

Abstract
Formyl peptide receptors (FPRs) play an essential role in the regulation of endogenous inflammation and immunity. In the present studies, a large series of pyridazin-3(2H)-one derivatives bearing an arylacetamide chain at position 2 was synthesized and tested for FPR agonist activity. The pyridazin-3(2H)-one ring was confirmed to be an appropriate scaffold to support FPR agonist activity, and its modification at the 4 and 6 positions led to the identification of additional active agonists, which induced intracellular Ca2+ flux in HL-60 cells transfected with either FPR1, FPR2, or FPR3. Seven formyl peptide receptor 1 (FPR1)-specific and several mixed FPR1/FPR2 dual agonists were identified with low micromolar EC50 values. Furthermore, these agonists also activated human neutrophils, inducing intracellular Ca2+ flux and chemotaxis. Finally, molecular docking studies indicated that the most potent pyridazin-3(2H)-ones overlapped in their best docking poses with fMLF and WKYMVM peptides in the FPR1 and FPR2 ligand binding sites, respectively. Thus, pyridazinone-based compounds represent potential lead compounds for further development of selective and/or potent FPR agonists.
Keywords: pyridazin-3(2H)-one, formyl peptide receptor, dual agonist, human neutrophils, molecular docking
1. Introduction
Formyl peptide receptors (FPRs) are chemoattractant receptors belonging to the seven transmembrane domain G protein-coupled receptor (GPCR) family known to couple to pertussis toxin (PTX)-sensitive Gi proteins [1]. FPRs play an important role in the regulation of inflammatory reactions and cellular dysfunction and are expressed in the majority of white blood cells, such as neutrophils, monocytes and dendritic cells. In humans, there are three FPR isoforms [2], including FPR1 (formyl peptide receptor 1) and FPR2 (formyl peptide receptor 2), which exhibit 69% amino acidic homology to each other, and FPR3 (formyl peptide receptor 3) with 56% amino acid sequence identity to FPR1 and 83% to FPR2. FPR1 was first identified in the early 1970s as a high-affinity binding site on the surface of neutrophils for N-formyl peptides, such as the prototypic fMLF (formyl-methionine-leucine-phenylalanine) [3]. In addition to FPR1, neutrophils also express the closely related FPR2, which is a low-affinity receptor for fMLF, despite the relatively high level of sequence homology with FPR1. These two receptors have a similar distribution in a variety of different tissues and cells involved in inflammation, such as endothelial cells, platelets and dendritic cells. Moreover, FPR2 is also expressed in a range of other cell types, including phagocytic leukocytes, hepatocytes, epithelial cells, T lymphocytes, neuroblastoma cells, astrocytoma cells, and microvascular endothelial cells [4]. Unlike FPR1 and FPR2, FPR3 does not bind N-formyl peptides and is expressed in neutrophils, but it is specifically expressed on monocytes and dendritic cells [5]. FPRs interact with a large number of structurally distinct ligands, and their activation induces a variety of complicated downstream signalling responses involved in inflammatory processes, such as chemotaxis, degranulation, and superoxide generation [6]. FPR activation has also been associated with various diseases, including ischemia-reperfusion injury [7], prion disease [8], amyloidosis, Alzheimer's disease [9], HIV [10], stomach ulcer [11], some cancers [12], and nociception associated with inflammatory processes [13].
Numerous receptors modulate the host inflammatory response, and resolution of inflammation involves the formation of endogenous anti-inflammatory mediators, which are essential for the proper functioning of host cells. FPRs interact with some resolving mediators (e.g., lipoxins) and have clearly emerged as crucial players in important endogenous anti-inflammation processes essential to ensure regulation and control of inflammation in order to avoid self-induced damage [14]. Thus, FPRs represent a promising new target for the discovery of novel anti-inflammatory and pro-resolving drugs able to selectively stimulate the innate immune response or endogenous anti-inflammation systems [6]. Indeed, there is evidence that bioactive ligands acting as FPR agonists might play a role as useful therapeutics in host defense and as immunomodulatory activators that are able to enhance selective innate immune responses in order to reduce detrimental effects associated with inflammation and infectious diseases [15].
Based on our previous results demonstrating the suitability of pyridazin-3(2H)-one as an appropriate scaffold for FPR agonists [16], as well as evidence from the literature that both FPR-specific [17,18] and mixed FPR1/FPR2 agonists [19,20] are of potential interest for treatment of diseases involving neutrophilic inflammation, we performed further modifications on this chemotype in order to evaluate if these modifications would increase the potency and/or selectivity of the agonists. Since we already showed that an acetamide side chain as well as the presence of lipophilic and/or electronegative substituents in the aryl group at the end of the chain were essential for activity at position 2 of the pyridazin-3(2H)-one [16], we performed several modifications at the C-4, C-5, and C-6 positions of the ring, as these modifications have not been well studied previously (Figure 1). For example, we eliminated the methyl group at position 6 and then increased the steric hindrance and/or modified the electronic characteristics of the substituents by introducing (substituted) aromatic and heteroaromatic systems. In addition, we evaluated the role of position 4 by changing or removing the substituents at the benzyl fragment and changing the phenyl ring with various heterocyclic rings. These new agonists were evaluated for their ability to activate intracellular Ca2+ mobilization and chemotaxis in human neutrophils and their binding to FPR1 and FPR2 was evaluated by molecular docking studies.
Figure 1.

Representative modifications performed on the pyridazin-3(2H)-one derivatives.
2. Chemistry
As extensively reported in the literature, γ-keto acids are the common starting material to easily generate dihydropyridazinone scaffolds [21,22], which are the key building blocks for the synthesis of the pyridazinone class of compounds outlined in the Schemes 1 and 2. With the exception of compounds 1a–c, which were synthesized following published procedures [23–25], all other γ-keto acids (1d–k) were purchased from commercial sources.
Scheme 1.

Synthesis of pyridazin-3(2H)-one derivatives 6a–w. Reagents and conditions: a) NH2NH2 (1 equiv), EtOH, 1–3 h, 60 °C; b) 3 or 4-methoxybenzaldehyde (1 equiv), KOH 5% (w/v) in anhydrous EtOH, 1–3 h, reflux; c) ethyl bromoacetate (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 1–3 h, reflux; d) NaOH 6N, 1–2 h, 60–80 °C; e) ethyl chloroformate (1.1 equiv), Et3N (3.5 equiv), substituted aniline (2 equiv), anhydrous THF, 12 h, -5 °C/rt; (f) 0.5 h, 160 °C.
Scheme 2.

Synthesis of pyridazin-3(2H)-one derivatives 11a–q and 15a,b. Reagents and conditions: a) substituted benzaldehyde (1 equiv), KOH 5% (w/v) in anhydrous EtOH, 1–5 h, reflux; b) ethyl bromoacetate (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 2–4 h, reflux; c) NaOH 6N, 1–5 h, 80 °C; d) ethyl chloroformate (1.1 equiv), Et3N (3.5 equiv), substituted aniline (2 equiv), anhydrous THF, 12 h, -5 °C/rt; e) N-(4-bromophenyl)-2-chloroacetamide [38] (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 2–3 h, reflux; f) POCl3, 3 h, 60 °C; g) SeO2 (3 equiv), anhydrous EtOH, 5–7 h, reflux.
Condensation of the appropriate γ-keto acid with hydrazine hydrate resulted in the previously described C-6 substituted dihydropyridazinones 2a–h,j,k [21,26–32] and the new 2i in good yields (Scheme 1). A further synthetic step was required to obtain compound 2l, which was generated by melting and spontaneous decarboxylation of the carboxylic acid 2k at 160 °C [33]. The C-6 modified pyridazinones 6a–w (Scheme 1) were then prepared in four steps starting from the dihydropyridazinones 2a–j,l. The previously described intermediates 3h,j,l [34] and the new 3ag, i,k,m–p were obtained through a Knoevenagel condensation with the commercially available 3- or 4-methoxybenzaldehyde in the presence of KOH. Subsequent alkylation with ethyl bromoacetate gave esters 4a–p, which were transformed into the corresponding carboxylic acids 5a–p by alkaline hydrolysis under standard conditions. Reaction of the intermediates 5a–p with ethyl chloroformate in the presence of triethylamine in THF resulted in the intermediate mixed anhydrides, which were transformed in good yields into the final amides 6a–w by treatment with the appropriate aryl amines (Scheme 1).
Scheme 2 shows the synthesis of compounds 11a–q, where the 3- or 4-methoxybenzyl moiety at the C-4 position of the pyridazinone ring was eliminated or replaced by several heterocyclic and substituted benzyl groups. The dihydropyridazinone 7 [16] was converted into the previously described derivatives 8h–j [35–37] and the new 8a–g,k,l by Knoevenagel condensation with the appropriate aromatic aldehyde in the presence of KOH. Subsequently, compounds 8a–j were firstly alkylated with ethyl bromoacetate to give the esters 9a–j, of which 9j was previously reported [37], and then hydrolyzed under basic conditions to give the new carboxylic acid derivatives 10a–j. These intermediates were transformed into the final amides 11a–l (Scheme 2) from the corresponding mixed anhydrides by performing the same reaction as described above for intermediates 6a–w. A different synthetic procedure was required to obtain compounds 11m–o (Scheme 2). Intermediates 8k,l and 8m, obtained from 8l by dehydration of the amide with phosphorus oxychloride, were unstable to strong base treatment and were directly converted into compounds 11m–o by alkylation under standard condition with the previously described N-(4- bromophenyl)-2-chloroacetamide [38]. Furthermore, compounds 15a,b (Scheme 2) were obtained in good yields by carrying out the usual reaction sequence (alkylation/hydrolysis/amide bond formation) described above, starting alternatively with dihydropyridazinone 7 [16] and its pyridazinone derivative 12 [31,39], which was obtained by using selenium dioxide as the oxidizing agent. Moreover, the synthesis of compounds 11p,q, which contain a 4-iodophenylacetamide chain at position 2, was carried out starting from 15a and using Knoevenagel condensation with the appropriate aromatic aldehyde (Scheme 2).
Scheme 3 shows the synthesis of the 4-arylketone derivative 18, which was prepared from 16 [16] by oxidation of the benzylic methylene (17) with cerium ammonium nitrate (CAN) and subsequent alkylation under standard conditions with the previously described intermediate N-(4- bromophenyl)-2-chloroacetamide [38].
Scheme 3.

Synthesis of pyridazin-3(2H)-one derivative 18. Reagents and conditions: a) CAN (3 equiv), CH3COOH 50%, 1.5 h, 60 °C; b) N-(4-bromophenyl)-2-chloroacetamide38 (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 6 h, reflux.
The synthetic pathway for compounds 23, 27 and 28a–c is shown in Scheme 4. The 4- amino-5-acetyl pyridazinone 19 [40] and its derivative 24 [41], obtained by deacetylation of 19 using 48% hydrobromic acid under pressure at high temperature (140 °C) [42], were previously described. Starting from compound 19 and following alkylation with ethyl bromoacetate (20), coupling on the amine at C-4 was performed using 4-methoxyphenylboronic acid in the presence of Cu(Ac)2 in CH2Cl2. The intermediate 21 was then processed by standard alkaline hydrolysis and coupling with 4-bromoaniline, through the intermediate mixed anhydride, to obtain the final amide 23 in good yield. Alternatively, compound 24 was directly processed by standard alkylation, hydrolysis, and amidation to generate the final 4-amino derivative 27 (Scheme 4). Subsequently, coupling of the amine group of 27 with 4-methoxyphenylboronic acid in the presence of Cu(Ac)2 gave the C-4 arylamine 28a, while the C-4 arylamide analogues 28b,c were obtained from 27 by treatment with the opportune arylchloride and triethylamine in CH2Cl2 at 0 °C.
Scheme 4.

Synthesis of pyridazin-3(2H)-one derivatives 23, 27 and 28a–c. Reagents and conditions: a) ethyl bromoacetate (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 3 h, reflux; b) 4-methoxyphenylboronic acid (2 equiv), Cu(OAc)2 (1.5 equiv), Et3N (2 equiv), CH2Cl2, 12 h, rt; c) NaOH 6N, 1–2 h, 80 °C; d) ethyl chloroformate (1.1 equiv), Et3N (3.5 equiv), 4-bromoaniline (2 equiv), anhydrous THF, 12 h, -5 °C/rt; e) HBr 48%, 1 h, 140 °C; f) for 28a: 4- methoxyphenylboronic acid (1 equiv), Cu(OAc)2 (1.5 equiv), Et3N (2 equiv), CH2Cl2, 12 h, rt; for 28b,c: substituted benzoyl chloride (2.4 equiv), Et3N (catalytic), anhydrous CH2Cl2, 16 h, 0 °C/rt.
The synthesis of compound 33 with a 4-methoxyphenyl group directly bonded to C-4 of the pyridazinone ring is shown in Scheme 5. Condensation of the known γ-keto ester 29 [43] with hydrazine hydrate resulted in intermediate 30, which was subjected to the usual alkylation/alkaline hydrolysis/amidation steps to obtain the final amide 33 in good yield.
Scheme 5.

