

Abstract
The role of redox enzymes in establishing a microenvironment for parasite development is well characterized. Mimicking human glucose-6-phosphate dehydrogenase and glutathione reductase (GR) deficiencies by redox-cycling compounds thus represents a challenge to the design of new preclinical antiparasitic drug candidates. Schistosomes and malarial parasites feed on hemoglobin. Heme, the toxic prosthetic group of the protein, is not digested and represents a challenge to the redox metabolism of the parasites. Here, we report on old and new redox-cycling compounds – whose antiparasitic activities are related to their interference with (met)hemoglobin degradation and hematin crystallization. Three key-assays allowed probing and differentiating the mechanisms of drug actions. Inhibition of β-hematin was first compared to the heme binding as a possible mode of action. All tested ligands interact with the hematin π-π dimer with KD similar to those measured for the major antiparasitic drugs. No correlation between a high affinity for hematin and the capacity to prevent β-hematin formation was however deduced. Inhibition of β-hematin formation is consequently not the result of a single process but results from redox processes following electron transfers from the drugs to iron(III)-containing targets. The third experiment highlighted that several redox-active compounds (in their reduced forms) are able to efficiently reduce methemoglobin to hemoglobin in a GR/NADPH-coupled assay. A correlation between methemoglobin reduction and inhibition of β-hematin was shown, demonstrating that both processes are closely related. The ability of our redox-cyclers to trigger methemoglobin reduction therefore constitutes a critical step to understand the mechanism of action of our drug candidates.
Keywords: hematin, β-hematin, antimalarial, antischistosomal, naphthoquinone, xanthone, NADPH-dependent disulfide reductase, redox-cycler
Introduction
Malaria and schistosomiasis are two major tropical infectious diseases caused by Plasmodia and Schistosoma parasites, respectively. These diseases are ranked first and second among the world’s parasitic diseases, in terms of extent of endemic areas, number of infected people and economic burden. Despite numerous antimalarial and antischistosomal agents developed throughout the 20th century, the diseases have not yet been eradicated and no vaccine is available.
The severe form of P. falciparum malaria is responsible for the death of less than one million people per year, mainly children below 5 years of age. Multidrug-resistance of malarial strains toward broadly used antimalarial drug treatments (e.g. chloroquine, quinine, sulfadoxine-pyrimethamine) has spread all over the world in the last five decades. Schistosomiasis is a group of widespread and devastating tropical diseases caused by the platyhelminth Schistosoma which affects more than 200 million people worldwide, especially in Africa. Praziquantel has been the anthelmintic drug of choice against flatworms for more than 20 years; currently more than 100 million people per year are treated with this drug. If resistant worms develop, schistosomiasis treatment will face a crisis because of the absence of alternative effective drugs or of a sufficiently protective vaccine. Thus, concerning these two neglected diseases, there is an urgent need for new ethical drugs to emerge which should be efficient, safe, cheap to synthesize and which display mechanism(s) of action that counteract the development of resistance.
During the intraerythrocytic cycle of malarial parasites, a large amount of host cell hemoglobin (Table 1) is digested in acidic vesicles and in the final food vacuole. Hemoglobin digestion, as a source of essential nutrients, was also observed to occur in the gut of Schistosoma worms [1]. This catabolic process generates indigestible and toxic heme (iron(III) protoporphyrin IX or FeIIIPPIX, Table 1) with concomitant high fluxes of reactive oxygen species (ROS) inducing oxygen-derived radical reactions and over-oxidation of macromolecules. Both malarial and schistosomal parasites evade the toxicity of the released free heme by expressing two major detoxification pathways, the biocrystallization of heme into a dark brown pigment called hemozoin [2,3] (Hz; the synthetic crystal formed from hematin is called β-hematin, Table 1) and an efficient thiol network based on the regeneration of thiols catalyzed by NADPH-dependent disulfide reductases [4,5,6] (Figure 1). These flavoenzymes include both glutathione reductases (GR) of Plasmodium-infected human erythrocytes identified as targets of antimalarial drugs [7] and the unique multifunctional enzyme, the thioredoxin-glutathione reductase (TGR) [8], specifically found in Schistosoma. Schistosoma mansoni TGR, which was recently proved to be an essential enzyme for Schistosoma worms to maintain the reducing balance, is also a key drug target for antischistosomal drug design [9]. Therefore, throughout the years, the development of antiparasitic agents against blood-feeding parasites like Plasmodium parasites and Schistosoma worms has been focused on chemical series which target the heme biocrystallization process. Not surprisingly, numerous heme binders sharing common structural motifs – from 4-aminoquinolines [10], quinoline methanols or arylmethanols [11,12], endoperoxides [13], to xanthones [14,15,16] – kill both malarial and schistosomal parasites.
Table 1.
Definition and Abbreviation of the Heme-Containing Compounds Cited in this Work.a
| Heme compound | Abbreviation | Definition |
|---|---|---|
| Heme | Heme FeIIIPPIX | Prosthetic group that consists of an iron atom contained in the center of a heterocyclic porphyrin ring, namely the protoporphyrin IX (see Figure 2). It corresponds to the active protein-free unit of hemoglobin molecule. |
| Hemoglobin | Hb(FeII) or Hb | Oxygen-carrying Fe(II) hemoprotein of erythrocytes made up of four different polypeptide globin chains that contain between 141 and 146 amino acids and a FeIIPPIX active site. |
| Oxyhemoglobin | oxyHb(FeII) or oxyHb | Hemoglobin protein containing an oxygen molecule in its FeIIPPIX site |
| Methemoglobin | metHb(FeIII) or metHb | Hematogenous pigment formed from hemoglobin by oxidation of the iron atom from the ferrous to the ferric state. A small amount is found in the blood normally, but injury or toxic agents convert a larger proportion of hemoglobin into methemoglobin, which does not function as an oxygen carrier. |
| Hemin chloride | Hemin FeIIIPPIXCl | Heme containing a chloride anion axially bound in the fifth position of the iron(III) porphyrinic system. |
| Hematin | Hematin FeIIIPPIX(OH2) or FeIIIPPIX(OH) | Heme containing a water or a hydroxide molecule axially bound in the fifth position of the iron(III) porphyrinic system. |
| μ-Oxo hematin dimer | μ-oxo FeIIIPPIX dimer | μ-oxo bridged dimer of iron(III) protoporphyrin IX |
| π-π Hematin dimer | π-π FeIIIPPIX dimer | π stacked iron(III) protoporphyrin IX dimer |
| μ-Pr hematin dimer | μ-Pr FeIIIPPIX dimer | Dimer formed by symmetrical and mutual intermolecular iron(III)-propionate coordination bonds in the particular case of FeIIIPPIX |
| β-hematin | β-hematin | Synthetic hematin crystal standing for hemozoin pigment equivalent |
| Hemozoin or malarial pigment | Hz | Disposal pigment – biocrystals which result from self-association of Fe(III) protoporphyrin IX - formed during the digestion of methemoglobin in several blood-feeding parasites (the FeIIIPPIX are held together mainly through π-π and μ-Pr interactions). |
Figure 1.

Putative Model for Redox-Active Compounds Affecting Redox Homeostasis in P. falciparum-Infected Red Blood Cell Accounting for the Observed Inhibition of Hemozoin Formation Causing the Death of the Parasites.a
aHMS, hexose monophosphate shunt. RedOx means redox-active compound with ox and red in subscript to indicate if the compound acts in its oxidized or its reduced state. Double dashed arrows represent transport processes. The potential redox-active compounds are proposed to be taken up by the infected red blood cells (step 1), to be reduced in the cytosol of the human red blood cell by GR (step 2), and then transported in the acidic vesicles or in the food vacuole (step 3) where hemoglobin digestion occurs. Subsequently, the redox-active compounds are proposed to be cycled between the acidic compartments and the cytosols and reduced by GR of the human host cell (not shown) or of the parasite (step 4) in a continuous redox cycle. The reduced species of redox-active compounds are assumed to be transported through FeIII complexation into the acidic vesicles where the reduced RedOx species transfer the electrons to oxidants (hematin or metHb(FeIII), step 5). The final result is an inhibition of hemozoin formation (step 6) and the arrest of trophozoite development. Thus, the physico-biochemical properties of RedOx accounting for the antimalarial activity were investigated through the different processes under quasi-physiological conditions in the presence of relevant parasitic oxidant targets like hematin or metHb(FeIII). Three assays are described: drug:hematin affinity studies at pH 7.5; inhibition of hematin crystallization at pH ~ 5.0; metHb(FeIII) reduction assays at pH 6.9 coupled to the NADPH-based GR system.
The malarial parasite indeed prevents damage (oxidative stress) by a cytosolic antioxidant thiol network that is regenerated by NADPH-dependent disulfide oxido-reductases, glutathione reductase (GR; EC 1.8.1.7), thioredoxin reductase (TrxR; EC 1.8.1.9) [4,5], and possibly by lipoamide dehydrogenase (LipDH; EC 1.8.1. 4) [17]. Among the most effective ligands of NADPH-dependent disulfide reductases the most potent antiparasitic effects were indeed observed for redox-cyclers acting as subversive substrates of these flavoenzymes (reaction 1 in Scheme 1). Subversive substrates of disulfide reductases are exemplified by methylene blue, 1,4-naphthoquinones, nitrofurans [4,5]. These compounds are reduced via 1 or 2 electron(s)-transfer and regenerated in the presence of major oxidants in the parasites (reaction 2 in Scheme 1), as previously shown with the couple metHb(FeIII)/oxyHb(FeII) (Table 1) in the presence of methylene blue or 3-benzoyl menadione derivatives as redox molecules [18,19].
Scheme 1.

Redox-Active Compounds as Subversive Substrates of NADPH-Dependent Disulfide Reductases.a,b RedOx stands for the redox-active agents like methylene blue, 1,4-naphthoquinones (given as an example in this scheme) or xanthones. The labels ox and red stand for the oxidized and reduced states of these redox active agents, respectively.
a DR means disulfide reductase; b X is the series variable, Y means H2 or O.
In the majority of reported in vitro UV-Vis spectrophotometric studies about hematin (hydroxylated or aquo heme or [(FeIIIPPIX)(OH)] or [(FeIIIPPIX)(OH2)], Table 1) crystallization, investigations of hematin speciation have been carried out in the presence of the potent antimalarial 4-aminoquinolines [20]. This is partly due to the high solubility of these weak bases containing three nitrogen atoms with a broad range of pKa values. Upon protonation of the quinoline (pKa ~ 8) and of the terminal amino group (pKa ~ 11) of the side chain, the solubility of the compounds is drastically increased in aqueous media, rendering “comfortable” the physicochemical analysis of the hematin:drug complexes in vitro under varying pH conditions. However, structurally distinct chemical series with limited solubility were reported to display antimalarial activities and inhibition effects on hemozoin formation. Because of the broad range of drug solubility, many in vitro assays using hematin at different pH values can lead to misinterpretations if precipitation of the drug occurs, especially if the assays are based on hematin absorbance at one fixed wavelength [21]. Furthermore, as it has been clearly demonstrated in recent reports [18,19], inhibition of hemozoin formation is not the result of only one process, i.e. of ligand binding to the π-π hematin dimer. It can also originate from redox processes following electron transfer reactions from the drug to the iron(III) of two relevant targets hematin and methemoglobin (noted metHb or metHb(FeIII), Table 1). In particular, heme(FeII) is an inhibitor of hematin crystallization [22]. To illustrate metHb(FeIII) conversion by redox cyclers, we recently demonstrated the ability of reduced 3-benzoylnaphthoquinones (i.e. dihydronaphthoquinones) to generate oxyHb(FeII) (Table 1) as a mechanism of antimalarial action of 3-benzylnaphthoquinone derivatives [18,19].
Plasmodium parasites digest methemoglobin (metHb(FeIII)) faster than oxyhemoglobin (oxyHb(FeII)) [23] and hemoglobin oxidation to methemoglobin in the acidic vesicles/food vacuole is the basis of creating space for parasite growth. Consequently, the reduction of metHb(FeIII) to oxyHb(FeII) by redox-active agents is expected to slow down both metHb(FeIII) digestion and the growth rate of the parasite. Another reason for targeting metHb(FeIII) reduction is that ferric hemoglobin is not able to transport oxygen; metHb(FeIII) elevation in brain blood vessels of infected host, for instance, contributes to hypoxia and cerebral malaria [24]. Thus, the reduction of redox-active agents by NADPH in reactions mediated by disulfide reductases and their oxidation, from their reduced forms, in spontaneous reactions with ferric species of heme and hemoglobin might contribute both to the elevation of free heme level and to interference with hemozoin formation in the parasites. Both events – oxidizing the reductants and reducing the oxidants in a continuous redox cycle (Figure 1) – are expected to contribute to the elimination of the parasites [18,19]. This strategy is well illustrated by recent findings about the mode of action of the antimalarial drug methylene blue [25] and a novel series of 3-benzylmenadione derivatives involved in a cascade of redox reactions in the parasites as part of the putative mode of action [18,19].
In this paper, it is our goal to describe three key-assays that can be used as probes to differentiate between distinct mechanisms of antimalarial (Figure 1) or antischistosomal drug actions and that can be employed to identify hemozoin inhibitors both in vitro and in vivo. Quasi-physiological conditions applied to these assays would bring high benefit to rationalize the effects of well-controllable inhibitors. Several different assays based on the speciation of hematin in aqueous solutions with varying pH have been developed for studying the interaction capabilities of the drugs with metHb(FeIII) at pH 6.9 or hematin at pH ~5 and pH 7.4. Two low solubility-featured compound series, antimalarial 1,4-naphthoquinones and antischistosomal xanthones, were selected in this present report to illustrate the most frequently applied spectrophotometric assays.
Thermodynamics and Structural Aspects of Ferriprotoporphyrin
From Heme to Hemozoin in Blood-Feeding Plasmodium and Schistosoma Parasites
As part of its complex life cycle, the parasite invades, grows and multiplies within the host’s red blood cells. The parasite digests a high quantity of hemoglobin from the host cell cytoplasm (Figure 1) using a cytostome and transports the hemoglobin to its acidic digestive vacuole (pH 5.0–5.5) [26]. In the process of hemoglobin digestion, free heme (ferriprotoporphyrin, noted FeIIIPPIX, Figure 2) is produced, which is highly toxic for the parasite.
Figure 2.

