at P less than 0.05 are noted.
3. Results
3.1. Experimental exclusion
Nineteen animals were included in experimental protocols. One animal (DZX) could not be resuscitated following 30 min regional ischemia and 30 min of global ischemia due to sustained intractable bradycardia, and was killed, humanely. No data from this animal was included in the final analysis as infarct size could not be determined due to shortened reperfusion time.
3.2. Equilibrium hemodynamics
Hemodynamic data during 30 min cannulation (Equilibrium) are summarized in Table 1. No significant differences in heart rate (HR), LV peak developed pressure (LVPDP), LV end-diastolic pressure (LVEDP), +dP/dt or mean arterial pressure were observed between groups after 30 min of cannulation (Equilibrium, Table 1).
Table 1.
Left ventricular hemodynamicsa
| Variables | Equilibrium (30 min) | Regional ischemia (70 min) | Global ischemia
|
Reperfusion
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| 80 min | 100 min | 130 min | 160 min | 190 min | 220 min | ||||
| Heart rate (beats/min) | DSA | 162.3 ± 9.9 | 182.2 ± 5.0* | 0.0 ± 0.0 | 0.0 ± 0.0 | 164.8 ± 8.0 | 140.8 ± 17.9 | 161.3 ± 7.3 | 161.8 ± 14.2 |
| DZX | 146.3 ± 11.1 | 173.8 ± 4.0* | 0.0 ± 0.0 | 0.0 ± 0.0 | 146.8 ± 13.4 | 161.7 ± 7.5 | 158.3 ± 8.9 | 172.0 ± 12.1 | |
| Mean arterial pressure (mmHg) | DSA | 79.2 ± 8.9 | 78.7 ± 5.1 | 76.3 ± 4.3 | 75.7 ± 1.9 | 71.2 ± 5.6 | 72.2 ± 3.4 | 76.5 ± 4.3 | 71.2 ± 3.1 |
| DZX | 77.3 ± 4.7 | 70.2 ± 6.8 | 69.3 ± 5.7* | 70.8 ± 4.5 | 72.8 ± 3.4 | 71.0 ± 2.4 | 72.3 ± 2.5 | 78.5 ± 3.1 | |
| LV peak developed pressure (mmHg) | DSA | 70.1 ± 4.6 | 71.9 ± 5.8 | 0.0 ± 0.0 | 0.0 ± 0.0 | 62.4 ± 7.3 | 71.1 ± 7.2 | 77.1 ± 7.5 | 68.1 ± 3.5 |
| DZX | 68.8 ± 4.2 | 68.3 ± 4.2 | 0.0 ± 0.0 | 0.0 ± 0.0 | 63.5 ± 4.0 | 65.6 ± 4.6 | 72.5 ± 3.1 | 75.5 ± 4.0 | |
| LV end-diastolic pressure (mmHg) | DSA | 7.4 ± 1.1 | 7.4 ± 1.2 | 0.0 ± 0.0 | 0.0 ± 0.0 | 8.0 ± 1.6 | 8.0 ± 1.8 | 7.8 ± 1.2 | 7.9 ± 1.8 |
| DZX | 5.8 ± 0.7 | 6.8 ± 0.9 | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.5 ± 0.6 | 5.3 ± 0.7 | 5.0 ± 0.5 | 5.7 ± 0.4 | |
| Positive dP/dt (mmHg/s) | DSA | 1993 ± 277 | 2171 ± 213 | 0 ± 0 | 0 ± 0 | 2055 ± 371 | 1987 ± 285 | 2162 ± 341 | 1892 ± 370 |
| DZX | 1802 ± 144 | 1815 ± 106 | 0 ± 0 | 0 ± 0 | 2035 ± 285 | 2356 ± 460 | 2209 ± 305 | 2362 ± 340 | |
All results are shown as mean ± SEM for each group.
Significant differences are shown as *P < 0.05 versus equilibrium hearts.
3.3. Initial myocardial function
Initial myocardial function was assessed, to allow for baseline comparison following 30 min equilibrium (30–40 min). No significant differences in PRSW, Tau, SS, SB or PSS were observed within or between DSA and DZX hearts (Table 2).
3.4. Hemodynamics during normothermic regional ischemia
Following 30 min normothermic regional ischemia (70 min, Table 1), HR was significantly (P < 0.05) increased as compared to equilibrium value in both DSA and DZX hearts but no significant difference in HR was observed between DSA and DZX hearts. No significant differences in LVPDP, LVEDP, +dP/dt, or mean arterial pressure were observed within or between groups during regional ischemia.
