

Abstract
This review focuses on the syntheses of PI3K/Akt/mTOR inhibitors that have been reported outside of the patent literature in the last 5 years but is largely centered on synthetic work reported in 2011 and 2012. While focused on syntheses of inhibitors, some information on in vitro and in vivo testing of compounds is also included. Many of these reported compounds are reversible, competitive adenosine triphosphate (ATP) binding inhibitors, so given the structural similarities of many of these compounds to the adenine core, this review presents recent work on inhibitors based on where the synthetic chemistry was started, i.e. inhibitor syntheses which started with purines/pyrimidines are followed by inhibitor syntheses which began with pyridines, pyrazines, azoles, and triazines then moves to inhibitors which bear no structural resemblance to adenine: liphagal, wortmannin and quercetin analogs. The review then finishes with a short section on recent syntheses of phosphotidyl inositol (PI) analogs since competitive PI binding inhibitors represent an alternative to the competitive ATP binding inhibitors which have received the most attention.
Keywords: PI3K/Akt/mTor inhibitor, protein kinase inhibitor, anti-cancer drugs
1. Introduction
The PI3K/Akt/mTOR signaling pathways regulate cell proliferation, survival, and angiogenesis and hence are extremely important in cancer biology. A number of recent reviews of work 2 performed to understand and inhibit these signaling pathways have appeared and there is increasing evidence that inhibitors of at least 2 signals in the pathways may be most desirable.1 None of the recent reviews referenced above focus on how these inhibitors were prepared, therefore this review will concentrate on the organic chemistry of the syntheses of inhibitors and include some selected in vitro and in vivo screening results. The literature reviewed includes reports that have appeared outside of the patent literature in the last 5 years but will concentrate on studies reported in 2011 and 2012 since approximately 100 reports of PI3K inhibitor syntheses have appeared in the last two years alone. Many of these reported compounds are reversible competitive ATP binding inhibitors and their synthetic preparation relies on chemistry which is initiated from purine (diazolopyrimidine)/ pyrimidine, pyridine, pyrazine, triazine or azole core structures. The first sections of this review article were organized by looking at where the syntheses started. In many cases, this meant what heterocycle did the chemists prepare first or purchase and start with, and that was defined as the core structure under which to file that synthesis, ie pyrimidines, pyridines, triazines, etc. Many of these inhibitors contain multiple heterocyclic rings so they could conceivably be placed under several of these categories if one just asked does it contain one of the heterocycles under the category being discussed.
Synthetic work also continues on inhibitors based on the steroidal and terpenoidal cores found in wortmannin, quercetin, and liphagal. Therefore, this review will present recent work on inhibitors based on purines/pyrimidines, followed by pyridines, pyrazines, azoles, and triazines then move to liphagal, wortmannin and quercetin analogs. Some synthetic work also continues on phosphotidyl inositol binding inhibitors and that work is presented last.
2. Pyrimidines and Quinazolines
Synthesis of pyrimidine containing PI3K inhibitors continues to be an area of intense interest. Compounds in this class were some of the first that were found to be selective PI3K α inhibitors.2 These initial reports have been followed in the last few years with a number of additional reports of the synthesis and testing of pyrimidine derivatives and, in particular, morpholino pyrimidine derivatives.
In early 2010, a number of new 4-morpholinopyrrolopyrimidines were reported.3 This work reported routes to pyrrolo[3,2-d]pyrimidines and pyrrolo[2,3-d]pyrimidines. The pyrrolo[3,2-d] pyrimidine core syntheses were initiated using 2,4-dichloro-6-methyl-5-nitropyrimidine (1) as a starting material (Figure 1). The 4-chloro (ortho to the nitro) group was replaced first via a SNAr reaction and then aromatic substitutents were added to the pyrimidine core in the 2 position via Suzuki cross coupling reactions of aryl boronic acids via the 2nd chloride (2-chloro) to produce 2. The pyrrolo[3,2-d]pyrimidine core was then formed via treatment with dimethylformamide dimethylacetal. This reagent forms methoxide and the iminium salt when heated so would be expected add a formyl group to the 6 methyl position. Reduction of the nitro group to an aniline then provided a substrate which cyclized to the pyrrolo[3,2-d]pyrimidine core (3). The enamine functional group within that core structure was then used to condense with aldehydes and ketones to add substituents to the 7 position (4).
Figure 1.

Pyrrolo[3,2-d]pyrimidine Syntheses.
The pyrrolo[2,3-d]pyrimidine core was synthesized via condensation of 6-amino uracil (5) with chloroacetaldehyde (6) (Figure 2). Conversion of the hydroxyl groups to chlorides with POCl3 was then followed by sequential addition of morpholine and aryl boronic acids as described above for the regioisomeric nucleus to produce 8. The pyyrole nitrogen was alkylated with alkyl halides and when 4-aminophenyl boronic acid was used for the Suzuki coupling then that 4-amino group was further converted into a variety of ureas (9) via treatment with triphosgene followed by amines.
Figure 2.

Pyrrolo[2,3-d]pyrimidine Syntheses.
These urea derivatives were synthesized to improve water solubility. These compounds inhibited PI3Kα and mTOR at low nanomolar concentrations. In vivo testing of 9 (R’ = CF3, R1 = -Ph-4’-C(O)N(Me)CH2CH2NMe2) in MDA-MB-361 breast cancer xenografts showed substantial inhibition of both p70S6 and Akt phosphorylation – signaling pathways downstream of PI3K, 8h after iv injection of 25mg/kg. This dose produced tumorostatic effects on MDA-MB-361 xenografts, whereas 50 mg/kg decreased tumor size.
2010 also saw the report of the syntheses of a number of triazoles that were PI3K and Akt inhibitors (Figure 3).4 These syntheses started with 4-chloro-6-methylpyrrolo[2,3-d]pyrimidine (10). The most active new compounds were prepared by displacement of the chloride with 3-pyrrolidinol followed by Parikh-Doering oxidation of the alcohol with SO3-pyridine. The pyrrolidinone was then used in a modified Strecker reaction where both dimethoxybenzylamine and TMS-CN were added to the ketone. These additions were followed by nitrile reduction with lithium aluminum hydride (LAH) to produce 11. The primary amine was then converted to different amides using hydroxybenzotriazole (HOBt) and a carbodiimide and the dimethoxybenzyl protecting group was removed with trifluoroacetic acid (TFA) to produce the final products (12).
Figure 3.

Syntheses of Pyrrolidine Substituted Pyrrolopyrimidines.
Twelve final compounds were evaluated for selectivity for Akt versus PKA kinases, and one compound was chosen for subsequent testing. Akt inhibitor (12) (R1 = Et, R = 2,4-difluorophenyl) reportedly inhibited Akt phosphorylation and decreased PC3 tumor growth by 25, 51 and 75% when administered orally bid for 10 days at 25, 75 or 100mg/kg, yet figures illustrating these effects were not presented.
Following these reports of the preparation and screening of pyrrolopyrimidines, a report appeared on preparation of dihydrothieno- and dihydrofuranopyrimidines which were more selective as Akt inhibitors but also functioned as PI3K inhibitors.5 The synthetic scheme used here started with the syntheses of the dihydrothiophene or dihydrofuran cores followed by construction of the pyrimidine (Figure 4). The five member rings were synthesized by Michael addition of 2-hydroxy or 2-mercapto methyl acetate (13) onto methyl crotonate (14) followed by Dieckmann condensation to yield the keto ester (16). Reaction of the keto ester with formamidine yielded the pyrimidinol which was converted to the chloride with POCl3. The chloride was replaced with a Boc protected piperazine. The NBoc was then removed and the free amine (18) was acylated with a variety of different amino acids to produce the targets (19).
Figure 4.

Preparation of Dihydrothieno- and Dihydrofuranopyrimidines.
One compound (19) (X = S, R = (S)-CH2NHisopropyl, Ar = 4-ClPh) with favorable PK/PD parameters was selected for in vivo studies. In tissue culture of LNCaP cells 50% nhibition of Akt at 137nM of 19 was reported. In U87 xenografts at dose –dependent inhibition of phosphorylation of Akt substrate PRAS40 was reported that platoed at 75–200mg/kg oral dose for 4 hours based on. Inhibition of PC3 prostate cancer xenografts growth was reported at a daily dose of 200 mg/kg. These reports were not supported by original data showing PRAS40 phosphorylation. It is also somewhat surprising that effects on Akt activity and tumor growth were not reported in the same models.
Pecchi and co-workers reported both solid and solution phase syntheses of a number of 2-morpholino-6-arylpyrimidines in 2010 (Figure 5).6 The solution phase syntheses started with aryl malonate esters (20) which were cyclized with morpholino guanidine (21) to provide the pyrimidine core. The hydroxyl group on the pyrimidine was converted to a triflate and then subjected to Pd catalyzed C-N cross coupling to produce the final products (23).
Figure 5.

Syntheses of Morpholino Aryl Pyrimidines.
The optimized compounds (23) were tested in the A2780 cell line (ovarian carcinoma, PTEN deleted), and inhibition of Akt at 7nM and proliferation at 0.37µM was reported (23, R = 3’-OH, R2 = H, R3 = 4’-OMe-3’-pyridyl).
In the summer of 2010, McDonald and co-workers reported the syntheses of a number of trisubstituted pyrimidines by a route very similar to that reported by Pecchi and co-workers (Figure 6).7 6-Aryl substituted pyrimidines were prepared via condensation of aryl β-ketoesters (24) with arylamidines (25) to generate pyrimidinones in which the hydroxyl was converted to a chloride then displaced with morpholine to produce the products (27). In this same report, alcohol and amine substituted pyrimidines were prepared starting with 2,4,6-trichloropyrimidine which was treated with the alcohol or amine of interest followed by morpholine. The monochloro pyrimidine products of these 2 steps were then cross coupled with aryl boronic acids using Pd(dppf)Cl2, Pd(PPh3)4, or in several cases a Pd(II) palladacyle catalyst. Tests in IGROV-1 ovarian cancer cells showed inhibition of S473 phosphorylation after 24 h of exposure.
Figure 6.

Additional Syntheses of Morpholino Aryl Pyrimidines.
Additional 4,6-disubstituted-2-morpholinopyrimidines were reported by Burger, Pecchi, and co-workers at Novartis Institutes for Biomedical Research in early 2011 (Figure 7).8 One synthetic route started from tribromopyrimidine (28) and the amine was added first and the morpholine second. Suzuki cross coupling of the mono bromo pyrimidine (29) with heteroaromatic boronates then produced the final 4,6-disubstituted-2-morpholino pyrimidines (30). Alternatively, morpholine formamidine hydrobromide (32) was condensed with diethyl malonate (31) and the pyrimidinol thus produced was converted to the dichloropyrimidine (33) using POCl3. The chlorines were then sequentially replaced using Suzuki cross coupling and Buchwald-Hartwig C-N bond formation to produce products (35).
Figure 7.

Syntheses of Amino Morpholino Aryl Pyrimidines.
Additional morpholino amino pyrimidinyl pyrimidines were also reported in early 2011 by Ohwada and co-workers (Figure 8).9 The morpholino pyrimidine core structures of these compounds were also prepared via a condensation of morpholino amidine (32) with a β-ketoester, in this case a β-ketolactone (36). Pyrimidinol chlorination was followed by a Pd catalyzed C-N bond formation to produce 38. In the cases where the syntheses originated with a morpholino dichloropyrimidine (39) rather than a morpholino monochloropyrimidine, the C-N bond formation could be performed first via SNAr chemistry and then the heteroaromatic could be added last via a Suzuki cross coupling to yield 40. Optimized compound CH5132799 (40, Ar = 2’-amino-5’-pyrimidinyl, R = SO2Me) showed inhibition of KPL-4 breast cancer xenografts at 12.6 mg/kg. In the follow-up experiments with KPL-4 cells in tissue culture Akt phosphorylation at S473 and T308 as well as of Akt substrate PRAS40 was inhibited at 10nM and of FOXO transcriptional factors at 100nM.10
Figure 8.

Syntheses of Bicyclic Morpholino Pyrimidines.
Early 2011 also saw the report of syntheses of some benzothiazole substituted pyrimidines which are PI3K/mTOR dual inhibitors (Figure 9).11 Syntheses of all of these compounds started with the use of dihalopyrimidines or dihalopyridines (41) which were cross coupled to the pinacol boronate ester or tetrafluoroborate of the benzothiazole (42). The pyrimidine or pyridine cores were further modified by a base catalyzed SNAr reaction with sulfur or oxygen nucleophiles or a Pd catalyzed C-N bond formation in the case of nitrogen nucleophiles whereas the cross coupling yielded the final products for the pyridines (43).
Figure 9.

Preparation of Benzothiazole Substituted Pyrimidines and Pyridines.
A pyridine benzothiazole biaryl (43, R = 4-F) with optimized pharmacokinetics that inhibited S473Akt phosphorylation in U87-MG cells with IC50 6,3nM was selected for in vivo tests. Inhibition of the PI3K/Akt pathway induced in liver by iv injection of HGF was observed starting at 0.1 mg/kg with maximal inhibition at 1mg/kg. A time course study showed that at 3mg/kg maximal inhibition lasted for 6h and significant inhibition lasted for 24 hours. Experiments in U87-MG (PTEN null) glioblastoma, A549 (KRAS mutant) lung adenocarcinoma and HCT11618,57 (KRAS and PI3KR mutant) colon adenocarcinoma tumor xenografts showed dose-dependent tumorostatic effect with ED 0.26 mg/kg. However subsequent in vivo experiments demonstrated accumulation in liver of a deacylated metabolite that inhibits PI3Kα at nM concentrations, which precluded further development of this compound.
In early 2011, Hong, Hong, and co-workers reported the syntheses of some PI3K inhibitors based on a xanthine core (Figure 10).12 This core was picked to produce inhibitors in which the sub cellular localization in live cells could be monitored by fluorescence microscopy. The fluorescent inhibitors screened here were prepared by a Cu(II) catalyzed N-arylation yielding 46 using aryl boronic acids followed by a Pd and Cu catalyzed C8 arylation (47) of the xanthine core.
Figure 10.

Preparation of Substituted Xanthines.
A 2011 follow up publication13 on furano and thienopyrimidines that showed good PI3Kα to β selectivity used the same synthetic scheme which had been reported in 20082b for the preparation of these new analogs. The amino furanyl or thioenyl ester was converted to the furano or thieno morpholino mono chloro pyrimidine then the chlorine was replaced via Suzuki coupling of a boronic acid or ester. In addition to these extensions of previous work, this paper outlined the syntheses of some thienobenzoxepin PI3K inhibitors (Figure 11). To prepare these compounds a keto benzoxepin core (48) was condensed with the β-chlorovinylaldehyde generated via treatment of DMF with POCl3 and that condensation product was treated with methyl thioglycolate to produce 49 then with the appropriate aniline to produce 50.
Figure 11.