Chemical Structure of the Deprotonated Form of FeIIIPPIX.
Indeed, heme is an ubiquitous and essential molecule playing key roles in biological processes like oxygen transport, respiration, photosynthesis and drug detoxification. However, due to its intrinsic properties, heme can cause numerous deleterious effects within the cell [27]. Due to its amphiphilic features, heme binds to phospholipid membranes altering their permeability and selectivity, and ultimately leading to cell lysis [28]. Because of its pro-oxidant character, heme is also capable of generating free reactive radicals through the decomposition of organic peroxides. These reactive oxygen species (ROS) mediate lipids peroxidation resulting in protein cross-linking and nucleic acid modifications. To overcome this free FeIIIPPIX-induced toxicity, efficient biomineralization of heme into a highly insoluble and relatively “unreactive” crystal called hemozoin (Hz or malaria pigment) is essential for its detoxification. The overlapping of the iron(III) porphyrins decrease the active surface and consequently significantly decreases the pro-oxidant capacity [29]. Nevertheless, far from being an “inert” pigment, hemozoin has also a redox activity, consistent with previous studies showing that hemozoin could induce lipid peroxidation in human monocytes [30,31]. Hemozoin is nowadays widely considered as the main key target of many antimalarial agents, in particular 4-aminoquinolines. Indeed, 4-aminoquinoline antimalarials such as chloroquine (Table 2) have been demonstrated to inhibit hemozoin formation by binding either to free FeIIIPPIX preventing biomineralization reactions [32].
Table 2.
Apparent Association Constants of Quinoline and Xanthone Based Drugs with Hematin or Hemin in Aqueous Solution.a
| Compound | Apparent association constant log KA | |||
|---|---|---|---|---|
| Aqueous 5.64 M DMSO/ Drug:Heme | Water/ Drug:Heme | Water [57]/ Drug:Heme | ||
 Methylene blue |
5.5b 1:2 [25] |
|||
|
| ||||
 Chloroquine |
5.52 ± 0.03b 1:1 [58] |
5.6c 1:4 [60] |
3.02d,f 1:1 |
5.11b,f 1:1 |
| 4.8 ± 0.1b 1:1 [59] |
5.51d,f 2:1 |
6.06b,f 2:1 |
||
|
| ||||
 Amodiaquine |
5.39 ± 0.04b 1:1 [58] |
4.97c 1:4 [60] |
3.26d,f 1:1 |
|
| 6.38d,f 2:1 | ||||
|
| ||||
 Quinine |
4.10 ± 0.02b 1:1 [58] |
4.32c 1:5 [60] |
3.0d,f 1:1 |
|
| 5.10d,f 2:1 | ||||
|
| ||||
 Mefloquine |
3.90 ± 0.08b 1:1 [58] |
4.08c 1:3 [60] |
3.70d,f 1:1 |
|
| 4.43d,f 2:1 | ||||
|
| ||||

|
4.95 ± 0.05b 1:1 [61] |
|||
|
| ||||
| Ferroquine | 5.52 ± 0.03b 1:1 [62] |
|||
|
| ||||
 Primaquine |
Not detectable [58] | 4.2c 1:7 [60] |
||
|
| ||||
 4,5-dihydroxyxanthones |
5.29e 1:2 |
|||
In aqueous 5.64 M DMSO (40% v/v) at pH 7.43, the predominant species for [FeIIIPPIX]tot < 10−2.5 M is most likely the (OH/H2O)-monomer (Figure 4b), while the hematin π-π dimer predominates in water (Figure 4a).
pH 7.43–7.5.
pH 6.5.
pH 5.6.
pH 5.8.
Interactions with hemin chloride. T = 22.5–28.5 °C.
FeIIIPPIX Speciation in Aqueous Solution and Relevance to Hemozoin Formation
The deeper understanding of the ferriprotoporphyrin (heme) speciation in solution is therefore of essential interest to unravel its role in biomineralization processes (formation of the hemozoin crystal) in malarial parasites and Schistosoma worms. The evaluation of the nature, the stoichiometry and the strength of ferriprotoporphyrin FeIIIPPIX complexes with antimalarial and antischistosomal redox biosensors, a focus of our ongoing research work and of this present report, is of fundamental importance. The nature of the hematin dimer in solution (μ-oxo [33] / ππ [34] / μ-Pr [35]), which has been established to be an important building block during the formation of hemozoin, was (and is still) a subject of interest and debates for more than sixty years. Iron(III) metalloporphyrins can undergo various equilibria in solution (self-association or dimerization, oligomerization) and protolytic reactions depending on the physico-chemical conditions of the medium (solvent, pH, ionic strength, temperature…). In 1947, J. Shack and W. M. Clarke [36] were among the first scientists to demonstrate that FeIIIPPIX displays intricate speciation behavior in aqueous solution. More than twenty years later, S. B. Brown et al. [37] showed, thanks to absorption spectrophotometry, that FeIIIPPIX can undergo dimerization in aqueous solution and first proposed this dimer to be a μ-oxo species (Figure 3) [38].
Figure 3.

Structural Aspects of Hemozoin and its Analogues (such as β-Hematin, the Synthetic Equivalent of Hemozoin).
(A) μ-oxo (μ-oxo bridged dimer of iron(III) protoporphyrin IX). (B) π-π dimer (π stacked iron(III) protoporphyrin IX dimer - for instance hemin dimer [FeIIIPPIX(Cl)]2). (C) μ-Pr (dimer formed by symmetrical and mutual intermolecular iron(III)-propionate coordination bonds in the particular case of FeIIIPPIX).
Since then, heme behavior in aqueous solutions was a matter of several contradictory reports on the nature and the speciation properties of the FeIIIPPIX dimer. T. J. Egan and his coworkers [20] recently established that FeIIIPPIX(H2O/HO−) does not spontaneously form a μ-oxo dimer in aqueous mixtures of protic solvent, but rather a cofacial π-π dimer (Figure 3B). The μ-oxo dimer (Figure 3A) is rather a predominant species in aqueous mixtures of aprotic solvents or in detergent solutions (e.g. with 10% pyridine in an aqueous solution of FeIIIPPIX at 0.1 M NaOH or in aqueous mixtures containing aprotic solvent such as 5.64 M DMSO, acetone or DMF at pH 10). The influence of experimental conditions (nature of the pH, protic or aprotic character of the solvent…) has been clearly demonstrated and reviewed in the report published by T. J. Egan and his coworkers [20]. In addition, this μ-oxo dimer is characterized by an absorption band in the visible region centered at about 575 nm (together with a shoulder at about 600 nm).
The thermodynamics of the (FeIIIPPIX)2 dimer formation (both under a π-π [39] or μ-oxo [20,40] state) has been recently revisited and thoroughly described. For instance, at pH ~ 7.4, which corresponds to acidity which was considered in our thermodynamic investigations, the (FeIIIPPIX)2 π-π dimer constitutes the major species in a wide range of [FeIIIPPIX]tot (Figure 4A), while the (FeIIIPPIX)2O μ-oxo dimer is a minor species for [FeIIIPPIX]tot below 10−2.5 M in aqueous 5.64 M DMSO (40% v/v) solutions (Figure 4B). These physico-chemical considerations support the fact that the (FeIIIPPIX)2 π-π dimer is by far a more stable bio-edifice under quasi-physiological conditions with respect to the (FeIIIPPIX)2O μ-oxo dimer except if not physiological conditions are used ([FeIIIPPIX]tot ≫ 3.16 mM, presence of aprotic solvents such as DMSO, high pH values – pH > 10…).
Figure 4.

Speciation Diagrams of the Ferriprotoporphyrin IX (FeIIIPPIX) Species as a Function of [FeIIIPPIX]tot at 25 °C and pH 7.4.
(A) Pure water; (B) aqueous 5.64 M DMSO (40 % v/v). Charges, solvent molecules, protons and hydroxides have been omitted in the equilibria given in the insets.
These physico-chemical results pointed out the intricacy of the behavior of Fe(III) protoporphyrin IX in aqueous solution [20,39]. The nature and the arrangement of the species were demonstrated to be closely dependent on numerous physico-chemical parameters such as the medium acidity, the FeIIIPPIX concentrations, the temperature, the electrolyte concentration and/or the nature of the solvents (polarity, proticity…). These physico-chemical factors determine whether FeIIIPPIX monomers, π-π, μ-oxo (and at lesser extend μ-Pr – Figure 3C) dimers or larger aggregates are formed in solution.
Figure 5 displays the distribution diagrams of the protonated species of the (FeIIIPPIX)2 π-π dimer as a function of pH. The dimerization constants as well as the protonation constant of [(FeIIIPPIX)(OH)] monomer and of [(FeIIIPPIX)2(H2O)2] π-π dimer [25,39] have been considered (equations 1–6) to calculate these speciation diagrams.
Figure 5.

Thermodynamics of Ferriprotoporphyrin IX under Monomeric and π-π Dimeric States. Distribution Diagrams as a Function of pH of FeIIIPPIX Species Calculated for [FeIIIPPIX]tot = 40 μM.
Thermodynamic data taken from reference [18] and [39]. H2O and OH correspond to the Fe(III) axially coordinated molecules. Charges have been omitted for the sake of clarity.
| Equation 1 |
| Equation 2 |
| Equation 3 |
| Equation 4 |
| Equation 5 |
| Equation 6 |
For the (FeIIIPPIX)2 π-π dimer, the two iron(III) protoporphyrins adopt a face to face cofacial arrangement (FeIIIPPIX…FeIIIPPIX interplane distance of 4.42(9) Å – see the example of hemin π-π dimer in Figure 3B) [41] and the two ferric cations are pentacoordinated with awater molecule completing the Fe(III) coordination sphere. The bisaquo complex [(FeIIIPPIX)(OH2)2] can undergo two successive deprotonations to afford the mono- or the bis(hydroxo) species (equations 4 and 5). Therefore, the (FeIIIPPIX)2 π-π dimer exists in aqueous solution under three different protonated states, namely [(FeIIIPPIX)(OH2)]2, [(FeIIIPPIX)(OH2).(FeIIIPPIX)(OH)] and [(FeIIIPPIX(OH)]2 (the charges have been omitted for the sake of clarity). The distribution diagrams presented in Figure 5 show that the biomineralization processes may be related to the formation below pH 6 of the bisaquo π-π species [(FeIIIPPIX)(OH2)]2. The water molecules are more labile than the hydroxyl groups and are hence more easily displaced by the propionate groups of the side arms of an adjoining ferriporphyrinic unit ((i) at pH ~ 6, the propionic acids are deprotonated; (ii) the corresponding pKa values have been calculated to be ~ 3.5 [42] in anionic (SDS) or neutral (TX-100) surfactants). This feature is expected to be one of the key parameters in biomineralization process of hematin. These speciation and protolytic properties of Fe(III) protoporphyrin IX also demonstrate that there is a narrow pH window (3.5 <pH < 6) suitable for biomineralization (e.g. the pH of the acidic (pH ~ 5.0–5.5) digestive vacuole of P. falciparum and of the worms gut [1] is well suited for hemozoin nucleation and growth). Competition in favor of protonation of the propionate units (pH < 3) or deprotonation of the bisaquo [(FeIIIPPIX)(OH2)]2 species (pH > 6) prevent hemozoin formation. These speciation curves also indicate that aquo/hydroxo complex is the predominant species at pH 7.4, the acidity which was chosen for the binding studies reported in this work. Lastly, these studies confirmed that FeIIIPPIX spontaneously and exclusively dimerizes in solution even under non physiological conditions ([FeIIIPPIX]tot > 0.4 mM) but does not form higher aggregates in aqueous solution above pH 5–6.
Hemozoin and β-Hematin X-Ray Structural Data
Hemozoin and models of hemozoin (β-hematin) [35,41] have been extensively analyzed in the past few years, and led recently to new sights about their biomineralization properties. Until very recently, hemozoin [35] and β-hematin were both regarded as repetitive occurrence of μ-Pr dimers (see vide infra for β-hematin) and the π-π stabilizing interactions were considered only in the context of crystal packing [35].
The repeat unit (triclinic unit cell) of β-hematin can be regarded as a centro-symmetric heme cyclic dimer, where the two FeIIIPPIX units are linked through iron-carboxylate bonds (RCOO−…FeIIIPPIX distance of 1.886(2) Å)) between the propionate side-chain of one molecule and the central Fe atom of the other (μ-Pr mode, Figure 3C). The dimers are π-stacked in a similar fashion to that typical of molecular crystals of porphyrins (large interplanar distance and small lateral overlap between the porphyrin moieties). Additional stabilization is gained from the dispersion interactions between the methyl and the vinyl side-chains of the adjacent dimers. Lastly, these structural studies also revealed hydrogen-bonding interactions, involving the remaining propionate side chains (averaged O…O = 2.8 A°), which link the dimers into a symmetrical two-dimensional matrix.
The crystal structure data of hemozoin from Plasmodium falciparum was solved at a resolution of 2.4 Å by powder diffraction data [41,43]. In this study, it was proposed that the driving forces for the FeIIIPPIX self-association are constituted of two different modes of interactions. The first one involves strong π-π interactions (FeIIIPPIX…FeIIIPPIX interplane distance of 4.42(9) Å) which initiate the crystal formation. In addition, the π-π dimers are interacting intermolecularly through iron(III)-carboxylate linkages (μ-Pr interaction - Figure 3C –RCOO−…FeIIIPPIX distance of 1.91(8) Å)) stabilizing the crystal structure.
Analyses of these two X-ray data revealed that the structures of hemozoin and β-hematin are very similar even though they were analyzed differently (μ-Pr versus π-π mode). However, many open issues still remain regarding the mechanisms by which hemozoin nucleates and growths. Hemozoin appears to be chemically and spectroscopically identical to β-hematin which is formed by heating FeIIIPPIX in concentrated acid at 60°C [43], or at physiological temperatures if a suitable lipid-water interface is provided [44].
Major Antimalarial Drugs Inhibiting Hemozoin Formation
Several series of antimalarial drugs were reported to exert to different extents their antimalarial activity by enhancing free heme toxicity through the inhibition of hemozoin formation. However, no direct correlation between the antimalarial activity (in vitro and in vivo) and the inhibition of hemozoin or β-hematin formation was clearly established unless the lipophilicity and the protonation constants (pKa) of the tested compounds have been taken into account [45]. The pKa values are important to consider in the case of antimalarial drugs which mainly interact with the Fe(III) protoporphyrin through drug-Fe(III) fifth axial coordination bond. The binding constants usually follow the order of the pKa values that are tuned by substituent-induced electronic effects [46]. Major families of antimalarial drugs inhibiting hemozoin formation involve methylene blue and their derivatives, quinolones, xanthones, azoles, isonitriles [47,48].
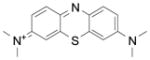
The first synthetic antimalarial drug, methylene blue (Table 2), a tricyclic phenothiazine blue cationic dye and some of its derivatives, have been shown to interact with heme and their antimalarial activities correlated well with the abilities to inhibit β-hematin formation [49]. It was also known as an effective subversive substrate of various NADPH-dependent disulfide reductases [7b]. We recently reported that methylene blue firmly binds to the hematin π-π dimer forming a (hematin)2: MB complex (MB = methylene blue) under quasi-physiological conditions (pH 7.4) [25]. This effect is likely to contribute to the inhibition of hemozoin formation in Plasmodium falciparum and to the increase of the concentration of parasite-toxic heme. On the other hand, the reduction of metHb(FeIII) to oxyHb(FeII) by NADPH is efficiently catalyzed by methylene blue and glutathione reductase (metHb reduction rate constant of 991 ±44 M−1 s−1). Thus, the redox properties of methylene blue can also affect the digestion of metHb by the malarial parasites and inhibit P. falciparum growth.
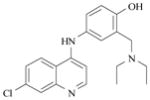
The 4-aminoquinolines are certainly the most important and studied class of antimalarial drugs considered until now that act by targeting the parasite specific hemoglobin digestion pathway. For example, chloroquine (Table 2), a famous member of this 4-aminoquinoline family of compounds, accumulates in the acidic digestive vacuole of the parasite [50]. It was shown to interact with the μ-oxo dimer of oxidized heme, thereby preventing hemozoin formation [51]. Chloroquine strongly binds to FeIIIPPIX leading to a 1:1 complex with association constants log KA ~ 5–6 (KD ~ 2–15 μM) at pH 7.5 [12,51,57,58,59,60]. The π-π interaction between chloroquine and the conjugated π-system of hematin is believed to govern and stabilize the formation of adducts. Free heme and heme-chloroquine complexes may kill parasites by inducing severe oxidative stress [52], which successively leads to peroxidation of parasite membrane lipids, damage of DNA, oxidation of protein and ultimately, death of the parasite. Quinine (Table 2), a quinoline methanol antimalarial (another important member of the quinoline family) was also shown to interact with FeIIIPPIX but less efficiently than chloroquine by forming a 1:1 complex characterized by association constants log KA ~ 4.1–4.3 (KD ~ 50–80 μM) at pH 7.5 [51]. Quinine was reported to inhibit hemozoin formation [53]. Even though many spectroscopic reports have been published [54], the lack of structural data related to the interactions of 4-aminoquinoline antimalarial drugs with heme-containing targets renders difficult the design and the development of novel compounds. The recent elucidation of the crystal structure of the complex of halofantrine with FeIIIPPIX by T. J. Egan and his coworkers [12] shed light on the existing interactions with antimalarials, especially the arylmethanols. A three-point interaction coordination complex that involves π-π stacking, O-FeIII coordination and intramolecular hydrogen bonding was shown. In addition to the π-stacking of the phenanthrene ring of halofantrine over the porphyrin ring system, the drug coordinates to the iron(III) centre of FeIIIPPIX through its alcohol unit. This arylmethanol/FeIIIPPIX complex is also stabilized through an additional intermolecular hydrogen bond occurring between the protonated terminal nitrogen atom of halofantrine and a propionate side arm of the FeIIIPPIX monomer. This salt bridge formation is suggested to interrupt the formation of the hemozoin dimer precursor produced during the heme detoxification pathway and satisfactorily accounted for the strong activity of the two active antimalarial drugs quinidine and quinine. By contrast, formation of comparable salt bridges with the inactive 9-epiquinidine and 9-epiquinine epimers induces more constrained species and necessitates much higher energy costs.
Xanthones have been identified as another potent class of antimalarial compounds [55]. Xanthones are supposed to act in a unique fashion to kill Plasmodium parasites through the formation of soluble complexes with heme, thereby inhibiting the process of hemozoin formation [15]. In particular, xanthones bearing hydroxyl groups at both the 4- and 5-positions, especially when paired with neighboring 3- and 6-substituted hydroxyls, demonstrated the most potent activity to inhibit hemozoin formation. To unravel the mechanism by which these xanthones inhibit Plasmodium growth, spectroscopic and physico-chemical analyses were performed on the 4,5-dihydroxyxanthone (Table 2). The heme:xanthone complex stoichiometry in aqueous solution was found to be 1:2. This interaction is non-cooperative and exhibited a heme complex dissociation constant KD of 5.1 μM at pH 5.8. The strength of the heme:xanthone interaction is comparable to those measured for the quinoline-based antimalarial drugs chloroquine and quinine (Table 2) [56]. The heme:xanthone complex formation was found to be both pH and solvent dependent, with strong evidences that the xanthone carbonyl moiety firmly interacts with the iron(III) metallic center of the hematin dimer. Hydrogen bonds between the hydroxyl groups of the xanthone (in positions 4 and 5) and the propionate side chains of heme, as well as π-π stacking between both aromatic π systems appeared to contribute to the overall stability of the xanthone-heme complex. It was concluded that this interaction is responsible for the activity of the 4,5-dihydroxyxanthone against Plasmodium through effective inhibition of hemozoin formation.
Synthesis and Physico-Biochemical Studies of Xanthone and 1,4-Naphthoquinone Derivatives targeting Iron(III)-Hemoglobin catabolites
Objectives
We now described a thorough physico-biochemical study of six compounds (Figure 6), which was conducted to gain a deeper understanding of the mechanism of action by which xanthone [16,63] and 1,4-naphthoquinone substrates could act as potential inhibitors of β-hematin crystallization through redox-cycling and exert antimalarial or antischistosomal activities. Plasmodium and Schistosoma parasites digest methemoglobin, resulting from endogenous oxidation of hemoglobin, as a source of essential nutrients (aminoacids) and detoxify the toxic free heme released during methemoglobin digestion via its biocrystallization into an insoluble and “inert” crystal called hemozoin (or malaria pigment or Schistosoma pigment, Figure 1). Considering that the process of hemoglobin (or methemoglobin) digestion is common to both Plasmodium and Schistosoma parasites, it was first of interest to study the ability of our compounds to interact with heme in solution, which is usually anticipated to be a first prerequisite when targeting hemozoin formation.
Figure 6.