Synthesis of pyridazin-3(2H)-one derivative 33. Reagents and conditions: a) NH2NH2 (2 equiv), anhydrous toluene, 2 h, reflux; b) ethyl bromoacetate (1.5 equiv), K2CO3 (2 equiv), anhydrous CH3CN, 1.5 h, reflux; c) NaOH 6N, 0.5 h, 60 °C; d) ethyl chloroformate (1.1 equiv), Et3N (3.5 equiv), 4-bromoaniline (2 equiv), anhydrous THF, 12 h, -5 °C/rt.
3. Pharmacology
In the present study, we synthesized a library of 49 pyridazinone-based compounds, which were screened in order to identify novel small-molecule FPR agonists. The compounds were evaluated for their ability to induce intracellular Ca2+ flux in human HL-60 cells transfected with FPR1, FPR2, or FPR3 [44,45]. Since undifferentiated HL-60 cells do not express any FPRs and do not respond to FPR agonists, we evaluated all compounds in non-transfected HL-60 cells to verify that the observed activity was specific for the individual FPRs expressed. Both EC50 values and relative efficacy were determined (Tables 5–7) and compared to the peptide agonists fMLF and WKYMVm, as well as to reference compounds A–D, which were previously designated as 14m, 14x, 14e, and 14a, respectively [16].
Table 5.
Activity of compounds 6a–w (Scheme 1) in HL-60 cells expressing human FPR1, FPR2,or FPR3
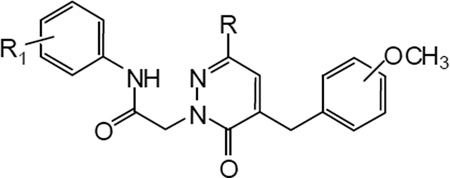 | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R | OCH3 | R1 | Ca2+ Mobilization EC50 (µM) and Efficacy (%) a | |||
| FPR1 | FPR2 | FPR3 | W.T.b | ||||
| A16 | CH3 | m | 3,4-methylenedioxy | 2.3 (50) | N.A. | N.A. | N.A. |
| B16 | CH3 | p | Br (p) | N.A. | 2.4 | N.A. | N.A. |
| C16 | CH3 | m | I (p) | 2.8 (90) | 6.8 (40) | 13.0 (30) | N.A. |
| D16 | CH3 | m | Br (p) | 3.4 (75) | 3.8 (70) | N.A. | N.A. |
| 6a | H | m | Br (p) | 6.1 (125) | 7.7 (60) | 14.6 (25) | N.A. |
| 6b | CH2CH3 | m | I (p) | 4.2 (70) | 5.5 (65) | N.A. | N.A. |
| 6c | CH(CH3)2 | m | Br (p) | 4.5 (135) | 7.2 (90) | 17.4 (30) | N.A. |
| 6d | C6H11 | m | Br (p) | 10.8 (80) | N.A. | N.A. | N.A. |
| 6e | C6H5 | m | Br (p) | 9.0 (110) | 4.3 (25) | N.A. | N.A. |
| 6f | C6H5 | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6g | C6H5 | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6h | 2-thienyl | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 6i | 2-thienyl | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6j | 2-thienyl | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6k | C6H4-OCH3 (p) | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 6l | C6H4-OCH3 (p) | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6m | C6H4-OCH3 (p) | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6n | C6H4-Cl (p) | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 6o | C6H4-Cl (p) | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6p | C6H4-Cl (p) | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6q | C6H4-CH3 (p) | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 6r | C6H4-CH3 (p) | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6s | C6H4-CH3 (p) | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6t | C6H4-F (p) | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 6u | C6H4-F (p) | m | 3,4-methylenedioxy | N.A. | N.A. | N.A. | N.A. |
| 6v | C6H4-F (p) | m | F (p) | N.A. | N.A. | N.A. | N.A. |
| 6w | CH2-C6H5 | p | Br (p) | N.A. | N.A. | N.A. | N.A. |
N.A., no activity (no response was observed during first 2 min after addition of compounds under investigation) considering the limits of efficacy > 20 % and EC50 < 50 µM.
W.T., wild-type nontransfected HL-60 cells. The EC50 values are presented as the average mean of three independent experiments, in which EC50 values were determined by nonlinear regression analysis of the dose-response curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p< 0.05). Efficacy (in brackets) is expressed as % of the response induced by 5 nM fMLF (FPR1) or 5 nM WKYMVm (FPR2 and FPR3).
Table 7.
Activity of compounds 18, 23, 27, 28a-c, 33 (Schemes 3–5) in HL-60 cells expressing human FPR1, FPR2, or FPR3.
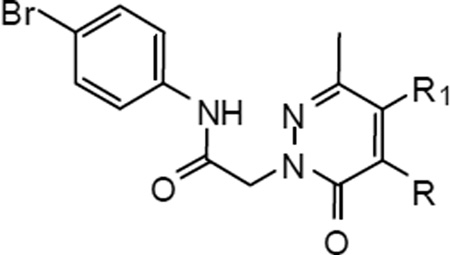 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R | R1 | Ca2+ Mobilization EC50 (µM) and Efficacy (%)a |
|||
| FPR1 | FPR2 | FPR3 | W.T.b | |||
| B16 | CH2-C6H4-OCH3 (p) | H | N.A. | 2.4 | N.A. | N.A. |
| D16 | CH2-C6H4-OCH3 (m) | H | 3.4 (75) | 3.8 (70) | N.A. | N.A. |
| 18 | CO-C6H4-OCH3 (m) | H | 3.0 (140) | 1.0 (110) | N.A. | N.A. |
| 23 | NH-C6H4-OCH3 (p) | COCH3 | 13.5 (75) | 1.7 (90) | N.A. | N.A. |
| 27 | NH2 | H | 8.1 (115) | 29.4 (85) | N.A. | N.A. |
| 28a | NH-C6H4-OCH3 (p) | H | 12.8 (100) | 7.8 (110) | N.A. | N.A. |
| 28b | NHCO-C6H4-OCH3 (m) | H | 9.3 (120) | 2.8 (100) | N.A. | N.A. |
| 28c | NHCO-C6H4-Br (p) | H | N.A. | N.A. | N.A. | N.A. |
| 33 | C6H4-OCH3 (p) | H | 11.2 (55) | N.A | N.A. | N.A. |
N.A., no activity (no response was observed during first 2 min after addition of compounds under investigation) considering the limits of efficacy > 20 % and EC50 < 50 µM.
W.T., wild-type nontransfected HL-60 cells. The EC50 values are presented as the average mean of three independent experiments, in which EC50 values were determined by nonlinear regression analysis of the doseresponse curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p< 0.05). Efficacy (in brackets) is expressed as % of the response induced by 5 nM fMLF (FPR1) or 5 nM WKYMVm (FPR2 and FPR3).
4. Results and discussion
4.1. Biological results
SAR analysis at position C-6 of the pyridazinone ring was used to investigate modification of the methyl group of the previously reported series of compounds [16] (Figure 1), including (a) elimination or replacement with superior homologues; (b) introduction of the cyclohexyl group; (c) introduction of the 2-thienyl group; (d) introduction of (substituted) aryl groups; and (e) introduction of the benzyl group (Table 5). As shown in Table 5, elimination of the methyl group led to the active but relatively non-selective compound 6a, which was able to activate all three FPR subtypes. The ethyl analogue (6b) displayed increased selectivity in comparison with reference compound C (Table 5) and exhibited very similar behaviour to the reference bromo-derivative B (Table 5), whereas the isopropyl analogue (6c) resulted a mixed agonist for FPR1, FPR2 and FPR3. In contrast, introduction of a cyclohexyl group at position C-6 (6d) was associated with FPR1 selectivity (EC50 = 10.8 µM). Among compounds having a phenyl group at C-6 (6e–g), only 6e, which contains Br in the phenylacetamide chain, was active and was a mixed FPR1/FPR2 agonist. The 2-thienyl derivatives 6h–j were completely inactive, as were the benzyl derivative 6w and all compounds containing OCH3 (6k–m), Cl (6n–p), CH3 (6q–s), and F (50t–v) in the para position of the phenyl ring at C-6 of the pyridazinone nucleus. It is worth noting that the complete inactivity of the 4-F analogue 6t, in comparison with compound 6e (EC50 = 9.0 µM at FPR1 and 4.3 µM at FPR2), suggests that for these derivatives, electronic features play a more important role than steric properties.
SAR studies at position C-4 were performed by modifying the reference compounds A–D [16] using the following criteria: (a) elimination of the OCH3 substituent on the benzyl group; (b) introduction of different substituents in the meta or para positions of the benzyl group; (c) replacement of the methoxybenzyl with heterocycles; (d) complete elimination of the C-4 substituent; (e) introduction of different functionalized linkers; and (f) elimination of the CH2- spacer of the benzyl group (Tables 6 and 7). In comparison to reference compound D [16], when OCH3 group was eliminated from the benzyl fragment at position C-4 of the pyridazinone scaffold (11a), activity was decreased, particularly as an FPR2 agonist (Table 6). The concomitant presence of an unsubstituted benzyl at C-4 and F (11b) or methylenedioxy (11c) in the aryl acetamide side chain was associated in the first case with complete loss of activity and in the latter case with selective FPR1 agonist activity (EC50 = 6.9 µM) (Table 6). On the other hand, replacement of the 3-methoxy group on the benzyl at position C-4 with different substituents was beneficial. In fact, replacement of OCH3 with F or Cl (Table 6, reference compound D) in the meta position resulted in compounds 11d and 11e, which were selective FPR1 agonists (EC50 = 6.6 and 10.5 µM, respectively). Conversely, the chloro analogue (11q) of reference compound C, which contains a 4- iodophenyl in the acetamidic spacer of position 2, had decreased activity and efficacy as a mixed FPR1/FPR2 agonist. Unexpectedly, introduction of a Br (11f) in place of the OCH3 group on the benzyl at C-4 was associated with complete loss of activity. These data suggest that the presence of a substituent of limited hindrance in the meta position is an essential requirement for binding to FPR1 and FPR2. Moreover, insertion of an additional methoxy group in reference compound D was associated with complete loss of activity (11g), whereas the 3,5-dimethoxybenzyl derivative 11p, which contains a 4-iodophenyl in the acetamidic spacer at position 2, exhibited weak, but specific FPR1 activity (Table 6, reference compound C). Introduction of CF3 (11h), CONH2 (11n), and CN (11o) in the para position was generally detrimental (Table 6).
Table 6.
Activity of compounds 11a-q and 15a,b (Scheme 2) in HL-60 cells expressing human FPR1, FPR2, or FPR3
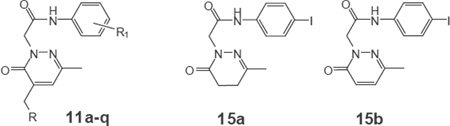 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R | R1 | Ca2+ Mobilization EC50 (µM) and Efficacy (%)a |
|||
| FPR1 | FPR2 | FPR3 | W.T.b | |||
| A16 | C6H4-OCH3 (m) | 3,4-methylenedioxy | 2.3 (50) | N.A. | N.A. | N.A. |
| B16 | C6H4-OCH3 (p) | Br (p) | N.A. | 2.4 | N.A. | N.A. |
| C16 | C6H4-OCH3 (m) | I (p) | 2.8 (90) | 6.8 (40) | 13.0 (30) | N.A. |
| D16 | C6H4-OCH3 (m) | Br (p) | 3.4 (75) | 3.8 (70) | N.A. | N.A. |
| 11a | C6H5 | Br (p) | 5.5 (50) | 11.6 (20) | N.A. | N.A. |
| 11b | C6H5 | F (p) | N.A. | N.A. | N.A. | N.A. |
| 11c | C6H5 | 3,4-methylenedioxy | 6.9 (55) | N.A. | N.A. | N.A. |
| 11d | C6H4-F (m) | Br (p) | 6.6 (110) | N.A. | N.A. | N.A. |
| 11e | C6H4-Cl (m) | Br (p) | 10.5 (100) | N.A. | N.A. | N.A. |
| 11f | C6H4-Br (m) | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 11g | C6H3-(OCH3)2 (m, m) | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 11h | C6H4-CF3 (p) | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 11i | 3-furyl | Br (p) | 5.8 (100) | 6.3 (75) | N.A. | N.A. |
| 11j | 3-thienyl | Br (p) | 4.5 (100) | 14.1 (65) | N.A. | N.A. |
| 11k | 2-thienyl | Br (p) | 8.1 (140) | 11.4 (60) | 10.2 (25) | N.A. |
| 11 l | 1-naphthyl | Br (p) | 13.8 (20) | N.A. | N.A. | N.A. |
| 11m | 3-pyridyl | Br (p) | 9.3 (85) | 2.8 (90) | N.A. | N.A. |
| 11n | C6H4-CONH2 (p) | Br (p) | 29.3 (40) | 27.2 (80) | N.A. | N.A. |
| 11o | C6H4-CN (p) | Br (p) | N.A. | N.A. | N.A. | N.A. |
| 11p | C6H3-(OCH3)2 (m, m) | I (p) | 7.6 (80) | N.A. | N.A. | N.A. |
| 11q | C6H4-Cl (m) | I (p) | 9.5 (55) | 16.9 (35) | N.A. | N.A. |
| 15a | - | - | N.A. | N.A. | N.A. | N.A. |
| 15b | - | - | N.A. | N.A. | N.A. | N.A. |
N.A., no activity (no response was observed during first 2 min after addition of compounds under investigation) considering the limits of efficacy > 20 % and EC50 < 50 µM.
