

Abstract
From the initial description of platelets in 1882, their propensity to aggregate and to contribute to thrombosis was apparent. Indeed, excessive platelet aggregation is associated with myocardial infarction and other thrombotic diseases whereas Glanzmann thrombasthenia, in which platelet aggregation is reduced, is a bleeding syndrome. Over the last half of the 20th century, many investigators have provided insights into the cellular and molecular basis for platelet aggregation. The major membrane protein on platelets, integrin αIIbβ3, mediates this response by rapidly transiting from its resting to an activated state in which it serves as a receptor for ligands that can bridge platelets together. Monoclonal antibodies, natural products, and small peptides were all shown to inhibit αIIbβ3 dependent platelet aggregation, and these inhibitors became the forerunners of antagonists that proceeded through preclinical testing and into large patient trials to treat acute coronary syndromes, particularly in the context of percutaneous coronary interventions. Three such αIIbβ3 antagonists, abciximab, eptifibatide, and tirofiban, received Food and Drug Administration approval. Over the past 15 years, millions of patients have been treated with these αIIbβ3 antagonists and many lives have been saved by their administration. With the side effect of increased bleeding and the development of new antithrombotic drugs, the use of αIIbβ3 antagonists is waning. Nevertheless, they are still widely used for the prevention of periprocedural thrombosis during percutaneous coronary interventions. This review focuses on the biology of αIIbβ3, the development of its antagonists, and some of the triumphs and shortcomings of αIIbβ3 antagonism.
Keywords: acute coronary syndromes, αIIbβ3 antagonists, integrin, percutaneous coronary intervention
Every year, since 1900, cardiovascular disease (CVD) has accounted for more deaths in the United States than any other disease. According to 2012 American Heart Association statistics, CVD claims more lives each year than cancer, chronic lung/respiratory disease, and accidents combined.1 Despite these grim statistics, dramatic progress has been made in the treatment of CVD, as evidenced by a 30.6% decline in death rates attributable to CVD between 1998 to 2008.1 Many factors contributed to this reduction, including improved diagnostic and interventional procedures, healthier lifestyles, and the emergence of new drugs. With the well-established evidence for the central role of platelet aggregation in thrombus formation, the inhibition of this response has long been recognized an attractive target for drugs to reduce morbidity and mortality arising from acute coronary syndromes (ACSs) and other CVDs. Throughout the late 1970s/early1980s, an understanding of the molecular basis of the platelet aggregation emerged and focused attention on the pivotal role on a single receptor, αIIbβ3, on the platelet surface in orchestrating the aggregation response, and further suggested that this receptor represented a rationale target for antithrombotic therapy. Throughout the late 1980s/1990s, most major bio-pharmaceutical companies and many fledgling biotechnology start-ups had aggressive programs in place to develop αIIbβ3 antagonists. In fact, these programs were successful. Many αIIbβ3 antagonists were identified, and 3 such drugs—abciximab, eptifibatide, and tirofiban—ultimately received Food and Drug Administration (FDA) approval. These drugs have been used extensively; it is estimated that at least 8 000 000 people have been treated with αIIbβ3 antagonists.2 Importantly, the rational targeting of αIIbβ3 and the clinical efficacy of αIIbβ3 antagonists established the central role of platelets in periprocedural thrombosis in the context of percutaneous coronary interventions (PCI).
Although the use of αIIbβ3 antagonists has waned since their peak years in the mid-2000s, the inhibition of the platelet aggregation response still remains a centerpiece in the treatment of ACS patients, and the development of newer antithrombotic strategies has very much benefited from the knowledge and experience gained in the development of αIIbβ3 antagonists. Furthermore, following the lead that αIIbβ3, an integrin, could be antagonized, researchers now consider at least 4 other integrin family members (α4β1, α4β7, αvβ3, αLβ2) as drug targets.3–6 Thus, the development of αIIbβ3 antagonists demonstrates how biomedical research can be harnessed for rational drug design and translated into clinical success. Here, we provide a brief summary of the story behind their development.
αIIbβ3: Historical, Functional, and Structural Perspectives
A time line depicting some of the key events in the development of αIIbβ3 agonists is depicted in Figure 1. The discovery of platelets is usually credited to the Italian physician Giulio Bizzozero. In his 1882 article, Bizzozero described platelets as a new element in the blood. Furthermore, he noted that platelets could aggregate, and suggested that this propensity might contribute to thrombosis.7 Almost 40 years later, the Swiss physician Eduard Glanzmann described a group of patients in whom abnormal platelet aggregation was associated with a bleeding tendency.8 Over the next half century, great strides were made in characterizing the composition of cell membranes, and these analyses were greatly accelerated by the application of gel electrophoresis technologies to separate the membrane proteins of various cell types. When applied to platelet membranes, a number of protein bands differing in their mobility were discerned.9,10 After establishing the patterns of the platelet membrane proteins from healthy individuals, Phillips et al11 showed that 2 glycoprotein bands, glycoprotein IIb (αIIb) and glycoprotein IIIa (β3), were missing from the surface of platelets from patients with Glanzmann thrombasthenia. Subsequent biochemical studies showed that these 2 polypeptides were the noncovalently associated subunits of a single membrane protein, glycoprotein IIb–IIIa (integrin αIIbβ3).12,13
Figure 1. Highlights of the chronology of key discoveries in αIIbβ3 receptor antagonists.

FDA indicates Food and Drug Administration; PLA1/PLA2, platelet alloantigen 1, 2; KGD, lysine-glycine-aspartic acid sequence; and RGD, arginine-glycine-aspartic acid sequence.
When αIIbβ3 is activated, it serves as a receptor for ligands that can bridge to other αIIbβ3 on adjacent platelets.14,15 Ligands of αIIbβ3 that can mediate this cross-bridging function are fibrinogen and von Willebrand factor,16,17 whereas other ligands of αIIbβ3, such as fibronectin, vitronectin, thrombospondin, and CD40 ligand, can modulate platelet aggregation.18–20 As the molecular details establishing the essential role of αIIbβ3 in platelet aggregation emerged, it became clear that inhibition of its ligand binding function would inhibit platelet aggregation and thereby limit thrombus formation. This rationale became the foundation of the new antithrombotic strategy, αIIbβ3 antagonism. The feasibility of αIIbβ3 antagonism was illustrated with antibodies and peptides that bound to αIIbβ321–23; and these became the forerunners of αIIbβ3 antagonists that were ultimately used to treat patients.
Another aspect of αIIbβ3 biology that is relevant to its antagonism is its familial relationships. As noted above, αIIbβ3 is an integrin, a large family of structurally related and broadly distributed membrane proteins.24 All integrins are noncovalent heterodimers composed of an α and a β subunit. In mammals, there are 18 α subunits and 8 β subunits, which interact to form the 24 different integrins.25 Each α and β subunit consists of a large extracellular segment, which is composed of a series of structural domains that are conserved throughout the integrin family (Figure 2A).26 Each subunit has a single pass transmembrane domain and, with a single exception, a short cytoplasmic tail (see Figure 2A). By virtue of their abilities to bind to ligands of the extracellular matrix on the surface of other cells, via their extracellular regions, and to link to the cytoskeletal matrix within cells via their cytoplasmic tails, integrins serve as a conduit for flow of information between the interior of the cell and its exterior environment. This communication is bidirectional and is used to control a variety of cellular responses ranging from cell adhesion and migration to gene expression and proliferation.27
Figure 2.

A, Depiction of the domain organization of the αIIb and β3 subunits in an extended conformation. B, Pathway for activation of αIIbβ3. Inside-out signaling can be induced by different agonists, usually binding to a G-protein coupled receptor, which initiate signaling pathways that lead to the cytoplasmic tails of αIIbβ3. Inside-out signaling to activate the receptor begins with unclasping of the membrane proximal complex between the αIIb and β3 cytoplasmic tails. Talin and kindlin cooperate in this activation process. Their separation triggers dissociation of the transmembrane complex of the subunits. These changes then trigger a conformational switch in the integrin extracellular domain, resulting in its conversion from a bent conformation in its resting state to an extended conformation in which it becomes competent to bind soluble ligands. The subsequent outside-in signaling initiated by ligand binding is transmitted back to the cytoplasmic tails to trigger intracellular responses. Portions of this figure are adapted from reference 44. EGF indicates epidermal growth factor; MIDAS, metal ion–dependent adhesion site; and PSI, plexin-semaphorin-integrin.