Chemical Structures of the Xanthones and 1,4-Naphthoquinones Considered in this Work.
The ability to inhibit β-hematin crystallization was then evaluated and compared with the heme binding properties of our substrates as it is generally accepted as a plausible mode of action of the known antimalarial and antischistosomal drugs (vide supra, Table 2). Here, we investigated the ability of redox-active compounds – the naphthoquinones and xanthones – to inhibit β-hematin formation, not only on the sole basis of hematin binding but also on the basis of electron transfer reactions. Hence, we evaluated if, under their reduced states – the compounds could trigger metHb(FeIII) reduction into oxyHb(FeII) as a critical step for potential antiparasitic mechanism of actions.
Synthesis of Xanthones via Palladium-Catalyzed C-H Addition to Nitriles
The synthesis of four xanthones 1 to 4, and of two 1,4-naphthoquinone derivatives 5 and 6 was performed to provide key-compounds useful for the physico-biochemical studies.
A rapid and easy two-step synthetic route to xanthones with excellent yields involving palladium-catalyzed CH-addition to nitriles followed by aromatic nucleophilic substitution SNAr was followed to synthesize the xanthones (Scheme 2) [64].
Scheme 2.

A Palladium-Catalyzed Two-Step Strategy to Access Xanthones.
The two-steps Larock’s strategy to obtain simple xanthones was used in order to prepare useful compounds for further structure-activity relationship studies. The 2-methoxy-9H-xanthen-9-one (xanthone 3) was synthesized in three steps (Scheme 3). Commercially available 1,4-dimethoxybenzene (2 equiv.), 2-fluorobenzonitrile (1 equiv.), Pd(OAc)2 (0.1 equiv.), DMSO and TFA were used as starting materials and heated at 95 °C for 24 h. The (2,5-dimethoxyphenyl)(2-fluorophenyl)methanone 3a was obtained as colorless oil with 85% yield after flash chromatography. Compound 3a was then selectively demethylated by dropwise addition of BBr3 (1 equiv.) at 0 °C in DCM for 1 h to give the (2-fluorophenyl)(2-hydroxy-5-methoxyphenyl)methanone 3b with 86% yield. The 2-methoxy-9H-xanthen-9-one 3 was finally obtained with 61% yield after a SNAr type reaction in the presence of 2.0 equivalents of potassium carbonate. The synthesis of both benz[c]xanthen-7-ones (shorten as benzoxanthones) 1 and 2 started from the corresponding naphthol instead of the phenol and the naphthol intermediate was submitted to the aromatic nucleophilic substitution under basic conditions (K2CO3). Both benzoxanthones 1 and 2 were produced with 69 and 86 % yield, respectively.
Scheme 3.

Synthesis of 2-Methoxy-9H-xanthen-9-one 3.
Reagents and conditions: a) 0.1 equiv. Pd(OAc)2, DMSO/TFA, 95°C, 24h; b) 1.0 equiv. BBr3, DCM, 0°C, 1h; c) 2.0 equiv. K2CO3, acetone, 50°C, 2h.
To evaluate the influence of fluorinated substituents in the xanthone skeleton on the biological activity, a fluorine xanthone derivative was synthesized. Commercially available 4-methoxy-3,5-difluorophenol (2 equiv.), 2-fluorobenzonitrile (1 equiv.), Pd(OAc)2 (0.1 equiv.), DMSO and TFA were used as starting materials and heated at 95 °C for 24 h (Scheme 4). The (2,4-difluoro-6-hydroxy-3-methoxyphenyl)(2-fluorophenyl)methanone 4a was obtained with 14% yield after flash chromatography. This result suggested that the introduction of fluorine atoms in the phenol reagent dramatically decreased the yield of the reaction. Compound 4a underwent SNAr type reaction in the presence of 2.0 equivalents of potassium carbonate to obtain 2,4-difluoro-3-methoxy-9H-xanthen-9-one 4 with 53% yield.
Scheme 4.

Synthesis of 2,4-Difluoro-3-methoxy-9H-xanthen-9-one 4.
Reagents and conditions: a) 0.1 equiv. Pd(OAc)2, DMSO/TFA, 95°C, 24h; b) 2.0 equiv. K2CO3, acetone, 50°C, 2h.
UV-visible Absorption Spectrophotometric Titration of Hematin
Principle of the Assay
The affinity of our compounds for hematin was investigated by UV-visible absorption spectrophotometry at pH 7.5, a pH which prevents hematin crystallization (Figure 5). It is noteworthy that under these pH conditions, FeIIIPPIX predominantly exists as a π-π dimer with one Fe(III) center being coordinated by a water solvent (fifth axial ligand) molecule and the second one with a hydroxyl ligand ([(FeIIIPPIX)(OH2).(FeIIIPPIX)(OH)] species).
An absorption (300 nm < λ < 800 nm) spectrophotometric titration of an aqueous hematin solution (~ 3 × 10−5 M in 0.2 M sodium hepes buffer) at pH 7.5 was carried out by adding microvolumes of a stock solution of the substrate (~5–9 × 10−4 M in DMSO). Aliquots of 5 to 20 μL of the stock substrate solution were added to the reaction mixture and the UV-visible absorption spectra were recorded after each addition. Special care was taken to ensure that equilibrium was attained after each addition.
Spectrophotometric Properties of Hematin
The high-spin FeIIIPPIX possesses a spin of S = 5/2 with all half-filled d-orbitals in close proximity with the porphyrin HOMO and LUMO. This results in a strong overlap between the porphyrin π-π* (eg π* excited state orbitals) and the FeIII d transitions (dyz, dxz orbitals). This is consequently reflected by broad Q bands in the visible absorption region. Similarly to ferric high spin derivatives of myoglobin and hemoglobin, the formation of a characteristic charge transfer band CT (also called band III (a2u → dyz) [65,66]) centered at ~ 613 nm (Figure 7) can be observed. These spectroscopic characteristics are markedly different from those of the μ-oxo FeIIIPPIX dimer (absorption in the visible at ~ 575 nm together with a shoulder at ~ 600 nm) thus allowing to precisely probe their occurrence in solution. In addition, exciton transfer occurs from electronic interaction by direct overlap of the π orbitals of the porphyrins (face to face cofacial arrangement for the two FeIIIPPIX subunits within the π-π dimer) and electron exchange between the individual chromophores. This excitonic coupling in the ground state breaks the degeneracy of the B (or Soret absorption band) excited state, giving rise to two Soret bands lying at 383 nm and 364 nm (Figure 7). This broad and split Soret band is therefore an apparent spectroscopic signature of the presence of the FeIIIPPIX π-π dimer in solution (KDim= 106.8 M−1 at pH ~ 7.5, Equations 1–5).
Figure 7.

UV-visible Electronic Spectrum of Free Hematin under π-π Dimeric State Measured in 0.2 M Sodium Hepes Buffer pH 7.5 and at T = 25 °C.
Upon addition of the substrate, concomitant bathochromic shifts and narrowing of the Soret band were observed for all the substrates emphasizing the formation of complexes with hematin π-π dimer. The association constants KA (M−1), the dissociation constants (μM) and the stoichiometries of the species at equilibrium have been determined by processing the spectrophotometric data with the Specfit program [67,68,69,70,71,72]. Specfit adjusts the stability constants and the corresponding molar extinction coefficients (M−1 cm−1) of the species at equilibrium.
Spectrophotometric Titrations of the Hematin π-π Dimer by the Substrates at pH 7.5
Xanthones and Benzoxanthones
The series of the four xanthones (compounds 1 to 4, Figure 6) was studied to evaluate their ability to interact with hematin π-π dimer in 0.2 M sodium hepes buffer pH 7.5 and at T = 25 °C. The spectrophotometric titrations of the benzoxanthone 2 (Figure 8) and of xanthone 4 (Figure 9) are given as representative examples.
Figure 8.

(A) Absorption Spectrophotometric Titration of Hematin (under a π-π Dimeric State) by Benzoxanthone 2.
Solvent: 0.2 M sodium hepes buffer pH 7.5; l = 1 cm; [FeIIIPPIX]tot = 3.07 × 10−5 M; (1) [2]tot/[FeIIIPPIX]tot = 0; (2) [2]tot/[FeIIIPPIX]tot = 3.7. (B) Absorption electronic spectra of the complexes formed between FeIIIPPIX and substrate 2. (C) Distribution diagrams as a function of [2]tot of the FeIIIPPIX complexes formed with substrate 2 calculated for [FeIIIPPIX]tot = 3.07 × 10−5 M.
Figure 9.

(A) Absorption Spectrophotometric Titration of Hematin (under a π-π Dimeric State) by Xanthone 4. Solvent: 0.2 M sodium hepes buffer pH 7.5; l = 1 cm; [FeIIIPPIX]tot = 3.0 × 10−5 M; (1) [4]tot/[FeIIIPPIX]tot = 0; (2) [4]tot/[FeIIIPPIX]tot = 5.33. (B) Absorption electronic spectra of the complexes formed between FeIIIPPIX and substrate 4. (C) Distribution diagrams as a function of [4]tot of the FeIIIPPIX complexes formed with substrate 4 calculated for [FeIIIPPIX]tot = 3.0 × 10−5 M.
Benzoxanthone 2 is structurally similar to 1, the only difference being the substitution by a trifluoromethyl group in 2. In addition, benzoxanthones 1 and 2 both possess a hydroxyl unit in position 2 which is methylated. Although this 2-hydroxyl site is not capable to coordinate the Fe(III) center of hematin, our absorption spectrophotometric titrations (Figure 8A), however, revealed the formation of complexes. Upon addition of the substrates 1 or 2, a narrowing of the broad and split Soret band, characteristic of the hematin π-π dimer, is observed. In addition, a concomitant bathochromic shift of the Soret band is observed and the intensity of the CT absorption of the hematin π-π dimer is decreasing. These data strongly suggest a loss of the excitonic coupling which characterizes the (FeIIIPPIX)2 π-π dimer (vide supra) and the formation a π-π complexes (or Charge Transfer CT) complexes with benzoxanthones 1 and 2. For benzoxanthone 1, the statistical processing of the absorption data revealed the formation of a [(FeIIIPPIX)(1)] complex characterized by an association constant of log K11 = 5.9 ± 0.01 (Equation 11, KD = 1.26 μM). The formation of this 1:1 species is in agreement with the dissociation of the hematin π-π dimer and the subsequent alteration of the broad and split Soret band to a sharp and intense absorption being bathochromically shifted. By contrast, two species, namely [(FeIIIPPIX)2(2)] (log β21 = 12.5 ± 0.1, Equation 7) and [(FeIIIPPIX)(2)2] (log β12 = 10.2 ± 0.2, Equation 9) were characterized with benzoxanthone 2 which bears an additional trifluoromethyl group. Taking into account the apparent dimerization constant of FeIIIPPIX measured at pH 7.5 (log KDim, = 6.82, Equations 1–5, 12), we can accordingly calculate the successive association constant (log K21 = 5.7 ± 0.1, Equation 8) as well the corresponding dissociation constant (KD = 2 μM).
| Equation 7 |
| Equation 8 |
| Equation 9 |
| Equation 10 |
| Equation 11 |
| Equation 12 |
A possible explanation for this striking dissimilar binding behavior observed for benzoxanthones 1 and 2 is that, because of steric hindrance and electronic constrains (i.e. electron-withdrawing effects) induced by the CF3 group and its position of substitution, a single binder 2 will not be sufficiently efficient to effectively dissociate the π-π dimer of hematin. Therefore, one molecule 2 might first associate on one side of the π-π dimer, thereby destabilizing it (e.g. marked variations of the broad and split Soret band, see Figure 8B). A second benzoxanthone molecule 2 can then intercalate within the weakened π-π dimer, leading to its dissociation and the formation a [(FeIIIPPIX)(2)2] “sandwich” like complex in agreement with the loss of exciton coupling characterizing the hematin π-π dimer. The two complexes formed between hematin and benzoxanthone 2 were characterized by their electronic spectra (Figure 8B). With respect to (FeIIIPPIX)2 dimer, the [(FeIIIPPIX)2(2)] species possesses a less intense, broad and split Soret band which is still confirming the dimeric nature of hematin. By contrast, [(FeIIIPPIX)(2)2] displays a much sharper Soret band lying at lower energies (λmax = 401 nm, Δλ = 18 nm with respect to (FeIIIPPIX)2), in agreement with the dissociation of the hematin π-π dimer and the presence of a single FeIIIPPIX heme surrounded by two benzoxanthones 2. The thermodynamic and spectroscopic data are gathered in Table 3.
Table 3.
Summary of the Binding Studies Between the Substrates Considered in this Work and Hematin π-π Dimer at pH 7.5 and T = 25°C (sh = shoulder).
| Substrate | Association constant Heme:drug | Dissociation constant KD (μM) | λmax (εmax) nm (× 104 M−1 cm−1) |
|---|---|---|---|
 1 |
1:1 log K11 = 5.9 ± 0.1 |
1.26 | 400 (5.18) sh 355 (2.9) |
|
| |||
 2 |
2:1 log β21 = 12.5 ± 0.1 |
2.00 | 386 (10.0) sh 359 (9.3) |
| 1:2 log β12 = 10.2 ± 0.2 |
401 (8.82) | ||
|
| |||
 3 |
1:1 log K11 = 5.78 ± 0.03 |
1.66 | 399 (4.4) sh 357 (3.41) |
|
| |||
 4 |
2:1 log β 21= 11.4 ± 0.1 |
25.1 | 395 (8.18) sh 357 (6.51) |
| 1:2 log β12= 9.2 ± 0.2 |
401 (10.9) sh 357 (3.37) |
||
|
| |||
 5 |
2:1 log β21= 12.6 ± 0.2 |
1.58 | 386 (9.76) sh 361 (9.3) |
| 1:2 log β12= 10.6 ± 0.2 |
400 (5.55) sh 342 (3.08) |
||
|
| |||
 6 |
2:1 log β21= 12.4 ± 0.3 |
2.63 | 386 (9.90) 353 (11.38) |
| 1:2 log β12= 10.6 ± 0.3 |
397 (4.76) 353 (6.02) |
||
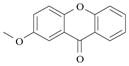
Xanthone 3 possesses similar association properties with hematin to benzoxanthone 1 and forms, under our experimental condition, a 1:1 complex associated to an apparent association constant of log K11 = 5.78 ± 0.03 (KD = 1.66 μM). The [(FeIIIPPIX)(3)] species is characterized by a red shift of the hematin dimer Soret band to λmax ~ 399 nm. The introduction of two fluorine atoms also markedly modified the binding properties of xanthone 4 with hematin π-π dimer. The speciation behavior of compound 4 with hematin is indeed comparable to that of benzoxanthone 2 with two species being successively formed, namely the [(FeIIIPPIX)2(4)] (log β21= 11.4 ± 0.1, Equation 7) and [(FeIIIPPIX)(4)2] (log β12 = 9.2 ± 0.2, Equation 9) species (Figure 9). Similar spectroscopic signatures can also be observed. [(FeIIIPPIX)2)(4)] is characterized by a broad and split Soret band in agreement with the π-π dimeric nature of heme, while the Soret absorption band of the [(FeIIIPPIX)(4)2] species is much sharper and bathochromically shifted of 18 nm (Figure 9) with respect to (FeIIIPPIX)2 suggesting the presence of a single heme molecule encircled by two xanthone molecules. The successive apparent association constant of [(FeIIIPPIX)2(4)] complex was calculated to be log K21 = 4.6 (KD = 25.1 μM, Equation 8) and is one order of magnitude lower than the association constants determined for the other substrates considered in this work. The introduction of fluorine atoms in the benzoxanthone or xanthone skeletons seemingly influence the electronic properties of these molecules and thereby disfavor efficient π-π interactions with the FeIIIPPIX macrocycle. In addition, increasing bulkiness brought by these substituents likely play a critical role in the binding processes.
The xanthones and benzoxanthones considered in this work demonstrated their capacities to firmly interact with heme and to efficiently dissociate the (FeIIIPPIX)2 π-π dimer. In the context of hemozoin targeting, this property is of fundamental importance and is usually anticipated to be a first pre-requisite to allow interaction with hematin and subsequent inhibition of hemozoin formation. Introduction of fluorine or CF3 substituents, however, alters the binding properties and leads to other stoichiometries such as 1:2 and 2:1 π-π complexes with hematin. The apparent association constants measured at pH 7.5 and at T = 25 °C range from 5 to 6 (dissociation constants KD in the μM range) and are comparable to those reported in the literature for antimalarial drug targeting hematin crystallization (Table 2). The main thermodynamic and spectroscopic results of the xanthone- and benzoxanthone-heme binding studies are gathered in Table 3.
1,4-Naphthoquinones
Based on a drug design and selection project, some of us recently demonstrated [18] that several benzyl-1,4-naphthoquinones were active at the nM concentrations against human pathogen Plasmodium falciparum in culture and against Plasmodium berghei in infected mice. It was therefore of interest to examine the binding properties of two model 1,4-naphthoquinones 5 and 6 (Figure 6). The benzylnaphthoquinone 5 and the benzoylnaphthoquinone 6 display a similar speciation behavior with hematin (Figure 10). They both lead to 1:2 or 2:1 heme complexes, and are characterized by global association constants ranging from 1010–1013 M−2 in 0.2 M sodium hepes buffer at pH 7.5 (Table 3). From the electronic spectra depicted in Figure 10B for the benzoylnaphthoquinone 6, a substrate first aggregates with the dimeric unit of hematin to afford a stable 2:1 complex (successive association constant of log K21 = 5.6, KD ~ 2.63 μM, Equation 8). The presence of a split and broad Soret band, but less intense, suggests that this heme species is not related to a “sandwich”-like arrangement but rather involve a substrate which is firmly associated on one face of the hematin π-π dimer (as already observed for several xanthones and benzoxanthones, vide supra). The strong absorptivity centered at about 350 nm originates from the phenolate subunit of substrate 6. It is noteworthy that desolvation of at least one water molecule from an iron(III) center has to take place to allow π-π contacts between the heme (in the π-π dimeric state) and the substrate (i.e. the predominant iron(III) protoporphyrinic species at pH 7.5 is [(FeIIIPPIX(OH2)).(FeIIIPPIX(OH))]. Addition of excess of the substrate will favor the formation of a 2:1 species with one protoporphyrin core being encircled by two substrates as suggested by the sharp and strong bathochromic shift (Δλ = 17 nm) of the Soret band (Figure 10). The loss of the charge transfer at ~ 610 nm is also indicative of the presence of a single heme unit within [(FeIIIPPIX)(S)2] complexes.
Figure 10.