Preparation of Thienobenzoxepins.
Following on earlier reports of the activity of morpholino pyridinyl or pyrimidinyl pyrimidines, the synthesis of a very active bismorpholino pyridinyl pyrimidine (BKM-120) (53) was reported in the late summer of 2011 (Figure 12).14 The synthesis of this compound involved treatment of 2,4,6-trichloropyrimidine (51) with excess morpholine to generate the chloro dimorpholino pyrimidine (52) which was separated from the isomeric 2-chloro-4,6-dimorpholino pyrimidine and then cross coupled with the pyridinyl pinacol boronic ester to generate the final product (53).
Figure 12.

Synthesis of BKM-120.
In vitro kinase assays, demonstrated inhibition of all class I PI3K (isoforms α β δ γ at 52, 166, 116 and 262 nM respectively) with over 50 fold specificity over mTOR and vps34 and high µM range against other tested kinases. In vitro tests were supported by tissue culture experiments in cells with ectopical expression of myristoylated (constitutively active) PI3K α β and δ that show inhibition of Akt with IC50 at 100, 200 and 500nM. Further tests in tissue culture showed convincing inhibition of constitutive Akt phosphorylation in PTEN-deficient U87 cells at 1 µM and PDGF-induced Akt phosphorylation at 250nM in SF268 cells. Experiments in Rat-1 xenografts that ectopically express myr-PI3K α showed Akt inhibition starting at 1h and lasted for 16h after oral administration of 60mg/kg of BKM120. Inhibition of Rat-1 xenografts that ectopically express myr-PI3K α was demonstrated with 40mg/kg. This dose also delayed growth of HCT116 or BT474 xenografts whereas combination with either MEK inhibitor AZD6244 or heceptin dramatically improved anti-tumor efficacy. Substantial inhibition of tumor neo vascularisation and reduction of tumor vacular permeability was also noted.
Favorable pharmacokinetics and pharmacodynamics profiles made BKM120 a component of choice for experiments that address the consequences of PI3K inhibition in mouse models of cancers. Phase I clinical trials showed that overall BKM120 was well tolerated (MTD-100mg half-life 40h with little interpatient variability). Dose-limiting toxicities were associated with hyperglycemia, mood alterations and skin rash.15 Phase II clinical trials are currently ongoing.
Several recent publications emphasized the enhanced antitumor effects in mouse models when BKM120 was combined with inhibitors of other signaling pathways, whereas when used alone or in combination with inhibitors of the PI3K/mTOR pathway it showed tumorostatic effects.16 Somewhat surprisingly, BKM120 showed better efficacy against metastases compared to primary xenograft tumors MDA-MB-453 and BT474.17
New analogs of the amino pyrimidinyl thieno pyrimidine (GDC 0941) were reported in late 2011 (Figure 13).18 These compounds were prepared using a method similar to this group’s earlier report.2b Boc protected piperazine was condensed with the aldehyde and the intermediate iminium ion was reduced to the amine using Na(OAc)3BH to produce 54. The aminopyrimidine was added via a Suzuki reaction, the Boc amino protecting group was removed and the terminal nitrogen of the piperazine was acylated to obtain 55.
Figure 13.

Preparation of Amino Pyrimidinyl Thieno Pyrimidines.
The most active new compound GDC-0980 (55, R = (S)-2-hydroxypropanoyl) inhibits both class I PI3K isoforms with IC50s of 5, 27, 7, and 14 nM for PI3Kα, β, δ, and γ, respectively; and inhibits mTOR with a Ki of 17 nM. At 7.5mg/kg daily dose compound GDC-0980 showed tumorostatic effects in PC3 and MCF-7neo/HER2. Subsequent experiments in tissue culture models demonstrated inhibition of Akt and PRAS40 phosphorylation at 0.1–0.5 mcM.19 Experiments in xenograft models showed delay or inhibition of tumor growth at 5 mg/kg. Analysis of pharmacodynamics in PTEN deficient PC3 xenografts showed inhibition of Akt phosphorylation by 75% at 24h after administration of 10 mg/kg, which caused tumor regression. Tumor regression was also demonstrated in breast cancer models MX-1 and MCF7-neo/HER2 and in NSCLC model A549. Currently GDC-0980 is in phase II clinical trials.
In late 2011, Blanchard and co-workers reported a series of 2-anilino-4-aryl-purine derivatives as inhibitors of PDK1 which is downstream of PI3K in cell signaling pathways (Figure 14).20 The preparation of these compounds involved protection of the NH in 2,4-dichloropurine (56) followed by Suzuki coupling at the 4 position (58) and a Buchwald-Hartwig coupling at the 2 position. The benzylated nitrogen was then deprotected using trifluoroacetic acid (TFA) to produce 59.
Figure 14.

Synthesis of 2-Anilino-4-Aryl Purines.
The last kinase of the PI3K/Akt/mTOR pathway is the ribosomal protein s6 kinase (p70S6K) and Bussenius and co-workers reported a series of pyrazolopyrimidines as inhibitors of this kinase in late 2011 (Figure 15).21 The target inhibitors were prepared via base catalyzed condensation of 3-substituted-5-chloro-2-methylphenylpiperazines (60) with 4-chloropyrazolopyrimidines (61). This same general synthetic sequence was also used to prepare an optimized dual Akt/p70S6K inhibitor reported in early 2012.22
Figure 15.

Synthesis of Pyrazolopyrimidines.
Meng and Yang and co-workers reported some morpholino pyrido pyrrolo triazines in late 2011/early 201223 using Piramid Pharma’s PI-103 (67, X = O, Y = C, furano rather than pyrrolo) as a lead compound (Figure 16).11 The compounds were prepared via initial condensation of chloro nitro pyridine (63) with the sodium salt of diethyl malonate. That product was completely decarboxylated and then deprotonated and condensed with diethyl oxalate to produce 64. Reduction of the nitro group and intramolecular condensation yielded the azaindole (65). Reaction of the indole nitrogen with NH2Cl followed by condensation with a 3-methoxybenzimidate produced the base of the target (66). Chlorination of the pyrimidinol and replacement of the Cl with morpholine followed by methyl ether cleavage with BBr3 completed the core preparation (67). The phenol was then acylated to produce several new compounds. Analysis of phosphorylation of Akt and p70S6 kinase (downstream substrates of PI3K and mTOR) showed that new compounds inhibited mTOR with similar potency yet inhibition of Akt phosphorylation was less potent compared to PI-103.
Figure 16.

Preparation of Morpholino Pyrido Pyrrolo Triazines.
As a follow up to earlier reports on the PI3K inhibiting ability of pyridopyrimidones, Lin and co-workers at GSK reported a series of imidazo pyrimidinones which were selective for PI3K β inhibition (Figure 17).24 These compounds were made from 2-aminoimidazoles (68) which were condensed with diethyl malonate to make imidazopyrimidinediones (69) which were then converted to the dichlorides (70) with POCl3. The dichlorides were hydrolyzed back to the mono chloro imidazopyrimidones (71) which were N-alkylated and then the remaining chloride was replaced with morpholine via simple microwave heating to yield 72. Similarly to TGX-221 these compounds preferentially targeted PI3K β and δ. Experiments in MDA-MB468 cells showed inhibition of Akt phosphorylation at 0.1µM.
Figure 17.

Preparation of Imidazopyrimidinones.
Synthesis of a family of triazolopyrimidinones (73) was reported by another GSK group in early 2012 and the synthetic route used to prepare these compounds was identical to the one described above except it started with a triazole rather than an imidazole (Figure 18).25
Figure 18.

A Triazolopyrimidinone.
These compounds demonstrated improved inhibition of PI3Kβ, thus, compound 73 (R = Me, R1 = 2-CH3-3-CF3benzyl, R2 = Me) inhibited PI3Kβ at 0.3 nM in vitro and inhibited Akt phosphorylation in MDA-MB-468 cells with IC50=5nM. However, high clearance rates precluded testing these new compounds in cancer models in mice.
In April 2012, sulfonyl-morpholino-pyrimidines (77) were reported as mTOR selective kinase inhibitors (Figure 19).26 The compounds were synthesized via an initial condensation of an amidine or urea with a chloro β-keto ester (74). The pyrimidinols thus produced were chlorinated with POCl3 and then the chloride was displaced with morpholine. The sulfonyl groups were added via sulfinic acid salt displacement of the primary chloride or thiolate displacement of the primary chloride followed by oxidation of the resulting sulfide with oxone to produce 77 and 79.
Figure 19.

Preparaton of Sulfonyl Morpholino Pyrimidines.
Morpholino pyrimidine derivatives which were selective PI3K β inhibitors were also reported in April27 and October 2012.28 The derivatives were prepared via condensation of aromatic diamines or aminophenols with the morpholino pyrimidine carboxylate salt (81)(Figure 20). The morpholino pyrimidine (80) was prepared via reaction of excess ethyl 3-amino-3-ethoxyacrylate with morpholine.
Figure 20.

Preparation of a Morpholino Pyrimidine Salt.
A condensation product of 80 (compound 82) selected for in vivo studies showed selective inhibition of PI3K β and inhibited Akt in cell based assays with IC50 76nM (8-fold less than TGX221, IC50 10nM). Oral administration of 300 mg/kg inhibited Akt in PC3 xenografts y over 50% for 6h. Effective intratumoral concentration was calculated to be 2.1µM. When administered twice per day this compound inhibited Akt for 24h and delayed PC3 xenograft growth without evident toxicity.
Thienopyrimidines which are selective for PI3K δ inhibition were reported in mid 2012 (Figure 21).29 Their syntheses started from a morpholine substituted thieno chloro pyrimidinyl aldehyde which had been reported earlier.18 Reductive amination of the aldehyde using Na(OAc)3BH produced amino morpholino thieno pyrimidinyl chlorides (83). Depending on the aryl substituent desired, three different coupling sequences were used on the chloride. In some cases, the thienopyrimidinyl chloride (83) was converted to a SnBu3 reagent and used in a Stille coupling with aryl bromides. In some cases the chloride (83) was used in Suzuki cross couplings with aryl boronic acids or esters. With indole nitrogen nucleophiles C-N bond formation could be accomplished under base catalyzed conditions whereas if displacement of the Cl by a benzimidazole was desired then Buchwald-Hartwig conditions were used. Lead compounds produced here demonstrated inhibition of Akt phosphorylation in cell-based assays with IC50 between 0.1 µM and 0.4 µM and at least 19 fold specificity over α and β PI3K isoforms.
Figure 21.

Preparation of Aryl Thieno Pyrimidines from Chloro Thieno Pyrimidines.
Also in mid 2012, Zhai, Gong, and co-workers reported a series of new 4-morpholinothienopyrimidines bearing arylmethylenehydrazones at the 2-position (Figure 22).30 These compounds were prepared from a 2,4-dichlorothienopyrimidine and the morpholine was added first at the 4 position followed by the addition of hydrazine to the 2 position. Condensation of the hydrazines with aldehydes then lead to the target hydrazones (85).
Figure 22.

Preparation of Morpholino Thieno Pyrimidines Bearing Aryl Methylene Hydrazones.
The importance of the indole or benzimidazole substituent on the morpholinopyrimidine core for PI3K δ selectivity was first reported in July of 2012 (Figure 23).31 The core structure used here was a pyridopyrimidine (86) which was then substituted by an amine. The indole or benzimidazole (linked to the pyrimidine via carbon) was then added via its boronic acid or ester via a Suzuki coupling to produce 87. A compound (87, amine = α,α-dimethyl-4-piperidinemethanol, R1 = 5-F) that inhibited Akt phosphorylation at 40nM in cell based assays and demonstrated over 200 fold selectivity toward PI3Kδ compared to other PI3K isoforms was chosen for in vivo testing. Since PI3Kδ is necessary for antigen-receptor signaling in B cells, selective inhibitors could be therapeutically useful for treatments of autoimmune conditions. Tests in rodents showed over 80% oral bioavailability and plasma half-life over 4h, which justified further tests in animal models with the ultimate goal of developing new drugs for rheumatoid arthritis and other autoimmune diseases.
Figure 23.

Preparation of Indole Substituted Pyrido Pyrimidines.
Murray, Sweeney and co-workers reported the preparation of 4-morpholinopurines bearing benzimidazoles at the 2 position in the summer of 2012 (Figure 24).32 Initially, they noted that pyrimidines fused to nitrogen heterocycles rather than sulfur heterocycles provided much better selectivity for PI3Kδ inhibition over PI3Kα inhibition. A variety of nitrogen heterocycles were added to a formyl group at the 8 position of the purine nucleus and a compound bearing a tetrahydropyranylazetidine was taken on into screening in rats and dogs.
Figure 24.

Syntheses of 4-Morpholino Purines Bearing Benzimidazoles.
In the late summer of 2012, Blake and co-workers reported the preparation of a series of piperazine substituted cyclopentapyrimidines which were selective ATP competitive Akt inhibitors (Figure 25).33 The 6-chlorocyclopentapyrimidines were prepared first via condensation of semicarbazide with α-carboethoxycyclopentanones followed by conversion of the pyrimidinol to the pyrimidinyl chloride (93). The pyrimidinyl chloride was reacted with Boc protected piperazine followed by Boc deprotection and amine condensation with a variety of substituted phenylalanine amino acids to produce the targets (95).
Figure 25.

Syntheses of Piperazine Substituted Cyclopentapyrimidines.
Pyridonyl pyrimidines which were PI3Kα and mTOR dual inhibitors were prepared and reported in August 2012 (Figure 26).34 Synthesis of the optimum new compound reported commenced with 1-amino-3-chloro-5-methyl pyrimidine (96). This compound was brominated and then the free amine was cyclized to a pyyrole (97) using 2,4 hexanedione. The chloride was displaced with hydrazine and the free NH2 was doubly alkylated with 1,4-dibromobutane to produce 98. The pyridine ring was installed using Heck chemistry and this first required N-acylation with acryloyl chloride. The intramolecular Heck reaction proceeded in a fairly low yield but it produced the desired pyridinone (99). The pyrrole was then deprotected, the pyridine brominated, and the N-Boc protected pyrazole was added via Suzuki cross coupling to produce 101. One wonders here if Buchwald conditions35 might not have worked better since those conditions are known to be successful with heterocyclic substrates which tend to deactivate palladium catalysts and these authors had to take a low yield at the end of this synthetic scheme with these conditions.
Figure 26.