αIIbβ3 is one of the 2 members of the β3 subfamily of integrins. The 2 members, αIIbβ3 and αVβ3,24,28,29 share the same β3 subunit and their α subunits exhibit 36% amino acid sequence identity. Integrin αIIbβ3 is found on the surface of platelets, megakaryocytes, basophils, mast cells, and some tumor cells.30 Integrin αVβ3 is expressed on many cell types, where it influences diverse physiological responses, such as cell adhesion, migration, and bone resorption, and pathological responses, such as angiogenesis, restenosis, tumor cell invasion, and atherosclerosis.29,31,32 These 2 integrins share several macromolecular ligands, including fibrinogen, fibronectin, thrombospondin, von Willebrand factor, and vitronectin.18,33,34 Common to these ligands is the presence of ≥1 arginine-glycine-aspartic acid sequence (RGD) sequences. RGD containing peptides bind to both β3 integrins, as well as several other integrins, and can inhibit the binding of the aforementioned ligands to αIIbβ3 and platelet aggregation.35,36 RGD peptides were used as a starting point in the design of several αIIbβ3 antagonists, including the FDA-approved drug, tirofiban.37 However, sequences other than RGD also bind to αIIbβ3. Particularly notable in this regard is the sequence at the C-terminus of one of the fibrinogen-constituent chains, the -chain, which is integrally involved in fibrinogen binding to αIIbβ3 and platelet aggregation.23 Glanzmann thrombasthenia can arise from mutations in either the αIIb or β3 genes, and mutations in either gene can lead to deficits in cell surface expression and function of αIIbβ3 and to episodic bleeding arising from a failure of platelets to aggregate.38,39 Mutations in β3 can also prevent expression of αVβ3. The symptoms of patients with mutations in the β3 subunit do not differ consistently from those of patients with αIIb mutations.40,41 This information is relevant to αIIbβ3 antagonism because some of the antagonists cross-react with αVβ3, whereas others show specificity exclusively for αIIbβ3. However, the absence of distinguishing symptoms in subjects lacking only αIIbβ3 compared with those lacking both β3 integrins does not imply a lack of biological function for αVβ3, and may just reflect the limited number of patients analyzed for other symptoms. For αIIbβ3 to bind its high–molecular weight ligands, it must undergo activation, a transition from a low- to high-affinity state (Figure 2B). On circulating platelets in blood, αIIbβ3 exists in its resting, low-affinity conformation. Stimulation of platelets with a variety of agonists, including ADP, thrombin, collagen, and epinephrine, triggers the transformation of αIIbβ3 to its higher affinity state.15,42 This transition is very rapid and depends on a series of intracellular signaling events from the agonist receptors that culminate in the binding of the cytoskeletal proteins, talin, and kindlin-3, to the cytoplasmic tail of the β3 subunit.15,27 Conformational changes initiated by interaction with these binding partners, are transmitted across the transmembrane domain to change the conformation of the ligand binding site in the extracellular domain. It is now broadly accepted that a central event in the activation process is a shift in the equilibrium from the bent conformation that predominates in the resting state to an extended conformation where ligands can more readily bind to the activated state (Figure 2B).43–45 Within the headpiece formed as a complex of the amino terminal domains of the α and β subunits, there are movements of helices and loops and a swing out of the hybrid domain in the vicinity of the ligand binding site, which provide greater access of ligands.30 Ligand binding to αIIbβ3 not only enables platelet–platelet interaction but also transmits an array of signals from the occupied and clustered receptor into the platelet (outside-in signaling), which are important for normal platelet responses, such as the stability and retraction of clots.46,47 Conflicting results have been reported as to whether αIIbβ3 receptor antagonists can also induce outside-in signaling and whether such signaling would be detrimental.48–50
The global structure of αIIbβ3 and the basis of the inside-out and outside-in signaling processes that lead to activation of αIIbβ3 and other integrins have been the subject of extensive investigations and reviews.14,15,43 Seminal insights were provided by the publication in 2001 of the crystal structure of much of the extracellular domain of αVβ3 by Xiong et al,51 and this was followed a year later by the structure of αVβ3 with a bound RGD peptide.52 This latter structure revealed how a ligand peptide nestles into a groove in the headpiece formed between the α and β subunits, with the Asp of the RGD peptide providing a coordination site to a divalent cation bound in the metal ion–dependent adhesion site of the receptor.52 Since then, several crystal structures of αIIbβ3 with or without various agonists and antagonists bound to the receptor have been reported.53,54 These crystal structures reveal that small-molecule antagonists bind to a pocket on top of the inte-grin head formed by the β3 A domain and loops from the αIIb β-propeller.53 The extent to which these antagonists interact with the αIIb β-propeller determines their relative specificity for αIIbβ3 versus αVβ3. With additional structural determinations of the transmembrane55,56 and cytoplasmic domains,57–60 a complete picture of the entire αIIbβ3 can be cobbled together. Although these structures became available some time after the design and clinical development of approved αIIbβ3 antagonists, we now have much better insights into their mechanisms of action and have information that can and has been used in the design of new αIIbβ3 antagonists with distinct modes of action.61 A next key advance in the understanding of the structure and function of αIIbβ3 is likely to come from approaches that allow for high-resolution visualization of the entire molecule as a single, intact entity in resting, active, and ligand-occupied conformers.
Great excitement was raised in 1996 when it was first suggested that a particular single-nucleotide polymorphism in αIIbβ3 was associated with acute myocardial infarction.62 The single-nucleotide polymorphism, referred to as platelet allo-antigen 1, 2 (PLA1/PLA2), leads to a single amino acid substitution at position 33, Leu in PLA1, or Pro in PLA2.63 This finding was replicated in several other studies. The PLA1/PLA2 polymorphism was reported to influence platelet activation, aggregation, and postoccupancy signaling by αIIbβ3.63,64 However, functional differences between the polymorphic forms of β3 were not supported in other studies.65 Furthermore, the first meta-analysis of several separate studies that combined data from 10 638 individuals concluded that the PLA2 was not an inherited risk factor for ACS.66 In 2 subsequent meta-analyses, Burr suggested67 the PLA2 variant was only weakly associated with ACS and restenosis whereas Le et al68 suggested that the PLA1/A2 polymorphism is not a major pathophysiological factor in patients who underwent coronary artery stenting. Indeed, 1 recent study even suggested that it was the PLA1 genotype that is disease associated.69 This polymorphism deserves mention because it was suggested that platelets of 1 genotype may be more sensitive to certain αIIbβ3 antagonists than the other.70 In vitro, blockade of aggregation by abciximab was reduced in platelets of the PLA2 genotype.71 Overall, the influence of the PLA1/A2 polymorphism on platelet aggregation seems to be modest as is its impact on the response to αIIbβ3 antagonists.
αIIbβ3 Antagonists
At one time >1 dozen αIIbβ3 antagonists were in development, either for intravenous and oral administration, and ultimately 3 received FDA approval for specific indications. These 3, abciximab (ReoPro), tirofiban (Aggrastat), and ep-tifibatide (Integrilin) are quite distinct in design from one another (Table), and all 3 are mechanistically distinct from other platelet inhibitors such as aspirin or P2Y12 inhibitors. Each of the 3 αIIbβ3 antagonists is discussed below. We do not intend to systematically summarize all the numerous clinical trials that supported the development of the 3 αIIbβ3 antagonists as there have been numerous comprehensive reviews of this topic.72–76 Rather, we touch on some of the key trials and highlights surrounding the development of the αIIbβ3 antagonists.
Table 1.
Table Integrin αIIbβ3 Antagonists
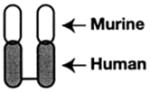
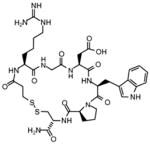
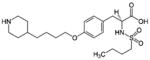
| αIIbβ3 Antagonist | Abciximab (REOPRO) | Eptifibatide (INTEGRILIN) | Tirofiban (Aggrastat) |
|---|---|---|---|
| Molecular design | Fab fragment of a chimeric human-murine monoclonal antibody to αIIbβ3. | Cyclic heptapeptide (6 amino acids+1 mercaptopropionyl (des-amino cysteinyl) | Small nonpeptide (N(butylsulfonyl)- |
| Monoclonal antibody 7E3 used as starting point for its development | On the basis of the sequence of the snake venom, barbourin | O-[4-(4-piperidinyl)butyl]-L-tyrosine monohydrochloride monohydrate) RGD peptide used as template | |
| Structure |
|
|
|
| Molecular weight | 47 615 Da | 832 Da | 495 Da |
| Initial FDA approval | 1994 | 1998 | 1998 |
| Licensed indication | cardiac ischemia, PCI | ACS | ACS |
| Specificity/selectivity | αIIbβ3, αvβ3, αMβ2 (Mac-1) | αIIbβ3 (αvβ3) | αIIbβ3 |
| Affinity for αIIbβ3 | KD=5 nmol/L | KD=120 nmol/L | KD=15 nmol/L |
| Platelet-bound half-life | 12–24 h | Seconds | Seconds |
| Plasma half-life | 2.5 h | Minutes | 2 h |
| Route of administration | IV (bolus+infusion) | IV (bolus+infusion) | IV (bolus+infusion) |
| Manufacturer and distributor | Eli Lilly (Centocor) | Merck and Co. (Schering Plough, Millennium Pharmaceuticals Inc COR Therapeutics) | Merck and Co |
| Clinical trials | 52 | 21 | 21 |
| 2000–2013 | |||
| 2012 Sales ($millions) | |||
| Worldwide | $147 | $213 | $5.6 |
| United States | $48 | $193 | $5.0 |
| Peak sales year | 1999 | 2007 | 2000 |
| Worldwide | $447 | $332 | $130 |
| United States | $322 | $312 | $100 |
ACS indicates acute coronary syndrome; FDA, Food and Drug Administration; PCI, percutaneous coronary interventions; and RGD, arginine-glycine-aspartic acid sequence.
Abciximab (ReoPro)
In the early 1980s, several monoclonal antibodies directed against αIIbβ3 were developed, and some inhibited binding of fibrinogen to platelets and platelet aggregation.21,77 One of these, monoclonal antibody 7E3, proceeded into clinical development.78 This monoclonal antibody was described as inducing a thrombasthenic-like state to platelets,21,79 which was regarded as a favorable property because bleeding is episodic in Glanzmann’s patients and is medically manageable. To limit Fc-mediated platelet clearance and reduce immunogenicity, the antibody was engineered to create a mouse/human chimeric antibody fragment, c7E3 Fab, which was dubbed abcix-imab.80 The epitope of abciximab in αIIbβ3 was mapped and shown to be dependent on a nonlinear amino acid sequence that resides near the cation binding metal ion–dependent adhesion-site motif in the β3 subunit.81 After displaying great potency in inhibiting platelet aggregation in ex vivo testing and showing remarkable efficacy in canine82 and subhuman primate models,83 the first clinical trial, EPIC, was launched in 1991 in patients at high risk of developing ischemic complications after PCI. Abciximab reduced the risk of the primary end points, death, myocardial infarction, repeat angioplasty, or bypass surgery, at 30 days by 35%: from 12% to 8% in the placebo group to 8% to 3% in patients treated with abciximab. However, bleeding was increased significantly.84 The EPILOG trial followed and sought to establish more effective administration regimen to maintain efficacy but reduce bleeding complications by weight adjusting the heparin anticoagulant dose. On the basis of these trials, abciximab, under the trade name ReoPro, won FDA approval in 1994 for use in the setting of PCI. Hence, abciximab became the first in class, and the clinical use of αIIbβ3 antagonists became a reality.
The GUSTO IV trial was launched with the anticipation that ReoPro would also be efficacious in high-risk ACS patients under medical management, but the results did not provide evidence of benefit.85 With abciximab, as well as with the other αIIbβ3 antagonists, optimal dosing was always challenging; the window between the therapeutically efficacious doses to achieve extensive blunting of platelet aggregation and higher doses that can lead to bleeding was narrow.48 Confounding the problem was the variability in this window among patients and the lack of assays to reliably monitor efficacy. It should be stressed that this problem was not unique to abciximab but confronted development of the entire class of αIIbβ3 antagonists. Recent studies reveal that these agents may have additional effects on platelet aggregates that have already formed, causing them to disengage and disperse, and such effects are more pronounced at doses of the αIIbβ3 antagonists higher than the conventional doses administered intravenously.86 Hence, the on-label use of ReoPro remains restricted to the setting of PCI. Nevertheless, millions of patients worldwide have been treated with ReoPro; and, based on the benefits seen in the PCI patients in clinical trials, many lives have been saved by this αIIbβ3 antagonist.