(A) Absorption Spectrophotometric Titration of Hematin (in a π-π Dimeric State) by the Benzoylnaphthoquinone 6.
Solvent: 0.2 M sodium hepes buffer pH 7.5; l = 1 cm; [FeIIIPPIX]tot = 2.93 × 10−5 M; (1) [6]tot/[FeIIIPPIX]tot = 0; (2) [6]tot/[FeIIIPPIX]tot = 2.96. (B) Absorption electronic spectra of the complexes formed between FeIIIPPIX and substrate 6. (C) Distribution diagrams as a function of [6]tot of the FeIIIPPIX complexes formed with substrate 6 calculated at [FeIIIPPIX]tot = 2.93 × 10−5 M.
Our spectroscopic and thermodynamic approach demonstrate the ability of xanthones, benzoxanthones or 1,4-naphthoquinones to strongly interact (KD in the μM scale, see Table 3) with hematin π-π dimer and to lead to its dissociation via the formation of [(FeIIIPPIX)(S)] or [(FeIIIPPIX)(S)2] complexes. These apparent association values measured at pH 7.5 are comparable to those reported in the literature for the most efficient antimalarial agents, such as chloroquine or quinine, which were shown to efficiently target and prevent hemozoin biocrystallization (Table 2). In the next section, we will evaluate the potential of these compounds to inhibit β-hematin crystallization under quasi-physiological conditions (concentration, temperature, pH…) which mimic the acidic digestive vacuoles and guts of the Plasmodium parasites and Schistosoma worms, respectively. The aim of this study is to correlate the heme binding properties of our substrates to inhibition of hemozoin formation.
Inhibition of β-Hematin Formation
Principle of the Assay
The host’s hemoglobin digestion by Plasmodium parasites and Schistosoma worms leads to the accumulation of free toxic heme. The parasites detoxify this exogenous deleterious species through biomineralization processes leading to an insoluble and much less active crystal named hemozoin. Inhibition of heme oligomerization would lead to death of the parasite by accumulation of toxic free heme in the membranes and subsequent oxidative stress [56,73]. Many antiparasitic agents against blood-feeding parasites were consequently designed to target the heme biocrystallization process. After a first evaluation of the heme binding capacities of our compounds, we then undertook a biochemical investigation of our compounds as inhibitors of hematin crystallization to β-hematin, the synthetic equivalent of hemozoin. We used a biochemical assay previously developed by K. K. Ncokazi and T. J. Egan [74]. This assay, performed first at a pH of ca. 5–5.5 (i.e. pH of the digestive vacuole at which hemozoin biomineralization efficiently occurs), is based on a classical hematin quantification method known as the pyridine ferrihemochrome method [75]. Aqueous pyridine (used in a very large excess – 5% by volume) forms a hexacoordinated low-spin complex with free heme [(FeIIIPPIX)(Pyr)2], but not with β-hematin. This red colored complex has a very high absorptivity of the Soret band in the visible region (e405 = 1.04 x 105 M−1 cm−1, Figure 11). Therefore, it allows a straightforward quantification of the residual free hematin in the presence of our substrates as reporter activity for inhibition of the hematin oligomerization (carried out at pH about 4.5–5.0). The final pH value of the assay was 7.5 after addition of the buffers containing pyridine (0.2 M sodium hepes + 5% (v/v) pyridine buffer at pH 8.2 and 0.02 M sodium hepes + 5% pyridine (v/v) buffer at pH 7.5) used to stop the reaction. The experimental conditions optimized by T. J. Egan and his coworkers were modified for our compounds (order of the addition of the reagents) and our assays were monitored by UV-visible absorption spectrophotometry (absorption spectra recorded from 300 nm to 750 nm). IC50 values for inhibition of β-hematin formation were determined from the absorbance changes at 405 nm versus the drug (equiv.) / hematin (equiv.) ratio in the following plots (vide infra).
Figure 11.

(A) Absorption Spectrophotometric Titration of Hematin (in a π-π Dimeric State) by Pyridine (noted Pyr).
Solvent: 0.2 M sodium hepes buffer pH 7.5; l = 1 cm; [FeIIIPPIX]tot = 1.68 × 10−5 M; (1) [Pyr]tot/[FeIIIPPIX]tot = 0; (2) [Pyr]tot/[FeIIIPPIX]tot = 51667. (B) Absorption electronic spectra of the complexes formed between FeIIIPPIX and pyridine. (C) Distribution diagrams as a function of [Pyr]tot of the FeIIIPPIX complexes formed with pyridine calculated at [FeIIIPPIX]tot = 1.68 × 10−5 M. log βFeIIIPPIX(Pyr)2 = 2.25 ± 0.05 calculated in this study.
Xanthones and Benzoxanthones
The four xanthone and benzoxanthone derivatives were evaluated for their ability to inhibit β-hematin crystallization. Despite their ability to strongly interact with hematin (Table 3), all these compounds displayed no significant inhibitory activity of hematin crystallization and displayed similar spectral variations to those depicted as examples in Figure 12 for benzoxanthone 1 and xanthone 3. The maximum of inhibition reached for the four xanthones or benzoxanthones is about 20–30% at 5 equivalents which can be clearly interpreted as the absence of any inhibitory potential for hematin crystallization of these substrates. Indeed, these inhibition values are usually reached for control experiments at 0 mM drug concentration. In addition, nor the addition of the drug simultaneously to hematin prior incubation at 60°C for one hour (typical conditions used to prepare β-hematin, see section above), neither the addition of the drug to preformed β-hematin led to significantly inhibitory activities. These data clearly demonstrate that these xanthone and benzoxanthone derivatives did not display any inhibitory potential for hematin crystallization either by prevention or by dissociation. To further confirm that most of the antimalarial drugs mainly prevent β-hematin formation rather than dissociating it, we carried out inhibition assays with amodiaquine under identical conditions. As expected, amodiaquine prevents β-hematin formation while it possesses weaker capacity to dissociate pre-formed β-hematin crystals as shown in Figure 13.
Figure 12.

Inhibition of β-Hematin Crystallization by (A) Benzoxanthone 1 and (B) Xanthone 3 Measured by UV-Visible Absorption Spectrophotometry using Pyridine (5% by volume) as Reporting Reagent. The squares (■) indicate the inhibition curve (a) obtained by addition of the drug prior to formation of β-hematin while the circles (●) represent the inhibition curve (b) obtained by addition of the drug after formation of β-hematin (hematin incubated at 60°C with 12.7 M acetate buffer at pH 4.5).
Solvent: 0.2 M + 0.02 M sodium hepes buffer pH 7.5, T = 25 °C.
Figure 13.

Inhibition of β-Hematin Crystallization by Amodiaquine Measured by UV-Visible Absorption Spectrophotometry using Pyridine (5% by volume) as Reporting Reagent. The squares (■) indicate the inhibition curve (a) obtained by addition of amodiaquine prior to formation of β-hematin while the circles (●) represent the inhibition curve (b) obtained by addition of amodiaquine after formation of β-hematin (hematin incubated at 60°C with 12.7 M acetate buffer at pH 4.5).
Solvent: 0.2 M + 0.02 M sodium hepes buffer pH 7.5, T = 25 °C.
Figure 14 displays the IR spectral characteristics of β-hematin crystals in the absence (Figure 14A) and in the presence of benzoxanthone 1 (Figure 14B) and of xanthone 3 (Figure 14D). Infrared spectroscopy was herein used to characterize the reaction products and to provide further information about the inhibitory activities of compounds 1–4 with respect to β-hematin. This technique unambiguously distinguishes between hematin and β-hematin [58]. The latter has distinct and intense sharp bands at 1661 and 1204 cm−1 which are absent in the former. Three equivalents of benzoxanthone 1 (Figure 14B) or of xanthone 3 (Figure 14D) did not inhibit the reaction in agreement with the previous β-hematin inhibition data. No drug association with hematin can be also demonstrated since the IR peaks of xanthone derivatives 1 or 3 observed in combination with β-hematin possess identical characteristics as in the absence of β-hematin. These data demonstrate that even though benzoxanthones 1 and 2 and xanthones 3 and 4 are able to bind to ferriprotoporphyrin, they do exhibit any inhibitory activity toward β-hematin. π-π interactions are mainly expected to drive the complex formation which is by far not sufficient to prevent β-hematin formation as observed, for instance, for arylmethanols antimalarials such as quinine, mefloquine or halofantrine [12,47].
Figure 14.

Infrared Spectra of β-Hematin (A) and of Hematin Reaction Products with 3 Equivalents of Benzoxanthone 1 (B) and Xanthone 3 (D). (A) β-hematin prepared according to refs. [58] and [76] (1709(s), 1661(s), 1620(sh), 1467(w), 1444(w), 1406(s), 1377(s), 1356(s), 1338(sh), 1294(s), 1279(s), 1224(w), 1204(s), 1147(m), 1120(m), 1088(w), 1076(m), 1049(w), 1008(w), 985 (m), 937(s), 902(s), 836(s), 751(m), 712(s)). The peaks for β-hematin at 1661 cm−1 and 1204 cm−1 are indicated with arrows. Peaks due to the drug are marked with stars (*). IR spectra of benzoxanthone 1 (C) and xanthone 3 (E) are given for comparison purposes.
1,4-Naphthoquinones
By contrast with the xanthone- and benzoxanthone-based substrates, the benzyl-1,4-naphthoquinone 5 and the benzoyl-1,4-naphthoquinone 6 displayed significant inhibitory potential to hinder β-hematin formation. The benzyl-naphthoquinone 5 was shown to inhibit the formation of β-hematin by a maximum of 80% at a drug (equiv.) / hematin (equiv.) ratio of 5. The IC50 value which was accordingly determined is equal to 4.2 drug equiv./ hematin equiv. (Figure 15). Among the substrates examined in this work, the benzoyl-naphthoquinone 6 is by far the most efficient system which efficiently prevents the formation of β-hematin (IC50 = 1.3 drug (equiv.)/ hematin (equiv.) with a maximum of inhibition of 90 %, Figure 15).
Figure 15.