Synthesis of Pyridonyl Pyrimidines.
Thienopyrimidines were known pan PI3K inhibitors and pyrimidinones were known to be more selective for PI3K β so researchers from GSK combined these 2 observations and reported preparation of thiazolopyrimidinones which were selective PI3K β inhibitors (Figure 27).36 These thiazolopyrimidinones were made by one of two routes. One route used a 4-amino-2-(4-morpholinyl)-1,3-thiazole-5-carboxylate (102) and the other used the 5-carbonitrile (104). For the route which utilized the methyl ester (102), the amine and ester were cyclized by condensation with formamide. The N-H was then deprotonated and alkylated with a benzyl halide. The pyrimidinone was hydrolyzed to the amino amido thiazole which was acylated with propionyl chloride and then subjected to base catalyzed ring closure to provide the final substituted pyrimidinone (103). In the route which utilized the carbonitrile (104), the amine was first acetylated then alkylated with a benzyl halide. The resulting N-acetyl carbonitrile (105) was then cyclized to the pyrimidinone (106) using basic aqueous sodium perborate.
Figure 27.

Preparation of Thiazolopyrimidinones.
Also in August 2012, Liu and co-workers at Pfizer reported the synthesis of cyclic sulfones fused to the pyrimidine nucleus and these compounds were highly selective for mTOR kinase over PI3K α (Figure 28).37 3, 3’-Thiobispropanoate (107) was used in a Dieckmann condensation to produce a thiopyranone (108) which was cyclized with urea to produce the bicyclic pyrimidine in low yield (One wonders here if this synthesis could have been accomplished more efficiently using a benzenecarboximidaamide). The pyrimidine diol was then chlorinated to produce 109 and the optically active 2-methyl morpholine was added. The sulfide was oxidized to the sulfone (110) with oxone and the phenyl urea was added via a Suzuki cross coupling of the pinacol boronate to yield 111. One also wonders here if the Buchwald conditions which work better for heterocyclic substrates (although admittedly usually used for C-N bond formation) might not have also significantly improved yield for this C-C bond formation with reactants containing a host of heteroatoms.35a,38
Figure 28.

Syntheses of Bicyclic Sulfonyl Pyrimidines.
September 2012 saw the publication of more morpholino thieno pyrimidines by Heffron and co-workers from Genentech and these thienopyrimidines were designed to be selective for PI3Kα and capable of penetrating the blood brain barrier (Figure 29).39 Almost all of the compounds in this study were prepared from the previously reported morpholino thieno pyrimidine monochloride (112).18 To prepare the most active compounds that contained azetidine or ether substituents off the thiophene ring, the thiophene was first metalated with BuLi. Quenching the Li salt with iodine and cross coupling with an iodo azetidine yielded a pyrimidinyl chloride (113) which was subsequently cross coupled with a pinacol borate of the appropriate amino pyrimidine to produce 114. Quenching the Li salt with ketones yielded alcohols containing chloropyrimidines that were directly cross coupled to pinacol borates of amino pyridines or the alcohols were converted to ethers then cross coupled to yield 115.
Figure 29.

Syntheses of Morpholino Thienopyrimidines Designed to Cross the Blood Brain Barrier.
A lead compound (115, R1 = R3 = Me, R2 = -CH2OCH2-) demonstrated 62% inhibition of pAkt in brain 1h afer 50mg/kg dose that achieved 3.6µM in mouse brain. Although the compound was not tested on intracranially implanted xenografts, the authors extrapolated that it will inhibit tumor growth based on tumorostatic effects in subcutaneously implanted U87 xenografts in which concentrations were at 2µM level.
In September 2012, Giordanetto and co-workers at AstraZeneca reported a series of morpholino-pyrimidinones which were selective PI3K β inhibitors (Figure 30).40 They started this synthetic work by treating 2,4,6-trichloropyrimidine (116) with p-methoxy benzylalcohol (117) to generate 2 PMBO isomers (118–119). They then treated that mixture of isomers with morpholine to replace a 2nd chloride and separated the three monochloro isomers (120–122) that were produced by silica chromatography. Buchwald-Hartwig Pd catalyzed C-N bond formation was then performed on each of the monochloro isomers followed by TFA catalyzed PMB ether cleavage to liberate the pyrimidinones (123–125).
Figure 30.

Syntheses of Morpholino Pyrimidinones.
Following this work a little later in 2012, Giordanetto and co-workers published a follow up report of synthetic routes to two specific classes of pyrimidinones that are selective PI3K β inhibitors (Figure 31).41 One family of compounds was prepared by starting from 4,6-dichloropyrimidin-2-amine (126). The amine was first alkylated with 1-(bromomethyl)naphthalene and then the dichloride was hydrolyzed to the monochloropyrimidinone (128) using aqueous base. The chloride was then replaced with amines in SNAr reactions or in some cases cross coupled with aryl boronic acids to yield targets (129). In either case yields for this last step were generally poor making one wonder if the order of these last two steps couldn’t have been reversed to synthetic advantage.
Figure 31.

Syntheses of Diamino Substituted Pyrimidinones.
The second family of compounds reported here were prepared in a one pot reaction from 4,6-dichloropyrimidin-2-ol (130) (Figure 32). One chlorine was replaced by addition of a napthyl amine and then the second chlorine was replaced by an addition of excess morpholine to produce 131. In vivo testing in dogs and rats did not show increased bleeding or inhibition of glucose uptake typical for PI3Kα inhibitors. However inhibition of PI3Kβ activity was not directly tested in vivo.
Figure 32.

Syntheses of Amino Morpholino Pyrimidinones.
A one pot multicomponent synthesis of quinazolinones which were designed to be selective PI3K-δ inhibitors was also reported in Fall 2012 (Figure 33).42 To prepare these compounds a substituted 2-aminobenzoic acid (132) was first condensed with 2-chloroacetylchloride and then the quinazolinone core (134) was formed via reaction with an aniline. Adenine was then used to displace the primary chloride that remained thus producing a family of purine quinazolinone derivatives (135).
Figure 33.

Syntheses of Quinazolinones.
In 2010, Lin and co-workers at Pfizer reported a series of quinazolines which we include here rather than with the pyrazines even though they were based on a pteridinone core (which contains a pyrazine) and use an intramolecular hydrogen bonding scaffold to mimic the pteridinones (Figure 34).43 2-Bromoaniline (136) was condensed with trichloroacetaldehyde and hydroxylamine to give isonitrosoacetanilide which was subjected to intramolecular Friedel-Crafts acylation. Oxidative decarboxylation of the ketoamide (137) yielded the amino bromo methyl benzoate. The ester was hydrolyzed to 138 and treated with acetic anhydride and ammonium acetate to prepare the quinazoline core (139). One wonders here if the reaction of acetamidine with 2,3-dibromobenzoic acid could also be used to prepare the desired core. The hydroxyl group was then converted to the chloride with POCl3 and then displaced with ammonia. Carbonylation of the bromide lead to an ester (140) which was hydrolyzed and condensed with amines to generate the final target amides (141).
Figure 34.

Synthesis of Pteridinone Mimics.
3. Pyridines, Quinolines and Indoles
Most compounds reported since 2010 with the pyridine/quinoline core structure have been designed to be selective for some target other than PI3Kα in the PI3K/mTOR kinase family. A compound containing a bis quinoline core (Torin 1) was reported in 2010 as a selective mTOR inhibitor.44 The Torin 1 core structure was further refined and Torin 2 which was based on a pyridinyl quinoline core and was about 1000 fold more selective for mTOR over PI3Kα was reported in early 2011 (Figure 35).45 The syntheses of all the compounds reported in this work commenced with 6-bromo-3-carboethoxy-4-chloro-quinoline (142). The chloride was replaced via a SNAr reaction with anilines to produce 143 and then the ethyl ester was reduced to the alcohol via a nitrogen assisted/directed NaBH4 reduction. The alcohol was oxidized to the aldehyde (144) and the carbon chain extended via Horner-Wadsworth-Emmons olefination. The ester generated by that reaction underwent intramolecular cyclization to produce the pyridinone (145). Suzuki cross coupling of the remaining aromatic bromide provided the final products (146). No yields were reported for these transformations in the text or the supplementary material though.
Figure 35.

Syntheses of Torins.
This same group of investigators also reported additional compounds in early 2011 based on the original (Torin 1) bis quinoline core but also containing a biaryl off the pyridinone.46 The target compounds were prepared via sequential Suzuki cross couplings off the bromo chloro compound shown below (147, R1 = Br, R2 = Cl) (Figure 36). The bromide on the CF3 substituted aromatic ring being most reactive and cross coupled first followed by the chloride. A lead compound prepared from 147 demonstrated improved in vivo half-life and bioavailability compared to Torin1 and preserved high specificity toward mTor. Pharmacodynamic studies showed that this compound blocked 80–90% phosphorylation of S6K (T389) and pAkt (S473) in liver and lung tissues even after 6 h at a dosage of 20 mg/kg.
Figure 36.

A Bishalopyridinone.
Also in 2011, Nishimura and co-workers at Amgen published the preparation of some biaryls containing pyridine and benzothiazoles which were pan PI3K/mTor inhibitors (Figure 37).11 They refined those compounds and later in 2011 reported a series of pyridyl quinolones and pyridyl quinoxalines which are selective PI3K δ inhibitors.47 All of the compounds reported here (150) were prepared via Suzuki cross coupling reactions of pyridinyl halides or boronates (148) with quinolinyl or quinoxalinyl halides or boronates (149) as appropriate.
Figure 37.

Preparation of Pyridinyl Quinolones and Quinoxalines.
In mid April 2011, a second Amgen group reported a different series of pyridinyl biaryls with the coupling partners to the pyridines in these cases all being 6,5-heterocyclic ring systems such as benzimidazoles, benzoxazoles, benzoisothiazoles, and triazolopyridines.48 Again all these compounds were prepared by coupling pyridinyl halides or boronates to the appropriate 6,5-heterocyclic halide or boronate similar to the 148 + 149 route to 150 shown above.
In the fall of 2011, Kendall and co-workers at the University of Auckland reported a series of pyrazolopyridines which were selective PI3Kα inhibitors (Figure 38).49 The compounds screened here were prepared via an initial N-amination of the pyridine ring using hydroxylamines with O substituents which make them good NH2 transfer reagents. These N-aminated pyridines were then used in 3 + 2 cycloaddition reactions with ethyl propiolate to produce 151. The ester was decarboxylated and then replaced with an aldehyde using a Vilsmeier reaction. Reaction of the aldehyde (152) with a hydrazine produced a hydrazone which was N-sulfonylated with an aromatic sulfonyl chloride to yield 153.
In late 2011, Hong and co-workers reported pyrimidinyl substituted pyridines which were selective PI3K β inhibitors (Figure 39).50 All the compounds in this study were prepared from 3-amino-5-bromopyridine (154) which was N-sulfonylated and then converted to the pinacol boronic ester (155). The pinacol boronate was then cross coupled to a variety of aromatic bromides, most of which were pyrimidinyl bromides to produce 156. Tissue culture assays showed inhibition of cell growth, but effects on the PI3K/Akt pathway were not presented.
Figure 39.

Preparation of Pyrimidinyl Pyridines.
Likewise in late 2011, 11C and 18F labeled versions of GSK2126458 (R1 = R2 = F) were synthesized and used for imaging PI3K and mTOR in cancers (Figure 40).51 The 2-18F and 4-18F compounds were prepared as well as the OMe compound substituted with a 11C labeled CH3. The synthesis involved sulfonylation of the amine in the amino pyridine (157) followed by Suzuki coupling of 159 with the quinolinyl boronate to produce 160.
Figure 40.

Synthesis of Labelled Pyridinyl Quinolines.
In work published in April52 and May of 201253, Ellard and co-workers at Cellzome disclosed a series of triazolopyridines (165) which were dual PI3K γ/δ inhibitors (Figure 41). To prepare these compounds, 5-bromopyridine-3-sulfonamides (161) were first cross coupled with amino pyridine boronic acid pinacol esters. The resulting amino pyridines (162) were then treated with ethoxycarbonyl isothiocyanate to produce a thiourea (163). Reaction of the thiourea with hydroxylamine produces an aminotriazole (164) and the free amine was converted into a number of different ureas (165).
Figure 41.

Preparation of Triazolopyridines.
These efforts resulted in identification of CZC24832 (164, R = t-Bu, R1 = F), a first selective inhibitor of PI3Kγ that showed significant dose-dependent effects in the mouse model of arthritis upon oral administration at 3 and 10 mg/kg for two weeks. Further elaborations on these structures produced lead compounds that showed rapid in vivo clearance, and were evaluated in a mouse model of LPS-induced pulmonary neutrophilia, as preliminary surrogate for asthma. At 0.5 mg dose as intratracheal inhalant compounds, they showed significant inhibition (37% and 34%) of neutrophil accumulation. Compound CZC24758 (164, R = t-Bu, R1 = H) showed significant dose-dependent effects in a clinical arthritis mouse model (30% and 42% improvement at oral doses of 3 and 10 mg/kg).
In the summer of 2012, a number of triazolopyridines which are selective PI3K γ inhibitors were reported (Figure 42).54 The starting material used here was 2-amino-5-bromopyridine (166). This compound was treated with ethoxy carbonyl isothiocyanate to produce an intermediate thiourea (167) which was cyclized to the triazolopyridine (168) using hydroxylamine. The free NH2 was then doubly acetylated and then hydrolyzed back to the monoacetate (169). The bromide was then cross coupled with aryl boronic acids or converted to the pinacol boronic ester and cross coupled with aryl bromides to yield the desired products (170). Two compounds with core structure (170) were given orally at 10mg/kg and showed 53% and 38% reduction of clinical symptoms in the mouse model of collagen-induced arthritis, however degree of inhibition and PI3K isoform selectivity was not assessed in vivo.
Figure 42.

Preparation of Triazolopyridines.
In work that was initiated with a starting material similar to 166, Choi, Hong and co-workers reported the preparation of imidazopyridine derivatives with enhanced selectivity for PI3Kα over Akt1 (Figure 43).55 To prepare these compounds the 5-bromopyridine-2-amine starting materials (171) (X = F in the most selective inhibitor) were converted to the amidine (172) which was alkylated and cyclized with bromoacetonitrile. The nitrile (173) was then converted to the oxadiazole (174) via treatment with hydroxylamine followed by acetic anhydride. The amino pyridine was then added by Suzuki coupling and the amine was sulfonylated with benzene sulfonyl chloride to produce 175.
Figure 43.