Of particular note is that patients treated with ReoPro showed a long-term and quite remarkable mortality benefit, even >5 years after initial treatment. Follow-ups at 7 years (EPIC), 4.5 years (EPILOG), or 3 years (EPISTENT)87 have shown that abciximab treatment reduced all-cause mortality by ≈20% during long-term follow-up after PCI. The ADMIRAL trial also reported favorable outcomes for abcix-imab treatment in stented patients from 30 days up to 3 years of follow-up.88 Not all trials detected the long-term benefit of abciximab; the ISAR-2 trial did not see a sustained clinical benefit at 5 years after the use of abciximab during coronary artery stenting in patients with acute myocardial infarction.89 One suggested explanation for the often-replicated long-term benefit of abciximab is its cross-reactivity with integ-rins other than αIIbβ3; abciximab also reacts with αVβ3 and αMβ2 (Mac-1, CD11b/CD18), a member of the β2 integrin subfamily of leukocyte integrins.90 This cross-reactivity contrasts with other αIIbβ3 antagonists, and may responsible for unique anti-inflammatory properties attributed to abciximab.91 Early data suggested that abciximab may reduce restenosis in patients undergoing PCI,92 especially in patients with diabetes mellitus, which was attributed to anti-inflammatory effects of the drug.93 Subsequent studies failed to substantiate the beneficial effect of abciximab in restenosis.94 Although the cross-reactivity of abciximab with other integrins distinguishes it from the other 2 FDA-approved αIIbβ3 antagonists, there is no clear evidence that such cross-reactivity was either benefi-cial or detrimental. Another unique feature of abciximab is its extended association with platelets. Whereas the free drug is rapidly eliminated from the circulation by the reticulo-endo-thelial system, abciximab circulates, bound to platelets for an extended time, and may dissociate and then reassociate with new target platelets for as long as 21 days after cessation of the drug.95
Eptifibatide (Integrilin)
Eptifibatide is a cyclic heptapeptide of <1 kDa. The starting point for its design was barbourin, a 73–amino acid disinteg-rin isolated from snake venom that was shown to have potent antiaggregating activity. Although several such snake venom proteins were isolated and characterized, most contained an RGD sequence, but the antiaggregating activity of barbourin was reliant on a lysine-glycine-aspartic acid sequence.96 This sequence provided a template for the development of synthetic peptide antagonists that contain a lysine-glycine-aspartic acid sequence.97 Potency was greatly enhanced by cyclizing the peptide via a disulfide bond (Table). The ultimate product, ep-tifibatide, is a highly potent inhibitor of fibrinogen binding to platelets and was reported to be specific for αIIbβ397 although there are some data to the contrary.98 For a peptide, eptifibatide has a relatively long half-life in plasma; its biological half-life is ≈2.5 hours.92,95,99 Eptifibatide is eliminated by the kidneys. Trials testing the safety and efficacy of eptifibatide were conducted in the mid-1990s in different clinical settings.100–103 Together, these trials showed that eptifibatide rapidly inhibited platelet aggregation attaining a maximum effect within 15 minutes after its bolus injection. A bolus injection followed by infusion inhibits platelet aggregation by 75% to 80%. Such potent inhibition is maintained during the infusion period, and platelet function recovers 2 to 4 hours after cessation of drug. Over multiple trials, eptifibatide showed only a slight tendency to prolong bleeding times with normalization within 30 minutes after infusion. Eptifibatide initial clinical trials were directed toward patients with ACS. Efficacy in the initial trial IMPACT II, of 4010 patients, was disappointing104 but in subsequent trials, PURSUIT (10 948 patients) and ESPRIT (2064 patients), using higher doses of eptifibatide, mortality was reduced by 25% to 35% in the eptifibatide-treated group compared with the placebo groups and only a modest increase in bleeding was observed.105–109 FDA approval was gained for eptifibatide in 1998 for treatment of ACS patients, including patients who were being managed medically and those undergoing PCI. Subsequent trials, such as PRIDE continued to optimize the dose of eptifibatide.110 Eptifibatide still remains the most widely used of the 3 FDA-approved αIIbβ3 antagonists (Table).
Tirofiban (Aggrastat)
Tirofiban is a low–molecular weight (<1 kDa) αIIbβ3 inhibitor. The starting point for its design was an RGD peptide. Peptide bonds were ultimately eliminated from the structure to create a potent and specific nonpeptide αIIbβ3 antagonist (Table). The small-molecule antithrombotic, originally designated MK-0383, was very effective in inhibiting αIIbβ3 function in animal models,111 and led to the development of tirofiban for clinical usage.112 Tirofiban has a plasma half-life (1.5–2 hours) and a shorter biological half-life (seconds), which reflect its reversible and relatively low-affinity binding to αIIbβ3. After stopping administration of tirofiban, platelet aggregation recovers to 50% of the baseline value within 4 hours. Tirofiban is removed by both renal and biliary excretion.113 Patients with renal insufficiency require dose adjustment of tirofiban. Tirofiban does not interact with αVβ3 or αMβ2.92,95,98
The clinical trials that led to FDA approval of tirofiban in 1998 for treatment of patients with ACS (unstable angina/ non–Q-wave myocardial infarction) were RESTORE (2139 patients), PRISM (3232 patients), PRISM-PLUS (1915 patients), which differed in their drug regimen. The 30-day reduction in mortality in these trials was 16% relative reduction (P=0.160) in RESTORE and 36% (2.3 versus 3.6%; P=0.02) in PRISM and 27% (8.7 versus 11.9%; P=0.027) in PRISM-PLUS. In the PRISM trial there was no difference in bleeding times between the tirofiban and placebo groups, and bleeding increased only modestly in the PRISM-PLUS (1.4 versus 0.8%; P=0.23) tirofiban+heparin versus heparin-alone groups and in RESTORE (5.3% versus 3.7%; P=0.096) tirofiban versus placebo groups.114,115 Initial comparative studies showed abciximab and eptifibatide were more effective than the standard dose of tirofiban used in inhibiting platelet aggregation,116 and abciximab to be more effective than the standard dose of tirofiban in preventing ischemic events in the TARGET trial.117 However, in a subsequent PCI trial, ADVANCE, tirofiban was given at a higher dose and was not inferior to abciximab118,119 although the FATA trial failed to show equivalence of higher-dose tirofiban to abciximab as adjunctive therapy during primary PCI for ST-segment elevation myocardial infarction.120
Current Usage of αIIbβ3 Antagonists
For more than a decade after FDA approval of the first αIIbβ3 antagonist in 1994, the class of drugs was used broadly in the treatment of ACS patients. According to data in the clinicaltrials.gov 84, large-scale clinical trials involving >90 000 patients and controls were conducted to test the efficacy and safety of the αIIbβ3 antagonists in various settings. In comparison to the widespread use of αIIbβ3 antagonists that characterized their use in the decade after their initial approval, their use has waned in recent years. By current American College of Cardiology/ American Heart Association guidelines, αIIbβ3 antagonists are given a class IIa recommendation; that is, conflicting evidence and a divergent opinion exist as to their usefulness/efficacy, but the weight of evidence/opinion is favorable.121 An American College of Cardiology/American Heart Association class I recommendation for αIIbβ3 antagonists has been given for their use in unstable angina/non Stelevation myocardial infarction patients undergoing PCI, who cannot tolerate clopi-dogrel (the widely used and inexpensive oral P2Y12 antagonist).121 According to data in the EvaluatePharma 2013 report (www.evaluatepharma.com), the sales peak of abciximab was in 1999, integrilin in 2007, and tirofiban in 2000 (Table). Compared with abciximab and eptifibatide, the sales of tirofiban dropped dramatically in the United States between 2003 and 2007, declining to ≈$2 million in the United States in 2007 but remaining considerably stronger, ≈$87 million, in Europe in the same year. Although the overall sales for the class, ≈$365 million worldwide in 2012 (Table) are still impressive, they pale when compared with clopidogrel (Plavix), the P2Y12 antagonist, which had sales of >$9 billion in 2011 and estimated sales of >$5 billion in 2012. This growing preference in part reflects the greater efficacy of clopidogrel compared with αIIbβ3 inhibitors in the PCI-CURE, CREDO, PCI-CLARITY, and ISAR-REACT trials,122,123 and the cost of the drugs per se; abciximab treatment cost per day is ≈$1000, 3 to 4 times higher than that of eptifibatide and tirofiban, and 200 times higher than that of clopidogrel (≈$5 per day).
At this juncture, use of αIIbβ3 antagonists has become limited primarily to the setting of PCI, particularly in high-risk patients or in patients not adequately pretreated with P2Y12 antagonists. Clopidogrel and the newer P2Y12 inhibitors and anticoagulants all compete in a similar space.124 However, PCI is not a narrow setting; it is the most commonly performed revascularization procedure worldwide for the treatment of coronary artery disease.125 Although the benefits of αIIbβ3 antagonists have not always been consistent across all clinical trials, they remain a potent therapeutic adjunct in high-risk and unstable patients undergoing PCI. Bosch et al126 analyzed the results of 36 randomized control trials of these agents in 30 696 patients undergoing PCI. The rate of death or myo-cardial infarction at 30 days and 6 months was 5.10% versus 7.52% and 7.51% versus 10.45% in the treatment versus the control groups, respectively. In one of the most recent assessments of the effects of αIIbβ3 blockers, Winchester et al75 analyzed the results of 22 randomized studies with 10 123 patients undergoing PCI with stenting, who were treated routinely with ADP receptor antagonists (thienopyridines). This analysis showed that at 30 days, patients receiving αIIbβ3 antagonists had a significant reduction in myocardial infarction, 5.1% versus 8.3% in the control group without a significant increase in major bleeds, 1.2% versus 0.9%. Minor bleeding was increased 3.0% versus 1.7%. However, overall mortality was not reduced.75 Ongoing trials using αIIbβ3 inhibitors are focused primarily on reduction of side effects (eg, reduction of bleeding, dosage optimization for patients with renal dysfunction, alternative ways of infusion, treatment of severe sepsis in pneumonia patients).
Side Effects of αIIbβ3 Inhibitors
During the early days of testing in animal models, it was suggested that particular αIIbβ3 antagonists could block platelet aggregation without prolonging bleeding times. In retrospect, such claims seem unrealistic; extensive inhibition of platelet aggregation will be associated with increased risk of bleeding. Indeed, bleeding is the major complication associated with αIIbβ3 antagonism, and is more frequent with αIIbβ3 antagonists than with other platelet inhibitors. In retrospective analyses, in the most severe forms, intracranial bleeds occurred in 2% of patients treated with αIIbβ3 antagonists and gastrointestinal bleeds in 15% of patients.72 Groin hematoma at sites of catheter insertion accounted for 60% to 80% of the major bleeding events and retroperitoneal bleeds for 5% to 10% of major bleeding events. In the initial clinical trials of αIIbβ3 antagonists, bleeding severity was evaluated based on the need for blood transfusions but was later replaced by physician assessment. The greater experience in dealing with αIIbβ3 antagonists may have tempered such assessments of bleeding.