Inhibition of β-Hematin Crystallization by the Benzoyl-1,4-naphthoquinone 6 Measured by UV-Visible Absorption Spectrophotometry using Pyridine (5% by volume) as Reporting Reagent.
Solvent: 0.2 M + 0.02 M sodium hepes buffer pH 7.5. The cross (×) and the square (■) labels correspond to two independent replicates, while the solid line corresponds to the averaged values.
The main results of the hematin crystallization inhibition by xanthones and benzoxanthones are gathered in Table 4. It clearly appears from the binding studies and the inhibition of β-hematin formation assays that no clear and direct correlation exists. For our systems, their capacities to prevent formation of the β-hematin (or hemozoin) are therefore not only related to their ability to firmly interact with the π-π hematin dimer (used as a model of β-hematin and hemozoin). Consequently, the mechanism by which the formation of hemozoin is prevented is most likely induced by another process. To get further insight into this mechanism of action, we then examined the ability of our substrates to reduce heme-containing targets in the presence of disulfide reductases.
Table 4.
Summary of the Hematin Crystallization Inhibition Assay with the Xanthone, Benzoxanthone and 1,4-Naphthoquinone Derivatives. (ni = no inhibition)
| Substrate | IC50 (equivalent inhibitor) | %max Inhibition |
|---|---|---|
 1 |
ni | ~ 28% at 5 equiv.a ~ 28% at 5 equiv.b |
 2 |
ni | ~ 23% at 5 equiv. |
 3 |
ni | ~ 25% at 5 equiv.a ~ 44% at 5 equiv.b |
 4 |
ni | ~ 26% at 5 equiv. |
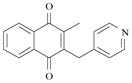 5 |
4.3 equiv. | ~ 80% |
 6 |
1.3 equiv. | ~ 90% |
 Amodiaquine |
0.4 equiv. | ~ 86% at 5 equiv.a ~ 40% at 5 equiv.b |
With no β-hematin preformed.
With β-hematin preformed (hematin incubated at 60°C for 60 mins with 12.7 M acetate buffer at pH 4.5)
Spectroscopic and Kinetic Studies of the Reduction of Methemoglobin to Hemoglobin
Principle of the assay
Plasmodium parasites and Schistosoma worms use methemoglobin (hereafter noted metHb(FeIII)) as a source of aminoacids for their own growth and digest it faster than hemoglobin (hereafter noted oxyHb(FeII)). At pH 5.0 to 6.0 (acidic digestive vacuoles or guts of Plasmodium parasites and Schistosoma worms, respectively), hemoglobin (HbFeII) or oxyhemoglobin (oxyHb or oxyHb(FeII)) is quickly oxidized to methemoglobin (metHb or metHb(FeIII)). Consequently, the reduction of metHb(FeIII) to oxyHb(FeII) can significantly slow down methemoglobin digestion.
The ability of our compounds to inhibit disulfide reductases (P. falciparum GR – noted pfGR- and of human erythrocytes – noted hGR or S. mansoni – noted SmTGR) combined to their potency to reduce metHb(FeIII) to oxyHb(FeII) (or heme-containing targets) could contribute to rise oxidative stress and interfere with hemozoin formation; both phenomena possibly leading to the death of the parasites. The pernicious and continuous use of NADPH flux under the catalysis of these disulfide reductases in the presence of our redox-active compounds (which behave as substrates or electron acceptors) can dramatically increase the flux of toxic reduced species (hydronaphthoquinones in the case of starting naphthoquinones; leucomethylene blue from methylene blue) within the parasite and lead to its death by shifting the equilibrium (FeIII) to (FeII) heme species. In addition, efficient reduction of the other ferric centers of porphyrinic targets such as hematin π-π dimers can also significantly contribute to the prevention of hemozoin (or β-hematin) crystallization.
In order to evaluate the potency of our compounds to reduce metHb(FeIII) to oxyHb(FeII), we used a reduction assay coupled to the hGR/NADPH system in vitro which regenerate the reduced species of our substrates continuously. This assay was recently established as a relevant in vivo model in our laboratory [18]. The UV-visible absorption spectrum of metHb(FeIII) between 300 nm and 700 nm is characterized by a maximum absorbance centered at 405 nm (Soret band of the Fe(III) heme and a broad band centered at 603 nm, see vide supra). Upon metHb(FeIII) reduction by our redox-active compounds (T = 37°C; pH = 6.9), which were pre-reduced by hGR, the formation of the oxyHb(FeII) species is associated to a bathochromic shift of the maximum of absorption of the Soret band from 405 nm to about 410 nm (in fact it corresponds to oxyHb(FeII) with the metal cation being hexacoordinated in a low spin state, see vide supra), as well as the formation of two new less intense absorptions at ~ 536 nm and ~ 576 nm.
No shift of the absorption band centered at 405 nm and, consequently, no metHb(FeIII) reduction was observed in the presence of chloroquine, used as negative control [18]. No shift of the Soret band was also observed in the absence of the redox-active drugs (control experiment, vide infra), demonstrating that hGR/NADPH-base system is not the prevailing reducing system and confirming that the substrates, under their reduced states, correspond to the bioactive intermediates during the reduction of metHb(FeIII) into oxyHb(FeII) mediated by the glutathione reductase. The spectral changes as well as the kinetic parameters of the reduction reaction were thoroughly analyzed. Since the coupled assay based on metHb(FeIII) and the hGR/NADPH-based system was performed with all the compounds under identical experimental conditions, the apparent kinetic rates can be compared and discussed and picture the redox-cycling capacities of the xanthone, benzoxanthone and 1,4-naphthoquinone substrates.
Xanthones and Benzoxanthones
The fours members of the xanthone and benzoxanthone series, whose binding capacities with hematin and inhibition properties of β-hematin formation were described (vide supra), were evaluated for their ability to reduce methemoglobin Fe(III) into hemoglobin Fe(II) (in fact oxyHb(FeII)) in the coupled assay with glutathione reductase and NADPH (used as cofactor). Even though all these four compounds were found to strongly interact with hematin π-π dimer at pH 7.5 and T = 25 °C in water, most of them did not induce a significant and fast reduction of metHb(FeIII) into oxyHb(FeII) (pH = 6.9, T = 37 °C). Benzoxanthones 1 and 2 were not capable to reduce metHb(FeIII) and the very weak shift observed, if any, was no more than 1 nm. In addition, assays of metHb(FeIII) reduction performed with benzoxanthones 1 and 2 display similar spectrophotometric characteristics as illustrated in Figure 16. The decrease of absorption band at 340 nm can be ascribed to NADPH consumption by the GR, while the weak decrease of the absorption of the Soret band is most likely related to degradation of metHb(FeIII) [77].
Figure 16.

UV-Visible Absorption Spectra Recorded as a Function of Time and Showing the Absence of metHb(FeIII) Reduction in the Coupled Assay with hGR/NADPH System in the Presence of Benzoxanthone 2.
Solvent: water (hGR acetate buffer pH 6.9 + 47 mM K2PO4 + 200 mM KCl); T = 37.0 °C; 40 μM NADPH + 66.5 nM hGR + 2 μM metHb + 40 μM benzoxanthone 2; (1) t = 0; (2) t ~ 60 min.
According to the absorption spectra recorded within the timescale (one hour) which was considered, xanthones 3 and 4 apparently do not induce fast reduction of metHb(FeIII) into oxyHb(FeII). The processing of the absorption data set versus time, however, allowed us to evidence the presence of two rate-limiting steps, with the second one being most likely associated to a slow reduction process of metHb(FeIII) into oxyHb(FeII) (i.e. (i) a shift of only 2 nm of the Soret band takes place after one hour in the presence of 3 and 4; (ii) a new and weak absorption band at ~ 540 nm is gradually formed with time; Figure 17a). As previously suggested for benzoxanthones 1 and 2, the first step may correspond to slow degradation of metHb(FeIII), while the second slower one is associated to the substrate-mediated reduction of metHb(FeIII). We calculated the apparent first order rate constants related to both steps. For the metHb(FeIII) reduction rate limiting step, the kobs values are equal to kobs = (7 ± 2) x 10−5 s−1 for 3 (tred1/2 = 165 min) and kobs = (1.2 ± 0.2) x 10−4 s−1 for 4 (tred1/2 = 96.3 min).
Figure 17.

(A and B) UV-Visible Absorption Spectra Recorded as a Function of Time Showing the Slow metHb(FeIII) Reduction in the Coupled Assay with the hGR/NADPH System in the Presence of Xanthone 3. (C) Electronic Spectra of the Reactants and the Products of the metHb(FeIII) Reduction. (D) Absorbance Changes at λ = 405 nm and λ = 420 nm.
Solvent: water (hGR acetate buffer pH 6.9 + 47 mM K2PO4 + 200 mM KCl); T = 37.0 °C; 40 μM NADPH + 133 nM hGR + 7.95 μM metHb + 40 μM 3; (1) t = 0; (2) t = 60 min.
In contrast with benzoxanthones 1 and 2, xanthones 3 and 4 are capable of reducing metHb(FeIII) into oxyHb(FeII) but according to very slow processes (tred1/2 ≈ 1.5 – 3 h) which are by far not biological relevant. These data show that, even though these xanthone and benzoxanthone derivatives are able to interact with hematin π-π dimer in water at pH 7.5 (Table 3), only those with fine-tuned electrochemical characteristics would rapidly and efficiently reduce metHb(FeIII) into oxyHb(FeII), an important pre-requisite when targeting inhibition of hemozoin formation, redox homeostasis and death of the parasites. In the other hand, this weak capacity to efficiently and rapidly reduce metHb(FeIII) can be seemingly related to their inability to inhibit β-hematin formation. The major results of the spectroscopic and kinetic studies relative to the metHb(FeIII) reduction coupled-assay with hGR/NADPH with the benzoxanthones 1 and 2 and the xanthones 3 and 4 are summarized in Table 5.
Table 5.
Summary of the Kinetic Studies of metHb(FeIII) Reduction by Xanthone, Benzoxanthone and Naphthoquinone Substrates.
| Substrate | Kinetic parameters kobs (s−1)/tred1/2 (min) |
Spectroscopic characteristics ελmax (M−1 cm−1) |
|---|---|---|
 1 |
No metHb(Fe III) → oxyHb(FeII) reaction | |
|
| ||
 2 |
No metHb(FeIII) → oxyHb(FeII) reaction | |
|
| ||
 3 |
(7 ± 2) x 10−5 s−1 165 min |
metHb: ε405 = 9.97 x 104 M−1 cm−1 |
| oxyHb: ε412 = 7.11 x 104 M−1 cm−1 | ||
| oxyHb: ε541 = 7.40 x 103 M−1 cm−1 | ||
|
| ||
 4 |
(1.2 ± 0.2) x 10−4 s−1 96.3 min |
metHb: ε405 = 1.02 x 105 M−1 cm−1 |
| oxyHb: ε408 = 7.78 x 104 M−1 cm−1 | ||
| oxyHb: ε538 = 7.75 x 103 M−1 cm | ||
|
| ||
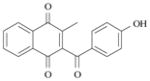 6 |
(2.5 ± 0.1) x 10−3 s−1 4.6 min |
metHb: ε406 = 1.01 x 105 M−1 cm−1 |
| oxyHb: ε409 = 9.20 x 104 M−1 cm−1 | ||
| oxyHb: ε540 = 8.82 x 103 M−1 cm | ||
|
| ||
 5 |
(1.95 ± 0.02) x 10−3 s−1 5.9 min |
metHb: ε405 = 1.11 x 105 M−1 cm−1 |
| oxyHb: ε409 = 9.49 x 104 M−1 cm−1 | ||
| oxyHb: ε541 = 1.00 x 104 M−1 cm | ||
1,4-Naphthoquinones
The benzyl- 5 and the benzoylnaphthoquinone 6 were then evaluated for their ability to reduce metHb(FeIII) into oxyHb(FeII) in the coupled assay with human glutathione reductase and NADPH. After pre-reduction (two electrons reduction process) with hGR/NADPH-based system under aerobic conditions [18], the dihydrogenated form of naphthoquinones 5 and 6 induce, in a much faster step (tred1/2 = 5–6 min), a significant bathochromic shift of the Soret band (~ 3–5 nm) of the metHb(FeIII) Soret transitions within the time range considered (20 to 45 min). These two compounds were shown to behave similarly as depicted in the UV-visible spectra provided in Figure 18. For example, a red shift from 405 to 410 nm was observed during the first rate-limiting step for compound 6 (kobs = (2.07 ± 0.03) x 10−3 s−1, tred1/2 = 5.55 min). For most of these substrates, slower steps (Figure 19) were also observed but concern either re-oxidation or degradation of oxyHb(FeII) once hGR/NADPH becomes inactive (i.e. when NADPH is consumed or when hGR becomes inactive). The kinetic and spectroscopic results obtained for the metHb(FeIII) reduction by substrates 5 and 6 are provided in Table 5.
Figure 18.

(A and B) UV-visible Absorption Spectra Recorded as a Function of Time Showing the fast metHb(FeIII) Reduction in the Coupled Assay with the hGR/NADPH System in the Presence of Benzoylnaphthoquinone 6. (C) Electronic Spectra of the Reactants and the Products of the metHb(FeIII) Reduction. (D) Absorbance Changes at λ = 404 nm and λ = 414 nm.
Solvent: water (hGR acetate buffer pH 6.9 + 47 mM K2PO4 + 200 mM KCl); T = 37.0 °C; 40 μM NADPH + 133 nM hGR + 7.95 μM metHb + 40 μM 3; (1) t = 0; (2) t ~ 19 min.
Figure 19.