W.T., wild-type nontransfected HL-60 cells. The EC50 values are presented as the average mean of three independent experiments, in which EC50 values were determined by nonlinear regression analysis of the dose-response curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p< 0.05). Efficacy (in brackets) is expressed as % of the response induced by 5 nM fMLF (FPR1) or 5 nM WKYMVm (FPR2 and FPR3).
Useful information originated by replacement of the methoxyphenyl group at C-4 with 5- and 6-membered heterocycles. Indeed, the furyl derivative 11i exhibited reasonable potency but weak selectivity for FPR1, as did both the thienyl (11j,k) and piridyl (11m) analogues, which had similar activity profiles without receptor subtype-selectivity (Table 6). In contrast, insertion of a naphtylmethyl (11l) in position C-4 was associated with decreased activity but increased selectivity for FPR1. The unsubstituted derivatives at C-4 in the pyridazinone ring 15a,b, which contains 4- iodophenyl in the acetamidic spacer at position 2, were completely devoid of activity (Table 6).
Introduction of a carbonyl group in the place of CH2 of the benzyl group at position 4 of the pyridazinone ring resulted in compound 18, which was a potent mixed FPR1/FPR2 agonist (Table 7, reference compound D). When CH2 was replaced by NH in both compound 23 and the 5-acetyl analogue 28a (reference compound B), agonist activity was found mainly at FPR2, whereas compound 27 was a less potent FPR2 agonist. Substitution of CH2 with an amidic group in 28b was detrimental for FPR1, but not FPR2 activity, whereas anologue 28c was totally inactive. Lastly, elimination of the methylenic linker resulted in a selective FPR1 agonist (33).
It should be noted that all FPR1/FPR2/FPR3 agonists were inactive in wild-type nontransfected HL-60 cells (Tables 5–7), supporting the specificity of our assays.
Twenty compounds that showed the best agonist profiles in transfected cells were selected for evaluation as chemotactic agents and for their ability to induce Ca2+ mobilization in human neutrophils. All compounds had EC50 values in the micromolar range and an efficacy range of 70- 130% (Table 8). The most active compounds were 6c and 6e, which had EC50 values of 0.85 and 0.6 µM for inducing Ca2+ flux and EC50 values of 0.70 and 0.60 µM for inducing chemotaxis, respectively. Representative kinetic curves of Ca2+ mobilization and dose-response for chemotactic activity for the phenyl derivative 6e are shown in Figure 2. It is interesting to note that among the tested compounds, there were mixed FPR1/FPR2/FPR3 agonists (6a, 6c, 11k), dual FPR1/FPR2 agonists (6b, 6e, 11i, 11j, 11m, 18, 23, 27, 28a, 28b) and selective FPR1 agonists (6d, 11c–e,11p, 33), suggesting that FPR1 is important for Ca2+ mobilization and chemotaxis in human neutrophils.
Table 8.
Ca2+ mobilization and chemotactic activity in human neutrophils treated with selected FPR1/FPRL1 agonists
| Compd. | Ca2+ flux | Chemotaxis |
|---|---|---|
| EC50 (µM) and Efficacy (%) a | ||
| 6a | 8.3 (100) | 15.9 |
| 6b | 1.0 (90) | 0.71 |
| 6c | 0.85 (110) | 0.70 |
| 6d | 2.4 (65) | 2.5 |
| 6e | 0.6 (115) | 0.60 |
| 11c | 6.1 (75) | 4.2 |
| 11d | 4.2 (130) | 1.9 |
| 11e | 6.0 (90) | 5.0 |
| 11i | 3.0 (125) | 1.5 |
| 11j | 3.9 (115) | 8.7 |
| 11k | 3.6 (80) | 3.9 |
| 11m | 4.9 (120) | 2.8 |
| 11p | 3.9 (75) | 2.1 |
| 11q | 2.4 (90) | 0.49 |
| 18 | 2.3 (115) | 0.87 |
| 23 | 3.9 (110) | 2.7 |
| 27 | 2.3 (100) | 0.8 |
| 28a | 2.1 (105) | 5.9 |
| 28b | 2.4 (110) | 1.9 |
| 33 | 3.0 (70) | 2.1 |
The data are presented as average mean of three independent experiments with cells from different donors, in which EC50 values were determined by nonlinear regression analysis of the dose-response curves (5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p<0.05).
Figure 2.

Analysis of chemotactic activity and Ca2+ mobilization in human neutrophils treated with compound 6e. Panel A. Representative kinetics of Ca2+ mobilization after treatment with compound 6e or fMLF. Neutrophils were treated with the compound 6e (210 and 620 nM), 5 nM fMLF (positive control), or DMSO vehicle (negative control), and Ca2+ flux was monitored for the indicated time. Panel B. Human neutrophil chemotaxis toward the indicated concentrations of compound 6e was determined, as described under Biological assays. The data are from one experiment that is representative of three independent experiments.
Although our functional assays suggested that all active compounds were relatively nontoxic, we evaluated the potential cytotoxic effects of the pyridazin-3(2H)-one derivatives (both active and inactive compounds) to determine if the results might be influenced by background toxicity. Using a cytotoxicity assay, we determined that none of the compounds significantly affected viability of wild-type HL-60 cells over a concentration range of 5–20 µM, verifying that these compounds were not toxic to these cells, at least during entire incubation period used in the Ca2+ flux and chemotaxis assays. As examples, effects of the 20 active compounds on cell viability are shown in Figure 3.
Figure 3.

Evaluation of selected FPR agonist cytotoxicity. Wild-type, non-transfected human promyelocytic leukemia HL-60 cells were incubated for 60 min at 37 0C with 5 µ;M (open bars), 10 µ;M (hatched bars), or 20 µ;M (solid bars) of the indicated compounds, and cell viability was determined using a luminescent cell viability assay kit, as described. Values are the mean ± S.D. of triplicate samples from one experiment, which is representative of two independent experiments.
4.2. Docking studies
To explore the possibility that the novel FPR1/FPR2 agonists could bind to the fMLF binding site of FPR1 and the WKYMVM binding site of FPR2, we used a previously published homology model of FPR1 [46,47] and also constructed a model of FPR2 by homology modeling using the Phyre2 server, since the crystal structure models of FPR1 and FPR2 are still unavailable. Both models are based on bovine rhodopsin (see Experimental section). It should be noted that slightly higher sequence identity between a given target and template alone does not always justify the choice of GPCR structure for the optimal homology modeling template, and additional considerations are important [48]. In the present modeling studies, the rhodopsin-based model of FPR2 was selected as the most predictive structure from 18 other homology models, based on 8 dissolved crystal structures of GPCRs, including bovine and squid rhodopsins, human adenosine receptor A2A, turkey β1 adrenoceptor, human β2 adrenoceptor, human histamine receptor H1, human dopamine D3 receptor, and human chemokine receptor CXCR4. Although the CXCR4 structure has a higher sequence identity with FPR2 (28%), this structure has lower resolution (3.2 Å) for the template, as compared to the rhodopsin structure (2.2 Å). Our recent modeling experiments showed that the geometric configuration of the FPR2 binding site in the rhodopsin-based model is in good agreement with the shape of the hydrophobic field obtained for the FPR2 agonist pharmacophore, which was obtained by independent modeling [49]. Importantly, the rhodopsin-based model of FPR1 effectively differentiated agonists from inactive compounds [47], and the binding structure of fMLF to the model is not in conflict with experimental evidence reported before by Mills et al. [50] and has a high similarity with the binding mode of AC-QAWF [46]. Thus, we feel that these features justify use of this model as a template for the docking of new FPR1 agonists vs. the other known GPCR crystal structures currently available.
The best docking poses of selected FPR1 agonists 6d, 11d, and 18, as well as previously published reference compounds A and D [16], to key sub-areas of the FPR1 binding site [47] are shown in Figure 4. Molecular skeletons within the binding site are located mainly along cavity B and directed to channels A and C or to cavity E, in accordance with binding modes for 2- arylacetamide pyridazin-3(2H)-ones. Noticeably, bromine-substituted compounds, in their best poses, had p-bromophenyl groups located in the vicinity of channel A and directed outside the binding site. Such an orientation could be required by the large volume and high hydrophobicity of bromo-substituted phenyl ring. Additionally, H-bonding interactions of Thr265 with the pyridazine nitrogen atom are found in agonists 6d, 18, and reference compound D, or with the carbonyl group of the pyridazinone moiety in 11m (Figure 4C). For compound 18, this carbonyl is also H-bonded to both Thr199 and Asn192. Reference compound A, which contains a benzodioxolane moiety instead of a p-bromophenyl group, had the opposite binding mode, with the benzodioxolane protruding into channel C. In its docking pose, this compound formed a H-bond between one of its endocyclic oxygen atoms and the hydroxyl groups in Thr199 and Asn192.
Figure 4.

Homology model of the FPR1 binding site with docked peptide fMLF and selected non-peptide FPR1 agonists with a pyridazin-3(2H)-one scaffold. Panel A. Docking poses of fMLF (atoms are highlighted by green spheres) and pyridazin-3(2H)-one 11d (atoms are not highlighted). Key sub-areas of the FPR1 binding site are indicated with arrows and include two channels (A and C), two cavities (B and E), and the bottom (D), as described previously and [47]. Channel A is located in the vicinity of Asn192, Thr199, Thr265, Ile268, and Leu271. Channel C is bounded by Leu101, Val105, Tyr257, Ser287, and Phe291. Cavity B is a curved groove, located behind the hydrophobic ledge formed by the isobutyl group of Leu198, and is restricted by Val160, Leu198, Arg201, Gly202, and Arg205. Large cavity E is located near Trp91, Trp95, Cys98, and Lys99, between channel C and a larger ledge. The bottom D of the binding site is bounded by channels A and C and is associated with Ala261, Ala264, and Val283. Surface coloring was made according to electrostatic properties, whereby negatively and positively charged areas are shown in red and blue, respectively. Panel B. Overlapping docking poses of fMLF (blue) and pyridazin-3(2H)-ones 6d (red), 11d (green), 18 (magenta), and reference compound A [16] (brown). Panel C. Interactions between Thr265 and compound 11m. H-bond is indicated with a dashed line.
The ligand binding site of FPR2 has a non-symmetric dumb-bell shape with two cavities different in size, where the smaller cavity lies deeply in the binding site and is surrounded by residues Val105, Asp106, Leu109, Phe110, Arg201, Trp254, and Gln258, as described previously [51] (Figure 5). The FPR2-specific peptide agonist WKYMVM in its best docking pose occupies this cavity with an indole moiety near the N-terminus of the peptide (Figures 5 and 6A). A narrow channel connecting the two cavities is bordered by residues Phe257, Val260, Ala261, Thr177, Phe178, and Phe180. The larger cavity of the docking site opens outside the receptor and has a complex shape. Four residues (Tyr, Val, and both Met) of the docked WKYMVM are located in this cavity (Figure 6A and Insert).
Figure 5.

Potential areas of FPR2 where ligands of could be docked. A PDB file of the homology model for FPR2, based on the bovine rhodopsin template, was loaded into MVD software, and the “Detect cavity” feature was applied with probe size 1.2 Å to identify potential areas of the receptor where ligands could be docked. Two cavities were found with volumes of 241 Å3 and 25 Å3 (green zones indicated by arrows). The binding pose of the FPR2 agonist WKYMVM is shown for comparison (red sticks). The search space with a sphere radius of 15 Å encompassed the WKYMVM molecule, eight residues belonging to the binding site [51] (His102, Val105, Asp106, Leu109, Trp254, Phe257, Ser288, Phe292; atoms of the residues are shown as green balls), and most of the large cavity (dark green sphere).
Figure 6.

Homology model of FPR2 with docked WKYMVM peptide and selected pyridazin- 3(2H)-ones. Panel A. Best docking pose of WKYMVM peptide (light-blue). The FPR2 model is shown in ribbon mode. The small docking cavity (green) lies deep within the binding site and is surrounded by residues Val105, Asp106, Leu109, Phe110, Arg201, Trp254, and Gln258. Insert: The larger cavity of the docking site showing the position of side chain tails of the tetrapeptide fragment (YMVM) of WKYMVM. Surface coloring was made according to electrostatic properties, whereby negatively and positively charged areas are shown in red and blue, respectively. Panel B. Overlapping docking poses of WKYMVM (atoms are indicated with green spheres) and pyridazin-3(2H)-ones (11m, 18, 23, and 28b) and reference compounds B and C [16]. FPR2 residues within 6 Å around the smaller cavity are shown. Panel C. Compound 18 docked into the FPR2 binding site. Residues lying within 3.5 Å of the pose are shown. H-bonds are indicated with dashed lines.
