

Abstract
In eukaryotic cells, chromatin serves as the physiological template for gene transcription, DNA replication, and repair. Chromatin assembly factor 1 (CAF-1) is the prime candidate protein to mediate assembly of newly replicated DNA into chromatin. To investigate the physiological role of CAF-1 in vivo, we used RNA interference (RNAi) to silence the 60-kDa subunit of CAF-1 (p60) in human cells. Transfection of a small interfering RNA (siRNA) directed against p60 resulted in efficient silencing of p60 expression within 24 h. This silencing led to an induction of programmed cell death in proliferating but not in quiescent human cells. Concomitantly, proliferating cells lacking p60 accumulated DNA double-strand breaks and increased levels of the phosphorylated histone H2A.X. Nuclear extracts from cells lacking p60 exhibited a 10-fold reduction of nucleosome assembly activity during DNA synthesis, which was restored upon addition of recombinant p60 protein. Nascent chromatin in cell nuclei lacking p60 showed significantly increased nuclease sensitivity, indicating chromatin assembly defects during DNA synthesis in vivo. Collectively, these data identify CAF-1 as an essential factor not only for S-phase-specific chromatin assembly but also for proliferating cell viability.
In eukaryotic cell nuclei, the genomic DNA is highly compacted into chromatin through an assembly with histone and nonhistone proteins. The structural subunit of chromatin is the nucleosome, containing 146 bp of DNA wrapped in just under two superhelical turns around an octamer of histones H2A, H2B, H3, and H4 (1). Nucleosome-packed genomic DNA assumes higher-order structures through internucleosomal contacts and association with linker histones and other proteins. This assembly serves as the natural substrate for DNA replication, repair, and recombination and for transcription in vivo.
In proliferating cells, the bulk of chromatin is assembled during DNA replication in the S phase of the cell cycle (19, 40, 43). New histones are synthesized during S phase and become incorporated into nucleosomes on the newly replicated DNA. Replication-specific nucleosome assembly is mediated by histone chaperones such as chromatin assembly factor 1 (CAF-1), which was initially purified from proliferating human cell nuclei (36). CAF-1 is a trimeric protein complex consisting of three subunits, p150, p60, and p48, which are required for replication-dependent nucleosome assembly in human cell extracts (10, 44). CAF-1 mediates the first step of nucleosome assembly, the deposition of new histone H3/H4 tetramers onto replicating DNA (37). The second step of nucleosome assembly is independent of CAF-1 and involves the association of histone H2A/H2B dimers to the precursor H3/H4 tetramer structure (37).
The two large subunits of CAF-1 physically colocalize with sites of chromosomal DNA replication throughout S phase in human cell nuclei (17), suggesting that CAF-1 is a physiologically relevant factor for nucleosome assembly during DNA replication in vivo. Furthermore, CAF-1 is substrate specific for DNA templates that undergo DNA replication or repair (7, 36). This specificity is achieved through the direct binding of the p150 subunit of CAF-1 to proliferating cell nuclear antigen (PCNA) (23, 33), a circular DNA clamp that provides a replication-dependent marking of DNA.
CAF-1 is evolutionarily conserved, and homologues have been described in yeast, insects, plants, and vertebrates. Despite the essential role of CAF-1 for chromatin assembly during DNA synthesis in human cell extracts in vitro, deletions of CAF-1 in the budding yeast Saccharomyces cerevisiae surprisingly did not yield a lethal phenotype but resulted in relatively mild physiological defects, such as UV sensitivity and impaired gene silencing at telomeres and at the mating-type loci (5, 6, 8, 11, 24). Moreover, when the two largest subunits of CAF-1 are deleted in the plant Arabidopsis thaliana, the resultant fasciata mutants are viable and show alterations only in postembryonic development, in particular a loss of cellular organization in shoot and root apical meristems (13). These results indicate the existence of redundant or alternative nucleosomal assembly pathways that provide for efficient chromatin assembly in the absence of CAF-1 in fungi and plants.
In higher eukaryotes, on the other hand, evidence for a more essential in vivo function of CAF-1 is accumulating. Severe early development defects were found in Xenopus laevis embryos upon microinjection of a dominant-negative mutant of the p150 subunit of CAF-1, which also blocked nucleosome assembly in vitro (26). A dominant-negative human p150 mutant also inactivated nucleosome assembly by CAF-1 in vitro, and its ectopic expression in vivo induced S-phase arrest, accompanied by DNA damage and S-phase checkpoint activation (45). Taken together, these studies suggest that CAF-1 is required for maintaining epigenetic information encoded in accurate chromatin assembly, and in vertebrates it has a central role in ensuring undisturbed chromatin replication in S phase. However, as the use of ectopically expressed dominant-negative mutant proteins could lead to secondary effects, it is important to investigate the in vivo roles of CAF-1 in human cells directly by knockout experiments.