UV-Visible Absorption Spectra Recorded as a Function of Time Showing the Influence of the Reduced Substrates on the metHb(FeIII) Reduction in the Coupled Assay with the hGR/NADPH System.
Solvent: water (hGR acetate buffer pH 6.9 + 47 mM K2PO4 + 200 mM KCl) (A) 7.95 μM metHb. (B) 7.95 μM metHb + 40 μM NADPH. (C) 7.95 μM metHb + 40 μM 3. (D) 40 μM NADPH + 133 nM hGR + 7.95 μM metHb + 40 μM 3. For A, B, C and D: (1) t = 0; (2) t = 60 min. (E) 100 μM NADPH + 133 nM hGR + 7.95 μM metHb, (1) t = 0; (2) t = 120 min.
Figure 19 illustrates the major mediation role of the hGR/NADPH system on the metHb(FeIII) reduction by our redox-cyclers (mainly 1,4-naphthoquinone substrates). Methemoglobin is stable in solution at pH 6.9 as depicted in Figure 19A and NADPH, which is the cofactor of hGR, has no influence on its reduction (Figure 19B). When mixing metHb(FeIII) with the 3-benzoyl-1,4-naphthoquinone 6 in the absence of hGR/NADPH (Figure 19C), no reduction occurs as well. Mixing metHb(FeIII) in the presence of the hGR/NADPH system, but in the absence of the redox-cycler 6, has also no effect on the reduction of metHb(FeIII). Disulfide reductases, such as the human glutathione reductase (hGR), are therefore not able by themselves to efficiently reduce these ferric targets. Importantly, the presence of hGR and its cofactor NADPH pre-reduced the redox-cycler substrate which is then able to trigger in its turn the reduction of metHb(FeIII) into oxyHb(FeII). These spectroscopic data enlightened that the bioactive forms of the substrates are the reduced species and clearly pointed out the crucial role of glutathione reductase in mediating metHb(FeIII) reduction by pre-reducing the substrate.
The thorough examination of the data obtained for the six substrates (Table 4 and Table 5) strikingly indicates that the redox-cyclers which are capable of efficiently reducing the metHb(FeIII) into oxyHb(FeII) are also those which are able to inhibit β-hematin (or hemozoin) formation, thus suggesting a closely related mode of action. Since all the compounds tested in this study were shown to interact with hematin π-π dimer (used as a model), no direct correlation can be establish between the ability to bind heme and bioactivity of these compounds toward the parasites, as it is usually anticipated for other series of drugs. However, our substrates (used as representative models of xanthone, benzoxanthone and 1,4-naphthoquinone redox-cyclers) operate according to a new mode of action which mainly targets the reduction of Fe(III)-containing species such as methemoglobin, ferric protoporphyrin or hemozoin-type biocrystals. The mechanism by which the inhibition of hemozoin (or β-hematin) is induced by these redox substrates is not related to the ability of the drugs to prevent hemozoin or β-hematin formation, but rather to their capacity to reduce the ferriprotoporphyrinic units, thereby weakening the hemozoin or β-hematin packing interactions. Their ability to rapidly and efficiently induce methemoglobin reduction in presence of disulfide reductases can also drastically slow down methemoglobin digestion by the parasites and will contribute to their overall bioactivities and to the prevention of the pathogen development.
Conclusion
In this study, we combined complementary bioanalytical methods such as absorption spectrophotometric binding titrations, hematin crystallization assay and methemoglobin reduction assay to evaluate the strength of the interactions between xanthone-, benzoxanthone- and 1,4-naphthoquine-based substrates with the iron(III)-containing targets hematin and methemoglobin in solution under quasi-physiological conditions. These physico-biochemical data allowed us to identify the key parameters related to the mechanism of action of redox substrates toward parasitic pathogens (Plasmodium and Schistosoma). All substrates were shown to firmly interact with hematin π-π dimer in hepes buffer at pH 7.5, which is frequently predicted to be a first prerequisite when targeting hemozoin formation. It appears from our studies that the capacity of our redox-active compounds to rapidly and efficiently reduce methemoglobin has also to be taken into account to rationalize their potential in preventing or inhibiting β-hematin (or hemozoin) formation. The inhibition of β-hematin crystallization was evaluated and correlated well to the binding properties and electron transfer capacities of our substrates.
Six low solubility-featured compounds which possess structures conferring high antiparasitic properties, namely the 1,4-naphthoquinone and the xanthone series, revealed valuable information for the better understanding of their mechanisms of action.
All the compounds were shown to display high affinities for hematin and lead to either 1:1 or 1:2 (and 2:1) charge-transfer complexes with apparent dissociation constant in the μM range (0.2 M sodium hepes buffer at pH 7.5). These binding constants are comparable to those reported for the major antimalarial drugs (Table 2) targeting hemozoin formation and demonstrate the propensity of our substrates to efficiently dissociate the hematin π-π dimer in model solutions. This binding study allowed us to draw structure-activity relationships such as substitution pattern, nature of the substituent, bulkiness of the substrate and redox properties. Even though all the tested substrates interact with hematin π-π dimer, only few of them displayed a potent inhibitory activity in the hematin crystallization assay. In contrast to some previous reports, no direct correlation between a high affinity for hematin π-π dimer and the capacity to prevent β-hematin formation can be deduced.
The ability of our substrates to reduce methemoglobin to hemoglobin was thus examined and discussed in the frame of β-hematin formation. Once again, only few compounds were shown to efficiently and rapidly reduce methemoglobin(FeIII) to hemoglobin(FeII). Very interestingly, a direct correlation between methemoglobin reduction and inhibition of hematin crystallization was observed, thereby demonstrating that the two processes are closely related. Compounds displaying an ability to reduce methemoglobin are also very potent inhibitors of β-hematin formation. The facility of our redox-active agents (mainly the naphthoquinone series) to be rapidly and efficiently reduced by NADPH with mediation of the disulfide reductases (i.e. glutathione reductase) and, in their reduced forms, to catalytically and specifically transfer electrons to ferric species of heme and hemoglobin contribute to interference with hemozoin biomineralization. Both events – oxidizing the reductants and reducing the oxidants in a continuous redox cycle – are expected to lead to the death of the parasites. These multifaceted properties of these hematin binders, efficient methemoglobin reductants after bioreduction by NADPH/GR couple and potent inhibitors of hemozoin/β-hematin oligomerization of several representatives of the naphthoquinone and xanthone/benzoxanthone series will guide the design and the synthesis of redox active antimalarial and antischistosomal drugs with optimized properties.
Experimental Section
Physico-Biochemical Studies
Materials and Methods
Distilled water was purified by passing it through a mixed bed of ion-exchanger (Bioblock Scientific R3-83002, M3-83006) and activated carbon (Bioblock Scientific ORC-83005). All stock solutions were prepared using an AG 245 Mettler Toledo analytical balance. The [FeIIIPPIX]tot and [metHb(FeIII)]tot were calculated from the molecular weight of the corresponding monomers. Hepes (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, Gerbu Biotechnik) buffer (0.2 M) was prepared in water and the pH (7.4) was adjusted with NaOH slurry. pH reading was done on a PHM240 MeterLab® millivoltmeter fitted with a combined glass electrode (Metrohm 6.0234.500, Long Life, 0.1 M NaCl). Calibration was done with commercial Merck® buffers (1.68, 4.00, 6.86, 7.41, and 9.18). Stock solutions of the substrates in DMSO for the different assays were freshly prepared in Eppendorf tubes just before the experiments. Hematin (FeIIIPPIX(OH)) solution was prepared from hemin equine Type III (FeIIIPPIXCl, Sigma-Aldrich) and 0.1 M NaOH solution vigorously stirred at room temperature (RT) for 1 h. Methemoglobin was prepared by oxidation of Human hemoglobin (Sigma-Aldrich) in solution by oxygen at RT (the molecular weight of the tetrameric hemoglobin is 64.458 kDa). NADPH (Biomol, Hambourg, Germany) solution was prepared in water and kept at 0°C during the experiments. The final concentration in the optical cell was calculated from the absorbance at 340 nm (ε340 = 6.22 mM−1 cm−1).
Spectrophotometric titration of hematin π-π dimer by the substrates
An aliquot of 10 μL of the FeIIIPPIX(OH) stock solution (~ 1.68 mM) were dissolved in 500 μL 0.2 M sodium hepes buffer at pH 7.5 in a 1 cm Hellma optical cell. The absorption spectrophotometric titrations were carried out by adding microvolumes of a stock solution of the substrate (~5–9 × 10−4 M in DMSO). Aliquots of 5–20 μL of the stock substrate solution were successively added to the reaction mixture and, after each addition, a UV-visible absorption spectrum (300 nm <λ< 800 nm) was recorded with a Uvikon 941 spectrophotometer. Special care was taken to ensure that equilibrium was attained. The association constants KA and the stoichiometries of the species at equilibrium were determined by processing the spectrophotometric data with the Specfit program [67,68,69,70,71,72]. Specfit uses factor analysis to reduce the absorbance matrix and to extract the eigenvalues prior to the multiwavelength fit of the reduced data set according to the Marquardt algorithm. The uncertainties on the log K values are given as 3σ with σ = standard deviation. Distribution curves of the various complexes and protonated species were calculated using the Hyss 2006 program [78]. Origin 5.0 was used to process the analytical results [79]. For the sake of simplicity charges are omitted in all the chemical equilibria.
β-Hematin Inhibition Assay
The β-hematin inhibition assay used in this work was inspired from that developed by the group of T. J. Egan [74] with some significant improvements in the order of the addition of the reactants, which was imposed by the low solubility of some of our compounds. Drug solutions (89.1 mM, 53.5 mM, 26.7 mM, 17.8 mM, 13.4 mM, 8.9 mM, 4.5 mM, 0 mM) were prepared by dissolving the drug in DMSO (Table 6). Hematin stock solution (1.68 mM) was prepared by dissolving equine hemin (1.05 mg) in 0.1 M NaOH (960 μL). The solution was incubated at room temperature for 1 h. In a series of 2 mL Eppendorf tubes, 2.0 μL of drug solution (or solvent for the blank reference) were dispensed. Then, 20.2 μL of hematin stock solution were added. The Eppendorf tubes were placed in an incubator at 60°C and then, 2 μL of 1 M HCl and 11.7 μL of 12.9 M sodium acetate solution (pH 4.5) pre-incubated at 60°C were added. The final hematin concentration was 1 mM. The final drug concentrations were 4.96 mM, 2.98 mM, 1.45 mM, 0.99 mM, 0.75 mM, 0.5 mM, 0.25 mM and 0 mM and the final pH of the solution was 4.5. The reaction mixtures were incubated at 60°C for 1 h. After incubation, the reaction mixtures were quenched at room temperature by adding 900 μL of 0.2 M sodium hepes buffer + 5% (v/v) pyridine (pH 8.2), to adjust the final pH of the mixture to a value between 7.2 and 7.5. Then, 1100 μL of 0.02 M sodium hepes buffer + 5% pyridine (v/v) (pH 7.5) was added. The Eppendorf tubes were shaken and the precipitate of β-hematin was scrapped from the walls of the Eppendorf tubes to ensure complete dissolution of hematin. The β-hematin was allowed to settle at room temperature for 1 h. The supernatant was carefully transferred to a Hellma optical cell (l = 1 cm) without disturbing the precipitate and the absorption spectra were measured between 300 and 750 nm.
Table 6.
Experimental conditions used for the β-Hematin inhibition assay.
| Drug (equiv.) /Hematin (equiv.) ratio | Concentration (mM) | Volume of stock solution (μL) | Volume of DMSO | Final [drug]0 in the Eppendorf tube (mM) |
|---|---|---|---|---|
| 5 | 89.1 | 15 | 0 | 4.96 |
| 3 | 53.5 | 9 | 6 | 2.98 |
| 1.5 | 26.7 | 4.5 | 10.5 | 1.45 |
| 1 | 17.8 | 3 | 12 | 0.99 |
| 0.75 | 13.4 | 2.25 | 12.75 | 0.75 |
| 0.5 | 8.9 | 1.5 | 13.5 | 0.5 |
| 0.25 | 4.5 | 0.75 | 14.25 | 0.25 |
| 0 | 0 | 0 | 15 | 0 |
MetHb reduction assay
Enzyme
Recombinant hGR was purified as previously reported [80]. One unit of GR activity is defined as the consumption of 1 μmol NADPH per min under conditions of substrate saturation. The enzyme stock solution (266 μM) used for kinetic determinations were > 98% pure as judged from silver stained SDS-PAGE and had specific activities of 200 U/mg (hGR). A hGR stock II solution was prepared by dilution (1/10) of the enzyme stock solution with hGR buffer (pH 6.9). [18]. The hGR buffer (pH 6.9) was prepared by dissolving KH2PO4 (2.79 g), K2HPO4, 3 H2O (6.04 g), EDTA (0.372 g), and KCl (14.91 g) in 1 L water. The pH was adjusted by dropwise addition of 5 M KOH.
Methemoglobin Reduction Coupled Assay with hGR/NADPH
Stock solutions of 2 mM of the drugs in DMSO were freshly prepared. Solutions of 397 μM methemoglobin in hGR buffer and ca. 4 mM NADPH in water were prepared and kept at 0°C during the experiment. The final NADPH concentration in the optical cell was calculated from the absorbance at 340 nm (ε340 = 6.22 mM−1 cm−1). In a Hellma optical cell (l = 1 cm), 945 μL of the hGR buffer, 20 μL of the methemoglobin solution (397 μM), 20 μL of the drug solution (2 mM) and 10 μL of the NADPH solution (ca. 4 mM) were introduced and pre-incubated at 37.5°C (Lauda E200/O11 thermostat and a Huber Minichiller cooler) for 5 min. Then, 5 μL of the hGR stock II solution was added. The reaction was monitored by UV-vis. absorption spectrophotometry on a Cary 300 spectrophotometer with the scanning kinetics method. A spectrum was measured every 30s between 250 and 500 nm over 1 to 2 h. The kinetic data were processed with the Specfit program [67,68,69,70,71,72].
Synthesis
Materials and Methods
Melting points were determined on a Büchi melting point apparatus and were not corrected. 1H (300 MHz), 13C (75 MHz) and 19F (282 MHz) NMR spectra were recorded on a Bruker DRX-300 spectrometer; chemical shifts were expressed in ppm relative to tetramethylsilane (TMS); multiplicity is indicated as s (singlet), d (doublet), t (triplet), q (quartet), sep (septet), m (multiplet), cm (centered multiplet), dd (doublet of a doublet), dt (doublet of a triplet), and td (triplet of a doublet). Cq indicates a quaternary carbon in the 13C NMR assignation. 19F NMR was performed using 1,2-difluorobenzene as external standard (δ = −139.0 ppm). IR spectra (cm−1) were recorded on a Perkin–Elmer Spectrum One Spectrophotometer. Intensities in the IR spectra are indicated as vs (very strong), s (strong), m (medium), w (weak), and b (broad). Elemental analyses were carried out at the Mikroanalytisches Laboratorium der Chemischen Fakultät der Universität Heidelberg. Electron impact mass spectrometry (EI-MS) was recorded at facilities of the Institut für Organische Chemie der Universität Heidelberg. Analytical TLC was carried out on pre-coated Sil G-25 UV254 plates from Macherey&Nagel. Flash chromatography was performed using silica gel G60 (230–400 mesh) from Macherey&Nagel.
Synthesis of Xanthones 1 to 4
General Procedure 1
A flask was purged with Argon and the following reagents were successively introduced under inert atmosphere: the arene (2.0 equiv.), the benzonitrile (1.0 equiv.), Pd(OAc)2 (0.1 equiv.), DMSO and TFA (Scheme 3 and Scheme 4). The flask was sealed and the reaction mixture was stirred and heated at 95°C for 24 h. The mixture was cooled and 8 mL of water was added to the flask. The resulting mixture was heated at 70°C for 2 h. Then, the mixture was cooled and extracted three times with 15 mL Et2O. The combined organic layers were dried over MgSO4 and evaporated. The resulting oil was purified through flash chromatography.