Docking of FPR2 agonists 11m, 18, 23, and 28b, as well as reference compounds B and D [16], to FPR2 showed that they adopt conformations that overlap well with the docking pose of WKYMVM. The bromo-substituted phenyl rings of 11m, 18, 28b, and reference agonists B and D lie within the smaller cavity found deep within the binding site (Figure 6B). The narrow channel contains an amide fragment adjacent to the p-bromophenyl groups of the molecules. In addition, the pyridazinone moieties are oriented similarly in the binding site with their carbonyl and methyl groups overlaid in the same positions. Such an orientation is conditioned by the possibility of H-bonding of the nitrogen atom in the pyridazine ring, as well as an oxygen or nitrogen atom in the amide moiety, with the hydroxyl group of Thr177. As an example, specific interactions of 18 with Thr177 and Ala261 are shown in Figure 6C. In contrast, compound 23 binds to FPR2 in a different manner. This ligand is positioned entirely in the larger cavity, possibly because of the presence of an acetyl group, which prevents passage of the molecule through the narrow channel (data not shown). However, compound 23 is also H-bonded to Thr177 by an acetyl oxygen atom. Thus, the formation of H-bonds between ligands and Thr177 seems to play an important role in positioning FPR agonists within the ligand binding site.
5. Conclusion
Our data confirm that pyridazinone-based compounds are a relevant chemotype to search for both selective and/or potent FPR agonists of human neutrophils. The biological results with C-4 and C-6 derivatives presented herein confirm our previously reported conclusion that an arylacetamide moiety at N-2 of the scaffold [16] is essential for agonist activity. Our SAR studies demonstrate that by manipulating the chemical structure of a series of pyridazinones (Figure 1), it is possible to achieve varying potency and selectivity towards the different FPR subtypes (Tables 5–7). Position C-6 (Table 6) of the pyridazinone ring was not very tolerant to modification, and a methyl group is still the substituent that supports the best results regarding agonist activity, while its elimination or substitution with a more hindered moiety resulted in a substantial loss in activity. Conversely, position C-4 (Tables 6 and 7) was more amenable to chemical manipulation. Indeed, substituted benzyl groups and heterocycles (e.g., thienyl, piridyl, furyl), as well as functionalized spacers (e.g., CO, NH), can be productively introduced at this level, retaining good agonist activity. Molecular docking experiments showed that the most potent pyridazin-3(2H)-ones overlap in their best docking poses with the docking poses for fMLF and WKYMVM in the FPR1 and FPR2 ligand binding sites, respectively. Overall, these pyridazin-3(2H)-ones appear to efficiently interact with the receptor through specific H-bonding with Thr265, Thr199 and Asn192 in FPR1 or Thr177 in FPR2.
In conclusion, the majority of pyridazinone-based compounds presented herein, which were modified at C-4 and C-6 positions, showed good FPR agonist activity, and some had selectivity among the three FPR isoforms. Thus, we were able to identify seven FPR1-selective agonists (6d, 11c–e, 11 l, 33) and several potent mixed FPR1/FPR2 dual agonists with EC50 values in the low micromolar range, including the 4-keto derivative 18 (EC50 = 3.0 µM at FPR1 and 1.0 µM at FPR2).
6. Experimental section
6.1. Chemistry
6.1.1. General
Reagents and starting materials were obtained from commercial sources. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. All reactions were monitored by thin layer chromatography (TLC) using commercial plates precoated with Merck silica gel 60 F-254. Visualization was performed by UV fluorescence (λmax = 254 nm) or by staining with iodine or potassium permanganate. Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck), flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck), silica gel preparative TLC (Kieselgel 60 F254, 20×20 cm, 2 mm), or a CombiFlash® Rf System (using RediSep® Rf Silica Columns, Teledyne Isco, Lincoln, Nebraska, USA). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. Compounds were named following IUPAC rules, as applied by Beilstein-Institut AutoNom 2000 (4.01.305) or CA Index Name. All melting points were determined on a microscope hot stage Büchi apparatus and are uncorrected. The identity and purity of intermediates and final compounds was ascertained through 1H NMR and TLC chromatography. 1H NMR spectra were recorded with Avance 400 instruments (Bruker Biospin Version 002 with SGU). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using solvent as the internal standard. Coupling constants (J values) are given in Hz and were calculated using ‘TopSpin 1.3’ software rounded to the nearest 0.1 Hz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Bruker Micro TOF), and reported mass values are within the error limits of ± 5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin-Elmer 260 elemental analyzer for C, H, and N, and they were within ± 0.4 % of the theoretical values.
6.1.2. General Procedures for 2a–k
To a stirred solution of the suitable γ-keto acid 1a–k (1.00 mmol) in EtOH (2 mL), hydrazine hydrate (1.00 mmol) was added dropwise. The mixture was heated at 60 °C for 1–3 h. After cooling, the precipitate was collected by suction and purified by recrystallization from either toluene or ethanol.
6.1.2.1. 6-(4-fluorophenyl)-4,5-dihydropyridazin-3(2H)-one [2i]
Yield = 95%; mp = 192−93 °C (EtOH); 1H NMR (CDCl3) δ 2.64 (dd, 2H, COCH2CH2, J = 1.2 Hz, J = 7.5 Hz), 3.00 (dd, 2H, COCH2CH2, J = 1.2 Hz, J = 7.5 Hz), 7.13 (qd, 2H, Ar, J = 1.8 Hz, J = 4.7 Hz), 7.72–7.76 (qd, 2H, Ar, J = 0.4 Hz, J = 3.1 Hz), 8.51 (exch br s, 1H, NH). Anal. C10H9FN2O (C, H, N).
6.1.3. General Procedures for 3a–p
To a solution of KOH in absolute EtOH (8 mL, 5%, w/v), the appropriate compound 2a–l (1.39 mmol) and 3- or 4-methoxybenzaldehyde (1.39 mmol) were added. The mixture was refluxed under stirring for 1–3 h. After cooling, the mixture was concentrated in vacuo, diluted with cold water (10–15 mL), and acidified with 2 N HCl. For compounds 3a–e,p, the suspension was extracted with CH2Cl2 (3×15 mL). Removal of the solvent resulted in the final compounds, which were purified by column chromatography using cyclohexane/ethyl acetate 1:3 (for 3a,p), 1:1 (for 3b,e) and 2:1 (for 3c,d) as eluents. After 1 h stirring in an ice-bath, compounds 3f–o were filtered off by suction from the acidic solutions and recrystallized from ethanol.
6.1.3.1. 4-(3-Methoxybenzyl)pyridazin-3(2H)-one [3a]
Yield = 27%; colorless oil (purified by column chromatography using cyclohexane/ethyl acetate 1:3 as eluent); 1H NMR (CDCl3) δ 3.83 (s, 3H, OCH3), 3.92 (s, 2H, C6H4-CH2), 6.81–6.89 (m, 4H, Ar), 7.29 (t, 2H, Ar, J = 7.8 Hz), 7.77 (d, 1H, Ar, J = 4.0 Hz). Anal. C12H12N2O2 (C, H, N).
6.1.4. General procedure for 4a–p
A mixture of the appropriate intermediate 3a–p (1.34 mmol), K2CO3 (2.68 mmol), and ethyl bromoacetate (2.01 mmol) in CH3CN (8 mL) was refluxed under stirring for 1–3 h. The mixture was then concentrated in vacuo, diluted with cold water, and extracted with CH2Cl2 (3×15 mL). The solvent was evaporated in vacuo, and compounds 4a–p were purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent.
6.1.4.1. Ethyl-2-[5-(3-methoxybenzyl)-6-oxopyridazin-1(6H)-yl]acetate [4a]
Yield = 47%; oil (purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent); 1H NMR (CDCl3) δ 1.31 (t, 3H, CH2CH3, J = 7.1 Hz), 3.82 (s, 3H, OCH3), 3.91 (s, 2H, CH2-Ar), 4.27 (q, 2H, OCH2CH3, J = 7.2 Hz), 4.92 (s, 2H, NCH2CO), 6.79–6.80 (m, 2H, Ar), 6.83 (d, 2H, Ar, J = 7.8 Hz), 7.28 (t, 1H, Ar, J = 7.8 Hz), 7.67 (d, 1H, Ar, J = 4.1 Hz). Anal. C16H18N2O4 (C, H, N).
6.1.5. General Procedures for 5a–p
A suspension of the appropriate intermediate 4a–p (1.33 mmol) in 6 N NaOH (10 mL) was stirred at rt up to 60–80 °C for 1–2 h. The mixture was diluted with cold water and acidified with 6 N HCl. After 1 h stirring in an ice-bath, the products 5a–p were filtered off by suction and recrystallized from ethanol.
6.1.5.1. 2-[5-(3-Methoxybenzyl)-6-oxopyridazin-1(6H)-yl]acetic acid [5a]
Yield = 99.9%; mp = 149−51 °C (EtOH); 1H NMR (CDCl3) δ 3.81 (s, 3H, OCH3), 3.91 (s, 2H, CH2-Ar), 4.98 (s, 2H, NCH2CO), 6.49 (exch br s, 1H, OH), 6.78 (s, 1H, Ar), 6.81–6.85 (m, 3H, Ar), 7.28 (t, 1H, Ar, J = 7.8 Hz), 7.72 (d, 1H, Ar, J = 4.1 Hz). Anal. C14H14N2O4 (C, H, N).
6.1.6. General Procedures for 6a–w
To a cooled (−5 °C) and stirred solution of compound 5a–p (0.60 mmol) in anhydrous tetrahydrofuran (6 mL), Et3N (2.10 mmol) was added. After 30 min, the mixture was allowed to warm up to 0 °C, and ethyl chloroformate (0.66 mmol) was added. After 1 h, the commercially (1.20 mmol) was added. The reaction was carried out at room temperature for 12 h, then the mixture was concentrated in vacuo, diluted with cold water (20–30 mL), and extracted with CH2Cl2 (3×15 mL). The solvent was evaporated to obtain final compounds 6a–w, which were purified by column chromatography using cyclohexane/ethyl acetate 2:1 for compounds 6b,g, cyclohexane/ethyl acetate 3:1 for compounds 6c,e, CH2Cl2/CH3OH/NH4OH 96:4:0.4 for compound 6d, cyclohexane/ethyl acetate 1:1 for compounds 6a,f,i,j,w, and n-hexane/ethyl acetate 3:2 for compound 6h,k–v as eluents.
6.1.6.1. N-(4-Bromophenyl)-2-[5-(3-methoxybenzyl)-6-oxopyridazin-1(6H)-yl]acetamide [6a]
Yield = 70%; mp = 138−40 °C (EtOH); 1H NMR (DMSO-d6) δ 3.73 (s, 3H, OCH3), 3.79 (s, 2H, CH2-Ar), 4.90 (s, 2H, NCH2CO), 6.83 (t, 3H, Ar, J = 9.5 Hz), 7.14 (d, 1H, Ar, J = 3.8 Hz), 7.23 (t, 1H, Ar, J = 7.8 Hz), 7.53 (q, 4H, Ar, J = 8.2 Hz), 7.85 (d, 1H, Ar, J = 3.8 Hz), 10.47 (exch br s, 1H, NH). ESI-MS calcd. for C20H18BrN3O3, 428.28; found: m/z 429.09 [M+H]+. Anal. C20H18BrN3O3 (C, H, N).
6.1.7. General Procedures for 8a–l
To 12 mL of KOH in absolute EtOH (5%, w/v), compound 7 [16] (4.46 mmol) and the appropriate substituted aromatic aldehyde (4.46 mmol) were added (4- cyanobenzaldehyde was used to obtain compound 8l). The reaction was refluxed under stirring for 3–5 h. After cooling, the mixture was concentrated in vacuo, diluted with ice-cold water (20–25 mL), and acidified with 2 N HCl. The suspension was extracted with CH2Cl2 (3×25 mL). Removal of the solvent resulted in compounds 8a–l, which were purified by crystallization in ethanol. For compounds 8e,k, an additional purification step was performed by flash column chromatography using cyclohexane/ethyl acetate 2:1 (for 8e) or NH4OH/EtOH/CH2Cl2/petroleum ether 4:25:150:50 (for 8k) as eluents.
6.1.7.1. 4-Benzyl-6-methylpyridazin-3(2H)-one [8a]
Yield = 99.9%; mp = 113−15 °C (EtOH); 1H NMR (CDCl3) δ 2.26 (s, 3H, CH3), 3.92 (s, 2H, CH2), 6.73 (s, 1H, Ar), 7.26–7.33 (m, 3H, Ar), 7.28 (t, 2H, Ar, J = 6.9 Hz). Anal. C12H12N2O (C, H, N).
6.1.8. 4-[(6-Methyl-3-oxo-2,3-dihydropyridazin-4-yl)methyl]benzonitrile [8m]
A suspension of compound 8l (0.82 mmol) in 5 mL of POCl3 was stirred at 60 °C for 3 h. After cooling, the mixture was concentrated in vacuo, diluted with ice-cold water (10 mL), and extracted with CH2Cl2 (3×15 mL). The organic layer was dried (Na2SO4) and concentrated in vacuo to obtain a yellow solid, which was purified by flash column chromatography using CH2Cl2/CH3OH 7:3 as eluent. Yield = 60%; mp = 213–215 °C (EtOH); 1H NMR (CDCl3) δ 2.29 (s, 3H, 6-CH3), 3.96 (s, 2H, CHCCH2), 6.79 (s, 1H, Ar), 7.41 (d, 2H, Ar, J = 7.0 Hz), 7.65 (d, 2H, Ar, J = 6.9 Hz), 10.61 (exch br s, 1H, NH). Anal. C13H11N3O (C, H, N).