Here, we used RNA interference (RNAi) (3, 4) to silence the p60 subunit of CAF-1 in human cells. Transfection of a small interfering RNA (siRNA) directed against p60 resulted in efficient silencing of p60 expression within 24 h. This silencing led to an induction of cell death in proliferating but not quiescent human cells. Concomitantly, transfected proliferating cells displayed accumulation of double-strand DNA breaks indicative of activated programmed cell death. Nuclear extracts derived from these cells exhibited a 10-fold reduction of nucleosome assembly activity during DNA synthesis, which was restored by adding back recombinant p60 protein to the in vitro replication reactions. Collectively, these data identify CAF-1 as an essential factor for S-phase-specific chromatin assembly and, most importantly, for proliferating cell viability.
MATERIALS AND METHODS
Cell culture.
HeLa-S3 and EJ30 cells were cultured as exponentially growing subconfluent monolayers in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen), supplemented with 10% fetal calf serum (Invitrogen), 10 U of penicillin (Sigma) per ml, and 0.1 mg of streptomycin (Sigma) per ml. EJ30 cells were made quiescent by cultivation in this medium supplemented with only 0.5% fetal calf serum for a minimum of 7 days (18). Cell cycle position was determined by flow cytometry of isolated nuclei. Nuclei were stained with propidium iodide (5 μg/ml in phosphate-buffered saline [PBS] containing 0.4% Triton X-100) and analyzed by FACScan (Becton Dickinson) with the CellQuest software. Data are presented as histograms showing relative DNA content (x axis) and cell number (y axis).
siRNA design and transfection.
Candidate siRNAs were designed according to criteria outlined elsewhere (4). Of two initial candidates, 286AATGATAACAAGGAGCCGGAG306 and 1577AAATTCAGTCAGAGACGCCTG1597, the former led to only a partial reduction of p60 levels (and induction of cell death in a reduced number of proliferating cells), while the latter was able to completely silence the p60 subunit. It targets the C-terminal coding region, nucleotide positions 1579 to 1597 relative to the first nucleotide of the start codon for p60 (10), and was used for all experiments discussed in this paper. The nontarget siRNA used as a negative control had the scrambled gene sequence 1AAGTCAGTCAGTCAGTCAGTC21. siRNAs were chemically synthesized with an Ambion Silencer siRNA construction kit according to the instructions of the manufacturer; 1 μM siRNA working stocks were prepared by diluting the main siRNA stock solution with resuspension buffer (0.2-μm-filtered sterile RNase-free water, 100 mM NaCl, 50 mM Tris-HCl [pH 7.5]) in a sterile RNase-free microcentrifuge tube.
Transfections were performed with the TransIT-TKO transfection reagent (Mirus) on 6-well and 24-well plates essentially as specified by the manufacturer. At 24 h prior to transfection, 3 × 104 to 4 × 104 HeLa S3 cells were seeded per well for a 24-well plate or 1.2 × 105 HeLa S3 cells were seeded per well for a 6-well plate. The final concentrations of siRNA in the culture media were 10 nM for proliferating HeLa cells and 25 nM for quiescent EJ30 cells. In the case of quiescent EJ30 cells, the transfection was performed in DMEM lacking serum and antibiotics.
Cell viability assay.
At appropriate time points posttransfection, 60 μl of CellTiter 96 Aqueous One Solution cell proliferation assay reagent (Promega) was added per well of a 24-well plate containing 300 μl of culture medium, mixed, and immediately returned to the 37°C incubator. After 5 min, 100 μl of 10% sodium dodecyl sulfate solution was added per well to stop the reaction. The supernatants of the samples were transferred to plastic cuvettes, and the absorbances were determined at 490 nm. All samples were tested and measured in quadruplicate, and mean values and standard deviations were determined. Absolute cell numbers per absorbance unit were calibrated by plating defined numbers of cells per well (counted prior to seeding with a hemocytometer), and a cell viability assay was immediately performed as described above. Background absorbance was determined with a blank solution consisting of 300 μl of DMEM culture medium and 300 μl of CellTiter 96 Aqueous One solution reagent, and this value was subtracted from all experimental values obtained.
TUNEL assay.