3.5. Effects of diazoxide
Following aortic crossclamping and initial administration of cardioplegia containing 50 μM diazoxide, mean arterial pressure decreased from 70.2 ± 6.8 to 65.5 ± 5.8 mmHg in DZX hearts and from 78.7 ± 5.1 to 75.5 ± 5.2 mmHg in DSA hearts. No significant difference in mean arterial pressure following the administration of cardioplegia was observed between DSA and DZX hearts.
3.6. Myocardial function during reperfusion in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1) and in the LV area subjected to 30 min hypothermic global ischemia (Region 2)
No significant differences in HR, LVPDP, LVEDP, +dP/ dt, mean arterial pressure, or LV ejection fraction were observed within or between DSA and DZX hearts during reperfusion following 30 min of normothermic regional ischemia and 30 min of hypothermic global ischemia.
Following 30 min reperfusion (130 min, Table 2), Tau was significantly increased in DSA hearts (P < 0.05) and PRSW was significantly decreased (p < 0.01) in both DSA and DZX hearts as compared to initial myocardial function (30–40 min, Table 2). At 90 min reperfusion (190 min) no significant difference in Tau was observed between DSA and DZX as compared to initial myocardial function (30–40 min, Table 2).
PRSW was significantly decreased (P < 0.05) in both DSA and DZX hearts at 90 min reperfusion (190 min, Table 2) as compared to initial myocardial function. No significant difference in PRSW was observed within or between DSA and DZX hearts during reperfusion. No significant difference in SS, SB or PSS in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) were observed between groups at the acquisition of initial myocardial function (30–40 min) or during 120 min reperfusion (Table 2). No significant differences in hematocrit and core temperature were observed throughout the experiment (results not shown).
3.7. Coronary flow following 30 min of normothermic regional ischemia and 30 min of hypothermic global ischemia
Coronary arterial flow during equilibrium, 30 min normothermic regional ischemia, 30 min hypothermic global ischemia and 120 min reperfusion is shown in Figs. 2A,B. No significant differences in proximal or distal LAD flow were observed within or between groups during equilibrium. Distal LAD flow was undetectable following snaring of the LAD in both DSA and DZX hearts. No significant differences in proximal LAD flow were observed within or between groups during 30 min normothermic regional ischemia. Proximal LAD flow was undetectable following the administration of cardioplegia, during 30 min hypothermic global ischemia, in both DSA and DZX hearts. A significant hyperemia (P < 0.05 versus equilibrium) was observed in proximal LAD flow throughout reperfusion in both DSA and DZX hearts. Distal LAD flow significantly increased after 20 min of reperfusion (120 min) in both DSA and DZX hearts (P < 0.01 versus equilibrium). No significant differences in proximal, distal LAD flow (Figs. 2A,B) or distal/proximal coronary arterial flow ratio (results not shown) were observed within or between DSA and DZX hearts during reperfusion.
Fig. 2.

Proximal (A), distal (B) coronary arterial during 30 min equilibrium, 30 min normothermic regional ischemia, 30 min hypothermic global ischemia, and 120 min reperfusion for DSA hearts and DZX hearts. A continuous hyperemia was observed throughout reperfusion in both DSA and DZX hearts. Results are shown as mean ± SEM for each group. Significant differences are shown as * = P < 0.05 versus equilibrium.
3.8. Myocardial infarct size following 30 min of normothermic regional ischemia and 30 min of hypothermic global ischemia
No significant difference in AAR was observed between DSA and DZX hearts (Fig. 3A). Myocardial infarct was observed only in the region of the heart subjected to normothermic regional ischemia in both DSA and DZX hearts. Infarct size expressed as a percentage of AAR is shown in Fig. 3B. Infarct size was significantly increased to 33.6 ± 2.9% in DSA hearts as compared to 16.8 ± 2.4% in DZX hearts (P < 0.01). Myocardial infarct in DZX hearts was centrally located within the AAR. In DSA hearts infarct was located centrally and was significantly increased at the borderline zone within the AAR (Fig. 3C). Apoptosis as measured by TUNEL positive staining of myocardial nuclei was 120.3 ± 48.8 in DSA hearts as compared to 21.4 ± 5.3 in DZX hearts (Fig. 4). No significant difference in the wet weight/dry weight ratio was observed between specimens from Regions 1 and 2, or within or between DSA and DZX hearts (results not shown).