6.1.9. General Procedures for 9a–j and 13a,b
A mixture of the suitable intermediate 8a–j, 7 [16] or 12 [31,39] (4.50 mmol), K2CO3 (9.00 mmol), and ethyl bromoacetate (6.75 mmol) in CH3CN (10 mL) was refluxed while stirring for 2–4 h. The mixture was then concentrated in vacuo, diluted with cold water, and extracted with CH2Cl2 (3×15 mL). The solvent was evaporated in vacuo, and compounds 9a–j and 13a,b were purified by crystallization from ethanol (compounds 9b–d,g) or by flash column chromatography using cyclohexane/ethyl acetate 1:1 (for 9a,e,f and 13a,b) and cyclohexane/ethyl acetate 2:1 (for 9h–j) as eluents.
6.1.9.1 Ethyl-2-[5-benzyl-3-methyl-6-oxopyridazin-1(6H)-yl]acetate [9a]
Yield = 98%; oil (purified by column chromatography using cyclohexane/ethyl acetate 1:1 as eluent); 1H NMR (CDCl3) δ 1.31 (t, 3H, CH2CH3, J = 7.2 Hz), 2.22 (s, 3H, 3-CH3), 3.90 (s, 2H, CHCCH2), 4.24 (q, 2H, CH2CH3, J = 7.2 Hz), 4.85 (s, 2H, NCH2CO), 6.66 (s, 1H, Ar), 7.23–7.28 (m, 3H, Ar), 7.33- 7.37 (m, 2H, Ar). Anal. C16H18N2O3 (C, H, N).
6.1.10. General Procedures for 10a–j and 14a,b
A suspension of the suitable intermediate 9a–j or 13a,b (4.4 mmol) in 6 N NaOH (10 mL) was stirred at rt up to 80 °C for 1–5 h. The mixture was diluted with ice-cold water and then acidified with 6 N HCl. Products 10a–j and 14a,b were filtered off by suction and recrystallized from ethanol.
6.1.10.1. 2-[5-Benzyl-3-methyl-6-oxopyridazin-1(6H)-yl]acetic acid [10a]
Yield = 92%; mp = 120−22 °C (EtOH); 1H NMR (CDCl3) δ 2.25 (s, 3H, 3-CH3), 3.93 (s, 2H, CHCCH2), 4.94 (s, 2H, NCH2CO), 5.50 (exch br s, 1H, OH), 6.70 (s, 1H, Ar), 7.24–7.32 (m, 3H, Ar), 7.35–7.39 (m, 2H, Ar). Anal. C14H14N2O3 (C, H, N).
6.1.11. General Procedures for 11a–l and 15a,b
To a cooled (−5 °C) and stirred solution of the appropriate compound 10a–j or 14a,b (2.06 mmol) in anhydrous tetrahydrofuran (6 mL), Et3N (7.21 mmol) was added. After 30 min, the mixture was allowed to warm up to 0 °C, and ethyl chloroformate (2.27 mmol) was added. After 1 h, the appropriately substituted arylamine (4.12 mmol) was added. The reaction was carried out at room temperature for 12 h. The mixture was then concentrated in vacuo, diluted with cold water (20–30 mL), and extracted with CH2Cl2 (3×15 mL). The solvent was evaporated to obtain final compounds 11a–l and 15a,b, which were purified by column chromatography using cyclohexane/ethyl acetate 2:1 (for 11a,b,d–f,l), cyclohexane/ethyl acetate 1:1 (for 11c,g,i,j), cyclohexane/ethyl acetate 1:2 (for 11h), CH2Cl2/CH3OH 9.8:0.2 (for 11k), cyclohexane/ethyl acetate 1:3 (for 15a) and CH2Cl2/CH3OH 9.9:0.1 (for compound 15b) as eluents.
6.1.11.1. 2-[5-Benzyl-3-methyl-6-oxopyridazin-1(6H)-yl]-N-(4-bromophenyl)acetamide [11a]
Yield = 47%; colorless oil (purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent); 1H NMR (CDCl3) δ 2.30 (s, 3H, 3-CH3), 3.93 (s, 2H, CHCCH2), 4.94 (s, 2H, NCH2CO), 6.82 (s, 1H, Ar), 7.24–7.28 (m, 2H, Ar), 7.30–7.38 (m, 7H, Ar), 9.18 (exch br s, 1H, NH). ESI-MS calcd. for C20H18BrN3O2, 412.28; found: m/z 413.06 [M+H]+. Anal. C20H18BrN3O2 (C, H, N).
6.1.12. General Procedures for 11m–o
A mixture of the appropriate compound 8k–m (0.79 mmol), K2CO3 (1.58 mmol) and N-(4-bromophenyl)-2-chloroacetamide [38] (1.19 mmol) in CH3CN (2 mL), was refluxed under stirring for 2–3 h. The mixture was then concentrated in vacuo and diluted with cold water. After 1 h stirring in an ice-bath, the precipitate was filtered off by suction and purified by flash column chromatography using CH2Cl2/CH3OH/NH4OH 9.5:0.5:0.05 (for 11m), CH2Cl2/CH3OH 9.5:05 (for 11n) or CH2Cl2/CH3OH 9.9:0.1 (for 11o) as eluents.
6.1.12.1. N-(4-Bromophenyl)-2-[3-methyl-6-oxo-5-(pyridin-3-ylmethyl)-pyridazin-1(6H)-yl] acetamide [11m]
Yield = 61%; mp = 216–218 °C (EtOH); 1H NMR (CDCl3) δ 2.30 (s, 3H, 3- CH3), 3.92 (s, 2H, CHCCH2), 4.93 (s, 2H, NCH2CO), 6.86 (s, 1H, Ar), 7.27–7.33 (m, 5H, Ar), 7.61 (d, 1H, Ar, J = 7.8 Hz), 8.54 (d, 2H, Ar, J = 6.2 Hz), 9.29 (exch br s, 1H, NH). ESI-MS calcd. for C19H17BrN4O2, 413.27; found: m/z 414.05 [M+H]+. Anal. C19H17BrN4O2 (C, H, N).
6.1.13. General Procedures for 11p,q
To 5 mL of KOH in absolute EtOH (5%, w/v), 15a (0.13 mmol) and the appropriate substituted aromatic aldehyde (0.13 mmol) were added. The mixture was refluxed under stirring for 1–5 h. After cooling, the suspension was concentrated in vacuo, diluted with ice-cold water (5–10 mL), acidified with 2 N HCl, and extracted with CH2Cl2 (3×25 mL). Removal of the solvent resulted in compounds 11p,q, which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent.
6.1.13.1. N-(4-Iodophenyl)-2-[5-(3,5-dimethoxybenzyl)-3-methyl-6-oxopyridazin-1(6H)- yl]acetamide [11p]
Yield = 30%; mp = 63–65 °C (EtOH); 1H NMR (CDCl3) δ 2.30 (s, 3H, 3-CH3), 3.79 (s, 6H, 2×OCH3), 3.87 (s, 2H, CHCCH2), 4.94 (s, 2H, NCH2CO), 6.40 (s, 3H, Ar), 6.82 (s, 1H, Ar), 7.28 (d, 2H, Ar, J = 5.7 Hz), 7.58 (d, 2H, Ar, J = 8.6 Hz), 9.01 (exch br s, 1H, NH). ESIMS calcd. for C22H22IN3O4, 519.33; found: m/z 520.07 [M+H]+. Anal. C22H22IN3O4 (C, H, N).
6.1.13.2. N-(4-Iodophenyl)-2-[5-(3-chlorobenzyl)-3-methyl-6-oxopyridazin-1(6H)-yl]acetamide [11q]
Yield = 25%; mp = 58–60 °C (EtOH); 1H NMR (CDCl3) δ 2.33 (s, 3H, 3-CH3), 3.90 (s, 2H, CHCCH2), 4.94 (s, 2H, NCH2CO), 6.86 (s, 1H, Ar), 7.13–7.16 (m, 1H, Ar), 7.20 (d, 2H, Ar, J = 7.0 Hz), 7.28 (d, 3H, Ar, J = 4.2 Hz), 7.52 (d, 2H, Ar, J = 8.7 Hz), 9.14 (exch br s, 1H, NH). ESI-MS calcd. for C20H17ClIN3O2, 493.73; found: m/z 494.05 [M+H]+. Anal. C20H17ClIN3O2 (C, H, N).
6.1.14. 4-(3-Methoxybenzoyl)-6-methylpyridazin-3(2H)-one [17]
To a stirred and heated (60 °C) suspension of compound 16 [16] (2.39 mmol) in 15 mL of acetic acid (50%, v/v), Ce(NH4)2(NO3)6 (7.17 mmol) was slowly added over 0.5 h, and the reaction is carried out at 60 °C for an additional 1 h. The mixture was then diluted with ice-cold water (10 mL) and extracted with CH2Cl2 (15 mL). After washing with H2O (3×10 mL), the organic layer was evaporated under vacuum, and the residue was purified by CombyFlash® (eluent: cyclohexane/ethyl acetate, gradient 1:1 to 1:3). Yield = 17%; oil (purified by CombyFlash® using a gradient 1:1 to 1:3 of cyclohexane/ethyl acetate as eluent); 1H NMR (CDCl3) δ 2.25 (s, 3H, 6-CH3), 3.92 (s, 3H, OCH3), 6.74–6.95 (m, 4H, Ar), 7.31 (t, 1H, Ar, J = 7.8 Hz), 11.32 (exch br s, 1H, NH). Anal. C13H12N2O3 (C, H, N).
6.1.15. N-(4-Bromophenyl)-2-[5-(3-methoxybenzoyl)-3-methyl-6-oxopyridazin-1(6H)- yl]acetamide [18]
A mixture of intermediate 17 (0.41 mmol), K2CO3 (0.82 mmol), and N-(4- bromophenyl)-2-chloroacetamide [38] (0.61 mmol) in CH3CN (5 mL) was refluxed under stirring for 6 h. The mixture was then concentrated in vacuo and extracted with CH2Cl2 (3×15 mL). After removal of the solvent under vacuum, the residue was purified by flash column chromatography using NH4OH/EtOH/CH2Cl2/petroleum ether 4:25:150:269 as eluent. Yield = 10%; mp = 176–177 °C (EtOH); 1H NMR (CDCl3) δ 2.32 (s, 3H, 3-CH3), 3.82 (s, 3H, OCH3), 4.92 (s, 2H, NCH2CO), 6.62 (s, 1H, Ar), 6.69 (t, 1H, Ar, J = 2.1 Hz), 6.74 (d, 1H, Ar, J = 7.5 Hz), 6.85 (dd, 1H, Ar, J = 6.3 Hz, J = 2.0 Hz), 7.29 (t, 1H, Ar, J = 8.2 Hz), 7.38–7.46 (m, 4H, Ar), 9.10 (exch br s, 1H, NH). ESIMS calcd. for C21H18BrN3O4, 456.29; found: m/z 457.05 [M+H]+. Anal. C21H18BrN3O4 (C, H, N).
6.1.16. General Procedure for 20 and 25
A mixture of the appropriate compound 19 [40] or 24 [41] (0.80 mmol), K2CO3 (1.60 mmol) and ethyl bromoacetate (1.20 mmol) in CH3CN (5 mL) was refluxed with stirring for 3 h. The solvent was removed under vacuum, then the crude mixture was diluted with cold water (10 mL) and extracted with CH2Cl2 (3×15 mL). The organic layer was dried over Na2SO4 and evaporated in vacuo. Compound 20 was purified by recrystallization from ethanol, whereas compound 25 was purified by column chromatography using CH2Cl2/CH3OH 95:5 as eluent.
6.1.16.1. Ethyl-2-[4-acetyl-5-amino-3-methyl-6-oxopyridazin-1(6H)-yl]acetate [20]
Yield = 44%; mp = 145−46 °C (EtOH); 1H NMR (CDCl3) δ 1.32 (t, 3H, CH2CH3, J = 7.2 Hz), 2.53 (s, 3H, 3-CH3), 2.60 (s, 3H, COCH3), 4.27 (q, 2H, OCH2CH3, J = 7.2 Hz), 4.83 (s, 2H, NCH2CO), 7.75 (exch br s, 1H, NH). Anal. C11H15N3O4 (C, H, N).
6.1.16.2. Ethyl-2-[5-amino-3-methyl-6-oxopyridazin-1(6H)-yl]acetate [25]
Yield = 71%; oil (purified by column chromatography using CH2Cl2/CH3OH 95:5 as eluent). 1H NMR (CDCl3) δ 1.30 (t, 3H, CH2CH3, J = 7.2 Hz), 2.22 (s, 3H, 3-CH3), 4.25 (q, 2H, OCH2CH3, J = 7.2 Hz), 4.84 (s, 2H, NCH2CO), 6.17 (s, 1H, Ar). Anal. C9H13N3O3 (C, H, N).