For the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay, HeLa cells were grown on glass coverslips in 24-well plates, transfected with siRNA as described above, and incubated for 48 h. Positive control cells were treated with 40 μg of etoposide per ml for 16 h before processing. Coverslips were removed, washed with PBS, and then fixed with 4% paraformaldehyde in PBS for 5 min. Coverslips were washed again with PBS and then incubated with PBS and 0.2% Triton for 5 min on ice. Another two washes with PBS were performed, followed by incubation of the coverslips with New England Biolabs buffer 4 containing 0.25 mM CoCl2. After this incubation, the coverslips were incubated for 1 h at 37°C in the above buffer containing 10 mM deoxynucleoside triphosphate mix (Roche), 0.5 mM digoxigenin-11-dUTP (Roche), and 14 U of terminal transferase (New England Biolabs). Incorporated digoxigenin was visualized by fluorescein-conjugated antidigoxigenin Fab fragments (Roche), and genomic DNA was visualized by staining with propidium iodide as detailed elsewhere (38). Imaging of the coverslips was performed with a Leica confocal microscope as detailed elsewhere (38).
Preparation of nuclear extracts.
HeLa S3 cells were grown in six-well plates and transfected at 50% confluency as described above. At 36 h after transfection, the cells were washed once with hypotonic buffer (20 mM HEPES-KOH, 5 mM KCl, 1.5 mM MgCl2 and 1 mM dithiothreitol). The plate was then placed on an ice bed, and 1 ml of ice-cold hypotonic buffer was added to each well for 10 min. The buffer was removed, and the cells were gently scraped from each well. The cell suspension was transferred to a fresh tube, and the volume of the cells was measured with a pipette. An equal volume of 2× hypertonic buffer (20 mM HEPES-KOH, 800 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, and 1 mM dithiothreitol) was added to the suspension. The cells were then homogenized by several passages through a 29-gauge syringe. The homogenate was incubated on ice for 30 min and centrifuged at 20,000 × g for 10 min. The supernatant was collected and dialyzed against 20 mM HEPES-KOH-5 mM potassium acetate-0.5 mM MgCl2-0.5 mM dithiothreitol for 1 h. This dialyzed nuclear extract solution was stored in small aliquots in liquid nitrogen. Nuclear extracts contained about 1 to 5 mg of total protein per ml as determined with the Bio-Rad protein assay with bovine serum albumin as a standard.
DNA synthesis-dependent nucleosome assembly reactions.
cDNA strand synthesis and nucleosome assembly reactions were performed essentially as described before (20). Standard reaction mixtures containing cytosolic extract from asynchronously proliferating HeLa cells (125 μg of protein) and 25 ng of single-stranded M13mp18 template DNA were supplemented with nuclear extracts from cells transfected with siRNA. Recombinant p60 was derived from baculovirus-infected insect Sf9 cells by using recombinant baculovirus particles (a gift from Bruce Stillman, Cold Spring Harbor, N.Y.) as described elsewhere (10).
Micrococcal nuclease assay.
For micrococcal nuclease digestions, transfected cells were harvested after 24 h via trypsinization. The cell pellet was resuspended in hypotonic buffer, and 1 μl of the cell suspension was used for determining the cell concentration with a hemocytometer. Equivalent numbers of cells were homogenized by passage through a 29-gauge syringe. This mixture was supplemented with a buffered mix of ribo- and deoxynucleoside triphosphates, including [α-32P]dATP as a tracer to allow nuclear run-on replication (17). After a 2-h incubation at 37°C, the nuclei were divided into five tubes, adjusted to 2 mM CaCl2, and treated with 1 μg of RNase A (Sigma) and 0.1, 0.3, 1, 3, or 10 U of micrococcal nuclease (Roche) per reaction in a 50-μl volume at 21°C for 30 min. Nuclease-resistant DNA was purified as described elsewhere (34). DNA purified from the stopped reactions was analyzed on 1.5% agarose gels and visualized by staining with ethidium bromide and phosphor imaging of the dried gel.
Visualization of DNA replication foci.
To detect the presence of active DNA replication foci in transfected cell nuclei, cells were homogenized at 24 h after transfection exactly as described for the micrococcal nuclease assay. DNA replication run-on reactions were performed for 3 h essentially as described before (17) but with digoxigenin-11-dUTP as the tracer and propidium iodide as the counterstain for nuclear DNA. Imaging of nuclei was performed with a Leica confocal microscope as detailed elsewhere (38).
RESULTS
Silencing of the p60 subunit of CAF-1 by RNAi.