Fig. 3.

(A) Area at risk (AAR; % of left ventricular mass); and (B) infarct size (% of AAR) in the left ventricular area subjected to 30 min normothermic regional ischemia, 30 min hypothermic global ischemia, and 120 min reperfusion (Region 1; Fig. 1) in DSA and DZX hearts. No detectable infarct was observed in the left ventricular area subjected to 30 min hypothermic global ischemia, and 120 min reperfusion (Region 2; Fig. 1) in either DSA or DZX hearts. Results are shown as mean ± SEM for each group. Significant differences are shown as * = P < 0.05 versus DSA hearts. (C) Representative photograph of pig heart slices from DSA and DZX hearts stained with 1% triphenyl tetrazolium chloride. Infarct size (% of AAR) was 43.0% in DSA heart and 19.2% in DZX heart. AAR is shown by the blue dotted line. Myocardial infarct (yellowish zone) was increased significantly at the borderline zones of AAR in DSA as compared to DZX hearts.
Fig. 4.

Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL)-positive cells/3000 myocytes in the left ventricular myocardium subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia and 120 min reperfusion (Region 1) in DSA and DZX hearts. Results are shown as the mean ± SEM of 10–15 high-powered (20 × ) fields in 5–6 slides from the six samples for each group. Significant differences are shown as * = P < 0.05 versus DSA hearts.
3.9. Effects of isoflurane anesthetic on the cardioprotection afforded by DSA cardioplegia and DSA cardioplegia with diazoxide
A separate study was performed using isoflurane as the anesthetic agent (n = 3 for each DSA and DZX heart). No significance in hemodynamics, PRSW, Tau or LV ejection fraction during equilibrium, regional ischemia, global ischemia or reperfusion was observed within or between DSA and DZX hearts in which isoflurane or sevoflurane were used as the anesthetic agent. Proximal coronary flow at the immediate start of reperfusion was significantly decreased in DZX hearts in which isoflurane was used as the anesthetic agent (P < 0.05) as compared with DSA hearts in which sevoflurane was used as the anesthetic agent at 30 min reperfusion (130 min) but no significant differences were observed at any other time point (results not shown).
As was the case with hearts in which sevoflurane was used as the anesthetic agent myocardial infarct was observed only in the region of the heart subjected to normothermic regional ischemia in both DSA and DZX hearts in which isoflurane was used as the anesthetic agent. Infarct size expressed as a percentage of AAR was 40.6 ± 5.9% in DSA hearts and 17.5 ± 2.1% in DZX hearts in which isoflurane was used as the anesthetic agent. There were no significant differences in infarct size observed between DSA and DZX hearts in which sevoflurane or isoflurane were used as the anesthetic agents.
4. Discussion
Myocardial ischemia occurs as the result of attenuation or cessation of coronary blood flow such that oxygen delivery to the myocardium is insufficient to meet oxygen requirements. In regional myocardial ischemia coronary blood flow can be variably obstructed resulting in limitation or termination of substrate delivery and the restriction or discontinuation of catabolite removal. The extent to which this obstruction is present and the time period over which oxygen delivery is severely obstructed can lead to irreversible myocardial ischemic injury.
Clinically the surgeon is confronted by a spectrum of increasing regional ischemia extending to acute occlusion from a previously obstructed vessel with the resultant rate and extent of ischemic injury dependent upon the formation and development of collateral coronary flow. In previous reports we and others have investigated modalities to alleviate myocardial ischemic injury using experimental models of stunning, regional and global ischemia to induce a spectrum of ischemic injury [8–12]. In the experimental model of regional ischemia a portion of the LV is subjected to complete coronary obstruction (dependent upon collateral coronary flow). This model provides data applicable to regional alterations but does not include global modifications resulting from surgical interventions including cardioplegic arrest and crossclamp time. In the experimental model of global ischemia the whole heart is at risk. In this report we have attempted to provide greater surgical relevance using a model of acute myocardial infarction. This model allows for the investigation of regional ischemic injury similar to that observed in the patient under conditions which more closely approximate those experienced during surgery. Our model mimics plaque rupture where a previously minimally obstructed vessel becomes acutely occluded without the formation and development of collateral coronary flow. In this model, a region of the LV is made ischemic, cardioplegic arrest and cross-clamping occurs and then the snare and the cross-clamp are released following bypass. We have used 30 min normothermic regional ischemia based on earlier studies which indicated that this period of ischemic time was needed to allow for statistical determination of experimental protocols [10]. The duration of cold blood cardioplegia arrest was based on approximation of the time required for single coronary bypass. The LAD snare was left in place throughout the 30 min normothermic regional ischemia and the 30 min hypothermic global arrest to ensure that no regional flow to the regional ischemic zone occurred and to mimic the effects of cardioplegia prior to distal anastomosis in a model in which no collateral coronary flow is present. This protocol resulted in the compartmentalization of the myocardium such that no cardioplegia was delivered to the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) during surgical arrest of the heart.