After bleeding, thrombocytopenia and severe reactions to readministration are the most serious side effects of αIIbβ3 antagonists. Thrombocytopenia may occur after use of all 3 αIIbβ3 antagonists, abciximab, tirofiban, and eptifibatide. On the basis of an analysis of clinical trials (EPIC, EPILOG, CAPTURE, RESTORE, IMPACT II) by Tcheng,127 mild thrombocytopenia (<100 000 platelets/mm3) occurred in 2% to 5% of patients and moderate thrombocytopenia (<50 000 platelets/mm3) in 2% of patients receiving abciximab and in <1% of patients treated with eptifibatide and tirofiban.127,128 Severe thrombocytopenia (<20 000 platelets/mm3) occurred rarely in patients treated with eptifibatide or tirofiban and in 0.7% of patients receiving abciximab. The thrombocytopenia is believed to be antibody mediated.129 Low levels of antibodies appear among 6% to 7% of patients receiving abciximab. The greatest concentration of such antibodies occurred between 1 week to 1 month after cessation of the αIIbβ3 antagonist and then gradually declined.127 Readministration of abciximab did not cause an increased risk of anaphylaxis, but 2.4% of patients did develop a severe thrombocytopenia.128,130 No data are available on the safety of tirofiban readministration, but high antibody titers have been found in some patients who developed thrombocytopenia after tirofiban treatment.131 It is believed that tirofiban binding induces a conformational change in αIIbβ3, and antibodies arise against the newly exposed epitopes in αIIbβ3.132,133 Antibodies may also mediate throm-bocytopenia associated with eptifibatide treatment. The rate of naturally occurring eptifibatide-dependent antibodies seems to be lower than seen with abciximab.131 Readministration of αIIbβ3 antagonists is not recommended after an episode of thrombocytopenia.128
Failure of Oral Inhibitors
Orally active αIIbβ3 antagonists were developed with the hope that they would provide long-term suppression of platelet aggregation and thereby secondary prevention of CVD. Four orally active αIIbβ3 antagonists reached the stage of testing in 5 major phase III trials,134 and several other orally active αIIbβ3 antagonists with encouraging preclinical profiles were in pharmaceutical pipelines. However, to the surprise and disappointment of many, none of the 5 large trials showed a beneficial effect of the oral αIIbβ3 antagonists; and, in fact, 4 of the trials were terminated prematurely because of adverse effects. A combined analysis confirmed this lack of efficacy and revealed a disturbing and highly significant (35% relative, or 0.7% absolute) increase in the risk of death in the combined 45 523 patients within these trials.135 These disappointing results halted further investigations into the use of these oral agents, and oral αIIbβ3 inhibition is regarded as a failed strategy.
The basis for lack of efficacy and increased mortality of the oral αIIbβ3 antagonists remains a topic of speculation with no definitive answers. It has been suggested that some of the drugs fell out of the therapeutic range between administrations, leaving patients at jeopardy between doses.134 Another popular hypothesis was that dissociation of drug from αIIbβ3 left the receptor in an activated and therefore prothrombotic state. This proposition was predicated on the long-standing observation that removal of bound RGD ligand from αIIbβ3 led to a brief activation of the receptor.134–136 Although some data supported this hypothesis,137 others did not.138 Some have even challenged the founding assertion that long-term suppression of αIIbβ3 would be beneficial. Although side effects (eg, bleeding and thrombocytopenia) associated with αIIbβ3 antagonists were manageable in the acute setting of PCI, with chronic administration, these effects may have become a life-threatening problem. Thrombocytopenia can increase the risk for bleeding and, in rare instances, may enhance blood clotting. In some studies, oral αIIbβ3 inhibitors facilitated, rather than inhibited thrombus formation; and, paradoxically, such effects were potentiated by concomitant administration of aspirin.139 Also, with chronic administration, nuisance bleeding may have impacted compliance with the drug regimen, and subjects may become vulnerable if they fell out of the therapeutic window of efficacy. Despite these conjectures, the explanation of the failure of oral αIIbβ3 remains equivocal.
Future Strategies Targeting αIIbβ3
As established by extensive clinical trials and usage, the clinical scenarios in which the current αIIbβ3 antagonists provide efficacy is more limited than originally hoped. Nevertheless, the essential role of αIIbβ3 in platelet aggregation and thrombus formation remains indisputable. Given the premise that targeting αIIbβ3 remains a fundamentally sound strategy, some investigators have sought to identify new αIIbβ3 antagonists, ones that might not induce conformational changes on association or dissociation from αIIbβ3 and might therefore contribute less to the bleeding and thrombocytopenia that occurs in some patients. Two possible approaches have been suggested to achieve this end: finding inhibitors that, like current antagonists, bind to the extracellular domain of the integrin but do so without promoting receptor activation140 or finding inhibitors that prevent receptor activation by binding to the intracellular domain of αIIbβ3.141 Both strategies are in early stages of development. Blue et al142 performed a high-throughput screen of >30 000 compounds and identified a novel low–molecular weight compound, RUC-1. RUC-1 selectively inhibited ligand binding to αIIbβ3 compared with αVβ3.143,144 A second congener RUC-2, was ≈100-fold more potent than RUC-161 and did not seem to induce major conformational changes in the protein β3 subunit or prime the receptor to bind ligand.140 RUC-2 is currently undergoing additional preclinical studies that will assess its suitability for use in patients with STEMI in the early prehospital setting.2
The precedent for intracellular approaches to inhibit αIIbβ3 came from studies in which membrane permeable derivatives of peptides corresponding to portions of the cytoplasmic tails of αIIb and β3 were shown to inhibit activation of the receptor.145 Koloka et al141 evaluated the role of the acidic extreme C-terminal region of the αIIb cytoplasmic tail, residues 1000 to 1008, and showed that a palmitoylated form of the peptide inhibited platelet activation. Supporting the notion that targeting αIIbβ3 activation from the inside would be advantageous, Petrich et al146 showed that a mutation in the cytoplasmic tail of the β3 subunit in mice, which prevented talin binding to the receptor and thereby platelet activation, inhibited thrombus formation with limited bleeding. Although such αIIbβ3 antagonists might have distinct advantages over current αIIbβ3 antagonists, the road to clinical development would be formable with potentially insurmountable obstacles, including the staggering expense of clinical trials with relatively small windows for improved efficacy over currently available antiplatelet agents.
Conclusions
The development and deployment of αIIbβ3 antagonists represent a success story: the estimates of >8 million patients who were treated with αIIbβ3 between 1999 and 2011 clearly point to how many lives have been saved with these drugs.2 On the basis of the report from the CathPCI registry of the National Cardiovascular Data Registry,147 integrin αIIbβ3 inhibitors were used overall in 28.7% of PCIs and slightly more frequently, 34.0%, among patients with an ACS. This report includes 1 110 150 patients undergoing only diagnostic cardiac catheterization and 941 248 undergoing PCI from January 1, 2010 until June 30, 2011. Thus, the development and deployment of αIIbβ3 antagonists does represent a success story. Nevertheless, it is also clear that the use of αIIbβ3 antagonists has declined in recent years as alternative antiplatelet and anticoagulant strategies have emerged, and αIIbβ3 antagonists have become confined to quite narrow settings. Nonetheless, newer approaches to antagonize αIIbβ3 may lead to superior drugs in this class. With the increase in radial-access PCI versus femoral-access PCI,148 or by introducing new atraumatic delivery methodologies bleeding has become less problematic and might allow return to the use of more potent antithrombotic strategies, such as αIIbβ3 antagonists. A recent clinical trial has suggested some benefit to direct intracoronary infusion of abciximab compared with systemic infusion in patients with a large anterior STEMI.149 Thus, novel routes of administration may open up particular subsets of patients to treatment with αIIbβ3 antagonists. Despite the dramatic reductions in deaths from coronary artery disease over the past few years, a coronary event still occurs once every 25 seconds, and there is a death from such events every 39 seconds in the United States.1 Antagonism of αIIbβ3 function on platelets, either directly or indirectly, remains a theoretically sound and practically proven approach to treat CVD in specific settings. Thus, the book on αIIbβ3 antagonism should be viewed as a success story, however, a book with chapters still to be written.
Translational Success Stories.
highlight how basic discoveries have led to clinical advances (such as the use of new drugs or diagnostic modalities in patients). This initiative reflects the renewed emphasis of our journal on translational research. It is hoped that these articles will stimulate efforts to translate basic insights into clinical practice.
Acknowledgments
We gratefully acknowledge Nadine Klimczak, who assisted with the preparation of this article. We acknowledge EvaluatePharma for providing sales data for αIIbβ3 inhibitors.
Sources of Funding: This work was supported in part by National Institutes of Health grants HL 073311 and HL 096062, to E.F. Plow, PhD, Department of Molecular Cardiology, Lerner Research Institute, Cleveland Clinic.