Syntheses of Imidazopyridines.
Turiso and co-workers reported a series of indolinyl pyridyl substituted quinolones which were dual PI3Kβ/δ inhibitors (Figure 44).56 These inhibitors were prepared from 2,4-dichloroquinolines (176). The pyridyl substituents were added via Stille coupling at the 2 position and then the indolines were added at the 4 position via NaH deprotonation followed by a SNAr reaction. The most active compound (179, X’ = morpholine, X= 7-F) was taken into a model of rheumatoid arthritis in rats.
Figure 44.

Synthesis of Indolinyl Pyridinyl Quinolines.
In late 2012, Zhong and co-workers at the University of Nebraska prepared a number of quinolone-carboxamides for use as PI3K α inhibitors (Figure 45).57 Most of the compounds reported (182) were prepared via a multicomponent coupling reaction of two equivalents of aniline (181) with triethyl methane tricarboxylate (180).
Figure 45.

Preparation of Quinolone-Carboxamides.
The purpose of this synthetic chemistry was to generate an inhibitor selective for PI3K with a H1047R mutation in kinase domain reported in 15–32% of breast, endometrial , colon, brain and upper digestive tract tumors that are characterized by a 2 fold increase in kinase activity. Comparison of Akt phosphorylation in colon carcinoma cells with wild type PI3K and H1047R mutant identified several compounds that inhibited mutant PI3K more effectively at 10–150 µM concentrations.
Pyridazino indoles were reported by Loge and co-workers as PI3Kα inhibitors in late 2012 (Figure 46).58 To prepare these compounds, indole 2-carboxylates (183) were subjected to Friedel-Crafts acylation followed by reaction with hydrazine to make the pyridazino indole core (185). The indole nitrogen was then alkylated with a variety of benzyl bromides under basic conditions to produce 186.
Figure 46.

Synthesis of Pyridazino Indoles.
4. Pyrazines and Quinoxalines
Given the known pan PI3K and mTOR inhibiting ability of a host of pyrimidines it is not surprising that a number of pyrazines and quinoxalines would also perform well as kinase inhibitors.
In 2011, Mortensen and co-workers at Celgene reported a series of imidazo pyrazinones which were selective mTOR inhibitors (Figure 47).59 The compounds screened here were made by one of two methods. One synthetic route began with 3,5-dibromopyrazin-2-amine (187) and used a selective nucleophilic aromatic substitution reaction followed by heating with carbonyl diimidazole or urea then Pd catalyzed cross coupling with the appropriate boronic acid to produce 190. For some substrates, a second route using 5-bromo-pyrazin-2-amine (191) was employed and that compound was cross coupled with the appropriate boronic ester. If the boronic ester aromatic group had sterically demanding substituents, then this route was used and the pyrazine 2-amine that was produced (192) was selectively brominated. The amine was then doubly protected with Boc groups to produce 193 and then cyclized to the imidazopyrazinone (190) by microwave heating with the appropriate amine. Inhibition of mTORC1 (IC 0.2 µM) and mTORC2 (IC 0.05µM) signaling was demonstrated in PC3 cell assays using phosphorylation of p70S6 and Akt as the readout (190, R = 4-methylenepyranyl, Ar = 4-(2’-imidazolyl)phenyl).
Figure 47.

Synthesis of Imidazopyrazinones.
Hu and co-workers reported preparation of a series of arylamino aryl sulfonyl quinoxalines (196) as PI3Kα inhibitors (Figure 48).60 The synthetic route used started with benzene-1,2-diamine (194) which was condensed with diethyl oxalate. A quinoxaline dione was produced which was chlorinated with SOCl2 to produce 195. Somewhat surprisingly, these authors then found that treatment of the dichloroquinoxaline with aryl sulfonyl hydrazides yielded mono chloro arylsulfonyl quinoxalines. The last chloride was replaced by nucleophilic aromatic substitution with aryl amines to yield 196. Inhibition of Akt phosphorylation by the most effective compound (196, Ar1 = Ph, Ar2 = 3,5-dimethoxyphenyl) in class 196 was demonstrated in PC3 cells at 10µM.
Figure 48.
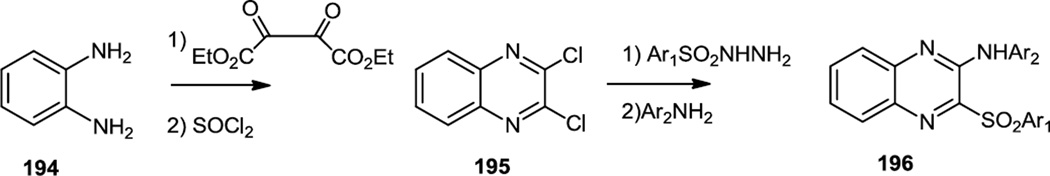
Preparation of Aryl Sulfonyl Quinoxalines.
Analogs of the selective PI3Kδ inhibitor AS605240 were prepared and tested in human and rat glioma cell lines in 2011 (Figure 49).61 To prepare these compounds, quinoxaline-6-carbaldehyde (197) was synthesized as described previously and subjected to base catalyzed aldol-dehydration reaction conditions with acetophenones to produce 198.
Figure 49.

Synthesis of Substituted Quinoxalines.
Crew and co-workers reported preparation of imidazopyrazines which were designed to be orally bioavailable mTOR specific inhibitors (Figure 50).62 The proof of concept compound, which was dosed orally in mice, was prepared starting from 1-(3-chloropyrazin-2-yl)methylamine (199). The pyrazinyl amine was treated with the carboxylic acid to effect amide formation then POCl3 to mediate cyclization. Treatment of the chloroimidazopyrazine (201) with NIS and ammonia generated the aminoiodoimidazopyrazine (202). Palladium catalyzed cross coupling was used to add the indole, then the Cbz protecting group was removed and the final carbamate (203) was prepared using carbamoyl chloride.
Figure 50.

Synthesis of Indolinyl Imidazo Pyrazines.
In late 2011, Pastor and co-workers at the Spanish National Cancer Research Center reported a series of imidazopyrazines (208) which were designed to be selective PI3K α and γ inhibitors (Figure 51).63 The synthetic route to these compounds started with bromination of 2-aminopyrazine (204) followed by replacement of the Br ortho to the amino group with morpholine. Reaction of that compound (205) with ethyl bromo pyruvate produced the imidazopyrazine core (206). The remaining aromatic bromide was substituted by various aromatic boronic acids via Suzuki couplings to produce 207 and then treatment with amines produced the target amides (208). Analysis in osteosarcoma U2OS cells showed inhibition of Akt phosphorylation at EC50 of 138nM by an active compound with structure 208 (Ar = 4-indazole, R = H), was still less active compared to GDC-0941, that was used as the prototype compound for the synthesis.
Figure 51.

Syntheses of Morpholino Imidazopyrazines.
Murphy and co-workers reported a series of pyrrolopyrimidinyl pyrazinyl methanones which were designed to be selective for PDK1 over PI3Kα Figure 52).64 The pyrrolopyrimidinyl pyrazinyl core was prepared using previously reported chemistry and the new compounds reported here (210) were prepared via SNAr reactions on a pyrazinyl chloride (209) so these compounds are included in the pyrazine section.
Figure 52.

Preparation of Pyrrolo Pyrimidinyl Pyrazinyl Methanones.
In early 2012, Hu and co-workers reported additional arylsulfonylquinoxalines (196).65 The amines used in this case were cyclic aliphatic amines rather than aromatic amines but the synthetic route used was analogous to their earlier report.60 The most potent compounds inhibited Akt phosphorylation in PC3 cells at 10µM.
Pastor and co-workers reported additional imidazopyrazines in 2012 (Figure 53).66 The compound selected for in vivo studies in this work was prepared by the same route they had reported earlier but using an amino pyrimidinyl boronate for the final coupling step.63 They also reported a new synthetic route here to some related compounds and this route commenced with amino chloro pyrazine (211) (Figure 53). This compound was iodinated with N-iodosuccinimide and then the imidazopyrazine core (212) was constructed by condensation with ethyl bromo pyruvate.
Figure 53.

Preparation of Additional Imidazopyrazines.
Compound ETP-46231 (213 prepared from 212) demonstrated inhibition of Akt phosphorylation in U2OS cells in tissue culture was selected for in vivo tests. Tests in BALB-c mice showed favorable pharmacokinetics. Analysis in the CEMM lung carcinoma model showed inhibition of Akt in tumor tissue by 75% 2 h after oral administration of 50 mg/kg of ETP-46231 and inhibition of tumor growth by 51% 3 weeks after same daily oral dose of ETP-46231.
In mid-2012 Johnson and co-workers ar Exelixis reported a series of sulfonyl piperazinyl pyrazines (216) and aryl sulfonamidyl pyrazines (220) which were selective PI3K γ inhibitors (Figure 54).67 The sulfonyl piperazinyl pyrazines were prepared via a base catalyzed substitution reaction of the sulfonyl piperazine and 2,3-dichloropyrazine (214) followed by a Pd(II) catalyzed cross coupling reaction with an aromatic boronic acid to replace the 2nd chloride and produce 216. The aryl sulfonamidyl pyrazines were prepared via an initial Sandmeyer reaction on bromo anilines (217). Reaction of the product sulfonyl chlorides (218) with amines followed by conversion to the boronate and Pd(II) catalyzed cross coupling with pyrazinyl bromides produced the final products (220). The lead compound (220, R1 = Cl, R2 = 2,3-difluorophenyl) was tested in vivo and demonstrated 50% inhibition of neutrophil migration when given orally 100mg/kg, which is dependent on PI3Kγ activity.
Figure 54.

Preparation of Aryl Sulfonamidyl Pyrazines.
In the summer of 2012, Pastor and co-workers published their third paper in a series, this time reporting aminocarbonyl imidazopyrazines (222) which were selective mTOR inhibitors (Figure 55).68 They used their earlier reported route to make the morpholino imidazopyrazinyl ester (221) then cross coupled it with an aminopyrimidine boronate followed by Lewis acid catalyzed conversion of the ester to a variety of amides (222).66
Figure 55.

Syntheses of Aminocarbonyl Imidazopyrazines.
In vivo studies with lead compound ETP-46992 (222, R1 = Me, R = CH2CH2OCH3) demonstrated 80% inhibition of Akt phosphorylation in mouse GEMM lung carcinoma at 10mg/kg given orally and inhibited tumor growth by 61% after 3 weeks with daily dose of 25mg/kg.
5. Azoles
Azoles, (diazoles, triazoles, imidazoles, thiazoles, oxadiazoles, etc.) are members of the five member ring heterocycle class and are all also known to be core structures of PI3K inhibitors.
Akt inhibitor IV contains a benzimidazole core and was reported in mid 2010 although its exact kinase targets were unclear at the time (Figure 56).69 The benzimidazole core (226) in this compound as well as a variety of analogs was prepared via a ZrCl4 mediated condensation of arylene diamines (225) with α, β-unsaturated aldehydes. The synthetic chemistry reported in this paper commenced from either 4-chloro-3-nitro benzaldehyde (223, R1 = CHO) or the corresponding benzoic acid. The aldehyde or acid was, in many cases, reacted with a nucleophile or nucleophiles to produce a substituted chloro nitro aromatic. The chloride was replaced by an amine to produce 224 and the nitro group reduced to a primary amine (225). The diamine (225) thus produced was then condensed with an aldehyde followed by oxidation or condensed with a benzoic acid derivative followed by N-alkylation to produce the target benzimidazole structures (227).
Figure 56.

Syntheses of Benzimidazole Salts.
Late in 2010, Huang and co-workers at Pfizer reported a large scale synthesis of some PI3K inhibitors which contained a triaryl-benzene-thiophene-triazole core (Figures 57 & 58).70 The critical part of these syntheses involved finding Suzuki coupling conditions that would convert the sterically hindered tetra substituted thiophene (228) into a biaryl benzene thiophene (229). Pd(P(tBu)3)2 and CsF proved to be the optimal catalyst/base combination for this cross coupling. The ester (229) was then hydrolyzed to the acid and the acid activated with 2-chloro-4,6-dimethoxy-[1,3,5]-triazine (CDMT) prior to treatment with ammonia to produce the amide (230). The amide was treated with N,N-dimethyl formamide dimethyl acetal (DMF-DMA) followed by slow addition of hydrazine to generate the target triazoles (231).
Figure 57.

Syntheses of Biaryl Benzene-Thiophenes.
Figure 58.

Syntheses of Triaryl Benzene-Thiophene-Triazoles.
In summer of 2011, Liu and co-workers at Pfizer used this optimized route reported above with Ar = 4-chlorophenyl (231) to prepare a PI3Kα inhibitor which was 7000 times more selective for PI3Kα over mTOR.71
In late summer 2011, Kendall and co-workers at the University of Auckland reported a series of pyrazolo pyridines (233) which were synthesized via condensation of a pyrazolopyridine aldehyde (232) with a series of benzene sulfonohydrazides (Figure 59).72 The resulting hydrazones (232) of the pyrazolopyridines were selective PI3K α inhibitors.
Figure 59.

Syntheses of Pyrazolopyridines.
In late 2011, Zhu and co-workers reported a series of oxadiazole derivatives which were PI3Kγ inhibitors (Figure 60).73 To prepare these compounds, aromatic hydrazides (234) were condensed with aromatic aldehydes to produce hydrazones which were cyclized to oxadiazoles (235) by treatment with acetic anhydride. A lead compound with structure 235 (R1 = OAc, R2 = Cl, R3 = H, R4 = Cl) was selected based on immunosuppressive effects on lymph node cells and was shown to inhibit PI3Kγ with IC50 1.75µM in vitro and inhibited Akt phosphorylation in vivo at 30µM.
Figure 60.

Syntheses of Oxadiazoles.
Zhu, Gong, and co-workers reported additional oxadiazoles (237) in early 2012, also targeting these compounds to PI3K γ (Figure 61).74 In this study the hydrazides (236) used were prepared from salicylic acids. Those hydrazides were then reacted with CS2 to produce oxadiazole thiols and the thiol was alkylated with a series of substituted benzyl bromides to produce 237. The most active compound with core structure 237 (R1 = Me, R2 = 4-NO2) with the highest immunosuppressive activity inhibited PI3Kγ at 0.31µM.
Figure 61.