6.1.17. Ethyl-2-[4-acetyl-5-(4-methoxyphenylamino)-3-methyl-6-oxopyridazin-1(6H)- yl]acetate [21]
To the suspension of 20 (0.91 mmol), copper acetate (1.36 mmol), and 4- methoxyphenylboronic acid (1.82 mmol) in CH2Cl2 (4 mL), Et3N (1.82 mmol) were added, and the mixture was stirred at room temperature for 12 h. The suspension was extracted with 15% aqueous ammonia (3×10 mL), and the organic layer was washed with water (10 mL) and dried over Na2SO4. After removal of the solvent in vacuo, the residue was purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 as eluent. Yield = 61%; mp = 115−17 °C (EtOH); 1H NMR (CDCl3) δ 1.34 (t, 3H, CH2CH3, J = 7.2 Hz), 1.88 (s, 3H, COCH3), 2.14 (s, 3H, 3- CH3), 3.81 (s, 3H, OCH3), 4.29 (q, 2H, OCH2CH3, J = 7.2 Hz), 4.88 (s, 2H, COCH2N), 6.85 (d, 2H, Ar, J = 6.7 Hz), 7.04 (d, 2H, Ar, J = 8.9 Hz), 7.62 (exch br s, 1H, NH). Anal. C18H21N3O5 (C, H, N).
6.1.18. General procedure for 22 and 26
A suspension of the suitable intermediate 21 or 25 (0.39 mmol) in 6 N NaOH (10 mL) was stirred at rt up to 80 °C for 1–2 h. The mixture was diluted with cold water and acidified with 6 N HCl. After 1 h stirring in an ice-bath, product 22 was filtered off by suction and recrystallized from ethanol. For compound 26, the aqueous phase was evaporated in vacuo. The crude residue was dissolved in ethanol, and the precipitate was filered off. Finally, evaporation of the solvent resulted in crude compound 26, which was purified by crystallization from cyclohexane.
6.1.18.1. 2-[4-Acetyl-5-(4-methoxyphenylamino)-3-methyl-6-oxopyridazin-1(6H)-yl]acetic acid [22]
Yield = 76%; mp = 104−06 °C (EtOH); 1H NMR (CDCl3) δ 1.88 (s, 3H, COCH3), 2.10 (exch br s, 1H, OH), 2.16 (s, 3H, 3-CH3), 3.82 (s, 3H, OCH3), 4.95 (s, 2H, COCH2N), 6.86 (d, 2H, Ar, J = 8.6 Hz), 7.05 (d, 2H, Ar, J = 8.6 Hz), 7.62 (exch br s, 1H, NH). Anal. C16H17N3O5 (C, H, N).
6.1.18.2. 2-[5-Amino-3-methyl-6-oxopyridazin-1(6H)-yl]acetic acid [26]
Yield = 99.9%; mp = 234−36 °C (cyclohexane); 1H NMR (CDCl3) δ 2.09 (s, 3H, 3-CH3), 4.62 (s, 2H, NCH2CO), 6.13 (s, 1H, Ar), 6.37 (exch br s, 2H, NH2), 6.55 (exch br s, 1H, OH). Anal. C7H9N3O3 (C, H, N).
6.1.19. General Procedures for 23 and 27
To a cooled (−5 °C) and stirred solution of 22 or 26 (0.30 mmol) in anhydrous tetrahydrofuran (3 mL), Et3N (1.06 mmol) was added. After 30 min, the mixture was allowed to warm up to 0 °C, and ethyl chloroformate (0.33 mmol) was added. After 1 h, 4-bromo aniline (0.60 mmol) was added, and the reaction was carried out at room temperature for 12 h. The mixture was then concentrated in vacuo, diluted with cold water (10 mL), and extracted with CH2Cl2 (3×15 mL). The organic layer was dried over Na2SO4 and the solvent was evaporated to obtain final compounds 23 or 27 respectively, which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent.
6.1.19.1. N-(4-Bromophenyl)-2-[4-acetyl-5-(4-methoxyphenylamino)-3-methyl-6-oxopyridazin- 1(6H)-yl]acetamide [23]
Yield = 62%; mp = 210−11 °C (EtOH); 1H NMR (CDCl3) δ 1.88 (s, 3H, COCH3), 2.19 (s, 3H, 3-CH3), 3.83 (s, 3H, OCH3), 4.97 (s, 2H, COCH2N), 6.87 (d, 2H, Ar, J = 8.9 Hz), 7.05 (d, 2H, Ar, J = 8.9 Hz), 7.44 (td, 4H, Ar, J = 2.8 Hz, J = 6.5 Hz), 7.65 (exch br s, 1H, NH), 8.64 (exch br s, 1H, NH). ESI-MS calcd. for C22H21BrN4O4, 485.33; found: m/z 486.11 [M+H]+. Anal. C22H21BrN4O4 (C, H, N).
6.1.19.2. N-(4-Bromophenyl)-2-[5-amino-3-methyl-6-oxopyridazin-1(6H)-yl]acetamide [27]
Yield = 50%; mp = 244−45 °C (EtOH); 1H NMR (CDCl3) δ 2.27 (s, 3H, 3-CH3), 4.93 (s, 2H, COCH2N), 6.24 (s, 1H, Ar), 7.42 (s, 4H, Ar), 8.83 (exch br s, 1H, NH), 8.64 (exch br s, 1H, NH). ESI-MS calcd. for C13H13BrN4O2, 337.17; found: m/z 338.02 [M+H]+. Anal. C13H13BrN4O2 (C, H, N).
6.1.20. N-(4-Bromophenyl)-2-[5-(4-methoxyphenylamino)-3-methyl-6-oxopyridazin-1(6H)-yl] acetamide [28a]
To a suspension of 27 (0.36 mmol), copper acetate (0.53 mmol) and 4- methoxyphenylboronic acid (0.36 mmol) in CH2Cl2 (3 mL), Et3N (0.72 mmol) was added and the mixture was stirred at room temperature for 12 h. The suspension was extracted with 15% aqueous ammonia (3×10 mL), and the organic layer was washed with 10 mL of water and dried over Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by flash column chromatography using CH2Cl2/CH3OH 9.5:0.5 as eluent. The analytical sample of compound 28a was obtained from further purification through silica gel preparative TLC (eluent: CH2Cl2/CH3OH 9.5:0.5). Yield = 10%; mp = 249−51 °C (EtOH); 1H NMR (CDCl3) δ 2.26 (s, 3H, 3- CH3), 3.86 (s, 3H, OCH3), 4.97 (s, 2H, COCH2N), 6.38 (s, 1H, Ar), 6.97 (d, 2H, Ar, J = 8.9 Hz), 7.18 (d, 2H, Ar, J = 8.9 Hz), 7.35 (exch br s, 1H, NH), 7.44 (dd, 4H, Ar, J = 5.5 Hz, J = 9.1 Hz), 8.77 (exch br s, 1H, NH). ESI-MS calcd. for C20H19BrN4O3, 443.29; found: m/z 444.10 [M+H]+. Anal. C20H19BrN4O3 (C, H, N).
6.1.21. General Procedures for 28b,c
A solution of compound 27 (0.21 mmol), Et3N (5 drops) and the appropriate substituted benzoyl chloride (0.25 mmol), in dry CH2Cl2 (5 mL), was stirred at 0 °C for 1 h. Extra benzoyl chloride (0.25 mmol) was added. The reaction was carried out at 0 °C for an additional 3 h, then at room temperature for 16 h. The mixture was extracted with 6 N NaOH (3×10 mL), and the organic layer was washed with water (10 mL) and dried over Na2SO4. After removal of the solvent in vacuo, crude compound 28b was purified by flash column chromatography using cyclohexane/ethyl acetate 1:3 as eluent. In the case of compound 28c, purification was performed by two consecutive silica gel preparative TLC steps using in both cases cyclohexane/ethyl acetate 1:2 as eluent.
6.1.21.1. 3-Metoxy-N-{2-[(4-bromophenylcarbamoyl)methyl]-6-methyl-3-oxo-2,3-dihydro pyridazin-4-yl}benzamide [28b]
Yield = 13 %; mp = 226−28 °C (EtOH); 1H NMR (CDCl3) δ 2.27 (s, 3H, 6-CH3), 3.89 (s, 3H, OCH3), 4.93 (s, 2H, COCH2N), 4.98 (exch br s, 1H, NH), 6.24 (s, 1H, Ar), 7.16–7.19 (m, 1H, Ar), 7.39–7.45 (m, 5H, Ar), 7.63 (t, 1H, Ar, J = 2.5 Hz), 7.73 (dd, 1H, Ar, J = 5.3 Hz, J = 1.0 Hz), 8.83 (exch br s, 1H, Ar). ESI-MS calcd. for C21H19BrN4O4, 471.30; found: m/z 472.08 [M+H]+. Anal. C21H19BrN4O4 (C, H, N).
6.1.21.2. 4-Bromo-N-{2-[2-(4-bromophenylcarbamoyl)methyl]-6-methyl-3-oxo-2,3-dihydro pyridazin-4-yl}benzamide [28c]
Yield = 10 %; colorless oil (purified by silica gel preparative TLC using cyclohexane/ethyl acetate 1:2 as eluent); 1H NMR (CDCl3) δ 2.43 (s, 3H, 6-CH3), 4.98 (s, 2H, COCH2N), 7.44 (s, 4H, Ar), 7.69 (d, 2H, Ar, J = 8.6 Hz), 7.82 (d, 2H, Ar, J = 8.6 Hz), 8.24 (s, 1H, Ar). ESI-MS calcd. for C20H16Br2N4O3, 520.17; found: m/z 521.00 [M+H]+. Anal. C20H16Br2N4O3 (C, H, N).
6.1.22. 4-(4-Methoxyphenyl)-6-methylpyridazin-3(2H)-one [30]
To an ice-cold solution of (E)- methyl 2-(4-methoxyphenyl)-4-oxopent-2-enoate 29 [43] (0.34 mmol) in dry toluene (3 mL), hydrazine hydrate (0.68 mmol) was added drop-wise. The solution was stirred at reflux temperature for 2 h. The solvent was evaporated, and ice-cold water (5 mL) was added to the residue. The aqueous layer was extracted with CH2Cl2 (3×15 mL), and the combined organic layers were dried over Na2SO4 and evaporated in vacuo. The crude product was purified by flash column chromatography (eluent: cyclohexane/ethyl acetate 1:3) to yield 30 as an amorphous white solid. Yield = 55%; mp = 155–158 °C (EtOH); 1H NMR (CDCl3) δ 2.39 (s, 3H, 6-CH3), 3.88 (s, 3H, OCH3), 6.99 (d, 2H, Ar, J = 8.9 Hz), 7.24 (s, 1H, Ar), 7.86 (d, 2H, Ar, J = 8.9 Hz), 10.51 (exch br s, 1H, NH). Anal. C12H12N2O2 (C, H, N).
6.1.23. Ethyl-2-[5-(4-methoxyphenyl)-3-methyl-6-oxopyridazin-1(6H)-yl]acetate [31]
A mixture of the intermediate 30 (0.18 mmol), K2CO3 (0.36 mmol), and ethyl bromoacetate (0.28 mmol) in CH3CN (2 mL), was refluxed under stirring for 1.5 h. The mixture was then concentrated in vacuo and extracted with CH2Cl2 (3×10 mL). After removal of the solvent under vacuo, the residue was purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 99.9 %; colorless oil (purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent); 1H NMR (CDCl3) δ 1.30 (t, 3H, CH2CH3, J = 7.0 Hz), 2.37 (s, 3H, 3-CH3), 3.92 (s, 3H, OCH3), 4.23 (q, 2H, CH2CH3, J = 7.0 Hz), 4.89 (s, 2H, NCH2CO), 6.94 (d, 2H, Ar, J = 8.8 Hz), 7.21 (s, 1H, Ar), 7.81 (d, 2H, Ar, J = 8.8 Hz). Anal. C16H18N2O4 (C, H, N).
6.1.24. 2-[5-(4-Methoxyphenyl)-3-methyl-6-oxopyridazin-1(6H)-yl]acetic acid [32]
A suspension of the intermediate 31 (0.20 mmol) in 6 N NaOH (1.5 mL) was stirred at 60 °C for 0.5 h. The mixture was diluted with ice-cold water (1 mL), acidified with 6 N HCl, and the final product 32 was then filtered off by suction and recrystallized from ethanol. Yield = 99.9%; mp = 202–203 °C (EtOH); 1H NMR (CDCl3) δ 2.41 (s, 3H, 3-CH3), 3.87 (s, 3H, OCH3), 4.99 (s, 2H, NCH2CO), 6.98 (d, 2H, Ar, J = 8.8 Hz), 7.26 (s, 1H, Ar), 7.82 (d, 2H, Ar, J = 8.8 Hz). Anal. C14H14N2O4 (C, H, N).