Diazoxide (50 μM) was included with DSA cardioplegia only during the initial administration of cardioplegia, but was not included when DSA was re-administered following 15 min of global ischemia based on the preliminary results which showed that re-administration of diazoxide resulted in a significant decrease in mean arterial pressure upon reperfusion (results not shown). These results are in agreement with that of Garlid et al. [7] who have shown that diazoxide decreases cell injury in a dose-dependent manner at concentrations between 1 and 30 μM, while concentrations from 30 to 100 μM diazoxide afford a similar level of cardioprotection. In this study we have used 50 μM diazoxide to investigate the role of mitoKATP channels in cardioprotection. Diazoxide was not administered during reperfusion due to preliminary results which indicated that this too resulted in a significant decrease in mean arterial pressure upon reperfusion (results not shown).
Our results indicate that the pharmacological opening of mitoKATP channels with diazoxide (50 μM) with DSA cardioplegia significantly decreased infarct size (P < 0.05 versus DSA) in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) as compared with DSA cardioplegia alone. No detectable infarct was observed in the left ventricular area subjected to 30 min hypothermic global ischemia (Region 2; Fig. 1) in either DSA or DZX hearts.
Diazoxide is used as anti-hypertensive drug and has vasodilatory properties and has a reported half life of 48 h [15,16]. We speculate that the significant decrease in infarct size observed in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) in DZX as compared with DSA hearts is attributable to the half life of diazoxide and that the presence of diazoxide during reperfusion. Support for this hypothesis comes in the distinct infarct patterns evident in DSA and DZX hearts in Region 1. The location of infarct within the AAR in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia (Region 1; Fig. 1) indicated that myocardial infarct in DZX hearts was centrally located within the AAR. In DSA hearts, infarct was located centrally and was significantly increased at the borderline zones within the area at risk (Fig. 3C). We speculate that the decrease in infarct at the borderzones of the AAR in DZX hearts is directly related to the prolonged action of diazoxide in the myocardium which would have affected these areas upon reperfusion presumably by infusion into the affect zone during reperfusion and reflects the infarct sparing properties of mitoKATP channel opening during reperfusion. These data are in agreement with our earlier studies in which we have shown that infarct size is modulated by mitoKATP channels both during ischemia and reperfusion but that infarct size modulation occurs primarily during ischemia [3].
Our results indicate that coronary arterial flow showed striking reactive hyperemia in both proximal and distal coronary artery flow throughout reperfusion in both DSA and DZX hearts with no significant differences being observed within or between DSA and DZX hearts. Further examination using distal/proximal coronary artery flow ratio indicated there was no significant difference between DSA and DZX hearts (results not shown). These data indicate that the reduction in infarct size in the LV area subjected to 30 min normothermic regional ischemia and 30 min hypothermic global ischemia in DZX hearts is not the result of increased coronary arterial flow in agreement with Grover [15] who has previously reported that increases in coronary flow are not necessary for cardioprotection.
Our results indicate that the addition of diazoxide (50 μM) to DSA cardioplegia had no effect on regional or global myocardial function as compared to DSA cardioplegia alone (Tables 1 and 2) in agreement with earlier reports in which we have shown that the opening of the mitoKATP channels modulates infarct size but has no effect on functional recovery [2,3].