Syntheses of Additional Oxadiazoles.
The same synthetic scheme reported above was also used to make oxadiazole derivatives from vanillic acid rather than salicylic acid and this work was reported in May of 2012.75 The resulting compounds selected based on in vitro immunosuppressive activity inhibited PI3Kγ at concentrations under 10µM.
Ding and co-workers reported the syntheses of a series of thiazole carboxamides where the thiazole also contained aminopyrimidinyl, pyrazolyl, or azaindolyl substituents (Figure 62).76 These syntheses utilized ethyl thiazole-5-carboxylate (241) or ethyl-2-bromo-thiazole carboxylate (238) as starting materials. When the bromo thiazole (238) was used as starting material, it was cross coupled to a nitrogen heterocycle and the ester was converted to the acid then the amide (240). When the carboethoxythiazole (241) was used, it was condensed with acetaldehyde and oxidized to prepare the acetyl thiazole (242). The acetyl group was then converted to the β-dimethylamino α,β-unsaturated acyl (243) using DMF-DMA. Treatment of that compound with hydrazine yielded the pyrazole substituted compounds (244) and treatment with methyl guanidine yielded the pyrimidinyl substituted thiazoles (245).
Figure 62.

Synthesis of Substituted Thiazole Carboxamides.
Similary, Bruce and co-workers reported syntheses of a number of different trisubstituted thiazoles which were designed to be selective for PI3K α, δ, or γ depending on the thiazole substituents (Figure 63).77 Some compounds were prepared from 2-amino-5-bromo-4-methylthiazole (246) via Suzuki coupling and N-acylation and some compounds were prepared from 3-aryl-2-propanones (248) which were halogenated and treated with thiourea to generate 2-amino-5-aryl-4-methylthiazoles which were also then N-acylated to yield 249.
Figure 63.


Synthesis of Trisubstituted Thiazoles.
In the fall of 2012, a series of biaryl thiazole-oxazoles were prepared as selective PI3K γ inhibitors (Figure 64).78 The thiazole-oxazole core (252 & 254) was prepared by one of three routes and all three used bromoacetyl thiazole (250) as the starting material.
Figure 64.

Preparation of Biaryl Thiazole-Oxazoles.
All three routes first used the reaction of azide anion with the bromoacetyl (250). The azide (251) could be reduced in situ with phosphine and reacted with an acid chloride to form the oxazole (252) or reduced to the amine (253) which was subsequently reacted with acid chloride. Condensation of the amine (253) with CS2yielded a thiol substituted oxazole (254) which was also converted into a number of sulfur substituted derivatives.
We also include here a report of the preparation of some thienobenzoxepin PI3K inhibitors since the most active compound in the series contained a thiophene in the core structure.79 The preparation of this most active compound was not reported but we assume it was prepared via the bromo keto benzoxepin (48) similar to how the compounds in ref [20] were prepared. The most active compound (255, GNE-614) inhibited PI3Kα,δ, and γ with all IC50s under 5mM. Tests in MCF7-neo/HER2 xenografts showed intratumoral concentrations of 1 µM one hour after oral dose 50 mg/kg, and inhibition of phosphorylations of Akt PRAS40 and S6PR comparable to GDC-0941, a compound that is currently in 13 clinical trials for advanced cancers.
5. Triazines
2-(Difluoromethyl)-1-[4,6-di(4-morpholinyl)-1,3,5-trazin-2-yl]-1H-benzimidazole (ZSTK 474) (256) was first reported as a potent ATP competitive pan class I PI3K inhibitor in 2006 (Figure 66).80 Not surprisingly, synthetic work on PI3K inhibitors based on the triazine or triazine-benzimidazole core structure has continued recently though not at the same pace as new reports based on purine/pyrimidine core structures.
Figure 66.

ZSTK 474.
In 2010, a series of bis(morpholino)triazines bearing bis aryl urea groups (261) were synthesized and shown to inhibit all class I PI3K isoforms as well as mTOR (Figure 67).81 The most potent compound in the series was 1-(4-phenyl)-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea (PKI-587). This compound was extensively tested in tissue culture and xenografts cancer models.82 In tissue culture of MDA-MB361, PKI-587 completely inhibited Akt phosphorylation at 30nM. Experiments in MDA-MB361 xenografts showed complete inhibition of Akt phosphorylation for 36 h by 25 mg/kg of PKI-587 injected iv. Dose-response analysis showed maximal anti-tumor effects at 10 mg/kg. Inhibition of Akt phosphorylation and xenograft tumor growth was also observed in HCT116 colon carcinoma, H1975 (non-small cell lung cancer) and U87 (glioma) models.
Figure 67.

Syntheses of Bis(morpholino) Bisarylurea Triazines.
Synthesis of these bis aryl urea substituted bis (morpholino) triazines starts with SNAr displacement of 2 of the chlorines in commercially available cyanuric chloride (257). Whereas there are prior reports of effecting these reactions of cyanuric chloride in DMF or CH2Cl2 using K2CO3 or amine bases, those reactions conditions require an extraction for workup.83 The preparation used here in 1:5 acetone: ice water with an amine base provided the product (258) by a simple precipitation and filtration. The last chlorine was then replaced with a 4-aminophenyl group via a Suzuki cross coupling. The resulting aniline (259) was converted to a urea through a nucleophilic addition to methyl 4-isocyanatobenzoate. The methyl ester was hydrolyzed and the carboxylic acid (260) was converted to the final amide (261) by treatment with 4-NMe2piperidine, Hunig’s base and HBTU (the uronium salt of 1-hydroxy-benzotriazole (HOBt) with a nonnucleophilic counterion (PF6−) in NMP.
Synthesis of triazine-benzimidazoles (265) which were selective for mTOR inhibition over PI3K inhibition were reported in 2011 (Figure 68).84 In these syntheses, cyanuric chloride (257) was originally treated with MeMgBr which results in a monomethylated dichloro triazine (262) in very high yield. This dichloride was then subjected to a SNAr reaction using 2-chloro benzimidazole (263). The mono chloro triazine mono chloro benzimidazole product (264) was then treated with ammonia or a primary amine followed by an aromatic amine. The most promising lead compound (265) was prepared using methylamine to displace the last chloride on the triazine core followed by 3-aminopyrazole to react with the chloro benzimidazole. In tissue culture experiments this compound inhibited proliferation of 18 cancer cell lines with IC50 uner 1 µM. In vivo tests demonstrated inhibition of HGF-stimulated Akt phosphorylation in liver at 30 mg/kg and phosphorylation of S6RP at 100 mg/kg.
Figure 68.

Syntheses of Triazine Benzimidazoles.
After the report of PKI-587 in 2010, some 2-oxatriazines with similar pan PI3K/mTOR dual inhibitor profiles were synthesized (Figure 69).85 These compounds were synthesized using a reaction sequence similar to that reported for PKI-587. Cyanuric chloride (257) was treated with morpholine followed by treatment with a lithium alkoxide to generate mono chloride (266). Microwave assisted cross coupling with 4-aminophenylboronic acid pinacol ester generated triazinyl aniline (267). Treatment of that aniline with triphosgene was then followed by addition of the second aniline for urea formation (268). A lead compound with structure 268 (R = 3-tetrahydrofuranyl, R1 = Me, R2 = CH2CH2NMe2) was compared with PKI-587 in vivo and showed less efficacy in inhibiting phosphorylations of Akt and S6 and p70S6K in MDA-MB361 xenografts when administered at either 10 or 25 mg/kg.
Figure 69.

Syntheses of 2-Oxatriazines.
Also in 2011, the synthesis of a number of analogs of ZSTK 474 were reported (Figure 70).86 The analogs fell into 2 major classes which then affected the synthetic pathway used. One set of analogs were prepared by using benzimidazoles which were substituted at the 2,4,5, and/or 6 position. A second set of analogs was then prepared where a pyrimidine core was used in place of the triazine core. Once the benzimidazoles or pyrimidines of interest were prepared, the syntheses of the final products followed one of two routes: 1) The monomorpholine triazine (270) was treated with the benzimidazole (269) and K2CO3 in DMF followed by the addition of the second morpholine in DMF or 2) the dimorpholino triazine monochloride (258) was prepared and then the benzimidazole (269) was added using NaH or K2CO3 under more forcing conditions. Since the chlorine in the 2 position of 2,4,6-trichloropyrimidine is always replaced first by nucleophilic substitution reactions, the desired substituents in that position drove the synthetic scheme used to prepare those analogs.
Figure 70.
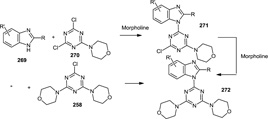
Syntheses of ZSTK 474 Analogs.
Two lead ZSTK474 derivatives (272) were identified by growth inhibition assays and inhibition of Akt phosphorylation in HCT116 cells and were given at 15, 40 and 50 mg/kg by ip injections and delayed growth of U87 xenografts in dose-response fashion yet showed significant toxicity.
In 2012, a series of pyridinyl substituted triazines was reported with compound (275, X = Y = H) being most active and most selective for PI3Kα over mTOR (Figure 71).38 The compounds in this study were prepared via sequential nucleophilic additions to cyanuric chloride (257) followed by a Pd catalyzed coupling of that mono halo triazine (273) to an aryl boronic acid or an aryl stannane. In the case of the most active compound (275), cyanuric chloride was treated with MeMgBr and NH3 followed by p-methoxybenzyl chloride to generate the PMB protected monohalide (273). That monohalide was then cross coupled with the boronic acid (274) using cat. Pd(Amphos)2Cl2 and KOAc in EtOH/H2). The free nitrogen was mesylated with methane sulfonyl chloride and the PMB protecting groups were then removed with TFA/TfOH to yield 275. Lead compound 275 (X = Y = H) provided orally demonstrated robust inhibition of HGF-induced Akt phosphorylation in liver at both 25 and 75 mg/kg for 8h. The higher dose maintained inhibition for 24h at 64%. Stabilization of U-87 tumor growth was achieved at 25mg/kg without significant toxicity. In late summer of 2012, an optimized candidate (275, X = F, Y = Me) with a superior pharmacokinetic profile and dual PI3Kα/Akt inhibition was reported.87
Figure 71.

Syntheses of Pyridinyl Triazines.
Peterson and co-workers followed their 2011 report of the syntheses of triazine-benzimidazoles with this related report of the synthesis of triazine-imidazopyridines and –imidazopyridazines (Figure 72).88 The synthetic strategy used here was very similar to that reported previously in that methyl dichlorotriazine (262) was subjected to Pd catalyzed cross coupling with 2-chloroimidazopyridine (276) and then the last chloride on the triazine was replaced with NH3. Buchwald-Hartwig amination of the chloroimidazopyridine (277) with pyrazol-3-amine yielded the final product (278).
Figure 72.

Syntheses of Triazine Imidazopyridazines.
Lead compound 278 was tested in vivo and showed inhibition of HGF-stimulated Akt and S6PR in mouse livers at 30gm/kg 6h after oral administration. This dose given BID delayed U87 tumor growth by 82%.
Also in 2012, Wurz and co-workers reported new amino pyridinyl triazines (282) where the amino group in the amino pyridine had been substituted by a variety of pyridinyl sulfonamides (Figure 73).89 To prepare these compounds 4-chloro-6-methyl-1,3,5-triazin-2-amine (279) was cross coupled with 2-fluoropyridine boronic acids (280) and the fluorine was then displaced by a base promoted SNAr reaction using 5-amino-pyridinyl-3-sulfonamides.
Figure 73.

Synthesis of Amino Pyridinyl Triazines.
6. Liphagal and Analogs
Liphagal, a terpenoid metabolite isolated from a marine sponge has an unusual 6,7 ring system fused to a highly substituted benzofuran (286, R1 = R2 = OH, R3 = CHO) and it was isolated in 2006.90 It has attracted interest as a PI3K inhibitor because, like many of the synthetic inhibitors discussed above it is more potent than LY294002 and more selective than wortmannin and it has an unusual structure. Recently, an enantioselective total synthesis of Liphagal has been reported91 as well as the synthesis of a number of analogs.92
The enantioselective total synthesis featured a simultaneous acid catalyzed pinacol rearrangement, benzyl ether deprotection, and furan ring formation from trans clerodane diol (283) (Figure 74). This transformation was achieved using Pd on carbon and cationic Amberlyst resin under a hydrogen atmosphere in methanol to produce 284.
Figure 74.

Key Step of an Enantioselective Liphagal Synthesis.
The analog synthesis paper reported by Andersen and co-workers relied on a carbocation initiated cyclization of 285 using SnCl4 to construct the desired tetracyclic ring system (286) (Figure 75).
Figure 75.

Liphagal Analog Syntheses.
7. Wortmannin and Quercetin Analogs
Wortmannin (287) and its analogs are known to be irreversible pan-PI3K inhibitors (Figure 76).93 In 2010, succinic and adipic anhydride were used as linking groups to synthesize linked wortmannin-rapamycin conjugates.94 These ester linkers were then hydrolyzed in blood to release both compounds and provide a hybrid PI3K/mTOR inhibitor. In vivo testing of the lead compound demonstrated inhibition of AKT and S6K phosphorylation at 15 mg/kg iv and this same dose stabilized tumor growth in HT29 renal tumor model, whereas 10 mg/kg of rapamycin or 5mg/kg of a wortmannin derivative showed much weaker anti-tumor effects. Likewise stronger anti-tumor effects were observed in A498 renal xenograft model when the rapamycin-wortmannin conjugate compound or rapamycin were combined with bevacizumab.
Figure 76.

Wortmannin
LY-294002 is a quercetin analog (288, R = H). Welker and co-workers synthesized an analog of LY-294002 containing a primary alcohol linking group off the phenyl ring in the biaryl core (288, R = CH2OH) (Figure 77).95 The biaryl core was produced by a Pd catalyzed cross coupling reaction. That primary alcohol (288) was then used as a linking point for a peptide sequence which is recognized and cleaved by prostate specific antigen (PSA) so the final synthetic target of this work (289) was designed to be a prodrug for prostate cancer (a similar approach using peptide linkers and TGX-221 was reported by Cheng et.al96 and Forrest et. al97 previously).
Figure 77.

Syntheses of an LY 294002 Analog and Subsequent Prodrug.
8. Inositol Phosphotriester Analogs
While not nearly as heavily investigated as the synthesis of ATP competitive inhibitors, additional syntheses of phosphatidyl inositol analogs have also been reported recently (Kozikowski’s group reported several 3-modified phosptidyl inositol analogs in the late 90’s and early 2000’s and these compounds were screened as PI3K and Akt inhibitors.98).
Conway and co-workers reported syntheses of four 5-position derivatives of inositol 1, 4, 5-triphopsphate in 2011 (Figure 78).99 They first prepared the 2,3,5-tri O-benzyl-5-O-allyl regioisomer of inositol (290). The 1 and 4 positions were phosphitylated with (BnO)2PN(iPr)2 and then the phosphorus was oxidized with mCPBA. The allyl protecting group at the 5 position was then removed with PdCl2 to produce 291. To prepare a methyl phosphonate analog (294), the 5-OH compound was treated with bis[(6-trifluoromethyl)benzo-triazol-1-yl]methyl phosphonate and then the benzyl groups were removed by hydrogenolysis. Similar treatment with SO3 as the electrophile generated the sulfonate (293). Oxidation of the allyl group followed by hydrogenolysis generated the carboxymethyl derivative (292).
Figure 78.