Nonstandard Abbreviations and Acronyms
- ACS
acute coronary syndrome
- CVD
cardiovascular disease
- FDA
Food and Drug Administration
- PCI
percutaneous coronary interventions
- PLA1/PLA2
platelet alloantigen 1, 2
- RGD
arginine-glycine-aspartic acid sequence
Footnotes
Disclosures
None.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:188–197. doi: 10.1161/CIR.0b013e3182456d46. [DOI] [PubMed] [Google Scholar]
- 2.Coller BS. Translating from the rivers of Babylon to the coronary bloodstream. J Clin Invest. 2012;122:4293–4299. doi: 10.1172/JCI66867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanderslice P, Woodside DG. Integrin antagonists as therapeutics for inflammatory diseases. Expert Opin Investig Drugs. 2006;15:1235–1255. doi: 10.1517/13543784.15.10.1235. [DOI] [PubMed] [Google Scholar]
- 4.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 5.Millard M, Odde S, Neamati N. Integrin targeted therapeutics. Theranostics. 2011;1:154–188. doi: 10.7150/thno/v01p0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman SL, Picard M. Integrins as therapeutic targets. Trends Pharmacol Sci. 2012;33:405–412. doi: 10.1016/j.tips.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Bizzozero G. Uber einen neuen formbestandteil des blutes und dessen rolle bei der thrombose und der blutgerinnung. Virchow Arch Path Anat Physiol. 1882;90:267. [Google Scholar]
- 8.Glanzmann E. Hereditare haemorrhagische Thrombasthenie: ein beitrag zur patholigie der blutplattchen. Jahrb Kinderheilk. 1918;88:113–141. [Google Scholar]
- 9.Nurden AT, Caen JP. Specific roles for platelet surface glycoproteins in platelet function. Nature. 1975;255:720–722. doi: 10.1038/255720a0. [DOI] [PubMed] [Google Scholar]
- 10.Phillips DR, Agin PP. Platelet plasma membrane glycoproteins. Evidence for the presence of nonequivalent disulfide bonds using non-reduced-reduced two-dimensional gel electrophoresis. J Biol Chem. 1977;252:2121–2126. [PubMed] [Google Scholar]
- 11.Phillips DR, Agin PP. Platelet membrane defects in Glanzmann’s throm-basthenia. Evidence for decreased amounts of two major glycoproteins. J Clin Invest. 1977;60:535–545. doi: 10.1172/JCI108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunicki TJ, Pidard D, Rosa JP, Nurden AT. The formation of Ca++-dependent complexes of platelet membrane glycoproteins IIb and IIIa in solution as determined by crossed immunoelectrophoresis. Blood. 1981;58:268–278. [PubMed] [Google Scholar]
- 13.Jennings LK, Phillips DR. Purification of glycoproteins IIb and III from human platelet plasma membranes and characterization of a calcium-dependent glycoprotein IIb-III complex. J Biol Chem. 1982;257:10458–10466. [PubMed] [Google Scholar]
- 14.Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115:3363–3369. doi: 10.1172/JCI26989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma YQ, Qin J, Plow EF. Platelet integrin alpha(IIb)beta(3): activation mechanisms. J Thromb Haemost. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- 16.Marguerie GA, Plow EF, Edgington TS. Human platelets possess an inducible and saturable receptor specific for fibrinogen. J Biol Chem. 1979;254:5357–5363. [PubMed] [Google Scholar]
- 17.Timmons S, Kloczewiak M, Hawiger J. ADP-dependent common receptor mechanism for binding of von Willebrand factor and fibrinogen to human platelets. Proc Natl Acad Sci USA. 1984;81:4935–4939. doi: 10.1073/pnas.81.15.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plow EF, McEver RP, Coller BS, Woods VL, Jr, Marguerie GA, Ginsberg MH. Related binding mechanisms for fibrinogen, fibronectin, von Willebrand factor, and thrombospondin on thrombin-stimulated human platelets. Blood. 1985;66:724–727. [PubMed] [Google Scholar]
- 19.Reheman A, Gross P, Yang H, Chen P, Allen D, Leytin V, Freedman J, Ni H. Vitronectin stabilizes thrombi and vessel occlusion but plays a dual role in platelet aggregation. J Thromb Haemost. 2005;3:875–883. doi: 10.1111/j.1538-7836.2005.01217.x. [DOI] [PubMed] [Google Scholar]
- 20.André P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8:247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 21.Coller BS, Peerschke EI, Scudder LE, Sullivan CA. A murine monoclonal antibody that completely blocks the binding of fibrinogen to platelets produces a thrombasthenic-like state in normal platelets and binds to glycoproteins IIb and/or IIIa. J Clin Invest. 1983;72:325–338. doi: 10.1172/JCI110973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH. The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc Natl Acad Sci USA. 1985;82:8057–8061. doi: 10.1073/pnas.82.23.8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kloczewiak M, Timmons S, Lukas TJ, Hawiger J. Platelet receptor recognition site on human fibrinogen. Synthesis and structure-function relationship of peptides corresponding to the carboxy-terminal segment of the gamma chain. Biochemistry. 1984;23:1767–1774. doi: 10.1021/bi00303a028. [DOI] [PubMed] [Google Scholar]
- 24.Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 25.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 26.Springer TA, Wang JH. The three-dimensional structure of integrins and their ligands, and conformational regulation of cell adhesion. Adv Protein Chem. 2004;68:29–63. doi: 10.1016/S0065-3233(04)68002-8. [DOI] [PubMed] [Google Scholar]
- 27.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varner JA, Cheresh DA. Integrins and cancer. Curr Opin Cell Biol. 1996;8:724–730. doi: 10.1016/s0955-0674(96)80115-3. [DOI] [PubMed] [Google Scholar]
- 29.Byzova TV, Rabbani R, D’Souza S, Plow EF. Role of integrin aVb3 in vascular biology. Thromb Haemost. 1998;80:726–734. [PubMed] [Google Scholar]
- 30.Bledzka K, Pesho MM, Ma YQ, Plow EF. Integrin alpha IIb beta 3. In: Michelson A, editor. Platelets. 3. Elsevier Science; San Diego, CA: 2012. pp. 233–248. [Google Scholar]
- 31.Felding-Habermann B, Cheresh DA. Vitronectin and its receptors. Curr Opin Cell Biol. 1993;5:864–868. doi: 10.1016/0955-0674(93)90036-p. [DOI] [PubMed] [Google Scholar]
- 32.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin avb3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 33.Thiagarajan P, Kelly KL. Exposure of binding sites for vitronectin on platelets following stimulation. J Biol Chem. 1988;263:3035–3038. [PubMed] [Google Scholar]
- 34.Lawler J, Weinstein R, Hynes RO. Cell attachment to thrombospondin: the role of ARG-GLY-ASP, calcium, and integrin receptors. J Cell Biol. 1988;107:2351–2361. doi: 10.1083/jcb.107.6.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruoslahti E. Integrins. J Clin Invest. 1991;87:1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruoslahti E. Fibronectin and its receptors. Annu Rev Biochem. 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111. [DOI] [PubMed] [Google Scholar]
- 37.Egbertson MS, Chang CT, Duggan ME, Gould RJ, Halczenko W, Hartman GD, Laswell WL, Lynch JJ, Jr, Lynch RJ, Manno PD. Non-peptide fibrinogen receptor antagonists. 2. Optimization of a tyrosine template as a mimic for Arg-Gly-Asp. J Med Chem. 1994;37:2537–2551. doi: 10.1021/jm00042a007. [DOI] [PubMed] [Google Scholar]
- 38.Nurden AT, Pillois X, Nurden P. Understanding the genetic basis of Glanzmann thrombasthenia: implications for treatment. Expert Rev Hematol. 2012;5:487–503. doi: 10.1586/ehm.12.46. [DOI] [PubMed] [Google Scholar]
- 39.Nurden AT, Pillois X, Nurden P. Understanding the genetic basis of Glanzmann thrombasthenia: implications for treatment. Expert Rev Hematol. 2012;5:487–503. doi: 10.1586/ehm.12.46. [DOI] [PubMed] [Google Scholar]
- 40.Shpilberg O, Rabi I, Schiller K, Walden R, Harats D, Tyrrell KS, Coller B, Seligsohn U. Patients with Glanzmann thrombasthenia lacking platelet glycoprotein alpha(IIb)beta(3) (GPIIb/IIIa) and alpha(v) beta(3) receptors are not protected from atherosclerosis. Circulation. 2002;105:1044–1048. doi: 10.1161/hc0902.104676. [DOI] [PubMed] [Google Scholar]
- 41.Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. doi: 10.1182/blood-2011-07-365635. [DOI] [PubMed] [Google Scholar]
- 42.Marguerie GA, Plow EF. The fibrinogen-dependent pathway of platelet aggregation. Ann N Y Acad Sci. 1983;408:556–566. doi: 10.1111/j.1749-6632.1983.tb23272.x. [DOI] [PubMed] [Google Scholar]
- 43.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 44.Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2004;2:e169. doi: 10.1371/journal.pbio.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinn MJ, Plow EF, Topol EJ. Platelet glycoprotein IIb/IIIa inhibitors: recognition of a two-edged sword? Circulation. 2002;106:379–385. doi: 10.1161/01.cir.0000019581.22812.b2. [DOI] [PubMed] [Google Scholar]
- 49.Cox D, Smith R, Quinn M, Theroux P, Crean P, Fitzgerald DJ. Evidence of platelet activation during treatment with a GPIIb/IIIa antagonist in patients presenting with acute coronary syndromes. J Am Coll Cardiol. 2000;36:1514–1519. doi: 10.1016/s0735-1097(00)00919-0. [DOI] [PubMed] [Google Scholar]
- 50.Peter K, Schwarz M, Ylänne J, Kohler B, Moser M, Nordt T, Salbach P, Kübler W, Bode C. Induction of fibrinogen binding and platelet aggregation as a potential intrinsic property of various glycoprotein IIb/IIIa (alphaIIbbeta3) inhibitors. Blood. 1998;92:3240–3249. [PubMed] [Google Scholar]