The mechanism by which the pharmacological opening of the mitoKATP channels affords enhanced cardioprotection by significantly decreasing infarct size remains to be elucidated. Our results presented herein and in earlier reports suggest that stabilization of the mitochondrial membrane is a key prerequisite in this mechanism [2,3]. Previous reports suggest that mitochondrial membrane depolarization caused by K+ entry through the opening of mitoKATP channels would reduce mitochondrial Ca2+ ([Ca2+]mito) entry through the calcium uniporter and would reduce mitochondrial Ca2+ overload [7,17]. Subsequently, these events are believed to result in ATP production and cell salvage [17]. The modulation of infarct size would presumably occur through the modulation of [Ca2+]mito accumulation and preservation of mitochondrial function.
Increased [Ca2+]mito accumulation has been shown to destabilize the inner mitochondrial membrane, causing the inner membrane pore to open, which permits further movement of cations across the mitochondrial membrane [4]. The opening of these pores renders the mitochondrion incapable of synthesizing ATP and has been suggested to be a key event in the process leading to myocardial cell death. We speculate that these events are directly related to infarct size reduction. Support for this hypothesis comes from an earlier report by us [1] and the previous investigation of Fryer et al. [18] who have shown that mitoKATP channels play an important role in the preservation of mitochondrial function. We speculate that the opening of the mitoKATP channels during ischemia effectively reduces [Ca2+]mito accumulation.
A recent in vivo study from Ozcan et al. [19] has suggested that treatment of mitochondria with diazoxide during the anoxic period effectively preserves the structural and functional integrity of mitochondria, maintaining ATP generation after the anoxic insult. This would agree with our observations using transmission electron microscopy of tissue obtained from Regions 1 and 2, which showed extensive swelling of the mitochondrial matrix and disruption of cristae in Region 1 which was less pronounced in Region 2 in DSA hearts. Only a slight swelling of the intercristae matrix was observed in Regions 1 and 2 in DZX hearts (McCully, JD et al. personal communication, results not shown). These results also agree with that of Garlid [20] who has suggested that the primary function of mitoKATP channels is to participate in regulation of mitochondrial volume and maintenance of matrix volume.
Our results also indicate that diazoxide significantly decreases the myocardial cell apoptosis in Region 1 as compared to DSA hearts. These data are in agreement with recent reports have shown that mitochondria play a key role in the regulation of apoptotic myocardial cell death [2–7,14,21,22]. While it remains unclear how the opening of the mitoKATP channels modulate myocardial cell necrosis and apoptosis induced by ischemia-reperfusion injury we speculate that anti-apoptotic effects of diazoxide are the result of modulation of [Ca2+]mito accumulation and the stabilization of mitochondrial inner membrane permeability, which would prevent the efflux of cytochrome c and activation of pro-apoptotic proteins [21,22].
There is some evidence to suggest that anesthetic agents can provide at least partially to cardioprotection [23]. Isoflurane has been shown to be associated with a significant increase in ecto-5′-nucleotidase activity and decreases in both troponin I and CK-MB release in humans, however these decreases failed to reach statistical significance as compared to matched controls [24]. The generic formulation of isoflurane provides an attractive cost incentive and is the anesthetic agent of choice at the Beth Israel Deaconess Medical Center. In our investigations we have used sevo-flurane as the anesthetic agent based on reports indicating sevoflurane does not activate KATP channels [25].
In this report we have performed additional experiments using isoflurane as the anesthetic agent with DSA cardio-plegia (n = 3) and with DSA cardioplegia with 50 μM diaz-oxide (n = 3) using the same protocol as described for experiments in which sevoflurane was used as the anesthetic agent to determine if added cardioprotection could be achieved. Our results indicate that no significant differences were observed in regional or global myocardial functional recovery during reperfusion, nor was any significant difference observed in infarct size between DSA and DZX hearts in which isoflurane or sevoflurane was used as anesthetic agents. These data suggest that the anesthetic agent isoflurane, in the pig heart model of acute myocardial infarction, does not provide added cardioprotection.
In conclusion our results indicate that the addition of diazoxide to cardioplegia significantly decreases regional myocardial cell necrosis and apoptosis in a model of acute myocardial infarction and represents an additional modality for achieving myocardial protection.
Acknowledgments
This study was supported by the National Institutes of Health (HL 29077 and HL 59542) and the American Heart Association.
Appendix A. Discussion
Dr B. Walpoth (Bern, Switzerland): Do you think it is important to give it before ischemia or do you think you could give it just at the onset or after ischemia, which might have an implication on treating acute myocardial infarction?