Synthesis of Inositol 1,4,5-Triphosphate Derivatives.
In 2012 Xi and co-workers reported the synthesis of a number of mono hydroxyl inositol compounds so that the hydroxyl could then be converted to a phosphoester functional group and then these compounds were screened as PI3K inhibitors and in several cancer cell lines (Figure 79).100 Syntheses of all analogs started with inositol (295) and the hydroxyls at the 1, 3, and 5 positions were protected via reaction with triethylorthoformate (296). Treatment of the triol thus produced with varying amounts of NaH and benzyl bromide (1, 2 or 3 equivalents) was then used to produce mono, di, or tri O-benzyl-myo-inositol-1,3,5-O-orthoformates (297–299). Treatment of the mono O-benzyl compound (297) with TBDMSCl and imidazole in DMF provided the 2-OTBDMS protected inositol with a free hydroxyl at the 4 position (300). The tri O-benzyl-1,3,5-O-orthoformate (299) was then treated with trimethyl aluminum or diisobutylaluminum hydride to selectively open the orthoformate and provide free hydroxyls at the 1 (3) (301) or 5 positions (302) of the inositol. The mono hydroxyl compounds thus produced were mono phosphorylated with iPr2NP(OC10H21)2 and the phosphorus was oxidized with mCPBA. Silyl protecting groups were removed with methanolic HCl and the benzyl protecting groups were removed by hydrogenolysis.
Figure 79.

Preparaton of Monohydroxyl Inositol Derivatives.
The growth inhibition effects of these compounds were determined against a number of cancer cell lines and the most active compound (305) was a di-O-benzyl tetra phosphate ester which was prepared via the 2-phosphorylated analog (303) via removal of the orthoformate and phosphorylation with iPr2NP(OCH3)2 followed by mCPBA oxidation (Figure 80).
Figure 80.
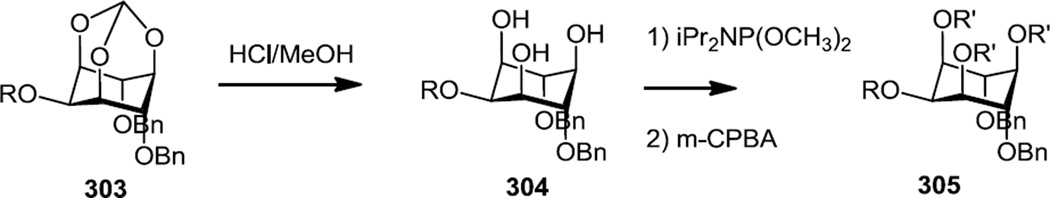
Preparation of a Tetra Phosphorylated Inositol Analog.
9. Conclusions
The vast majority of PI3K inhibitors currently available are reversible inhibitors. Since these reversible inhibitors must compete with ATP for the enzyme binding site many of these inhibitors contain core structures which bear some structural similarity to adenine. These adenine analog targets then dictate synthetic chemistry which relies heavily on combinations of nucleophilic aromatic substitution and Pd catalyzed cross coupling reactions. Since ATP is present at the micromolar level in cells that dictates that these reversible inhibitors will typically also have to be continually present at similar concentrations to be effective. Wortmannin is an irreversible PI3K inhibitor with the essentially irreversible step being a Michael addition of the Lys 833 NH2 in PI3K α onto an α, β-unsaturated lactone. Preparation of wortmannin is a difficult multi step synthesis so this makes one wonder if some of these reversible inhibitors with known PI3K binding geometries, easier synthesis, and better pharmacological properties (ie better water solubility and water stability than wortmannin) couldn’t also be converted into longer acting inhibitors by the addition of a Michael reaction acceptor or other soft electrophiles which would be positioned proximal to Lys 833 after inhibitor binding. If not, constant dosing with ATP mimics seems inevitable.
Figure 38.

Syntheses of Pyrazolopyridines.
Figure 65.