- 51.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extra-cellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of inte-grin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 53.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Ma YQ, Page RC, Misra S, Plow EF, Qin J. Structure of an integrin alphaIIb beta3 transmembrane-cytoplasmic heterocomplex provides insight into integrin activation. Proc Natl Acad Sci USA. 2009;106:17729–17734. doi: 10.1073/pnas.0909589106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 2009;28:1351–1361. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vinogradova O, Haas T, Plow EF, Qin J. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci USA. 2000;97:1450–1455. doi: 10.1073/pnas.040548197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 59.Ulmer TS, Yaspan B, Ginsberg MH, Campbell ID. NMR analysis of structure and dynamics of the cytosolic tails of integrin alpha IIb beta 3 in aqueous solution. Biochemistry. 2001;40:7498–7508. doi: 10.1021/bi010338l. [DOI] [PubMed] [Google Scholar]
- 60.Metcalf DG, Moore DT, Wu Y, Kielec JM, Molnar K, Valentine KG, Wand AJ, Bennett JS, DeGrado WF. NMR analysis of the alphaIIb beta3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc Natl Acad Sci USA. 2010;107:22481–22486. doi: 10.1073/pnas.1015545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu J, Choi WS, McCoy JG, Negri A, Zhu J, Naini S, Li J, Shen M, Huang W, Bougie D, Rasmussen M, Aster R, Thomas CJ, Filizola M, Springer TA, Coller BS. Structure-guided design of a high-affinity platelet integrin alphaIIbbeta3 receptor antagonist that disrupts Mg(2)(+) binding to the MIDAS. Sci Transl Med. 2012;4:125ra32. doi: 10.1126/scitranslmed.3003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss EJ, Bray PF, Tayback M, Schulman SP, Kickler TS, Becker LC, Weiss JL, Gerstenblith G, Goldschmidt-Clermont PJ. A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med. 1996;334:1090–1094. doi: 10.1056/NEJM199604253341703. [DOI] [PubMed] [Google Scholar]
- 63.Newman PJ, Derbes RS, Aster RH. The human platelet alloantigens, PlA1 and PlA2, are associated with a leucine33/proline33 amino acid polymorphism in membrane glycoprotein IIIa, and are distinguishable by DNA typing. J Clin Invest. 1989;83:1778–1781. doi: 10.1172/JCI114082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vijayan KV, Goldschmidt-Clermont PJ, Roos C, Bray PF. The Pl(A2) polymorphism of integrin beta(3) enhances outside-in signaling and adhesive functions. J Clin Invest. 2000;105:793–802. doi: 10.1172/JCI6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett JS, Catella-Lawson F, Rut AR, Vilaire G, Qi W, Kapoor SC, Murphy S, FitzGerald GA. Effect of the Pl(A2) alloantigen on the function of beta(3)-integrins in platelets. Blood. 2001;97:3093–3099. doi: 10.1182/blood.v97.10.3093. [DOI] [PubMed] [Google Scholar]
- 66.Zhu MM, Weedon J, Clark LT. Meta-analysis of the association of platelet glycoprotein IIIa PlA1/A2 polymorphism with myocardial infarction. Am J Cardiol. 2000;86:1000–5. A8. doi: 10.1016/s0002-9149(00)01136-x. [DOI] [PubMed] [Google Scholar]
- 67.Burr D, Doss H, Cooke GE, Goldschmidt-Clermont PJ. A meta-analysis of studies on the association of the platelet PlA polymorphism of glycoprotein IIIa and risk of coronary heart disease. Stat Med. 2003;22:1741–1760. doi: 10.1002/sim.1375. [DOI] [PubMed] [Google Scholar]
- 68.Le Hello C, Morello R, Lequerrec A, Duarte C, Riddell J, Hamon M. Effect of PlA1/A2 glycoprotein IIIa gene polymorphism on the long-term outcome after successful coronary stenting. Thromb J. 2007;5:19. doi: 10.1186/1477-9560-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Addad F, Elalamy I, Chakroun T, Abderrazek F, Dridi Z, Hamdi S, Hassine M, Ben-Farhat M, Gerotziafas G, Hatmi M, Gamra H. Platelet glycoprotein IIIa (platelet antigen 1/platelet antigen 2) polymorphism and 1-year outcome in patients with stable coronary artery disease. Blood Coagul Fibrinolysis. 2010;21:674–678. doi: 10.1097/MBC.0b013e32833e47c1. [DOI] [PubMed] [Google Scholar]
- 70.Michelson AD, Furman MI, Goldschmidt-Clermont P, Mascelli MA, Hendrix C, Coleman L, Hamlington J, Barnard MR, Kickler T, Christie DJ, Kundu S, Bray PF. Platelet GP IIIa Pl(A) polymorphisms display different sensitivities to agonists. Circulation. 2000;101:1013–1018. doi: 10.1161/01.cir.101.9.1013. [DOI] [PubMed] [Google Scholar]
- 71.Wheeler GL, Braden GA, Bray PF, Marciniak SJ, Mascelli MA, Sane DC. Reduced inhibition by abciximab in platelets with the PlA2 polymorphism. Am Heart J. 2002;143:76–82. doi: 10.1067/mhj.2002.119763. [DOI] [PubMed] [Google Scholar]
- 72.Hanna EB, Rao SV, Manoukian SV, Saucedo JF. The evolving role of glycoprotein IIb/IIIa inhibitors in the setting of percutaneous coronary intervention strategies to minimize bleeding risk and optimize outcomes. JACC Cardiovasc Interv. 2010;3:1209–1219. doi: 10.1016/j.jcin.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 73.De Luca G, Navarese EP, Cassetti E, Verdoia M, Suryapranata H. Meta-analysis of randomized trials of glycoprotein IIb/IIIa inhibitors in high-risk acute coronary syndromes patients undergoing invasive strategy. Am J Cardiol. 2011;107:198–203. doi: 10.1016/j.amjcard.2010.08.063. [DOI] [PubMed] [Google Scholar]
- 74.Tricoci P, Newby LK, Hasselblad V, et al. Upstream use of small-molecule glycoprotein iib/iiia inhibitors in patients with non-ST-segment elevation acute coronary syndromes: a systematic overview of randomized clinical trials. Circ Cardiovasc Qual Outcomes. 2011;4:448–458. doi: 10.1161/CIRCOUTCOMES.110.960294. [DOI] [PubMed] [Google Scholar]
- 75.Winchester DE, Wen X, Brearley WD, Park KE, Anderson RD, Bavry AA. Efficacy and safety of glycoprotein IIb/IIIa inhibitors during elective coronary revascularization: a meta-analysis of randomized trials performed in the era of stents and thienopyridines. J Am Coll Cardiol. 2011;57:1190–1199. doi: 10.1016/j.jacc.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 76.Kubica A, Kozinski M, Navarese EP, Grzesk G, Goch A, Kubica J. Intracoronary versus intravenous abciximab administration in STEMI patients: overview of current status and open questions. Curr Med Res Opin. 2011;27:2133–2144. doi: 10.1185/03007995.2011.621417. [DOI] [PubMed] [Google Scholar]
- 77.Bennett JS, Hoxie JA, Leitman SF, Vilaire G, Cines DB. Inhibition of fibrinogen binding to stimulated human platelets by a monoclonal antibody. Proc Natl Acad Sci USA. 1983;80:2417–2421. doi: 10.1073/pnas.80.9.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coller BS, Scudder LE. Inhibition of dog platelet function by in vivo infusion of F(ab’)2 fragments of a monoclonal antibody to the platelet glycoprotein IIb/IIIa receptor. Blood. 1985;66:1456–1459. [PubMed] [Google Scholar]
- 79.Wagner CL, Mascelli MA, Neblock DS, Weisman HF, Coller BS, Jordan RE. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood. 1996;88:907–914. [PubMed] [Google Scholar]
- 80.Suzuki K, Sakai Y, Hisamichi N, Taniuchi Y, Sato K, Terazaki C, Kaku S, Kawasaki T, Yano S, Inagaki O, Masuho Y. Comparison of the antiplate-let effect of YM337 and abciximab in rhesus monkeys. Eur J Pharmacol. 1997;336:169–176. doi: 10.1016/s0014-2999(97)01241-7. [DOI] [PubMed] [Google Scholar]
- 81.Artoni A, Li J, Mitchell B, Ruan J, Takagi J, Springer TA, French DL, Coller BS. Integrin beta3 regions controlling binding of murine mAb 7E3: implications for the mechanism of integrin alphaIIbbeta3 activation. Proc Natl Acad Sci USA. 2004;101:13114–13120. doi: 10.1073/pnas.0404201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gold HK, Coller BS, Yasuda T, Saito T, Fallon JT, Guerrero JL, Leinbach RC, Ziskind AA, Collen D. Rapid and sustained coronary artery recana-lization with combined bolus injection of recombinant tissue-type plas-minogen activator and monoclonal antiplatelet GPIIb/IIIa antibody in a canine preparation. Circulation. 1988;77:670–677. doi: 10.1161/01.cir.77.3.670. [DOI] [PubMed] [Google Scholar]
- 83.Yasuda T, Gold HK, Fallon JT, Leinbach RC, Guerrero JL, Scudder LE, Kanke M, Shealy D, Ross MJ, Collen D. Monoclonal antibody against the platelet glycoprotein (GP) IIb/IIIa receptor prevents coronary artery reocclusion after reperfusion with recombinant tissue-type plasminogen activator in dogs. J Clin Invest. 1988;81:1284–1291. doi: 10.1172/JCI113446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.The EPIC Investigators. Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angio-plasty. N Engl J Med. 1994;330:956–961. doi: 10.1056/NEJM199404073301402. [DOI] [PubMed] [Google Scholar]
- 85.Simoons ML GUSTO IV-ACS Investigators. Effect of glycoprotein IIb/ IIIa receptor blocker abciximab on outcome in patients with acute coronary syndromes without early coronary revascularisation: the GUSTO IV-ACS randomised trial. Lancet. 2001;357:1915–1924. doi: 10.1016/s0140-6736(00)05060-1. [DOI] [PubMed] [Google Scholar]
- 86.Speich HE, Furman RR, Lands LT, Moodie GD, Jennings LK. Elevating local concentrations of GPIIb-IIIa antagonists counteracts platelet thrombus stability. [Accessed March 22, 2013];J Thromb Thrombolysis. 2012 Oct 17; doi: 10.1007/s11239-012-0814-7. http://link.springer.com/article/10.1007%2Fs11239-012-0814-7. [DOI] [PMC free article] [PubMed]
- 87.Topol EJ, Lincoff AM, Kereiakes DJ, Kleiman NS, Cohen EA, Ferguson JJ, Tcheng JE, Sapp S, Califf RM. Multi-year follow-up of abciximab therapy in three randomized, placebo-controlled trials of percutaneous coronary revascularization. Am J Med. 2002;113:1–6. doi: 10.1016/s0002-9343(02)01145-2. [DOI] [PubMed] [Google Scholar]
- 88.Admiral Investigators. Three-year duration of benefit from abciximab in patients receiving stents for acute myocardial infarction in the randomized double-blind ADMIRAL study. Eur Heart J. 2005;26:2520–2523. doi: 10.1093/eurheartj/ehi620. [DOI] [PubMed] [Google Scholar]