6.1.25. N-(4-Bromophenyl)-2-[5-(4-methoxyphenyl)-3-methyl-6-oxo-pyridazin-1(6H)- yl]acetamide [33]
To a cooled (-5 °C) and stirred solution of compound 32 (0.20 mmol) in anhydrous tetrahydrofuran (2 mL), Et3N (0.70 mmol) was added. After 30 min, the mixture was allowed to warm up to 0 °C, and ethyl chloroformate (0.22 mmol) was added. After 1 h, 4- bromoaniline (0.40 mmol) was added, and the reaction was carried out at room temperature for 12 h. The mixture was then concentrated in vacuo, diluted with cold water (5 mL), and extracted with CH2Cl2 (3×10 mL). After removal of the solvent, the residue was purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 35%; mp = 251–253 °C (EtOH); 1H NMR (CDCl3) δ 2.43 (s, 3H, 6-CH3), 3.88 (s, 3H, OCH3), 5.00 (s, 2H, NCH2CO), 7.00 (d, 2H, Ar, J = 8.7 Hz), 7.27 (s, 1H, Ar), 7.40 (q, 4H, Ar, J = 9.2 Hz), 7.80 (d, 2H, Ar, J = 8.7 Hz), 9.07 (exch br s, 1H, NH). ESI-MS calcd. for C20H18BrN3O3, 428.28; found: m/z 429.05 [M+H]+. Anal. C20H18BrN3O3 (C, H, N).
6.2. Biological assays
6.2.1. Cell culture
Human promyelocytic leukemia HL-60 cells stably transfected with FPR1 (HL-60-FPR1), FPR2 (HL-60-FPR2), or FPR3 (HL-60-FPR3) were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 µg/ml streptomycin, 100 U/ml penicillin, and G418 (1 mg/mL), as described previously [16,19,47,49]. Specificity of FPR expression on these cells was demonstrated previously, and it has been shown in a number of studies that these cell lines respond only to receptor subtype-specific agonists [44,45,52]. Wild-type HL-60 cells were cultured under the same conditions, but without G418.
6.2.2. Isolation of human neutrophils
Blood was collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as previously described [53]. Isolated neutrophils were washed twice and resuspended in HBSS without Ca2+ and Mg2+ (HBSS-). Neutrophil preparations were routinely > 95 % pure, as determined by light microscopy, and > 98 % viable, as determined by trypan blue exclusion.
6.2.3. Ca2+ mobilization assay
Changes in intracellular Ca2+ were measured with a FlexStation II scanning fluorometer using a FLIPR 3 calcium assay kit (Molecular Devices, Sunnyvale, CA) for human neutrophils and HL-60 cells. All active compounds were evaluated in parent (wild-type) HL-60 cells for supporting that the agonists are inactive in non-transfected cells. Human neutrophils or HL-60 cells, suspended in HBSS- containing 10 mM HEPES, were loaded with Fluo-4 AM dye (Invitrogen) (1.25 µg/mL final concentration) and incubated for 30 min in the dark at 37 °C. After dye loading, the cells were washed with HBSS- containing 10 mM HEPES, resuspended in HBSS containing 10 mM HEPES and Ca2+ and Mg2+ (HBSS+), and aliquotted into the wells of a flat-bottomed, half-area-well black microtiter plates (2×105 cells/well). The compound source plate contained dilutions of test compounds in HBSS+. Changes in fluorescence were monitored (λex = 485 nm, λem = 538 nm) every 5 s for 240 s at room temperature after automated addition of compounds. Maximum change in fluorescence, expressed in arbitrary units over baseline, was used to determine agonist response. Responses were normalized to the response induced by 5 nM fMLF (Sigma Chemical Co., St. Louis, MO) for HL-60-FPR1 and neutrophils, or 5 nM WKYMVm (Calbiochem, San Diego, CA) for HL-60-FPR2 and HL-60-FPR3 cells, which were assigned a value of 100%. Curve fitting (5–6 points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose-response curves generated using Prism 5 (GraphPad Software, Inc., San Diego, CA).
6.2.4. Chemotaxis assay
Neutrophils were suspended in HBSS+ containing 2% (v/v) fetal bovine serum (FBS) (2×106 cells/mL), and chemotaxis was analyzed in 96-well ChemoTx chemotaxis chambers (Neuroprobe, Gaithersburg, MD), as previously described [53]. Briefly, lower wells were loaded with 30 µL of HBSS+ containing 2% (v/v) FBS and the indicated concentrations of test compound, DMSO (negative control), or 1 nM fMLF as a positive control. Neutrophils were added to the upper wells and allowed to migrate through the 5.0 µm porepolycarbonate membrane filter for 60 min at 37 °C and 5% CO2. The number of migrated cells was determined by measuring ATP in lysates of transmigrated cells using a luminescence-based assay (CellTiter-Glo; Promega, Madison, WI), and luminescence measurements were converted to absolute cell numbers by comparison of the values with standard curves obtained with known numbers of neutrophils. The results are expressed as percentage of negative control and were calculated as follows: (number of cells migrating in response to test compounds/spontaneous cell migration in response to control medium)×100. EC50 values were determined by nonlinear regression analysis of the dose-response curves generated using Prism 5 software.
6.2.5. Cytotoxicity assay
Cytotoxicity was analyzed with a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega), according to the manufacturer’s protocol. Briefly, wild-type non-transfected human promyelocytic leukemia HL-60 cells were cultured at a density of 3×104 cells/well with tested compounds for 60 min at 37°C and 5% CO2. Following treatment, the cells were allowed to equilibrate to room temperature for 30 min, substrate was added, and the samples were analyzed with a Fluoroscan Ascent FL.
6.3. Molecular docking analysis
The FPR1 homology model was created using the crystal structure of the bovine rhodopsin receptor, which has a sequence identity of 20% for 348 aligned residues that correspond to the seven transmembrane domains, as reported previously [46]. It should be noted, that the crystal structure of bovine rhodopsin was successfully applied previously to homology modeling of FPR1 [12,54]. The position of the ligand binding site in these models, based on cross-linking and mutagenesis studies, was found to be located in the upper region of a helical bundle comprising transmembrane domains (TM) 2, 5, 6, and 7 [50,55] and was recently supported by docking studies of FPR1 agonists, including peptide Ac-QAWF [46] and series of non-peptide 2-(benzimidazol-2- ylthio)-N-phenylacetamides [47].
A PDB file of the homology model for FPR1 was loaded into the Molegro Virtual Docker (MVD) program (MVD 2010.4.2, Molegro ApS). Docking search space was defined as a sphere centered at the carbonyl carbon of the Ala residue in Ac-QAWF [46]. The radius of the sphere was adopted to be 11 Å. This search space encompassed the whole Ac-QAWF molecule and included, at least partially, the following 36 residues of FPR1: Trp91, Trp95, Cys98, Lys99, Leu101, Phe102, Thr103, Val105, Asp106, Phe110, Leu156, Thr157, Leu158, Pro159, Val160, Ile161, Ile162, Asn192, Val193, Ala196, Met197, Leu198, Thr199, Val200, Arg201, Gly202, Arg205, Phe206, Tyr257, Ala261, Ala264, Thr265, Ile268, Arg269, Glu270, and Val283.
For homology modeling of FPR2, the primary amino acid sequence of FPR2 was submitted to the Phyre2 (Protein Homology/analogy Recognition Engine V2.0) protein fold recognition server (http://www.sbg.bio.ic.ac.uk/phyre2) [56]. The server used one template with known crystal structure for homology modeling per protein sequence. We obtained 18 predicted protein structure models with the highest level of confidence, based on dissolved crystal structures of several GPCRs, including bovine and squid rhodopsins, human adenosine receptor A2A, turkey β1 adrenoceptor, human β2 adrenoceptor, human histamine receptor H1, human dopamine D3 receptor, and human chemokine receptor CXCR4. Two FPR2 homology models were pre-selected from the set of predicted models. One model, based on the CXCR4 structure, had a maximal sequence identity of 28%, but with a low crystal structure resolution of 3.2 Å for the template. The second model has a template with sequence identity of 16%, but the highest resolution crystal structure (2.2 Å) known to date for a GPCR. Side chain conformations of eight residues in FPR2 (His102, Val105, Asp106, Leu109, Trp254, Phe257, Ser288, Phe292), which were previously identified as belonging to the binding site [51], were optimized in both models using the corresponding module of Molegro software. Since our pre-docking studies indicated that the rhodopsin-based model gave the best docking positions for FPR2 agonists that were used previously for pharmacophore modeling [49,57], we propose that these data justify use of the bovine rhodopsin structure as a template for the FPR2 homology model vs. the CXCR4 template. Thus, further modeling was based on the rhodopsin-based model of the FPR2.
Taking into account the lack of structural information about any ligand-receptor complex with FPR2, we sought to locate cavities in the macromolecule obtained by homology modeling in order to identify the search space for docking. Use of the MVD “Detect cavity” module with probe size 1.2 Å gave two cavities with volumes of 241 and 25 Å3 in the region of the ligand binding site. Positions of these two cavities obviously reflect the dumb-bell shape of the binding site. Hence, for FPR2, we also chose a spherical search space with a default radius of 15 Å centered at the terminus of the larger cavity directed to the smaller one (Figure 5).
Before docking, structures of the compounds were pre-optimized using HyperChem software with MM+ force field and saved in Tripos MOL2 format. The ligand structures were then imported into the MVD with the options “Create explicit hydrogens”, “Assign charges (calculated by MVD)”, and “Detect flexible torsions in ligands” enabled. Selected molecules were docked into FPR1 and FPR2 using the search spaces indicated above with a rigid receptor structure. Ligand flexibility was accounted for with respect to torsion angles auto-detected in MVD. MolDock score functions were used with a 0.3 Å grid resolution. The “Internal HBond” option was activated in the “Ligand evaluation” menu of Docking Wizard. Thirty docking runs were performed for each molecule, while 60 docking runs were performed for the peptide. The option “Return multiple poses for each run” was enabled, and the post-processing options “Energy minimization” and “Optimize H-bonds” were applied after docking. Similar poses were clustered at a RMSD threshold of 1 Å.
Supplementary Material
Highlights.
Arylacetamide moiety at N-2 of the pyridaninone scaffold is foundamental for activity.
Differently from C-4, position C-6 resulted not tolerant to modifications.
Molecular docking studies showed analogies with poses for fMLF and WKYMVM.
Compounds were evaluated on HL-60 cells and on human neutrophils.
We found FPR1-selective agonists and potent FPR1/FPR2 dual agonists.
Table 1.
Legend for intermediates 1,2a–k.
| Compds 1,2 | R |
|---|---|
| a | CH2CH3 |
| b | CH(CH3)2 |
| c | C6H11 |
| d | C6H5 |
| e | 2-thienyl |
| f | C6H4-CH3 (p) |
| g | C6H4-OCH3 (p) |
| h | C6H4-Cl (p) |
| i | C6H4-F (p) |
| j | CH2-C6H5 |
| k | COOH |
Table 2.
Legend for intermediates 3–5a–p.
| Compds 3–5 |
R | OCH3 |
|---|---|---|
| a | H | m |
| b | CH2CH3 | m |
| c | CH(CH3)2 | m |
| d | C6H11 | m |
| e | C6H5 | m |
| f | 2-thienyl | p |
| g | 2-thienyl | m |
| h | C6H4-OCH3 (p) | p |
| i | C6H4-OCH3 (p) | m |
| j | C6H4-Cl (p) | p |
| k | C6H4-Cl (p) | m |
| l | C6H4-CH3 (p) | p |
| m | C6H4-CH3 (p) | m |
| n | C6H4-F (p) | p |
| o | C6H4-F (p) | m |
| p | CH2-C6H5 | p |
Table 3.
Legend for intermediates 8a–l.
| Compds 8 | R |
|---|---|
| a | C6H5 |
| b | C6H4-F (m) |
| c | C6H4-Cl (m) |
| d | C6H4-Br (m) |
| e | C6H3-(OCH3)2 (m, m) |
| f | C6H4-CF3 (p) |
| g | 3-furyl |
| h | 3-thienyl |
| i | 2-thienyl |
| j | 1-naphthyl |
| k | 3-pyridyl |
| l | C6H4-CONH2 (p) |
Table 4.
Legend for intermediates 9,10a–j.
| Compds 9,10 | R |
|---|---|
| a | C6H5 |
| b | C6H4-F (m) |
| c | C6H4-Cl (m) |
| d | C6H4-Br (m) |
| e | C6H3-(OCH3)2 (m, m) |
| f | C6H4-CF3 (p) |
| g | 3-furyl |
| h | 3-thienyl |
| i | 2-thienyl |
| j | 1-naphthyl |
Acknowledgements
This work was supported in part by was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number GM103500 and the Montana State University Agricultural Experimental Station.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Yields, mp, and 1H NMR spectral data for all new compounds.