A Thienobenzoxepin
Acknowledgements
This work was supported by federal grant R01CA118329 from the National Cancer Institute to G. Kulik and by institutional grants from Wake Forest University Health Sciences to G. Kulik and from Wake Forest University to M. Welker.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mark E. Welker, Department of Chemistry, Wake Forest University, P.O. Box 7486, Winston-Salem, NC 27109, (USA), welker@wfu.edu, FAX: (336)-758-4656
George Kulik, Department of Cancer Biology, Wake Forest School of Medicine, Medical Center Blvd., Winston-Salem, NC 27157, USA, gkulik@wakehealth.edu.
References
- 1.(a) Wymann MP, Schultz C. ChemBioChem. 2012;13:2022. doi: 10.1002/cbic.201200089. [DOI] [PubMed] [Google Scholar]; (b) Sabbah DA, Brattain MG, Zhong H. Curr. Med. Chem. 2011;18:5528. doi: 10.2174/092986711798347298. [DOI] [PubMed] [Google Scholar]; (c) Ciraolo E, Morello F, Hirsch E. Curr. Med. Chem. 2011;18:2674. doi: 10.2174/092986711796011193. [DOI] [PubMed] [Google Scholar]; (d) McNamara CR, Degterev A. Future Med. Chem. 2011;3:549. doi: 10.4155/fmc.11.12. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Oncotarget. 2012;3:371. doi: 10.18632/oncotarget.477. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang P, Zhang L, Hao Q, Zhao G. Mini-Rev. Med. Chem. 2011;11:1093. doi: 10.2174/138955711797655380. [DOI] [PubMed] [Google Scholar]; (g) Shuttleworth SJ, Silva FA, Cecil ARL, Tomassi CD, Hill TJ, Raynaud FI, Clarke PA, Workman P. Curr. Med. Chem. 2011;18:2686. doi: 10.2174/092986711796011229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wu P, Hu YZ. MedChemComm. 2012;3:1337. [Google Scholar]; (i) de la Vega MD-CE, Alvarez RH. Future Med. Chem. 2012;4:893. doi: 10.4155/fmc.12.45. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Okada M, Ohta M, Tsukamoto S-i, Parker P, Workman P, Waterfield M. Bioorg.Med. Chem. 2006;14:6847. doi: 10.1016/j.bmc.2006.06.046. [DOI] [PubMed] [Google Scholar]; (b) Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJA, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. J. Med. Chem. 2008;51:5522. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 3.Chen Z, Venkatesan AM, Dehnhardt CM, Ayral-Kaloustian S, Brooijmans N, Mallon R, Feldberg L, Hollander I, Lucas J, Yu K, Kong F, Mansour TS. J. Med. Chem. 2010;53:3169. doi: 10.1021/jm901783v. [DOI] [PubMed] [Google Scholar]
- 4.Freeman-Cook KD, Autry C, Borzillo G, Gordon D, Barbacci-Tobin E, Bernardo V, Briere D, Clark T, Corbett M, Jakubczak J, Kakar S, Knauth E, Lippa B, Luzzio MJ, Mansour M, Martinelli G, Marx M, Nelson K, Pandit J, Rajamohan F, Robinson S, Subramanyam C, Wei L, Wythes M, Morris J. J. Med. Chem. 2010;53:4615. doi: 10.1021/jm1003842. [DOI] [PubMed] [Google Scholar]
- 5.Bencsik JR, Xiao D, Blake JF, Kallan NC, Mitchell IS, Spencer KL, Xu R, Gloor SL, Martinson M, Risom T, Woessner RD, Dizon F, Wu W-I, Vigers GPA, Brandhuber BJ, Skelton NJ, Prior WW, Murray LJ. Bioorg. Med. Chem. Lett. 2010;20:7037. doi: 10.1016/j.bmcl.2010.09.112. [DOI] [PubMed] [Google Scholar]
- 6.Pecchi S, Renhowe PA, Taylor C, Kaufman S, Merritt H, Wiesmann M, Shoemaker KR, Knapp MS, Ornelas E, Hendrickson TF, Fantl W, Voliva CF. Bioorg. Med. Chem. Lett. 2010;20:6895. doi: 10.1016/j.bmcl.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 7.Large JM, Torr JE, Raynaud FI, Clarke PA, Hayes A, Stefano Fd, Urban F, Shuttleworth SJ, Saghir N, Sheldrake P, Workman P, McDonald E. Bioorg. Med. Chem. 2011;19:836. doi: 10.1016/j.bmc.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Burger MT, Knapp M, Wagman A, Ni Z-J, Hendrickson T, Atallah G, Zhang Y, Frazier K, Verhagen J, Pfister K, Ng S, Smith A, Bartulis S, Merrit H, Weismann M, Xin X, Haznedar J, Voliva CF, Iwanowicz E, Pecchi S. ACS Med. Chem. Lett. 2010;2:34. doi: 10.1021/ml1001932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohwada J, Ebiike H, Kawada H, Tsukazaki M, Nakamura M, Miyazaki T, Morikami K, Yoshinari K, Yoshida M, Kondoh O, Kuramoto S, Ogawa K, Aoki Y, Shimma N. Bioorg. Med. Chem. Lett. 2011;21:1767. doi: 10.1016/j.bmcl.2011.01.065. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y. Clin. Cancer Res. 2011;17:3272. doi: 10.1158/1078-0432.CCR-10-2882. [DOI] [PubMed] [Google Scholar]
- 11.D’Angelo ND, Kim T-S, Andrews K, Booker SK, Caenepeel S, Chen K, D’Amico D, Freeman D, Jiang J, Liu L, McCarter JD, San Miguel T, Mullady EL, Schrag M, Subramanian R, Tang J, Wahl RC, Wang L, Whittington DA, Wu T, Xi N, Xu Y, Yakowec P, Yang K, Zalameda LP, Zhang N, Hughes P, Norman MH. J. Med. Chem. 2011;54:1789. doi: 10.1021/jm1014605. [DOI] [PubMed] [Google Scholar]
- 12.Kim D, Lee H, Jun H, Hong S-S, Hong S. Bioorg. Med. Chem. 2011;19:2508. doi: 10.1016/j.bmc.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 13.Heffron TP, Wei B, Olivero A, Staben ST, Tsui V, Do S, Dotson J, Folkes AJ, Goldsmith P, Goldsmith R, Gunzner J, Lesnick J, Lewis C, Mathieu S, Nonomiya J, Shuttleworth S, Sutherlin DP, Wan NC, Wang S, Wiesmann C, Zhu B-Y. J. Med. Chem. 2011;54:7815. doi: 10.1021/jm2007084. [DOI] [PubMed] [Google Scholar]
- 14.Burger MT, Pecchi S, Wagman A, Ni Z-J, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, Frazier K, Ng S, Smith A, Verhagen J, Haznedar J, Huh K, Iwanowicz E, Xin X, Menezes D, Merritt H, Lee I, Wiesmann M, Kaufman S, Crawford K, Chin M, Bussiere D, Shoemaker K, Zaror I, Maira S-M, Voliva CF. ACS Med. Chem. Lett. 2011;2:774. doi: 10.1021/ml1001932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QHC, Peters M, Goldbrunner M, Baselga J. J. Clin. Oncol. 2012;30:282. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 16.(a) Ren H, Chen MW, Yue P, Tao H, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. Cancer Lett. 2012;325:139. doi: 10.1016/j.canlet.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzman M, Grueso J, Rodriguez O, Calvo MT, Aura C, Diez O, Rubio IT, Perez J, Rodon J, Cortes J, Ellisen LW, Scaltriti M, Baselga J. Cancer Discov. 2012;2:1036. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, Scully R, Asara JM, Cantley LC, Wulf GM. Cancer Discov. 2012;2:1048. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanni P, Nicoletti G, Palladini A, Croci S, Murgo A, Ianzano ML, Grosso V, Stivani V, Antognoli A, Lamolinara A, Landuzzi L, di Tomaso E, Iezzi M, De Giovanni C, Lollini PL. PLoS One. 2012;7 doi: 10.1371/journal.pone.0039626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, Folks A, Friedman L, Goldsmith R, Gunzner J, Heffron T, Lesnick J, Lewis C, Mathieu S, Murray J, Nonomiya J, Pang J, Pegg N, Prior WW, Rouge L, Salphati L, Sampath D, Tian Q, Tsui V, Wan NC, Wang S, Wei B, Wiesmann C, Wu P, Zhu B-Y, Olivero A. J. Med .Chem. 2011;54:7579. doi: 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- 19.Wallin JJ, Edgar KA, Guan JE, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang JD, Salphati L, Olivero AG, Sutherlin DP, O’Brien C, Spoerke JM, Patel S, Lensun L, Kassees R, Ross L, Lackner MR, Sampath D, Belvin M, Friedman LS. Mol. Cancer Ther. 2011;10:2426. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard S, Soh CK, Lee CP, Poulsen A, Bonday Z, Goh KL, Goh KC, Goh MK, Pasha MK, Wang HS, Williams M, Wood JM, Ethirajulu K, Dymock BW. Bioorg. Med. Chem. Lett. 2012;22:2880. doi: 10.1016/j.bmcl.2012.02.058. [DOI] [PubMed] [Google Scholar]
- 21.Bussenius J, Anand NK, Blazey CM, Bowles OJ, Bannen LC, Chan DSM, Chen BL, Co EW, Costanzo S, DeFina SC, Dubenko L, Engst S, Franzini M, Huang P, Jammalamadaka V, Khoury RG, Kim MH, Klein RR, Laird D, Le DT, Mac MB, Matthews DJ, Markby D, Miller N, Nuss JM, Parks JJ, Tsang TH, Tsuhako AL, Wang Y, Xu W, Rice KD. Bioorg. Med. Chem. Lett. 2012;22:2283. doi: 10.1016/j.bmcl.2012.01.105. [DOI] [PubMed] [Google Scholar]
- 22.Rice KD, Kim MH, Bussenius J, Anand NK, Blazey CM, Bowles OJ, Canne-Bannen L, Chan DSM, Chen BL, Co EW, Costanzo S, DeFina SC, Dubenko L, Engst S, Franzini M, Huang P, Jammalamadaka V, Khoury RG, Klein RR, Laird AD, Le DT, Mac MB, Matthews DJ, Markby D, Miller N, Nuss JM, Parks JJ, Tsang TH, Tsuhako AL, Wang Y, Xu W. Bioorg. Med. Chem. Lett. 2012;22:2693. doi: 10.1016/j.bmcl.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Wang X, Chen Y, Chen S, Chen G, Tong L, Meng L, Xie Y, Ding J, Yang C. Bioorg. Med. Chem. Lett. 2012;22:339. doi: 10.1016/j.bmcl.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 24.Lin H, Erhard K, Hardwicke MA, Luengo JI, Mack JF, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Sanchez RM, Schaber MD, Schulz MJ, Spengler MD, Tedesco R, Xie R, Zeng JJ, Rivero RA. Bioorg. Med. Chem. Lett. 2012;22:2230. doi: 10.1016/j.bmcl.2012.01.092. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez RM, Erhard K, Hardwicke MA, Lin H, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Schaber MD, Spengler MD, Moore ML, Yu H, Luengo JI, Tedesco R, Rivero RA. Bioorg.Med.Chem. Lett. 2012;22:3198. doi: 10.1016/j.bmcl.2012.03.039. [DOI] [PubMed] [Google Scholar]
- 26.Finlay MRV, Buttar D, Critchlow SE, Dishington AP, Fillery SM, Fisher E, Glossop SC, Graham MA, Johnson T, Lamont GM, Mutton S, Perkins P, Pike KG, Slater AM. Bioorg. Med. Chem. Lett. 2012;22:4163. doi: 10.1016/j.bmcl.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 27.Certal V, Halley F, Virone-Oddos A, Delorme C, Karlsson A, Rak A, Thompson F, Filoche-Rommé B, El-Ahmad Y, Carry J-C, Abecassis P-Y, Lejeune P, Vincent L, Bonnevaux H, Nicolas J-P, Bertrand T, Marquette J-P, Michot N, Benard T, Below P, Vade I, Chatreaux F, Lebourg G, Pilorge F, Angouillant-Boniface O, Louboutin A, Lengauer C, Schio L. J. Med. Chem. 2012;55:4788. doi: 10.1021/jm300241b. [DOI] [PubMed] [Google Scholar]
- 28.Certal V, Halley F, Virone-Oddos A, Thompson F, Filoche-Rommé B, El-Ahmad Y, Carry J-C, Delorme C, Karlsson A, Abecassis P-Y, Vincent L, Bonnevaux H, Nicolas J-P, Morales R, Michot N, Vade I, Louboutin A, Perron S, Doerflinger G, Tric B, Monget S, Lengauer C, Schio L. Bioorg. Med. Chem. Lett. 2012;22:6381. doi: 10.1016/j.bmcl.2012.08.072. [DOI] [PubMed] [Google Scholar]
- 29.Safina BS, Baker S, Baumgardner M, Blaney PM, Chan BK, Chen Y-H, Cartwright MW, Castanedo G, Chabot C, Cheguillaume AJ, Goldsmith P, Goldstein DM, Goyal B, Hancox T, Handa RK, Iyer PS, Kaur J, Kondru R, Kenny JR, Krintel SL, Li J, Lesnick J, Lucas MC, Lewis C, Mukadam S, Murray J, Nadin AJ, Nonomiya J, Padilla F, Palmer WS, Pang J, Pegg N, Price S, Reif K, Salphati L, Savy PA, Seward EM, Shuttleworth S, Sohal S, Sweeney ZK, Tay S, Tivitmahaisoon P, Waszkowycz B, Wei B, Yue Q, Zhang C, Sutherlin DP. J. Med. Chem. 2012;55:5887. doi: 10.1021/jm3003747. [DOI] [PubMed] [Google Scholar]
- 30.Zhu WF, Zhai X, Fu QQ, Guo F, Bai M, Wang JQ, Wang HY, Gong P. Chem. Pharm. Bull. 2012;60:1037. doi: 10.1248/cpb.c12-00342. [DOI] [PubMed] [Google Scholar]
- 31.Sutherlin DP, Baker S, Bisconte A, Blaney PM, Brown A, Chan BK, Chantry D, Castanedo G, DePledge P, Goldsmith P, Goldstein DM, Hancox T, Kaur J, Knowles D, Kondru R, Lesnick J, Lucas MC, Lewis C, Murray J, Nadin AJ, Nonomiya J, Pang J, Pegg N, Price S, Reif K, Safina BS, Salphati L, Staben S, Seward EM, Shuttleworth S, Sohal S, Sweeney ZK, Ultsch M, Waszkowycz B, Wei B. Bioorg. Med. Chem. Lett. 2012;22:4296. doi: 10.1016/j.bmcl.2012.05.027. [DOI] [PubMed] [Google Scholar]
- 32.Murray JM, Sweeney ZK, Chan BK, Balazs M, Bradley E, Castanedo G, Chabot C, Chantry D, Flagella M, Goldstein DM, Kondru R, Lesnick J, Li J, Lucas MC, Nonomiya J, Pang J, Price S, Salphati L, Safina B, Savy PPA, Seward EM, Ultsch M, Sutherlin DP. J. Med. Chem. 2012;55:7686. doi: 10.1021/jm300717c. [DOI] [PubMed] [Google Scholar]
- 33.Blake JF, Xu R, Bencsik JR, Xiao D, Kallan NC, Schlachter S, Mitchell IS, Spencer KL, Banka AL, Wallace EM, Gloor SL, Martinson M, Woessner RD, Vigers GPA, Brandhuber BJ, Liang J, Safina BS, Li J, Zhang B, Chabot C, Do S, Lee L, Oeh J, Sampath D, Lee BB, Lin K, Liederer BM, Skelton NJ. J. Med. Chem. 2012;55:8110. doi: 10.1021/jm301024w. [DOI] [PubMed] [Google Scholar]
- 34.Le PT, Cheng H, Ninkovic S, Plewe M, Huang X, Wang H, Bagrodia S, Sun S, Knighton DR, LaFleur Rogers CM, Pannifer A, Greasley S, Dalvie D, Zhang E. Bioorg. Med. Chem. Lett. 2012;22:5098. doi: 10.1016/j.bmcl.2012.05.100. [DOI] [PubMed] [Google Scholar]
- 35.(a) Ikawa T, Barder TE, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2007;129:13001. doi: 10.1021/ja0717414. [DOI] [PubMed] [Google Scholar]; (b) Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew. Chem. Intl. Ed. 2006;45:6523. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 36.Lin H, Schulz MJ, Xie R, Zeng J, Luengo JI, Squire MD, Tedesco R, Qu J, Erhard K, Mack JF, Raha K, Plant R, Rominger CM, Ariazi JL, Sherk CS, Schaber MD, McSurdy-Freed J, Spengler MD, Davis CB, Hardwicke MA, Rivero RA. ACS Med. Chem. Lett. 2012;3:524. doi: 10.1021/ml300045b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu KKC, Bailey S, Dinh DM, Lam H, Li C, Wells PA, Yin M-J, Zou A. Bioorg. Med. Chem. Lett. 2012;22:5114. doi: 10.1016/j.bmcl.2012.05.104. [DOI] [PubMed] [Google Scholar]
- 38.Smith AL, D’Angelo ND, Bo YY, Booker SK, Cee VJ, Herberich B, Hong FT, Jackson CLM, Lanman BA, Liu LB, Nishimura N, Pettus LH, Reed AB, Tadesse S, Tamayo NA, Wurz RP, Yang K, Andrews KL, Whittington DA, McCarter JD, Miguel TS, Zalameda L, Jiang J, Subramanian R, Mullady EL, Caenepeel S, Freeman DJ, Wang L, Zhang N, Wu T, Hughes PE, Norman MH. J. Med. Chem. 2012;55:5188. doi: 10.1021/jm300184s. [DOI] [PubMed] [Google Scholar]
- 39.Heffron TP, Salphati L, Alicke B, Cheong J, Dotson J, Edgar K, Goldsmith R, Gould SE, Lee LB, Lesnick JD, Lewis C, Ndubaku C, Nonomiya J, Olivero AG, Pang J, Plise EG, Sideris S, Trapp S, Wallin J, Wang L, Zhang X. J. Med. Chem. 2012;55:8007. doi: 10.1021/jm300867c. [DOI] [PubMed] [Google Scholar]
- 40.Giordanetto F, Wållberg A, Cassel J, Ghosal S, Kossenjans M, Yuan Z-Q, Wang X, Liang L. Bioorg. Med. Chem. Lett. 2012;22:6665. doi: 10.1016/j.bmcl.2012.08.101. [DOI] [PubMed] [Google Scholar]
- 41.Giordanetto F, Wållberg A, Ghosal S, Iliefski T, Cassel J, Yuan Z-Q, von Wachenfeldt H, Andersen SM, Inghardt T, Tunek A, Nylander S. Bioorg. Med. Chem. Lett. 2012;22:6671. doi: 10.1016/j.bmcl.2012.08.102. [DOI] [PubMed] [Google Scholar]
- 42.Sawant SD, Srinivas M, Lakshma Reddy G, Venkateswar Rao V, Singh PP, Vishwakarma RA. Tetrahedron Lett. 2012;53:6195. [Google Scholar]