(2,5-dimethoxyphenyl)(2-fluorophenyl)methanone (3a)
Commercially available 1,4-dimethoxybenzene (500 mg, 3.62 mmol), 2-fluorobenzonitrile (220 mg, 1.81 mmol), DMSO (0.20 mL) and TFA (4.5 mL) were used as starting materials and treated according to general procedure. The resulting yellow oil was purified through flash chromatography (hexane:EtOAc 2:1). The product 3a was obtained as a colorless oil (400 mg, 1.54 mmol, 85%). Rf (hexane:EtOAc 2:1) = 0.44. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.76–7.85 (m, 2H), 7.61–7.73 (m, 1H), 7.34–7.45 (m, 2H), 7.19–7.25 (m, 1H), 7.05 (d, J = 8.79 Hz, 1H), 3.95 (s, 3H, OCH3), 3.77 (s, 3H, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 193.71 (C=O), 161.03 (d, 1JCF = 256 Hz, Cq-F), 153.41 (Cq), 153.05 (Cq), 133.94 (d, 3JCF = 9 Hz, CHar), 130.95 (d, 4JCF= 2 Hz, CHar), 129.11 (Cq), 128.0 (d, 2JCF = 12 Hz, Cq), 124.09 (d, 3JCF = 3.7 Hz, CHar), 120.00 (CHar), 115.96 (d, 2JCF = 22 Hz, CHar), 114.75 (CHar), 113.29 (CHar), 56.24 (OCH3), 55.92 (OCH3). 19F NMR (282 MHz, CDCl3): δ (ppm) = −112.81 (ddd, 3JHF = 11 Hz, 4JHF = 7 Hz, 4JHF = 5Hz). EI-MS (70 eV, m/z (%)): 260.2 ([M+], 100) for C15H13FO3 [64].
(2-fluorophenyl)(2-hydroxy-5-methoxyphenyl)methanone (3b)
A solution of (2,5-dimethoxyphenyl)(2-fluorophenyl)methanone 3a (100 mg, 0.384 mmol) in 8 mL of dichloromethane (DCM) was cooled at 0°C and kept stirring for 30 min. Then, BBr3 (0.38 mL, 1M in DCM) was added dropwise to the solution and the reaction mixture was stirred at 0°C for 1 h. The reaction mixture was quenched with 8 mL of methanol (MeOH). A saturated solution of NaCl was added to the mixture and it was extracted three times with 10 mL DCM and twice with 10 mL ethyl acetate. The organic layers were combined, dried over MgSO4 and evaporated. The product 3b was obtained as a yellow powder (82 mg, 0.33 mmol, 86%). Rf (cyclohexane:DCM 3:2) = 0.32. 1H NMR (300 MHz, CDCl3): δ (ppm) = 11.59 (s, 1H, Ph-OH), 7.45–7.58 (m, 2H, Har), 7.14–7.32 (m, 3H, Har), 7.01 (d, J = 9.1 Hz, 1H, Har), 6.86 (t, J = 7.5 Hz, 1H, Har), 3,68 (s, 3H, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 198.10 (C=O), 159.10 (d, 1JCF= 252 Hz, Cq-F), 157.54 (Cq), 151.77 (Cq), 132.96 (d, 3JCF = 8 Hz, CHar-F), 129.87 (d, 3JCF = 8 Hz, CHar-F), 126.21 (d, 2JCF = 16 Hz, Cq-F), 125.02 (CHar), 124.40 (CHar), 119.31 (Cq), 119.19 (CHar), 116.37 (d, 2JCF = 21 Hz, CHar-F), 115.17 (d, 4JCF = 2 Hz, CHar-F), 55.90 (OCH3). 19F NMR (282 MHz, CDCl3): δ (ppm) = −112.22 (cm, 3JHF=10 Hz, 4JHF=7 Hz, 4JHF=5 Hz, 5JHF=3 Hz). EI-MS (70 eV, m/z (%)): 246.1 ([M+], 100) for C14H11FO3 [64].
(2,4-difluoro-6-hydroxy-3-methoxyphenyl)(2-fluorophenyl)methanone (4a)
Commercially available 4-methoxy-3,5-difluorophenol (200 mg, 1.25 mmol), 2-fluorobenzonitrile (67 μL, 0.63 mmol), DMSO (0.06 mL) and TFA (1.6 mL) were used as starting materials and treated according to general the procedure 1. The resulting brown oil was purified through flash chromatography (cyclohexane:EtOAc 10:1). The product 4a was obtained as a yellow powder (25 mg, 0.09 mmol, 14%). Rf (cyclohexane:EtOAc 10:1) = 0.34. 1H NMR (300 MHz, CDCl3): δ (ppm) = 11.93 (s, 1H, Ph-OH), 7.50–7.57 (m, 2H), 7.27 (ddd, 3JHH =8 Hz, 4JHF = 7 Hz, 4JHH = 1 Hz, 1H), 7.13 (ddd, 3JHF = 10 Hz, 3JHH = 8 Hz, 4JHH = 1 Hz, 1H), 6.61 (dd, 3JHF = 12 Hz, 5JHF = 2 Hz, 1H), 3.85 (s, 3H, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 193.70 (C=O), 164.90 (Cq), 161.62 (dd, 1JCF = 259 Hz, 3JCF = 9 Hz, Cq-F), 161.24 (Cq), 159.57 (d, 1JCF = 252 Hz, Cq-F), 156.81 (dd, 1JCF = 259 Hz, 3JCF = 9 Hz, Cq-F), 159.23 (dd, 2JCF = 16 Hz, Cq-O), 156.36 (Cq), 133.57 (d, 3JCF = 8 Hz, CHar), 129.38 (d, 3JCF = 8 Hz, CHar), 124.40 (d, 4JCF = 3 Hz, CHar), 115.84 (d, 2JCF = 22 Hz, CHar), 101.50 (dd, 2JCF = 22 Hz, 4JCF = 3 Hz, CHar), 62.49 (t, 4JCF = 3 Hz, OCH3). 19F NMR (282 Hz, CDCl3): δ (ppm) = −113.87 (dd, 4JFF = 15 Hz, 3JHF = 12 Hz), −114.47 (cm, 3JHF = 13 Hz, 4JHF = 7 Hz), −122.21 (dd, 4JFF = 15 Hz, 5JHF =7Hz). EI-MS (70 eV, m/z (%)): 282.1 ([M+], 24) for C14H9F3O3.
General procedure 2
The benzophenone derivative (1.0 equiv.) and K2CO3 (2.0 equiv.) were placed in a round-bottom flask. The flask was sealed under Argon and 10 mL of dry Acetone were added. The reaction mixture was stirred at 50°C for 2 h. The suspension was then filtered through a pad of celite and washed with 25 mL of diethylether (Et2O). The filtrate was concentrated under vacuum.
2-methoxy-9H-xanthen-9-one (3)
(2-fluorophenyl)(2-hydroxy-5-methoxyphenyl)methanone 3b (80 mg, 0.325 mmol) was used as a starting material and treated according to general procedure 2. The 2-methoxy-9H-xanthen-9-one (3) was obtained as white, “cotton-like” flakes (45 mg, 0.20 mmol, 61%). Rf (cyclohexane:DCM 3:2) = 0.20. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.35 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H), 7.69–7.75 (m, 2H), 7.31–7.50 (m, 4H), 3.92 (1H, 3H, OCH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 177.09 (C=O), 156.14 (Cq-O), 156.02 (Cq), 151.02 (Cq), 134.60 (CHar), 126.69 (CHar), 124.91 (CHar), 123.73 (CHar), 122.14 (Cq), 121.27 (Cq), 119.43 (CHar), 113.23 (CHar), 105.86 (CHar), 55.95 (CH3). EI-MS (70 eV, m/z (%)): 226.1 ([M+], 42), 211.1 ([M-CH3]+, 30) for C14H10O3. Elemental analysis calcd for C14H10O3 (%): C, 74.33; H, 4.46; found: C, 74.04; H, 4.38. m.p. 126–128°C.
2,4-difluoro-3-methoxy-9H-xanthen-9-one (4)
(2,4-difluoro-6-hydroxy-3-methoxyphenyl)(2-fluorophenyl)methanone 4a (200 mg, 0.71 mmol) was used as a starting material and treated according to general procedure 2. 2,4-difluoro-3-methoxy-9H-xanthen-9-one (4) was obtained as a beige powder (100 mg, 0.38 mmol, 53%). Rf (cyclohexane:EtOAc 10:1) = 0.21. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.30 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H), 7.72 (dd, 3J = 8 Hz, 4J = 1 Hz, 1H), 7.40–7.45 (m, 2H), 7.06 (dd, 3JHF = 10 Hz, 5JHF = 2 Hz, 1H), 4.04 (1H, 3H, OCH3); 13C NMR (75 MHz, CDCl3): δ (ppm) = 174.53 (C=O), 159.61 (Cq), 158.94 (Cq), 159.46 (dd, 1JCF = 260 Hz, 3JCF = 8 Hz, Cq-F), 155.37 (Cq), 154.99 (dd, 1JCF = 257 Hz, 3JCF = 8 Hz, Cq-F), 151.96 (dd, 3JCF = 16 Hz, Cq), 134.92 (CHar), 126.68 (CHar), 124.50 (CHar), 121.75 (Cq), 117.47 (CHar), 101.35 (dd, 2JCF = 23 Hz, 4JCF = 4 Hz, CHar), 62.47 (dd, 4JCF = 3 Hz, OCH3). 19F NMR (282 MHz, CDCl3): δ (ppm) = − 116.61 (dd, 4JFF = 15 Hz, 3JHF = 11 Hz), − 126.69 (d, 4JFF = 15 Hz). EI-MS (70 eV, m/z (%)): 262.0 ([M+], 73), 247.0 ([M-CH3]+, 15). Elemental analysis calcd for C14H10O3 (%): C, 64.13; H, 3.08; found: C, 64.36; H, 3.02. m.p. 125–127°C.
Characterization Data of Benzoxanthones 1 and 2
5-methoxy-6-methyl-7H-benzo[c]xanthen-7-one (1) was obtained as an orange powder (96 mg, 0.33 mmol, 69%). Rf (cyclohexane:EtOAc 10:1) = 0.36. 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.61–8.64 (m, 1H), 8.31–8.34 (m, 1H), 8.13–8.16 (m, 1H), 7.69–7.75 (m, 2H), 7.58–7.65 (m, 2H), 7.37–7.42 (cm, 1H), 3.91 (s, 3H, OCH3), 2.96 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 178.68 (C=O), 154.81 (Cq), 151.84 (Cq), 149.81 (Cq), 133.93 (CHar), 130.99 (Cq), 129.87 (CHar), 126.57 (CHar), 126.29 (CHar), 125.86 (Cq), 124.22 (CHar), 123.79 (Cq), 123.28 (CHar), 123.21 (Cq), 122.17 (CHar), 117.46 (CHar), 117.35 (Cq), 61.45 (OCH3), 14.51 (CH3). EI-MS (70 eV, m/z (%)): 290.1 ([M+], 53) for C19H14O3. Elemental analysis calcd for C19H14O3 (%): C, 78.61; H, 4.86; found: C, 78.81; H, 4.92. m.p. 175–177°C.
5-methoxy-6-methyl-11-(trifluoromethyl)-7H-benzo[c]xanthen-7-one (2) was obtained as a light yellow, cotton-like solid (120 mg, 0.33 mmol, 86%). 1H NMR (300 MHz, CDCl3): δ (ppm) = 8.66–8.68 (m, 1H), 8.54–8.57 (m, 1H), 8.17–8.20 (m, 1H), 8.02–8.05 (m, 1H), 7.76–7.82 (cm, 1H), 7.67–7.73 (cm, 1H), 7.47–7.53 (cm, 1H), 3.92 (s, 3H, OCH3), 2.97 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ (ppm) = 177.39 (C=O), 151.79 (Cq), 151.52 (Cq), 150.50 (Cq), 131.27 (q, 3JCF= 4 Hz, CHar), 130.99 (CHar), 130.30 (CHar), 126.92 (CHar), 125.56 (Cq), 124.05 (Cq), 123.70 (Cq), 123.49 (CHar), 123.34 (CHar), 123.13 (q, 1JCF = 273 Hz, CF3), 122.15 (CHar), 119.70 (Cq), 119.08 (q, 2JCF = 32 Hz, Cq-CF3), 117.32 (Cq), 61.54 (OCH3), 14.46 (CH3). 19F NMR (282 MHz, CDCl3): δ (ppm) = −61.31 (s, CF3). EI-MS (70 eV, m/z (%)): 358.0 ([M+], 60), 343.0 ([M-CH3]+, 100) for C20H13O3F3. Elemental analysis calcd for C20H13O3F3 (%): C, 67.04; H, 3.66; found: C, 66.95; H, 3.80. m.p. 198–200°C.
Acknowledgments
The Centre National de la Recherche Scientifique (CNRS) and the University of Strasbourg (UMR 7509 CNRS-UdS) partly supported this work. This work was also funded by the International Center for Frontier Research in Chemistry (ic-FRC) in Strasbourg. L.J. and D.A.L. are grateful to DFG via the SFB 544 (B14 project) and NIH/National Institute of Allergy and Infectious Disease (NIAID) (grant R01AI065622) for their salaries. The authors are grateful to Sokune Seng for her help in measuring IR spectra and β-hematin inhibition assay.
List of Abbreviations
- ε
molar absorptivity coefficient
- benzoxanthone
benz[c]xanthen-7-one
- λ
wavelength
- CT
charge transfer (complex or band)
- DCM
dichloromethane
- DMSO
dimethylsulfoxide
- EDTA
ethylenediaminetetraacetic acid
- EI-MS
electron impact mass spectrometry
- FeIIIPPIX
ferriprotoporphyrin
- FeIIIPPIXCl
ferriprotoporphyrin chloride
- Hb(FeII)
hemoglobin subunit
- hGR
human glutathione reductase
- KA
association constant
- KD
dissociation constant
- KDim
dimerization constant
- M
molar
- MeOH
methanol
- metHb(FeIII)
methemoglobin subunit
- NADPH
reduced form of nicotinamide adenine dinucleotide phosphate
- oxyHb(FeII)
hemoglobin subunit with O2 molecule coordinated on iron(II)
- ROS
reactive oxygen species
- TFA
trifluoroacetic acid
- tot
total
- UV-vis
ultraviolet/visible (spectroscopy)
References
- 1.Corrêa Soares JB, Maya-Monteiro CM, Bittencourt-Cunha PR, Atella GC, Lara FA, d’Avila JC, Menezes D, Vannier-Santos MA, Oliveira PL, Egan TJ, Oliveira MF. Extracellular lipid droplets promote hemozoin crystallization in the gut of the blood fluke Schistosoma mansoni. FEBS Lett. 2007;581:1742–50. doi: 10.1016/j.febslet.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 2.Oliveira MF, d’Avila JC, Torres CR, Oliveira PL, Tempone AJ, Rumjanek FD, Braga CM, Silva JR, Dansa-Petretski M, Oliveira MA, de Souza W, Ferreira ST. Haemozoin in Schistosoma mansoni. Mol Biochem Parasitol. 2000;111:217–21. doi: 10.1016/s0166-6851(00)00299-1. [DOI] [PubMed] [Google Scholar]
- 3.Chen MM, Shi L, Sullivan DJ., Jr Haemoproteus and Schistosoma synthesize heme polymers similar to Plasmodium hemozoin and beta-hematin. Mol Biochem Parasitol 2001; 113:1–8. b) Egan, TJ, Recent advances in understanding the mechanism of hemozoin (malaria pigment) formation. J Inorg Biochem 2008; 102: 1288–99. c) Weissbuch, I, Leiserowitz, L. Interplay between malaria, crystalline hemozoin formation, and antimalarial drug action and design. Chem Rev. 2008;108:4899–914. doi: 10.1016/s0166-6851(00)00365-0. [DOI] [PubMed] [Google Scholar]
- 4.Schirmer RH, Müller JG, Krauth-Siegel RL. Disulfide reductase inhibitors as chemotherapeutic agents: the design of drugs for trypanosomiasis and malaria. Angew Chem Int Ed. 1995;34:141–54. [Google Scholar]
- 5.Krauth-Siegel RL, Bauer H, Schirmer RH. Dithiol proteins as guardians of the intracellular redox milieu in parasites: old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew Chem Int Ed. 2005;44:690–715. doi: 10.1002/anie.200300639. [DOI] [PubMed] [Google Scholar]
- 6.a) Atamna H, Ginsburg H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J Biol Chem. 1995;270:24876–83. doi: 10.1074/jbc.270.42.24876. [DOI] [PubMed] [Google Scholar]; b) Meierjohan S, Walter RD, Müller S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. Biochem J. 2002;368:761–8. doi: 10.1042/BJ20020962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Färber PM, Arscott LD, Williams CH, Jr, Becker K, Schirmer RH. Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett 1998; 422: 311–4. b) Buchholz K, Schirmer RH, Eubel JK, Akoachere MB, Dandekar T, Becker K, Gromer S. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob Agents Chemother 2008; 52: 183–91. c) Davioud-Charvet E, Delarue S, Biot C, Schwöbel B, Böhme CC, Müssigbrodt A, Maes L, Sergheraert C, Grellier P, Schirmer RH, Becker K. A prodrug form of a Plasmodium falciparum glutathione reductase inhibitor conjugated with a 4-anilinoquinoline. J Med Chem. 2001;44:4268–76. [Google Scholar]
- 8.Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol Biochem Parasitol. 2002;121:129–39. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 9.Kuntz AN, Davioud-Charvet E, Dessolin J, Sayed AA, Califf LL, Arnér ESJ, Williams DL. Schistosome redox balance and thioredoxin glutathione reductase: An essential parasite enzyme and novel drug target. PLoS Medicine. 2007;4:e206, 1–16. doi: 10.1371/journal.pmed.0040206. Erratum in: PLoS Medicine 2007; 4: e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliveira MF, d’Avila JC, Tempone AJ, Soares JB, Rumjanek FD, Ferreira-Pereira A, Ferreira ST, Oliveira PL. Inhibition of heme aggregation by chloroquine reduces Schistosoma mansoni infection. J Infect Dis. 2004;190:843–52. doi: 10.1086/422759. [DOI] [PubMed] [Google Scholar]
- 11.Corrêa Soares JB, Menezes D, Vannier-Santos MA, Ferreira-Pereira A, Almeida GT, Venancio TM, Verjovski-Almeida S, Zishiri VK, Kuter D, Hunter R, Egan TJ, Oliveira MF. Interference with hemozoin formation represents an important mechanism of schistosomicidal action of antimalarial quinoline methanols. PLoS Negl Trop Dis. 2009;3:e477. doi: 10.1371/journal.pntd.0000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Villiers KA, Marques HM, Egan TJ. The crystal structure of halofantrine-ferriprotoporphyrin IX and the mechanism of action of arylmethanol antimalarials. J Inorg Biochem. 2008;102:1660–7. doi: 10.1016/j.jinorgbio.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Xiao SH, Catto BA. In vitro and in vivo studies of the effect of artemether on Schistosoma mansoni. Antimicrob Agents Chemother 1989; 33: 1557–62. b) Hong YL, Yang YZ, Meshnick SR. The interaction of artemisinin with malarial hemozoin. Mol Biochem Parasitol 1994; 63: 121–8. c) Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228:1049–55. [Google Scholar]
- 14.a) Xu Kelly J, Winter R, Riscoe M, Peyton DH. A spectroscopic investigation of the binding interactions between 4,5-dihydroxyxanthone and heme. J Inorg Biochem. 2001;86:617–25. doi: 10.1016/s0162-0134(01)00217-3. [DOI] [PubMed] [Google Scholar]; b) Nabih I. Structure and activity in the schistosomicidal thiaxanthone and xanthone derivatives. J Pharm Sci. 1966;55:221–2. doi: 10.1002/jps.2600550224. [DOI] [PubMed] [Google Scholar]
- 15.Ignatushchenko MV, Winter RW, Bächinger HP, Hinrichs DJ, Riscoe MK. Xanthones as antimalarial agents; studies of a possible mode of action. FEBS Lett. 1997;409:67–73. doi: 10.1016/s0014-5793(97)00405-5. [DOI] [PubMed] [Google Scholar]
- 16.Riscoe M, Kelly JX, Winter R. Xanthones as antimalarial agents: discovery, mode of action, and optimization. Curr Med Chem. 2005;12:2539–49. doi: 10.2174/092986705774370709. [DOI] [PubMed] [Google Scholar]
- 17.McMillan PJ, Stimmler LM, Foth BJ, McFadden GI, Müller S. The human malaria parasite Plasmodium falciparum possesses two distinct dihydrolipoamide dehydrogenases. Mol Microbiol. 2005;55:27–38. doi: 10.1111/j.1365-2958.2004.04398.x. [DOI] [PubMed] [Google Scholar]
- 18.Müller T, Johann L, Jannack B, Brückner M, Lanfranchi DA, Bauer H, Sanchez C, Yardley V, Deregnaucourt C, Schrével J, Lanzer M, Schirmer RH, Davioud-Charvet E. Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-naphthoquinones - A new strategy to combat malarial parasites. J Am Chem Soc. 2011;133:11557–71. doi: 10.1021/ja201729z. [DOI] [PubMed] [Google Scholar]