- 89.Ndrepepa G, Kastrati A, Neumann FJ, Schmitt C, Mehilli J, Schömig A. Five-year outcome of patients with acute myocardial infarction enrolled in a randomised trial assessing the value of abciximab during coronary artery stenting. Eur Heart J. 2004;25:1635–1640. doi: 10.1016/j.ehj.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 90.Simon DI, Xu H, Ortlepp S, Rogers C, Rao NK. 7E3 monoclonal antibody directed against the platelet glycoprotein IIb/IIIa cross-reacts with the leukocyte integrin Mac-1 and blocks adhesion to fibrinogen and ICAM-1. Arterioscler Thromb Vasc Biol. 1997;17:528–535. doi: 10.1161/01.atv.17.3.528. [DOI] [PubMed] [Google Scholar]
- 91.Coller BS. Binding of abciximab to alpha V beta 3 and activated alpha M beta 2 receptors: with a review of platelet-leukocyte interactions. Thromb Haemost. 1999;82:326–336. [PubMed] [Google Scholar]
- 92.Coller BS. Potential non-glycoprotein IIb/IIIa effects of abciximab. Am Heart J. 1999;138:S1–S5. doi: 10.1053/hj.1999.v138.99078. [DOI] [PubMed] [Google Scholar]
- 93.Hong YJ, Jeong MH, Lee SR, Hong SN, Kim KH, Park HW, Kim JH, Kim W, Ahn Y, Cho JG, Park JC, Kang JC. Anti-inflammatory effect of abciximab-coated stent in a porcine coronary restenosis model. J Korean Med Sci. 2007;22:802–809. doi: 10.3346/jkms.2007.22.5.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schühlen H, Kastrati A, Mehilli J, Hausleiter J, Dirschinger J, Dotzer F, Bollwein H, Schömig A. Abciximab and angiographic restenosis after coronary stent placement. Analysis of the angiographic substudy of ISAR-REACT–a double-blind, placebo-controlled, randomized trial evaluating abciximab in patients undergoing elective percutaneous coronary interventions after pretreatment with a high loading dose of clopi-dogrel. Am Heart J. 2006;151:1248–1254. doi: 10.1016/j.ahj.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 95.Kereiakes DJ, Runyon JP, Broderick TM, Shimshak TM. IIb’s are not IIb’s. Am J Cardiol. 2000;85:23C–31C. doi: 10.1016/s0002-9149(00)00876-6. [DOI] [PubMed] [Google Scholar]
- 96.Phillips DR, Charo IF, Scarborough RM. GPIIb-IIIa: the responsive integrin. Cell. 1991;65:359–362. doi: 10.1016/0092-8674(91)90451-4. [DOI] [PubMed] [Google Scholar]
- 97.Scarborough RM, Naughton MA, Teng W, Rose JW, Phillips DR, Nannizzi L, Arfsten A, Campbell AM, Charo IF. Design of potent and specific integrin antagonists. Peptide antagonists with high specificity for glycoprotein IIb-IIIa. J Biol Chem. 1993;268:1066–1073. [PubMed] [Google Scholar]
- 98.Lele M, Sajid M, Wajih N, Stouffer GA. Eptifibatide and 7E3, but not tirofiban, inhibit alpha(v)beta(3) integrin-mediated binding of smooth muscle cells to thrombospondin and prothrombin. Circulation. 2001;104:582–587. doi: 10.1161/hc3101.092199. [DOI] [PubMed] [Google Scholar]
- 99.Phillips DR, Teng W, Arfsten A, Nannizzi-Alaimo L, White MM, Longhurst C, Shattil SJ, Randolph A, Jakubowski JA, Jennings LK, Scarborough RM. Effect of Ca2+ on GP IIb-IIIa interactions with integ-rilin: enhanced GP IIb-IIIa binding and inhibition of platelet aggregation by reductions in the concentration of ionized calcium in plasma anticoagulated with citrate. Circulation. 1997;96:1488–1494. doi: 10.1161/01.cir.96.5.1488. [DOI] [PubMed] [Google Scholar]
- 100.Ohman EM, Kleiman NS, Gacioch G, et al. Combined accelerated tissue-plasminogen activator and platelet glycoprotein IIb/IIIa integrin receptor blockade with Integrilin in acute myocardial infarction. Results of a randomized, placebo-controlled, dose-ranging trial. IMPACT-AMI Investigators. Circulation. 1997;95:846–854. doi: 10.1161/01.cir.95.4.846. [DOI] [PubMed] [Google Scholar]
- 101.Harrington RA, Kleiman NS, Kottke-Marchant K, Lincoff AM, Tcheng JE, Sigmon KN, Joseph D, Rios G, Trainor K, Rose D. Immediate and reversible platelet inhibition after intravenous administration of a peptide glycoprotein IIb/IIIa inhibitor during percutaneous coronary intervention. Am J Cardiol. 1995;76:1222–1227. doi: 10.1016/s0002-9149(99)80345-2. [DOI] [PubMed] [Google Scholar]
- 102.Tcheng JE, Harrington RA, Kottke-Marchant K, Kleiman NS, Ellis SG, Kereiakes DJ, Mick MJ, Navetta FI, Smith JE, Worley SJ. Multicenter, randomized, double-blind, placebo-controlled trial of the platelet integ-rin glycoprotein IIb/IIIa blocker Integrelin in elective coronary intervention. IMPACT Investigators. Circulation. 1995;91:2151–2157. doi: 10.1161/01.cir.91.8.2151. [DOI] [PubMed] [Google Scholar]
- 103.Schulman SP, Goldschmidt-Clermont PJ, Topol EJ, et al. Effects of integrelin, a platelet glycoprotein IIb/IIIa receptor antagonist, in unstable angina. A randomized multicenter trial. Circulation. 1996;94:2083–2089. doi: 10.1161/01.cir.94.9.2083. [DOI] [PubMed] [Google Scholar]
- 104.The IMPACT II Investigators. Effects of competitive platelet glycopro-tein IIb/IIIa inhibition with integrilin in reducing complications of percutaneous coronary intervention. Lancet. 1997;349:1422–1428. [PubMed] [Google Scholar]
- 105.Lincoff AM, Harrington RA, Califf RM, Hochman JS, Guerci AD, Ohman EM, Pepine CJ, Kopecky SL, Kleiman NS, Pacchiana CM, Berdan LG, Kitt MM, Simoons ML, Topol EJ. Management of patients with acute coronary syndromes in the United States by platelet glyco-protein IIb/IIIa inhibition. Insights from the platelet glycoprotein IIb/ IIIa in unstable angina: receptor suppression using integrilin therapy (PURSUIT) trial. Circulation. 2000;102:1093–1100. doi: 10.1161/01.cir.102.10.1093. [DOI] [PubMed] [Google Scholar]
- 106.Harrington RA. Design and methodology of the PURSUIT trial: evaluating eptifibatide for acute ischemic coronary syndromes. Platelet glyco-protein IIb-IIIa in unstable angina: receptor suppression using integrilin therapy. Am J Cardiol. 1997;80:34B–38B. doi: 10.1016/s0002-9149(97)00575-4. [DOI] [PubMed] [Google Scholar]
- 107.The PURSUIT Investigators. Inhibition of the platelet glycoprotein IIb/ IIIa with eptifibatide in patients with acute coronary syndromes. Platelet glycoprotein IIb/IIIa in unstable angina: receptor suppression using Integrilin therapy. N Engl J Med. 1998;339:436–443. [Google Scholar]
- 108.The ESPRIT Investigators. Novel dosing regimen of eptifibatide in planned coronary stent implantation (ESPRIT): a randomised, placebo-controlled trial. Lancet. 2000;356:2037–2044. doi: 10.1016/S0140-6736(00)03400-0. [DOI] [PubMed] [Google Scholar]
- 109.O’Shea JC, Hafley GE, Greenberg S, Hasselblad V, Lorenz TJ, Kitt MM, Strony J, Tcheng JE ESPRIT Investigators (Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin Therapy trial) Platelet gly-coprotein IIb/IIIa integrin blockade with eptifibatide in coronary stent intervention: the ESPRIT trial: a randomized controlled trial. JAMA. 2001;285:2468–2473. doi: 10.1001/jama.285.19.2468. [DOI] [PubMed] [Google Scholar]
- 110.Tcheng JE, Talley JD, O’Shea JC, Gilchrist IC, Kleiman NS, Grines CL, Davidson CJ, Lincoff AM, Califf RM, Jennings LK, Kitt MM, Lorenz TJ. Clinical pharmacology of higher dose eptifibatide in percutaneous coronary intervention (the PRIDE study) Am J Cardiol. 2001;88:1097–1102. doi: 10.1016/s0002-9149(01)02041-0. [DOI] [PubMed] [Google Scholar]
- 111.Lynch JJ, Jr, Cook JJ, Sitko GR, Holahan MA, Ramjit DR, Mellott MJ, Stranieri MT, Stabilito II, Zhang G, Lynch RJ. Nonpeptide glycoprotein IIb/IIIa inhibitors. 5. Antithrombotic effects of MK-0383. J Pharmacol Exp Ther. 1995;272:20–32. [PubMed] [Google Scholar]
- 112.Barrett JS, Murphy G, Peerlinck K, De Lepeleire I, Gould RJ, Panebianco D, Hand E, Deckmyn H, Vermylen J, Arnout J. Pharmacokinetics and pharmacodynamics of MK-383, a selective non-peptide platelet glyco-protein-IIb/IIIa receptor antagonist, in healthy men. Clin Pharmacol Ther. 1994;56:377–388. doi: 10.1038/clpt.1994.152. [DOI] [PubMed] [Google Scholar]
- 113.Vickers S, Theoharides AD, Arison B, Balani SK, Cui D, Duncan CA, Ellis JD, Gorham LM, Polsky SL, Prueksaritanont T, Ramjit HG, Slaughter DE, Vyas KP. In vitro and in vivo studies on the metabolism of tirofiban. Drug Metab Dispos. 1999;27:1360–1366. [PubMed] [Google Scholar]
- 114.Alexander JH, Harrington RA. Recent antiplatelet drug trials in the acute coronary syndromes. Clinical interpretation of PRISM, PRISM-PLUS, PARAGON A and PURSUIT. Drugs. 1998;56:965–976. doi: 10.2165/00003495-199856060-00002. [DOI] [PubMed] [Google Scholar]
- 115.The RESTORE Investigators. Effects of platelet glycoprotein IIb/IIIa blockade with tirofiban on adverse cardiac events in patients with unstable angina or acute myocardial infarction undergoing coronary angio-plasty. Circulation. 1997;96:1445–1453. doi: 10.1161/01.cir.96.5.1445. [DOI] [PubMed] [Google Scholar]
- 116.Herrmann HC, Swierkosz TA, Kapoor S, Tardiff DC, DiBattiste PM, Hirshfeld JW, Klugherz BD, Kolansky DM, Magness K, Valettas N, Wilensky RL. Comparison of degree of platelet inhibition by abciximab versus tirofiban in patients with unstable angina pectoris and non-Q-wave myocardial infarction undergoing percutaneous coronary intervention. Am J Cardiol. 2002;89:1293–1297. doi: 10.1016/s0002-9149(02)02329-9. [DOI] [PubMed] [Google Scholar]
- 117.Topol EJ, Moliterno DJ, Herrmann HC, Powers ER, Grines CL, Cohen DJ, Cohen EA, Bertrand M, Neumann FJ, Stone GW, DiBattiste PM, Demopoulos L TARGET Investigators. Do Tirofiban and ReoPro Give Similar Efficacy Trial. Comparison of two platelet glycoprotein IIb/ IIIa inhibitors, tirofiban and abciximab, for the prevention of ischemic events with percutaneous coronary revascularization. N Engl J Med. 2001;344:1888–1894. doi: 10.1056/NEJM200106213442502. [DOI] [PubMed] [Google Scholar]