Dr Levitsky: It certainly could. Diazoxide has a half-life of 24 h. It also causes hypotension when given directly into the coronary arteries. If the cardiologist got into trouble in the cardiac catheterization laboratory, or if a patient came in with an acute myocardial infarction, theoretically it might be wise, once we learned the correct dose, to give diazoxide because this may have some effect in decreasing the area of myocardial infarction necrosis. But more importantly, I think it is affecting apoptosis.
For the most part, infarction has already occurred by the time we see the patient in the operating room, but apoptosis, the prevention of myocardial cells deteriorating at a later period of time, is something that is surgically important. So the answer is yes. The other possibility is to give this drug to patients who are undergoing open heart surgery before they are induced, provided we don’t get significant hypotension, to see if we could protect the mitochondria.
I am going to divert for a moment to discuss the origin of these studies which is based on our looking at mitochondria in a series of patients undergoing CABG: we had three patients out of a group of about 15 who we needed inotropic support to get off bypass. When we looked at these patients in our molecular biology laboratory, we found that it was the cytochrome c in the mitochondria that were being adversely affected, and perhaps if we had used this drug, we might not have gotten into trouble. So that was the clinical incentive for doing these studies and led us to the trail of looking at the mitochondrial potassium channel openers.
Dr J. Wistbacka (Vasa, Finland): I have a question regarding this model. Was the LAD still occluded at the time of cross-clamping? I mean, did this diazoxide plegia get through the LAD to this ischemic area or did it not?
Dr Levitsky: That is a very important point that you raised. The LAD was occluded during the period of ischemia. We tried to mimic exactly what we do in the operating room. So in the ischemic area the only time that the diazoxide could get to the myocardium is during reperfusion, because during reperfusion we remove the tourniquet from the left anterior descending coronary artery.
Footnotes
Presented at the joint 15th Annual Meeting of the European Association for Cardiothoracic Surgery and the 9th Annual Meeting of the European Society of Thoracic Surgeons, Lisbon, Portugal, September 16–19, 2001.
References
- 1.Faulk EA, McCully JD, Tsukube T, Hadlow NC, Krukenkamp IB, Levitsky S. Myocardial mitochondrial calcium accumulation modulates nuclear calcium accumulation and DNA fragmentation. Ann Thorac Surg. 1995;60:338–344. doi: 10.1016/0003-4975(95)00446-r. [DOI] [PubMed] [Google Scholar]
- 2.Toyoda Y, Friehs I, Parker RA, Levitsky S, McCully JD. Differential role of sarcolemmal and mitochondrial KATP channels in adenosine enhanced ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000;279:H2694–H2703. doi: 10.1152/ajpheart.2000.279.6.H2694. [DOI] [PubMed] [Google Scholar]
- 3.Toyoda Y, Levitsky S, McCully JD. Opening of mitochondrial ATP-sensitive potassium channels enhances cardioplegic protection. Ann Thorac Surg. 2001;71:1281–1289. doi: 10.1016/s0003-4975(00)02667-9. [DOI] [PubMed] [Google Scholar]
- 4.Frolkis VV, Frolkis RA, Mkhitarian LS, Shevchuk VG, Fraifeld VE, Vakulenko LG, Syrovy I. Contractile function and Ca2+ transport system of myocardium in ageing. Gerontology. 1988;34:64–74. doi: 10.1159/000212932. [DOI] [PubMed] [Google Scholar]
- 5.Ferrari R. The role of mitochondria in ischemic heart disease. J Cardiovasc Pharmacol. 1996;28:S1–S10. doi: 10.1097/00005344-199600003-00002. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 7.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell MP, Hearse DJ, Yellon DM. Species variation in the coronary collateral circulation during regional myocardial ischaemia: a critical determinant of the rate of evolution and extent of myocardial infarct. Cardiovasc Res. 1987;21:737–746. doi: 10.1093/cvr/21.10.737. [DOI] [PubMed] [Google Scholar]