References
- 1.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 2.Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses, Cytok. Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quehenberger O, Prossnitz ER, Cavanagh SL, Cochrane CG, Ye RD. Multiple domains of the N-formyl peptide receptor are required for high-affinity ligand binding. Construction and analysis of chimeric N-formyl peptide receptors. J. Biol. Chem. 1993;268:18167–18175. [PubMed] [Google Scholar]
- 5.Migeotte I, Riboldi E, Franssen JD, Grégoire F, Loison C, Wittamer V, Detheux M, Robberecht P, Costagliola S, Vassart G, Sozzani S, Parmentier M, Communi D. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 2005;201:83–93. doi: 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dufton N, Perretti M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol. Ther. 2010;127:175–188. doi: 10.1016/j.pharmthera.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Gavins FNE. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol. Sci. 2010;31:266–276. doi: 10.1016/j.tips.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui Y, Le Y, Yazawa H, Gong W, Wang JM. Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer's disease. J. Leukoc. Biol. 2002;72:628–635. [PubMed] [Google Scholar]
- 9.Cattaneo F, Guerra G, Ammendola R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010;35:2018–2026. doi: 10.1007/s11064-010-0301-5. [DOI] [PubMed] [Google Scholar]
- 10.Kilby JM, Hopkins S, Venetta TM, Di Massimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambertm D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 11.De Paulis A, Montuori N, Prevete N, Fiorentino I, Rossi FW, Visconte V, Rossi G, Marone G, Ragno P. Urokinase induces basophil chemotaxis through a urokinase receptor epitope that is an endogenous ligand for formyl peptide receptor-like 1 and -like 2. J. Immunol. 2004;173:5739–5748. doi: 10.4049/jimmunol.173.9.5739. [DOI] [PubMed] [Google Scholar]
- 12.Edwards BS, Bologa C, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 13.Pieretti S, Di Giannuario A, De Felice M, Perretti M, Cirino G. Stimulus-dependent specificity for annexin 1 inhibition of the inflammatory nociceptive response: the involvement of the receptor for formylated peptides. Pain. 2004;109:52–63. doi: 10.1016/j.pain.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Falla TJ. Host defense peptides for use as potential therapeutics. Curr. Opin. Invest. Drugs. 2009;10:164–171. [PubMed] [Google Scholar]
- 16.Cilibrizzi A, Quinn MT, Kirpotina LN, Schepetkin IA, Holderness J, Ye RD, Rabiet MJ, Biancalani C, Cesari N, Graziano A, Vergelli C, Pieretti S, Dal Piaz V, Giovannoni MP. 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones: a novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem. 2009;52:5044–5057. doi: 10.1021/jm900592h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsman H, Kalderén C, Nordin A, Nordling E, Jensen AJ, Dahlgren C. Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem. Pharmacol. 2011;81:402–411. doi: 10.1016/j.bcp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Frohn M, Xu H, Zou X, Chang C, McElvain M, Plant MH, Wong M, Tagari P, Hungate R, Bürli RW. New 'chemical probes' to examine the role of the hFPRL1 (or ALXR) receptor in inflammation. Bioorg. Med. Chem. Lett. 2007;17:6633–6637. doi: 10.1016/j.bmcl.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 19.Cilibrizzi A, Schepetkin IA, Bartolucci G, Crocetti L, Dal Piaz V, Giovannoni MP, Graziano A, Kirpotina LN, Quinn MT, Vergelli C. Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists. Bioorg. Med. Chem. 2012;20:3781–3792. doi: 10.1016/j.bmc.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sogawa Y, Ohyama T, Maeda H, Hirahara K. Formyl peptide receptor 1 and 2 dual agonist inhibits human neutrophil chemotaxis by the induction of chemoattractant receptor cross-desensitization. J. Pharmacol. Sci. 2011;115:63–68. doi: 10.1254/jphs.10194fp. [DOI] [PubMed] [Google Scholar]
- 21.Reddy RS, Saravanan K, Kumar P. An efficient approach to γ-alkylidene γ-butyrolactones: Application to the syntheses of pyridazinones and diazocinones. Tetrahedron. 1998;54:6553–6564. [Google Scholar]
- 22.Druey J. Pyridazine in der Arzneimittelsynthese. Angew. Chem. 1958;70:5–13. [Google Scholar]
- 23.Barberis M, Pérez-Prieto L. Enantioselective synthesis of sabina ketone. Tetrahedron Lett. 2003;44:6683–6685. [Google Scholar]
- 24.Shinkai H, Ozeki H, Motomura T, Ohta T, Furukawa N, Uchida I. 4-(trans-4- Methylcyclohexyl)-4-Oxobutyric Acid (JTT-608). A new class of antidiabetic agent. J. Med. Chem. 1998;41:5420–5428. doi: 10.1021/jm9804228. [DOI] [PubMed] [Google Scholar]
- 25.Singh S, Verma M, Singh KN. Superoxide ion induced oxidation of γ-lactones to γ- ketocarboxylic acids in aprotic medium. Synthetic Commun. 2004;34:4471–4475. [Google Scholar]
- 26.Grundmann C. Über sulfanilamido-pyridazine (heterocyclische sulfonamide, I.Mitteil) Chem. Ber. 1948;81:1–11. [Google Scholar]
- 27.Gouault N, Cupif JF, Picard S, Lecat A, David M. Synthesis of diverse 4,5-dihydro- 3(2H)-pyridazinones on Wang resin. J. Pharm.. Pharmacol. 2001;53:981–985. doi: 10.1211/0022357011776225. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee PS, Sharma PK, Nema RK. Synthesis and anticonvulsant activity of pyridazinone derivatives. Int. J. Chem. Tech. Res. 2009;1:522–525. [Google Scholar]
- 29.Steiner G, Gries J, Lenke D. Synthesis and antihypertensive activity of new 6-heteroaryl-3- hydrazinopyridazine derivatives. J. Med. Chem. 1981;24:59–63. doi: 10.1021/jm00133a013. [DOI] [PubMed] [Google Scholar]
- 30.Kaupp G, Schmeyers J. Solid-state reactivity of the hydrazine–hydroquinone complex. J. Phys. Org. Chem. 2000;13:388–394. [Google Scholar]
- 31.Hu W, Ralay Ranaivo H, Roy SM, Behanna HA, Wing LK, Munoz L, Guo L, Van Eldik LJ, Watterson DM. Development of a novel therapeutic suppressor of brain proinflammatory cytokine up-regulation that attenuates synaptic dysfunction and behavioral deficits. Bioorg. Med. Chem. Lett. 2007;17:414–418. doi: 10.1016/j.bmcl.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steck EA, Brundage RP, Fletcher LT. Pyridazine Derivatives. I. Some amebacidal 3- pyridazones. J. Am. Chem. Soc. 1953;75:1117–1119. [Google Scholar]
- 33.Castleman ER, Wiselogle FY. Studies in the pyridazine series. The absorption spectrum of pyridazine. J. Am. Chem. Soc. 1945;67:60–62. [Google Scholar]
- 34.Kaddah AM, Khalil AM. Reactions of 3-pyridazinones with aldehydes and Grignard reagents. Indian J. Chem. 1977;15B:1025–1028. [Google Scholar]
- 35.Ismail MF, El Khamry AA, Shams NA, El Sawy OM. Base-catalyzed condensation of aromatic aldehydes with 4,5-dihydro-6-methylpyridazin-3(2H-one) Indian J. Chem. 1980;19B:203–205. [Google Scholar]
- 36.Powell P, Sosabowski MH. Preparation and reactions of some 2-thienyl- and 3- thienylpyridazinones and –pyridazines. J. Chem. Res. (S) 1995;8:306–307. [Google Scholar]
- 37.Kandile NG, Ahmed EA. Synthesis of some new pyridazinones. Acta Chim. Hung. 1990;127:829–835. [Google Scholar]
- 38.Baraldi PG, Preti D, Tabrizi MA, Fruttarolo F, Saponaro G, Baraldi S, Romagnoli R, Moorman AR, Gessi S, Varani K, Borea PA. N6-[(Hetero)aryl/(cyclo)alkyl-carbamoylmethoxy- phenyl]-(2-chloro)-5′-N-ethylcarbox-amido-adenosines: The first example of adenosine-related structures with potent agonist activity at the human A2B adenosine receptor. Bioorg. Med. Chem. 2007;15:2514–2527. doi: 10.1016/j.bmc.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 39.Overend WG, Wiggins LF. The conversion of sucrose into pyridazine derivatives. Part I. 3-Sulfanilamido-6-methylpyridazine. J. Chem. Soc. 1947:239. doi: 10.1039/jr9470000239. [DOI] [PubMed] [Google Scholar]
- 40.Costantino L, Rastelli G, Gamberoni MC, Giovannoni MP, Dal Piaz V, Vinello P, Barlocco D. Isoxazolo-[3,4-d]-pyridazin-7-(6H)-one as a potential substrate for new aldose reductase inhibitors. J. Med. Chem. 1999;42:1894–1900. doi: 10.1021/jm981107o. [DOI] [PubMed] [Google Scholar]
- 41.Coates WJ, McKillop A. Preparation of 4-amino-3(2H)-pyridazinones by direct amination of 3(2H)-pyridazinones with hydrazine. Heterocycles. 1989;29:1077–1090. [Google Scholar]
- 42.Biancalani C, Giovannoni MP, Pieretti S, Cesari N, Graziano A, Vergelli C, Cilibrizzi A, Di Gianuario A, Colucci M, Mangano G, Garrone B, Polenzani L, Dal Piaz V. Further studies on arylpiperazinyl alkyl pyridazinones: discovery of an exceptionally potent, orally active, antinociceptive agent in thermally induced pain. J. Med. Chem. 2009;52:7397–7409. doi: 10.1021/jm900458r. [DOI] [PubMed] [Google Scholar]
- 43.Lerche H, Koenig D, Severin T. Reactions with nitroenamines. XII. Reaction of esters and lactones with nitroenamines. Chem. Ber. 1974;107:1509–1517. [Google Scholar]
- 44.Christophe T, Karlsson A, Rabiet MJ, Boulay F, Dahlgren C. Phagocyte activation by Trp-Lys-Tyr-Met-Val-Met, acting through FPRL1/LXA4R, is not affected by lipoxin A4 . Scand. J. Immunol. 2002;56:470–476. doi: 10.1046/j.1365-3083.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 45.Forsman H, Andréasson E, Karlsson J, Boulay F, Rabiet MJ, Dahlgren C. Structural characterization and inhibitory profile of formyl peptide receptor 2 selective peptides descending from a PIP2-binding domain of gelsolin. J. Immunol. 2012;189:629–637. doi: 10.4049/jimmunol.1101616. [DOI] [PubMed] [Google Scholar]
- 46.Movitz C, Brive L, Hellstrand K, Rabiet MJ, Dahlgren C. The annexin I sequence gln(9)- ala(10)-trp(11)-phe(12) is a core structure for interaction with the formyl peptide receptor 1. J. Biol. Chem. 2010;285:14338–14345. doi: 10.1074/jbc.M109.080465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khlebnikov AI, Schepetkin IA, Kirpotina LN, Brive L, Dahlgren C, Jutila MA, Quinn MT. Molecular docking of 2-(benzimidazol-2-ylthio)-N-phenylacetamide-derived small-molecule agonists of human formyl peptide receptor 1. J. Mol. Model. 2012;18:2831–2843. doi: 10.1007/s00894-011-1307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deflorian F, Jacobson KA. Comparison of three GPCR structural templates for modeling of the P2Y12 nucleotide receptor. J. Comput. Aided Mol. Des. 2011;25(4):329–338. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Leopoldo M, Lucente E, Lacivita E, De Giorgio P, Quinn MT. 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: molecular modeling of chiral recognition by FPR2. Biochem. Pharmacol. 2013;85(3):404–416. doi: 10.1016/j.bcp.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mills JS, Miettinen HM, Cummings D, Jesaitis AJ. Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of Met-Leu-Phe and Met-Met-Trp-Leu-Leu. J. Biol. Chem. 2000;275:39012–39017. doi: 10.1074/jbc.M003081200. [DOI] [PubMed] [Google Scholar]
- 51.Fujita H, Kato T, Watanabe N, Takahashi T, Kitagawa S. Stimulation of human formyl peptide receptors by calpain inhibitors: homology modeling of receptors and ligand docking simulation. Arch. Biochem. Biophys. 2011;516:121–127. doi: 10.1016/j.abb.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 52.Dahlgren C, Christophe T, Boulay F, Madianos PN, Rabiet MJ, Karlsson A. The synthetic chemoattractant Trp-Lys-Tyr-Met-Val-DMet activates neutrophils preferentially through the lipoxin A4 receptor. Blood. 2000;95:1810–1818. [PubMed] [Google Scholar]
- 53.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Quinn MT. High throughput screening for small-molecule activators of neutrophils: identification of novel N-formyl peptide receptor agonists. Mol. Pharmacol. 2007;71:1061–1074. doi: 10.1124/mol.106.033100. [DOI] [PubMed] [Google Scholar]
- 54.Ferrari C, Macchiarulo A, Costantino G, Pellicciari R. Pharmacophore model for bile acids recognition by the FPR receptor. J. Comput. Aided Mol. Des. 2006;20:295–303. doi: 10.1007/s10822-006-9055-1. [DOI] [PubMed] [Google Scholar]
- 55.Miettinen HM, Mills JS, Gripentrog JM, Dratz EA, Granger BL, Jesaitis AJ. The ligand binding site of the formyl peptide receptor maps in the transmembrane region. J. Immunol. 1997;159:4045–4054. [PubMed] [Google Scholar]
- 56.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 57.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Jutila MA, Quinn MT. Gastrinreleasing peptide/neuromedin B receptor antagonists PD176252, PD168368, and related analogs are potent agonists of human formyl-peptide receptors. Mol. Pharmacol. 2011;79:77–90. doi: 10.1124/mol.110.068288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.