- 118.Schneider DJ, Herrmann HC, Lakkis N, Aguirre F, Lo MW, Yin KC, Aggarwal A, Kabbani SS, DiBattiste PM. Increased concentrations of tirofiban in blood and their correlation with inhibition of platelet aggregation after greater bolus doses of tirofiban. Am J Cardiol. 2003;91:334–336. doi: 10.1016/s0002-9149(02)03163-6. [DOI] [PubMed] [Google Scholar]
- 119.Valgimigli M, Campo G, Percoco G, et al. Multicentre Evaluation of Single High-Dose Bolus Tirofiban vs Abciximab With Sirolimus-Eluting Stent or Bare Metal Stent in Acute Myocardial Infarction Study (MULTISTRATEGY) Investigators. Comparison of angioplasty with infusion of tirofiban or abciximab and with implantation of sirolimus-eluting or uncoated stents for acute myocardial infarction: the MULTISTRATEGY randomized trial. JAMA. 2008;299:1788–1799. doi: 10.1001/jama.299.15.joc80026. [DOI] [PubMed] [Google Scholar]
- 120.Marzocchi A, Manari A, Piovaccari G, Marrozzini C, Marra S, Magnavacchi P, Sangiorgio P, Marinucci L, Taglieri N, Gordini G, Binetti N, Guiducci V, Franco N, Reggiani ML, Saia F FATA Investigators. Randomized comparison between tirofiban and abciximab to promote complete ST-resolution in primary angioplasty: results of the facilitated angioplasty with tirofiban or abciximab (FATA) in ST-elevation myocardial infarction trial. Eur Heart J. 2008;29:2972–2980. doi: 10.1093/eurheartj/ehn467. [DOI] [PubMed] [Google Scholar]
- 121.Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE, Jr, Ettinger SM, Fesmire FM, Ganiats TG, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP. 2012 ACCF/ AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2012;60:645–681. doi: 10.1016/j.jacc.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 122.Pannu R, Andraws R. Effects of glycoprotein IIb/IIIa inhibitors in patients undergoing percutaneous coronary intervention after pretreat-ment with clopidogrel: a meta-analysis of randomized trials. Crit Pathw Cardiol. 2008;7:5–10. doi: 10.1097/HPC.0b013e318163ebc9. [DOI] [PubMed] [Google Scholar]
- 123.Sabatine MS, Hamdalla HN, Mehta SR, Fox KA, Topol EJ, Steinhubl SR, Cannon CP. Efficacy and safety of clopidogrel pretreatment before percutaneous coronary intervention with and without glycoprotein IIb/ IIIa inhibitor use. Am Heart J. 2008;155:910–917. doi: 10.1016/j.ahj.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 124.Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discov. 2010;9:154–169. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- 125.Singh IM, Holmes DR., Jr Myocardial revascularization by percutaneous coronary intervention: past, present, and the future. Curr Probl Cardiol. 2011;36:375–401. doi: 10.1016/j.cpcardiol.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 126.Bosch X, Marrugat J, Sanchis J. Platelet glycoprotein IIb/IIIa blockers during percutaneous coronary intervention and as the initial medical treatment of non-ST segment elevation acute coronary syndromes. Cochrane Database Syst Rev. 2010;9:CD002130. doi: 10.1002/14651858.CD002130.pub2. [DOI] [PubMed] [Google Scholar]
- 127.Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J. 2000;139:S38–S45. doi: 10.1067/mhj.2000.103742. [DOI] [PubMed] [Google Scholar]
- 128.Tcheng JE, Kereiakes DJ, Lincoff AM, George BS, Kleiman NS, Sane DC, Cines DB, Jordan RE, Mascelli MA, Langrall MA, Damaraju L, Schantz A, Effron MB, Braden GA. Abciximab readministration: results of the ReoPro Readministration Registry. Circulation. 2001;104:870–875. doi: 10.1161/hc3301.094533. [DOI] [PubMed] [Google Scholar]
- 129.Curtis BR, Swyers J, Divgi A, McFarland JG, Aster RH. Thrombocytopenia after second exposure to abciximab is caused by antibodies that recognize abciximab-coated platelets. Blood. 2002;99:2054–2059. doi: 10.1182/blood.v99.6.2054. [DOI] [PubMed] [Google Scholar]
- 130.Weitz JI, Bates SM. Beyond heparin and aspirin: new treatments for unstable angina and non-Q-wave myocardial infarction. Arch Intern Med. 2000;160:749–758. doi: 10.1001/archinte.160.6.749. [DOI] [PubMed] [Google Scholar]
- 131.Bougie DW, Wilker PR, Wuitschick ED, Curtis BR, Malik M, Levine S, Lind RN, Pereira J, Aster RH. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood. 2002;100:2071–2076. [PubMed] [Google Scholar]
- 132.Madan M, Berkowitz SD. Understanding thrombocytopenia and antigenicity with glycoprotein IIb-IIIa inhibitors. Am Heart J. 1999;138:317–326. doi: 10.1053/hj.1999.v138.a100465. [DOI] [PubMed] [Google Scholar]
- 133.Bednar B, Cook JJ, Holahan MA, Cunningham ME, Jumes PA, Bednar RA, Hartman GD, Gould RJ. Fibrinogen receptor antagonist-induced thrombocytopenia in chimpanzee and rhesus monkey associated with preexisting drug-dependent antibodies to platelet glycoprotein IIb/IIIa. Blood. 1999;94:587–599. [PubMed] [Google Scholar]
- 134.Cox D. Oral GPIIb/IIIa antagonists: what went wrong? Curr Pharm Des. 2004;10:1587–1596. doi: 10.2174/1381612043384673. [DOI] [PubMed] [Google Scholar]
- 135.Chew DP, Bhatt DL, Sapp S, Topol EJ. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter randomized trials. Circulation. 2001;103:201–206. doi: 10.1161/01.cir.103.2.201. [DOI] [PubMed] [Google Scholar]
- 136.Du XP, Plow EF, Frelinger AL, 3rd, O’Toole TE, Loftus JC, Ginsberg MH. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 137.Peter K, Schwarz M, Bode C. Activating effects of GPIIb/IIIa blockers: an intrinsic consequence of ligand-mimetic properties. Circulation. 2002;105:E180–E181. doi: 10.1161/01.cir.0000017280.72394.1f. [DOI] [PubMed] [Google Scholar]
- 138.Frelinger AL, 3rd, Furman MI, Krueger LA, Barnard MR, Michelson AD. Dissociation of glycoprotein IIb/IIIa antagonists from platelets does not result in fibrinogen binding or platelet aggregation. Circulation. 2001;104:1374–1379. doi: 10.1161/hc3701.095950. [DOI] [PubMed] [Google Scholar]
- 139.Catella-Lawson F, Kapoor S, Moretti D, De Marco S, Vigilante GJ, Cucchiara AJ, Ramsey KE, Combe S, Rocca B, Theroux P, FitzGerald GA AFIRME Investigators. Antagonism of the Fibrinogen Receptor after Myocardial Events. Oral glycoprotein IIb/IIIa antagonism in patients with coronary artery disease. Am J Cardiol. 2001;88:236–242. doi: 10.1016/s0002-9149(01)01632-0. [DOI] [PubMed] [Google Scholar]
- 140.Negri A, Li J, Naini S, Coller BS, Filizola M. Structure-based virtual screening of small-molecule antagonists of platelet integrin alphaIIb-beta3 that do not prime the receptor to bind ligand. J Comput Aided Mol Des. 2012;26:1005–1015. doi: 10.1007/s10822-012-9594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Koloka V, Christofidou ED, Vaxevanelis S, Dimitriou AA, Tsikaris V, Tselepis AD, Panou-Pomonis E, Sakarellos-Daitsiotis M, Tsoukatos DC. A palmitoylated peptide, derived from the acidic carboxyl-terminal segment of the integrin alphaIIb cytoplasmic domain, inhibits platelet activation. Platelets. 2008;19:502–511. doi: 10.1080/09537100802266875. [DOI] [PubMed] [Google Scholar]
- 142.Blue R, Murcia M, Karan C, Jirousková M, Coller BS. Application of high-throughput screening to identify a novel alphaIIb-specific small- molecule inhibitor of alphaIIbbeta3-mediated platelet interaction with fibrinogen. Blood. 2008;111:1248–1256. doi: 10.1182/blood-2007-08-105544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Blue R, Kowalska MA, Hirsch J, Murcia M, Janczak CA, Harrington A, Jirouskova M, Li J, Fuentes R, Thornton MA, Filizola M, Poncz M, Coller BS. Structural and therapeutic insights from the species specificity and in vivo antithrombotic activity of a novel alphaIIb-specific al-phaIIbbeta3 antagonist. Blood. 2009;114:195–201. doi: 10.1182/blood-2008-08-169243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhu J, Zhu J, Negri A, Provasi D, Filizola M, Coller BS, Springer TA. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood. 2010;116:5050–5059. doi: 10.1182/blood-2010-04-281154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu XY, Timmons S, Lin YZ, Hawiger J. Identification of a functionally important sequence in the cytoplasmic tail of integrin beta 3 by using cell-permeable peptide analogs. Proc Natl Acad Sci USA. 1996;93:11819–11824. doi: 10.1073/pnas.93.21.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ, Ginsberg MH. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J Clin Invest. 2007;117:2250–2259. doi: 10.1172/JCI31024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dehmer GJ, Weaver D, Roe MT, Milford-Beland S, Fitzgerald S, Hermann A, Messenger J, Moussa I, Garratt K, Rumsfeld J, Brindis RG. A contemporary view of diagnostic cardiac catheterization and percutaneous coronary intervention in the United States: a report from the CathPCI Registry of the National Cardiovascular Data Registry, 2010 through June 2011. J Am Coll Cardiol. 2012;60:2017–2031. doi: 10.1016/j.jacc.2012.08.966. [DOI] [PubMed] [Google Scholar]
- 148.King A. Interventional cardiology: Radial access improves PCI outcomes. Nat Rev Cardiol. 2012;9:127. doi: 10.1038/nrcardio.2012.2. [DOI] [PubMed] [Google Scholar]
- 149.De Luca G, Verdoia M, Suryapranata H. Benefits from intracoronary as compared to intravenous abciximab administration for STEMI patients undergoing primary angioplasty: a meta-analysis of 8 randomized trials. Atherosclerosis. 2012;222:426–433. doi: 10.1016/j.atherosclerosis.2012.02.041. [DOI] [PubMed] [Google Scholar]