- 9.McCully JD, Uematsu M, Levitsky S. Adenosine enhanced ischemic preconditioning provides myocardial protection equal to that of cold blood cardioplegia. Ann Thorac Surg. 1999;67:699–704. doi: 10.1016/s0003-4975(98)01371-x. [DOI] [PubMed] [Google Scholar]
- 10.Toyoda Y, Di Gregorio V, Parker RA, Levitsky S, McCully JD. Anti-stunning and anti-infarct effects of adenosine enhanced ischemic preconditioning. Circulation. 2000;102:326–331. doi: 10.1161/01.cir.102.suppl_3.iii-326. [DOI] [PubMed] [Google Scholar]
- 11.Ovize M, Przyklenk K, Hale SL, Kloner RA. Preconditioning does not attenuate myocardial stunning. Circulation. 1992;85:2247–2254. doi: 10.1161/01.cir.85.6.2247. [DOI] [PubMed] [Google Scholar]
- 12.Marsch SC, Dalmas S, Philbin DM, Ryder WA, Foex P. Myocardial ischemia and reperfusion are associated with an increased stiffness of remote non-ischemic myocardium. Anesth Analg. 1996;82:695–701. doi: 10.1097/00000539-199604000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Coetzee A, Fourie P. Postsystolic shortening as an index of regional myocardial ischemia in an experimental model. J Cardiothorac Vasc Anesth. 1991;5:546–550. doi: 10.1016/1053-0770(91)90003-c. [DOI] [PubMed] [Google Scholar]
- 14.Stadler B, Phillips J, Toyoda Y, Federman M, Levitsky S, McCully JD. Adenosine-enhanced ischemic preconditioning modulates necrosis and apoptosis: effects of stunning and ischemia-reperfusion. Ann Thorac Surg. 2001;72:555–564. doi: 10.1016/s0003-4975(01)02665-0. [DOI] [PubMed] [Google Scholar]
- 15.Grover GJ. Pharmacology of ATP-sensitive potassium channel (KATP) openers in models of myocardial ischemia and reperfusion. Can J Physiol Pharmacol. 1997;75:309–315. doi: 10.1139/cjpp-75-4-309. [DOI] [PubMed] [Google Scholar]
- 16.Kirsten R, Nelson K, Kirsten D, Heintz B. Clinical pharmacokinetics of vasodilators. Part I Clin Pharmacokinet. 1998;34:457–482. doi: 10.2165/00003088-199834060-00003. [DOI] [PubMed] [Google Scholar]
- 17.Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol Heart Circ Physiol. 1998;275:H1567–H1576. doi: 10.1152/ajpheart.1998.275.5.H1567. [DOI] [PubMed] [Google Scholar]
- 18.Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ. Ischemic preconditioning in rats: role of mitochondrial KATP channel in preservation of mitochondrial function. Am J Physiol Heart Circ Physiol. 2000;278:H305–H312. doi: 10.1152/ajpheart.2000.278.1.H305. [DOI] [PubMed] [Google Scholar]
- 19.Ozcan C, Holmuhamedov EL, Jahangir A, Terzic A. Diazoxide protects mitochondria from anoxic injury: implication for myopreservation. J Thorac Cardiovasc Surg. 2001;121:298–306. doi: 10.1067/mtc.2001.111421. [DOI] [PubMed] [Google Scholar]
- 20.Garlid KD. Opening mitochondrial KATP in the heart – what happens, and what does not happen. Basic Res Cardiol. 2000;95:275–279. doi: 10.1007/s003950070046. [DOI] [PubMed] [Google Scholar]
- 21.Akao M, Ohler A, O’Rourke B, Marban E. Mitochondrial ATP-sensitive potassium channels inhibit apoptosis induced by oxidative stress in cardiac cells. Circ Res. 2001;88:1267–1275. doi: 10.1161/hh1201.092094. [DOI] [PubMed] [Google Scholar]
- 22.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- 23.Ross S, Foex P. Protective effects of anaesthetics in reversible and irreversible ischaemia-reperfusion injury. Br J Anaesth. 1999;82:622–632. doi: 10.1093/bja/82.4.622. [DOI] [PubMed] [Google Scholar]
- 24.Belhomme D, Peynet J, Louzy M, Launay JM, Kitakaze M, Menasche P. Evidence for preconditioning by isoflurane in coronary artery bypass graft surgery. Circulation. 1999;100(Suppl II):II340–II344. doi: 10.1161/01.cir.100.suppl_2.ii-340. [DOI] [PubMed] [Google Scholar]
- 25.Kersten JR, Schmeling T, Tessmer J, Hettrick DA, Pagel PS, Warltier DC. Sevoflurane selectively increases coronary collateral blood flow independent of KATP channels in vivo. Anesthesiology. 1999;90:246–256. doi: 10.1097/00000542-199901000-00031. [DOI] [PubMed] [Google Scholar]