- 19.Davioud-Charvet E, Lanfranchi DA. Subversive substrates of glutathione reductases from P. falciparum-infected red blood cells as antimalarial agents. In: Becker K, Selzer PM, editors. Apicomplexan parasites -Molecular approaches toward targeted drug development. Vol. 2. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2011. from the series Drug Discovery in Infectious Diseases. [Google Scholar]
- 20.Asher C, de Villiers KA, Egan TJ. Speciation of ferriprotoporphyrin IX in aqueous and mixed aqueous solution is controlled by solvent identity, pH, and salt concentration. Inorg Chem. 2009;48:7994–8003. doi: 10.1021/ic900647y. [DOI] [PubMed] [Google Scholar]
- 21.Ncokazi KK, Egan TJ. A colorimetric high-throughput beta-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal Biochem. 2005;338:306–19. doi: 10.1016/j.ab.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 22.Monti D, Vodopivec B, Basilico N, Olliaro P, Taramelli D. A novel endogenous antimalarial: Fe(II)-protoporphyrin IX alpha (heme) inhibits hematin polymerization to beta-hematin (malaria pigment) and kills malaria parasites. Biochemistry. 1999;38:8858–63. doi: 10.1021/bi990085k. [DOI] [PubMed] [Google Scholar]
- 23.Hogg T, Nagarajan K, Herzberg S, Chen L, Shen X, Jiang H, Wecke M, Blohmke C, Hilgenfeld R, Schmidt CL. Structural and functional characterization of Falcipain-2, a hemoglobinase from the malarial parasite Plasmodium falciparum. J Biol Chem. 2006;281:25425–37. doi: 10.1074/jbc.M603776200. [DOI] [PubMed] [Google Scholar]
- 24.Anstey NM, Hassanali MY, MLalasi J, Manyenga D, Mwaikambo ED. Elevated levels of methaemoglobin in Tanzanian children with severe and uncomplicated malaria. Trans R Soc Trop Med Hyg. 1996;90:147–51. doi: 10.1016/s0035-9203(96)90118-2. [DOI] [PubMed] [Google Scholar]
- 25.Blank O, Davioud-Charvet E, Elhabiri M. Interactions of the antimalarial methylene blue with methemoglobin and heme derivatives - A physico-biochemical study. Antioxid & Redox Signal. 2012;17:544–554. doi: 10.1089/ars.2011.4239. [DOI] [PubMed] [Google Scholar]
- 26.Lehane AM, Hayward R, Saliba KJ, Kirk K. A verapamil-sensitive chloroquine-associated H+ leak from the digestive vacuole in chloroquine-resistant malaria parasites. J Cell Sci. 2008;121:1624–32. doi: 10.1242/jcs.016758. [DOI] [PubMed] [Google Scholar]
- 27.Ryter SW, Tyrell RM. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt TH, Frezzatti WA, Jr, Schereier S. Heme-induced lipid membrane disorder and increased permeability: a molecular model for the mechanism of cells lysis. Arch Biochem Biophys. 1993;307:96–103. doi: 10.1006/abbi.1993.1566. [DOI] [PubMed] [Google Scholar]
- 29.Oliveira MF, Timm BL, Machado EA, Miranda K, Attias M, Silva JR, Dansa-Petretski M, de Oliveira MA, de Souza W, Pinhal NM, Sousa JJ, Vugman NV, Oliveira PL. On the pro-oxidant effects of haemozoin. FEBS Lett. 2002;512:139–44. doi: 10.1016/s0014-5793(02)02243-3. [DOI] [PubMed] [Google Scholar]
- 30.Green MD, Xiao L, Lal AA. Formation of hydroxyeicosatetraenoic acids from hemozoin-catalyzed oxidation of arachidonic acid. Mol Biochem Parasitol. 1996;83:183–8. doi: 10.1016/s0166-6851(96)02769-7. [DOI] [PubMed] [Google Scholar]
- 31.Schwarzer E, Bellomo G, Giribaldi G, Ulliers D, Arese P. Phagocytosis of malarial pigment haemozoin by human monocytes: a confocal microscopy study. Parasitology. 2001;123:125–31. doi: 10.1017/s0031182001008216. [DOI] [PubMed] [Google Scholar]
- 32.Hanscheid T, Egan TJ, Grobusch MP. Haemozoin: from melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect Dis. 2007;7:675–85. doi: 10.1016/S1473-3099(07)70238-4. [DOI] [PubMed] [Google Scholar]
- 33.Cheng L, Lee J, Powell DR, Richter-Addo GB. μ-Oxo-bis[(protoporphyrin IX dimethyl ester)-iron(III)] Acta Crystallogr, Sect E: Struct Rep Online. 2004;60:m1340–m1342. [Google Scholar]
- 34.Koenig DF. The structure of α-chlorohemin. Acta Crystallogr. 1965;18:663–73. doi: 10.1107/s0365110x65001536. [DOI] [PubMed] [Google Scholar]
- 35.a) Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. The structure of malaria pigment β-haematin. Nature. 2000;404:307–10. doi: 10.1038/35005132. [DOI] [PubMed] [Google Scholar]; b) Bohle DS, Dodd EL, Kosar AJ, Sharma L, Stephens PW, Suárez L, Tazoo D. Soluble synthetic analogues of malaria pigment: structure of mesohematin anhydride and its interaction with chloroquine in solution. Angew Chem Int Ed. 2011;50:6151–4. doi: 10.1002/anie.201100910. [DOI] [PubMed] [Google Scholar]
- 36.Shack J, Clarke WM. Metalloporphyrins: VI. Cycles of changes in systems containing heme. J Biol Chem. 1947;171:143–87. [Google Scholar]
- 37.Brown SB, Dean TC, Jones P. Aggregation of ferrihaems. Dimerization and protolytic equilibria of protoferrihaem and deuteroferrihaem in aqueous solution. Biochem J. 1970;117:733–9. doi: 10.1042/bj1170733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Little RG, Dymock KR, Ibers JA. Crystal and molecular structure of the ferrihemochrome bis(1-methylimidazole)(protoporphyrin IX)iron-methanol-water. J Am Chem Soc. 1975;97:4532–9. doi: 10.1021/ja00849a014. [DOI] [PubMed] [Google Scholar]
- 39.de Villiers KA, Kaschula CH, Egan TJ, Marques HM. Speciation and structure of ferriprotoporphyrin IX in aqueous solution: Spectroscopic and diffusion measurements demonstrate dimerization, but not μ-oxo dimer formation. J Biol Inorg Chem. 2007;12:101–17. doi: 10.1007/s00775-006-0170-1. [DOI] [PubMed] [Google Scholar]
- 40.Crespo MP, Tilley L, Klonis N. Solution behavior of hematin under acidic conditions and implications for its interactions with chloroquine. J Biol Inorg Chem. 2010;15:1009–22. doi: 10.1007/s00775-010-0661-y. [DOI] [PubMed] [Google Scholar]
- 41.Klonis N, Dilanian R, Hanssen E, Darmanin C, Streltsov V, Deed S, Quiney H, Tilley L. Hematin-hematin self-association states involved in the formation and reactivity of the malaria parasite pigment, hemozoin. Biochemistry. 2010;49:6804–11. doi: 10.1021/bi100567j. [DOI] [PubMed] [Google Scholar]
- 42.Das DK, Medhi OK. The role of heme propionate in controlling the redox potentials of hemes: square wave voltammetry of protoporphyrinato IX iron(III) in aqueous surfactant micelles. J Inorg Biochem. 1998;70:83–90. doi: 10.1016/s0162-0134(98)10002-8. [DOI] [PubMed] [Google Scholar]
- 43.a) Slater AF, Swiggard WJ, Orton BR, Flitter WD, Goldberg DE, Cerami A, Henderson GB. An iron-carboxylate bond links the heme units of malaria pigment. Proc Natl Acad Sci. 1991;88:325–29. doi: 10.1073/pnas.88.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fitch CD, Kanjananggulpan P. The state of ferriprotoporphyrin IX in malaria pigment. J Biol Chem. 1987;262:15552–5. [PubMed] [Google Scholar]
- 44.Egan TJ, Chen JYJ, de Villiers KA, Mabotha TE, Naidoo KJ, Ncokazi KK, Langford SJ, McNaughton D, Pandiancherri S, Wood BR. Haemozoin (β-haematin) biomineralization occurs by self-assembly near the lipid/water interface. FEBS Lett. 2006;580:5105–10. doi: 10.1016/j.febslet.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 45.a) Egan TJ, Hunter R, Kaschula CH, Marques HM, Misplon A, Walden JC. Structure-function relationships in aminoquinolines: effect of amino and chloro groups on quinoline-hematin complex formation, inhibition of beta-hematin formation, and antiplasmodial activity. J Med Chem. 2000;43:283–91. doi: 10.1021/jm990437l. [DOI] [PubMed] [Google Scholar]; b) Kuter D, Chibale K, Egan TJ. Linear free energy relationships predict coordination and π-stacking interactions of small molecules with ferriprotoporphyrin IX. J Inorg Biochem. 2011;105:684–92. doi: 10.1016/j.jinorgbio.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Ogoshi H, Mizutani T, Hayashi T, Kuroda Y. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. Academic Press; San Diego, CA: 2000. [Google Scholar]
- 47.de Villiers KA, Egan TJ. Recent advances in the discovery of haem-targeting drugs for malaria and schistosomiasis. Molecules. 2009;14:2868–87. doi: 10.3390/molecules14082868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burrows JN, Chibale K, Wells TNC. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11:1226–54. doi: 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- 49.Atamna H, Krugliak M, Shalmiev G, Deharo E, Pescarmona G, Ginsburg H. Mode of antimalarial effect of methylene blue and some of its analogues on Plasmodium falciparum in culture and their inhibition of P.vinckei petteri and P.yoelii nigeriensis in vivo. Biochem Pharmacol 1996; 51: 693–700. b) Kalkanidis M, Klonis N, Tilley L, Deady LW. Novel phenothiazine antimalarials: synthesis, antimalarial activity, and inhibition of the formation of beta-haematin. Biochem Pharmacol. 2002;63:833–42. [Google Scholar]
- 50.a) Egan TJ, Mavuso WW, Ncokazi KK. The mechanism of beta-hematin formation in acetate solution. Parallels between hemozoin formation and biomineralization processes. Biochemistry. 2001;40:204–13. doi: 10.1021/bi0013501. [DOI] [PubMed] [Google Scholar]; b) Egan TJ. Haemozoin formation as a target for the rational design of new antimalarials. Drug Design Rev. 2004;1:93–110. [Google Scholar]
- 51.Egan TJ, Mavuso WW, Ross DC, Marques HM. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J Inorg Biochem. 1997;68:137–45. doi: 10.1016/s0162-0134(97)00086-x. [DOI] [PubMed] [Google Scholar]
- 52.Loria P, Miller S, Foley M, Tilley L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem J. 1999;339:363–70. [PMC free article] [PubMed] [Google Scholar]
- 53.a) Egan TJ. Haemozoin (malaria pigment): a unique crystalline drug target. Targets. 2003;2:115–24. [Google Scholar]; b) Egan TJ, Ncokazi KK. Quinoline antimalarials decrease the rate of β-hematin formation. J Inorg Biochem. 2005;99:1532–9. doi: 10.1016/j.jinorgbio.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 54.Egan TJ. Interactions of quinoline antimalarials with hematin in solution. J Inorg Biochem. 2006;100:916–26. doi: 10.1016/j.jinorgbio.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 55.Winter RW, Ignatushchenko M, Ogundahunsi OAT, Cornell KA, Oduola AMJ, Hinrichs DJ, Riscoe MK. Potentiation of an antimalarial oxidant drug. Antimicrob Agents Chemother. 1997;41:1449–54. doi: 10.1128/aac.41.7.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly JX, Winter R, Riscoe MK, Peyton DH. A spectroscopic investigation of the binding interactions between 4,5-dihydroxanthone and heme. J Inorg Biochem. 2001;86:617–25. doi: 10.1016/s0162-0134(01)00217-3. [DOI] [PubMed] [Google Scholar]
- 57.Bachhawat K, Thomas CJ, Surolia N, Surolia A. Interaction of chloroquine and its analogues with heme: an isothermal titration calorimetric study. Biochem Biophys Res Commun. 2000;276:1075–79. doi: 10.1006/bbrc.2000.3592. [DOI] [PubMed] [Google Scholar]
- 58.Egan TJ, Hempelmann E, Mavuso WW. Characterisation of synthetic β-haematin and effects of the antimalarial drugs quinidine, halofantrine, desbutylhalofantrine and mefloquine on its formation. J Inorg Biochem. 1999;73:101–7. doi: 10.1016/S0162-0134(98)10095-8. [DOI] [PubMed] [Google Scholar]
- 59.Dascombe MJ, Drew MGB, Morris H, Wilairat P, Auparakkitanon S, Moule WA, Alizadeh-Shekalgourabi S, Evans PG, Lloyd M, Dyas AM, Carr P, Ismael FMD. Mapping antimalarial pharmacophores as a useful tool for the rapid discovery of drugs effective in vivo: design construction, characterisation and pharmacology of metaquine. J Med Chem. 2005;48:5423–36. doi: 10.1021/jm0408013. [DOI] [PubMed] [Google Scholar]
- 60.Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem Pharmacol. 1998;55:727–36. doi: 10.1016/s0006-2952(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 61.Biot C, Taramelli D, Forfar-Bares I, Maciejewski LA, Boyce M, Nowogrocki G, Brocard JS, Basilico N, Olliario P, Egan TJ. Insights on the mechanism of action of ferroquine. Relationship between physicochemical properties and antimalarial activity. Mol Pharmaceut. 2005;2:185–93. doi: 10.1021/mp0500061. [DOI] [PubMed] [Google Scholar]
- 62.Dubar F, Egan TJ, Pradines B, Kuter D, Ncokazi KK, Forge D, Paul JF, Pierrot C, Kalamou H, Khalife J, Buisine E, Rogier C, Vezin H, Forfar I, Slomianny C, Trivelli X, Kapishnikov S, Leiserowitz L, Dive D, Biot C. The antimalarial ferroquine: role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem Biol. 2011;6:275–87. doi: 10.1021/cb100322v. [DOI] [PubMed] [Google Scholar]
- 63.Sousa ME, Pinto MMM. Synthesis of xanthones: an overview. Curr Med Chem. 2005;12:2447–79. doi: 10.2174/092986705774370736. [DOI] [PubMed] [Google Scholar]
- 64.Zhou C, Larock RC. Synthesis of aryl ketones or ketimines by palladium-catalyzed arene C-H addition to nitriles. J Org Chem. 2006;71:3551–8. doi: 10.1021/jo060220g. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida S, Iizuka T, Nozawa T, Hatano M. Studies on the charge transfer band in high spin state of ferric myoglobin and hemoglobin by low temperature optical and magnetic circular dichroism spectroscopy. Biochim Biophys Acta (BBA) - Protein Structure. 1975;405:122–35. doi: 10.1016/0005-2795(75)90322-0. [DOI] [PubMed] [Google Scholar]
- 66.Eaton WA, Hanson LK, Stephens PJ, Sutherland JC, Dunn JBR. Optical spectra of oxy- and deoxyhemoglobin. J Am Chem Soc. 1978;100:4991–5003. [Google Scholar]
- 67.Gampp H, Maeder M, Meyer CJ, Zuberbühler AD. Calculation of equilibrium constants from multiwavelength spectroscopic data-I Mathematical considerations. Talanta. 1985;32:95–101. doi: 10.1016/0039-9140(85)80035-7. [DOI] [PubMed] [Google Scholar]
- 68.Rossoti FJC, Rossoti HS, Whewell RJ. The use of electronic computing techniques in the calculation of stability constants. J Inorg Nucl Chem. 1971;33:2051–65. [Google Scholar]
- 69.Gampp H, Maeder M, Meyer CJ, Zuberbühler AD. Calculation of equilibrium constants from multiwavelength spectroscopic data - II. SPECFIT: Two user-friendly programs in BASIC and standard FORTRAN 77. Talanta. 1985;32:257–64. doi: 10.1016/0039-9140(85)80077-1. [DOI] [PubMed] [Google Scholar]
- 70.Gampp H, Maeder M, Meyer CJ, Zuberbühler AD. Calculation of equilibrium constants from multiwavelength spectroscopic data - IV. Model-free least-squares refinement by use of evolving factor analysis. Talanta. 1986;33:943–51. doi: 10.1016/0039-9140(86)80233-8. [DOI] [PubMed] [Google Scholar]
- 71.Marquardt DW. An algorithm for least-squares estimation of nonlinear parameters. J Soc Indust Appl Math. 1963;11:431–41. [Google Scholar]
- 72.Maeder M, Zuberbühler AD. Nonlinear least-squares fitting of multivariate absorption data. Anal Chem. 1990;62:2220–4. [Google Scholar]
- 73.Vippagunta SR, Dorn A, Matile H, Bhattacharjee AK, Karle JM, Ellis WY, Ridley RG, Vennerstrom JL. Structural specificity of chloroquine-hematin binding related to inhibition of hematin polymerization and parasite growth. J Med Chem. 1999;42:4630–9. doi: 10.1021/jm9902180. [DOI] [PubMed] [Google Scholar]
- 74.Ncokazi KK, Egan TJ. A colorimetric high-throughput beta-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal Biochem. 2005;338:306–19. doi: 10.1016/j.ab.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 75.a) Partos S. The hemochrome of Herzfeld and Klinger. Biochem Zeit. 1922;129:89–100. [Google Scholar]; b) Marques HM, Munro OQ, Crawcour ML. Coordination of N-donor ligands by hematohemin. Inorg Chim Acta. 1992;196:221–9. [Google Scholar]
- 76.Egan TJ, Ross DC, Adams PA. Quinoline anti-malarial drugs inhibit spontaneous formation of π-haematin (malaria pigment) FEBS Letters. 1994;352:54–7. doi: 10.1016/0014-5793(94)00921-x. [DOI] [PubMed] [Google Scholar]
- 77.Kelder PP, De Molt NJ, Lamberth MJ. Mechanistic aspects of the oxidation of phenothiazine derivatives by methemoglobin in the presence of hydrogen peroxide. Biochem Pharmacol. 1991;42:1551–9. doi: 10.1016/0006-2952(91)90424-4. [DOI] [PubMed] [Google Scholar]
- 78.Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord Chem Rev. 1999;184:311–8. [Google Scholar]
- 79.MicrocalTM OriginTM. Microcal Software, Inc; Northampton, MA: 1997. [Google Scholar]
- 80.Nordhoff A, Bucheler US, Werner D, Schirmer RH. Folding of the four domains and dimerization are impaired by the Gly446→Glu exchange in human glutathione reductase. Implications for the design of antiparasitic drugs. Biochemistry. 1993;32:4060–6. doi: 10.1021/bi00066a029. [DOI] [PubMed] [Google Scholar]