- 43.Liu KKC, Huang X, Bagrodia S, Chen JH, Greasley S, Cheng H, Sun S, Knighton D, Rodgers C, Rafidi K, Zou A, Xiao J, Yan S. Bioorg. Med. Chem. Lett. 2011;21:1270. doi: 10.1016/j.bmcl.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 44.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS. J. Med. Chem. 2010;53:7146. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, Sabatini DM, Gray NS. J. Med. Chem. 2011;54:1473. doi: 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Choi HG, Waller DL, Sim T, Sabatini DM, Gray NS. Bioorg. Med. Chem. Lett. 2011;21:4036. doi: 10.1016/j.bmcl.2011.04.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimura N, Siegmund A, Liu L, Yang K, Bryan MC, Andrews KL, Bo Y, Booker SK, Caenepeel S, Freeman D, Liao H, McCarter J, Mullady EL, San Miguel T, Subramanian R, Tamayo N, Wang L, Whittington DA, Zalameda L, Zhang N, Hughes PE, Norman MH. J. Med. Chem. 2011;54:4735. doi: 10.1021/jm200386s. [DOI] [PubMed] [Google Scholar]
- 48.Stec MM, Andrews KL, Booker SK, Caenepeel S, Freeman DJ, Jiang J, Liao H, McCarter J, Mullady EL, San Miguel T, Subramanian R, Tamayo N, Wang L, Yang K, Zalameda LP, Zhang N, Hughes PE, Norman MH. J. Med. Chem. 2011;54:5174. doi: 10.1021/jm2004442. [DOI] [PubMed] [Google Scholar]
- 49.Kendall JD, O’Connor PD, Marshall AJ, Frédérick R, Marshall ES, Lill CL, Lee W-J, Kolekar S, Chao M, Malik A, Yu S, Chaussade C, Buchanan C, Rewcastle GW, Baguley BC, Flanagan JU, Jamieson SMF, Denny WA, Shepherd PR. Bioorg. Med . Chem. 2012;20:69. doi: 10.1016/j.bmc.2011.11.029. [DOI] [PubMed] [Google Scholar]
- 50.Kim J, Hong S, Hong S. Bioorg. Med. Chem. Lett. 2011;21:6977. doi: 10.1016/j.bmcl.2011.09.118. [DOI] [PubMed] [Google Scholar]
- 51.Wang M, Gao MZ, Miller KD, Sledge GW, Zheng QH. Bioorg. Med. Chem. Lett. 2012;22:1569. doi: 10.1016/j.bmcl.2011.12.136. [DOI] [PubMed] [Google Scholar]
- 52.Bergamini G, Bell K, Shimamura S, Werner T, Cansfield A, Müller K, Perrin J, Rau C, Ellard K, Hopf C, Doce C, Leggate D, Mangano R, Mathieson T, O’Mahony A, Plavec I, Rharbaoui F, Reinhard F, Savitski MM, Ramsden N, Hirsch E, Drewes G, Rausch O, Bantscheff M, Neubauer G. Nat. Chem.Biol. 2012;8:576. doi: 10.1038/nchembio.957. [DOI] [PubMed] [Google Scholar]
- 53.(a) Ellard K, Sunose M, Bell K, Ramsden N, Bergamini G, Neubauer G. Bioorg. Med. Chem. Lett. 2012;22:4546. doi: 10.1016/j.bmcl.2012.05.121. [DOI] [PubMed] [Google Scholar]; (b) Sunose M, Bell K, Ellard K, Bergamini G, Neubauer G, Werner T, Ramsden N. Bioorg. Med. Chem. Lett. 2012;22:4613. doi: 10.1016/j.bmcl.2012.05.090. [DOI] [PubMed] [Google Scholar]
- 54.Bell K, Sunose M, Ellard K, Cansfield A, Taylor J, Miller W, Ramsden N, Bergamini G, Neubauer G. Bioorg. Med. Chem. Lett. 2012;22:5257. doi: 10.1016/j.bmcl.2012.06.049. [DOI] [PubMed] [Google Scholar]
- 55.Jeong Y, Lee J, Lee Y, Ham K, Choi S, Hong S. Chemmedchem. 2012;7:1379. doi: 10.1002/cmdc.201200245. [DOI] [PubMed] [Google Scholar]
- 56.de Turiso FGL, Shin Y, Brown M, Cardozo M, Chen Y, Fong D, Hao XL, He X, Henne K, Hu YL, Johnson MG, Kohn T, Lohman J, McBride HJ, McGee LR, Medina JC, Metz D, Miner K, Mohn D, Pattaropong V, Seganish J, Simard JL, Wannberg S, Whittington DA, Yu G, Cushing TD. J. Med. Chem. 2012;55:7667. doi: 10.1021/jm300679u. [DOI] [PubMed] [Google Scholar]
- 57.Sabbah DA, Simms NA, Wang W, Dong Y, Ezell EL, Brattain MG, Vennerstrom JL, Zhong HA. Bioorg. Med. Chem. 2012;20:7175. doi: 10.1016/j.bmc.2012.09.059. [DOI] [PubMed] [Google Scholar]
- 58.Bruel A, Loge C, de Tauzia ML, Ravache M, Le Guevel R, Guillouzo C, Lohier JF, Santos JSD, Lozach O, Meijer L, Ruchaud S, Benedetti H, Robert JM. Eur. J. Med. Chem. 2012;57:225. doi: 10.1016/j.ejmech.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 59.Mortensen DS, Perrin-Ninkovic SM, Harris R, Lee BGS, Shevlin G, Hickman M, Khambatta G, Bisonette RR, Fultz KE, Sankar S. Bioorg. Med. Chem. Lett. 2011;21:6793. doi: 10.1016/j.bmcl.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 60.Wu P, Su Y, Liu X, Zhang L, Ye Y, Xu J, Weng S, Li Y, Liu T, Huang S, Yang B, He Q, Hu Y. Eur. J. Med. Chem. 2011;46:5540. doi: 10.1016/j.ejmech.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 61.Mielcke TR, Mascarello A, Filippi-Chiela E, Zanin RF, Lenz G, Leal PC, Chirardia LD, Yunes RA, Nunes RJ, Battastini AMO, Morrone FB, Campos MM. Eur. J. Med. Chem. 2012;48:255. doi: 10.1016/j.ejmech.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 62.Crew AP, Bhagwat SV, Dong H, Bittner MA, Chan A, Chen X, Coate H, Cooke A, Gokhale PC, Honda A, Jin M, Kahler J, Mantis C, Mulvihill MJ, Tavares-Greco PA, Volk B, Wang J, Werner DS, Arnold LD, Pachter JA, Wild R, Gibson NW. Bioorg. Med. Chem. Lett. 2011;21:2092. doi: 10.1016/j.bmcl.2011.01.139. [DOI] [PubMed] [Google Scholar]
- 63.Martínez González S, Hernández AI, Varela C, Rodríguez-Arístegui S, Alvarez RM, García AB, Lorenzo M, Rivero V, Oyarzabal J, Rabal O, Bischoff JR, Albarrán M, Cebriá A, Alfonso P, Link W, Fominaya J, Pastor J. Bioorg. Med. Chem. Lett. 2012;22:1874. doi: 10.1016/j.bmcl.2012.01.074. [DOI] [PubMed] [Google Scholar]
- 64.Murphy ST, Alton G, Bailey S, Baxi SM, Burke BJ, Chappie TA, Ermolieff J, Ferre R, Greasley S, Hickey M, Humphrey J, Kablaoui N, Kath J, Kazmirski S, Kraus M, Kupchinsky S, Li J, Lingardo L, Marx MA, Richter D, Tanis SP, Tran K, Vernier W, Xie Z, Yin MJ, Yu XH. J. Med. Chem. 2011;54:8490. doi: 10.1021/jm201019k. [DOI] [PubMed] [Google Scholar]
- 65.Wu P, Su Y, Liu X, Yang B, He Q, Hu Y. Bioorg. Med. Chem. 2012;20:2837. doi: 10.1016/j.bmc.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 66.Martínez González S, Hernández AI, Varela C, Rodríguez-Arístegui S, Lorenzo M, Rodríguez A, Rivero V, Martín JI, Saluste CG, Ramos-Lima F, Cendón E, Cebrián D, Aguirre E, Gomez-Casero E, Albarrán M, Alfonso P, García-Serelde B, Oyarzabal J, Rabal O, Mulero F, Gonzalez-Granda T, Link W, Fominaya J, Barbacid M, Bischoff JR, Pizcueta P, Pastor J. Bioorg. Med. Chem. Lett. 2012;22:3460. doi: 10.1016/j.bmcl.2012.03.090. [DOI] [PubMed] [Google Scholar]
- 67.Leahy JW, Buhr CA, Johnson HWB, Kim BG, Baik T, Cannoy J, Forsyth TP, Jeong JW, Lee MS, Ma S, Noson K, Wang L, Williams M, Nuss JM, Brooks E, Foster P, Goon L, Heald N, Holst C, Jaeger C, Lam S, Lougheed J, Nguyen L, Plonowski A, Song J, Stout T, Wu X, Yakes MF, Yu P, Zhang W, Lamb P, Raeber O. J. Med. Chem. 2012;55:5467. doi: 10.1021/jm300403a. [DOI] [PubMed] [Google Scholar]
- 68.Martínez González S, Hernández AI, Varela C, Lorenzo M, Ramos-Lima F, Cendón E, Cebrián D, Aguirre E, Gomez-Casero E, Albarrán MI, Alfonso P, García-Serelde B, Mateos G, Oyarzabal J, Rabal O, Mulero F, Gonzalez-Granda T, Link W, Fominaya J, Barbacid M, Bischoff JR, Pizcueta P, Blanco-Aparicio C, Pastor J. Bioorg. Med. Chem. Lett. 2012;22:5208. doi: 10.1016/j.bmcl.2012.06.093. [DOI] [PubMed] [Google Scholar]
- 69.Sun Q, Wu R, Cai S, Lin Y, Sellers L, Sakamoto K, He B, Peterson BR. J. Med. Chem. 2011;54:1126. doi: 10.1021/jm100912b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang Q, Richardson PF, Sach NW, Zhu J, Liu KKC, Smith GL, Bowles DM. Org. Proc. Res. Dev. 2011;15:556. [Google Scholar]
- 71.Liu KKC, Zhu J, Smith GL, Yin M-J, Bailey S, Chen JH, Hu Q, Huang Q, Li C, Li QJ, Marx MA, Paderes G, Richardson PF, Sach NW, Walls M, Wells PA, Zou A. ACS Med. Chem. Lett. 2011;2:809. doi: 10.1021/ml200126j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kendall JD, Giddens AC, Tsang KY, Frédérick R, Marshall ES, Singh R, Lill CL, Lee W-J, Kolekar S, Chao M, Malik A, Yu S, Chaussade C, Buchanan C, Rewcastle GW, Baguley BC, Flanagan JU, Jamieson SMF, Denny WA, Shepherd PR. Bioorg. Med. Chem. 2012;20:58. doi: 10.1016/j.bmc.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 73.Yan R, Zhang Z-M, Fang X-Y, Hu Y, Zhu H-L. Bioorg. Med. Chem. 2012;20:1373. doi: 10.1016/j.bmc.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Z-M, Zhang X-W, Zhao Z-Z, Yan R, Xu R, Gong H-B, Zhu H-L. Bioorg. Med .Chem. 2012;20:3359. doi: 10.1016/j.bmc.2012.03.064. [DOI] [PubMed] [Google Scholar]
- 75.Tang J-F, Lv X-H, Wang X-L, Sun J, Zhang Y-B, Yang Y-S, Gong H-B, Zhu H-L. Bioorg. Med. Chem. 2012;20:4226. doi: 10.1016/j.bmc.2012.05.055. [DOI] [PubMed] [Google Scholar]
- 76.Chang SH, Zhang Z, Zhuang XX, Luo JF, Cao XW, Li HL, Tu ZC, Lu XY, Ren XM, Ding K. Bioorg. Med. Chem. Lett. 2012;22:1208. doi: 10.1016/j.bmcl.2011.11.080. [DOI] [PubMed] [Google Scholar]
- 77.Bruce I, Akhlaq M, Bloomfield GC, Budd E, Cox B, Cuenoud B, Finan P, Gedeck P, Hatto J, Hayler JF, Head D, Keller T, Kirman L, Leblanc C, Le Grand D, McCarthy C, O’Connor D, Owen C, Oza MS, Pilgrim G, Press NE, Sviridenko L, Whitehead L. Bioorg. Med. Chem. Lett. 2012;22:5445. doi: 10.1016/j.bmcl.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 78.Oka Y, Yabuuchi T, Fujii Y, Ohtake H, Wakahara S, Matsumoto K, Endo M, Tamura Y, Sekiguchi Y. Bioorg. Med. Chem. Lett. 2012;22:7534. doi: 10.1016/j.bmcl.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 79.Staben ST, Siu M, Goldsmith R, Olivero AG, Do S, Burdick DJ, Heffron TP, Dotson J, Sutherlin DP, Zhu B-Y, Tsui V, Le H, Lee L, Lesnick J, Lewis C, Murray JM, Nonomiya J, Pang J, Prior WW, Salphati L, Rouge L, Sampath D, Sideris S, Wiesmann C, Wu P. Bioorg. Med. Chem. Lett. 2011;21:4054. doi: 10.1016/j.bmcl.2011.04.124. [DOI] [PubMed] [Google Scholar]
- 80.Yaguchi S-i, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, Hirono S, Yamazaki K, Yamori T. J. Nat. Cancer Inst. 2006;98:545. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- 81.Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen ZC, Dos Santos O, Ayral-Kaloustian S, Khafizova G, Brooijmans N, Mallon R, Hollander I, Feldberg L, Lucas J, Yu K, Gibbons J, Abraham RT, Chaudhary I, Mansour TS. J. Med. Chem. 2010;53:2636. doi: 10.1021/jm901830p. [DOI] [PubMed] [Google Scholar]
- 82.(a) Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Delos Santos E, Chen ZC, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Clin. Cancer Res. 2011;17:3193. doi: 10.1158/1078-0432.CCR-10-1694. [DOI] [PubMed] [Google Scholar]; (b) Gedaly R, Angulo P, Hundley J, Daily MF, Chen CG, Evers BM. J. Surg. Res. 2012;176:542. doi: 10.1016/j.jss.2011.10.045. [DOI] [PubMed] [Google Scholar]
- 83.Matsuno T, Kato M, Tsuchida Y, Takahashi M, Yaguchi S, Terada S. Chem. Pharm. Bull. 1997;45:291. doi: 10.1248/cpb.45.291. [DOI] [PubMed] [Google Scholar]
- 84.Peterson EA, Andrews PS, Be X, Boezio AA, Bush TL, Cheng AC, Coats JR, Colletti AE, Copeland KW, DuPont M, Graceffa R, Grubinska B, Harmange JC, Kim JL, Mullady EL, Olivieri P, Schenkel LB, Stanton MK, Teffera Y, Whittington DA, Cai T, La DS. Bioorg. Med. Chem. Lett. 2011;21:2064. doi: 10.1016/j.bmcl.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 85.Dehnhardt CM, Venkatesan AM, Chen ZC, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, Yu K, Hollander I, Feldberg L, Lucas J, Mallon R. Bioorg. Med. Chem. Lett. 2011;21:4773. doi: 10.1016/j.bmcl.2011.06.063. [DOI] [PubMed] [Google Scholar]
- 86.Rewcastle GW, Gamage SA, Flanagan JU, Frederick R, Denny WA, Baguley BC, Kestell P, Singh R, Kendall JD, Marshall ES, Lill CL, Lee WJ, Kolekar S, Buchanan CM, Jamieson SMF, Shepherd PR. J. Med. Chem. 2011;54:7105. doi: 10.1021/jm200688y. [DOI] [PubMed] [Google Scholar]
- 87.Norman MH, Andrews KL, Bo YXY, Booker SK, Caenepeel S, Cee VJ, D’Angelo ND, Freeman DJ, Herberich BJ, Hong FT, Jackson CLM, Jiang J, Lanman BA, Liu LB, McCarter JD, Mullady EL, Nishimura N, Pettus LH, Reed AB, Miguel TS, Smith AL, Stec MM, Tadesse S, Tasker A, Aidasani D, Zhu XC, Subramanian R, Tamayo NA, Wang L, Whittington DA, Wu B, Wu T, Wurz RP, Yang K, Zalameda L, Zhang N, Hughes PE. J. Med.Chem. 2012;55:7796. doi: 10.1021/jm300846z. [DOI] [PubMed] [Google Scholar]
- 88.Peterson EA, Boezio AA, Andrews PS, Boezio CM, Bush TL, Cheng AC, Choquette D, Coats JR, Colletti AE, Copeland KW, DuPont M, Graceffa R, Grubinska B, Kim JL, Lewis RT, Liu JZ, Mullady EL, Potashman MH, Romero K, Shaffer PL, Stanton MK, Stellwagen JC, Teffera Y, Yi SY, Cai T, La DS. Bioorg. Med. Chem. Lett. 2012;22:4967. doi: 10.1016/j.bmcl.2012.06.033. [DOI] [PubMed] [Google Scholar]
- 89.Wurz RP, Liu LB, Yang K, Nishimura N, Bo YX, Pettus LH, Caenepeel S, Freeman DJ, McCarter JD, Mullady EL, Miguel TS, Wang L, Zhang N, Andrews KL, Whittington DA, Jiang J, Subramanian R, Hughes PE, Norman MH. Bioorg. Med. Chem. Lett. 2012;22:5714. doi: 10.1016/j.bmcl.2012.06.078. [DOI] [PubMed] [Google Scholar]
- 90.Marion F, Williams DE, Patrick BO, Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R, Andersen RJ. Org. Lett. 2005;8:321. doi: 10.1021/ol052744t. [DOI] [PubMed] [Google Scholar]
- 91.M, Haidour A, Alvarez-Manzaneda Rn. Org. Lett. 2010;12:4450. doi: 10.1021/ol101173w. [DOI] [PubMed] [Google Scholar]
- 92.Pereira AR, Strangman WK, Marion F, Feldberg L, Roll D, Mallon R, Hollander I, Andersen RJ. J. Med. Chem. 2010;53:8523. doi: 10.1021/jm100531u. [DOI] [PubMed] [Google Scholar]
- 93.Zask A, Kaplan J, Toral-Barza L, Hollander I, Young M, Tischler M, Gaydos C, Cinque M, Lucas J, Yu K. J. Med. Chem. 2008;51:1319. doi: 10.1021/jm7012858. [DOI] [PubMed] [Google Scholar]
- 94.Ayral-Kaloustian S, Gu J, Lucas J, Cinque M, Gaydos C, Zask A, Chaudhary I, Wang J, Di L, Young M, Ruppen M, Mansour TS, Gibbons JJ, Yu K. J. Med. Chem. 2009;53:452. doi: 10.1021/jm901427g. [DOI] [PubMed] [Google Scholar]
- 95.Baiz D, Pinder TA, Hassan S, Karpova Y, Salsbury F, Welker ME, Kulik G. J. Med. Chem. 2012;55:8038. doi: 10.1021/jm300881a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tai W, Shukla RS, Qin B, Li B, Cheng K. Mol. Pharm. 2011;8:901. doi: 10.1021/mp200007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao Y, Duan S, Zeng X, Liu C, Davies NM, Li B, Forrest ML. Mol. Pharm. 2012;9:1705. doi: 10.1021/mp3000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Castillo SS, Brognard J, Petukhov PA, Zhang C, Tsurutani J, Granville CA, Li M, Jung M, West KA, Gills JG, Kozikowski AP, Dennis PA. Cancer Res. 2004;64:2782. doi: 10.1158/0008-5472.can-03-1530. [DOI] [PubMed] [Google Scholar]
- 99.Keddie NS, Ye Y, Aslam T, Luyten T, Bello D, Garnham C, Bultynck G, Galione A, Conway SJ. Chem. Commun. 2011;47:242. doi: 10.1039/c0cc03003a. [DOI] [PubMed] [Google Scholar]
- 100.Song F, Zhang J, Zhao Y, Chen W, Li L, Xi Z. Org. Biomol. Chem. 2012;10:3642. doi: 10.1039/c2ob00031h. [DOI] [PubMed] [Google Scholar]