We decided to investigate the role of CAF-1 in HeLa cells by selectively silencing the expression of the p60 subunit, which was previously shown to be essential for nucleosome assembly activity (10). Two independent siRNA molecules were designed and tested. Transfection of HeLa cells with these siRNAs resulted in significantly reduced expression of p60, albeit with different silencing kinetics (data not shown). Therefore, we chose only the most efficient siRNA, targeting a sequence near the 3′ end of the p60 mRNA, for all subsequent experimentation. With this siRNA, expression of p60 was specifically reduced by about 90% at 24 h and by more than 95% at 48 h posttransfection (Fig. 1A). These Western blot data were corroborated by immunofluorescent staining of HeLa cells at 24 h posttransfection. Expression of p60 was no longer detectable in more than 90% of the cells (Fig. 1B and data not shown). In contrast, expression of the p150 and p48 subunits of CAF-1 as well as actin was not reproducibly affected by the silencing of the p60 subunit at 24 h posttransfection. We observed a reduction of p150 protein levels of up to 20% in only two of five independent experiments (Fig. 1A and data not shown). However, at 48 h posttransfection we observed a reduction in the expression levels of p150 of up to 50% in only three of five independent silencing experiments (Fig. 1A and data not shown). In conclusion, efficient and rapid silencing of the p60 subunit by RNAi did not result in concomitant silencing of the p150 and p48 subunits.
FIG. 1.

RNAi of the p60 subunit of CAF-1. (A) Western blot analysis of whole-cell extracts of HeLa cells prepared at 24 h and 48 h posttransfection with a 10 nM concentration of a nontarget control siRNA (nt) and a p60-specific siRNA (p60). Untransfected cell extract was used as an additional control (−). Equal protein amounts were loaded and probed with antibodies specific for the CAF-1 subunits p150 (35), p60 (22), and p48 (25) and for actin. To detect p48, a dual-specificity antibody was used (25), which detects both p48 (upper band) and the related protein p46 (lower band). (B) Confocal immunofluorescence microscopy of HeLa cells after RNAi. Proliferating cells were transfected as above, and at 24 h posttransfection, DNA was visualized by propidium iodide staining (left column) and p60 was visualized by indirect immunofluorescence (right column).
Induction of cell death after silencing of p60.
Following transfection with the p60-specific siRNA, we routinely observed morphological changes such as nuclear fragmentation in the majority of cells by fluorescence microscopy (Fig. 1B), suggesting an induction of programmed cell death upon silencing of p60. Therefore, we directly investigated cell viability using a quantitative colorimetric assay based on the reduction of a tetrazolium salt in living cells. We quantified cell viability at the time of transfection and at 24 h and 48 h posttransfection (Fig. 2A). Untransfected control cells grew exponentially throughout this experiment. Cells transfected with a nontarget control siRNA showed a reduced growth curve compared to that of the control as a result of the transfection procedure. In contrast, HeLa cells transfected with the p60-specific siRNA exhibited a dramatic reduction in the number of living cells during this experiment, indicating the occurrence of cell death as a result of silencing the expression of p60 (Fig. 2A). This observation was corroborated by trypan blue staining of these cells at 48 h posttransfection, confirming large numbers of dead cells after silencing of p60 (data not shown).
FIG. 2.

Loss of cell viability upon silencing of p60 in proliferating HeLa cells. (A) Cell viability assay. Cells were grown in 24-well plates and transfected with nontarget (nt) and p60 siRNAs. Untransfected cells were used as a control. After 24 and 48 h, cell viability assay solution was added directly to the wells of all samples, and the number of viable cells was determined by spectrophotometry of the supernatants (see Materials and Methods). Mean values and standard deviations for four independent experiments are shown. (B) Direct visualization of these cells by phase-contrast light microscopy before the cell viability assay. Of the three samples, p60-silenced cells exhibited the most acute cytopathicity and cell death.
We also observed the morphology of these cells by phase contrast light microscopy (Fig. 2B). At 24 h posttransfection, all cell samples appeared generally healthy. After 48 h, HeLa cells transfected with the nontarget control siRNA still largely appeared healthy compared to the proliferating untransfected cells. In contrast, cells transfected with the p60-specific siRNA exhibited acute cytopathicity and cell death (Fig. 2B). We therefore conclude that CAF-1 is an essential protein for human cells and that silencing of its p60 subunit leads to cell death between 24 h and 48 h posttransfection.
Induction of DNA damage and mechanism of cell death after p60 silencing.
To identify the pathway of cell death, we asked whether silencing of p60 expression results in DNA breaks and the induction of apoptosis. Therefore, we first checked by Western blotting for the presence of the phosphorylated form of histone H2A.X (γ-H2A.X), which has been shown to become phosphorylated adjacent to sites of double-stranded DNA breaks (28, 30). Figure 3A shows that no detectable levels of γ-H2A.X were observed in untreated cells or in cells transfected with the nontarget control siRNA at 24 h or 48 h posttransfection. In contrast, transfection with the p60-specific siRNA caused a signal for γ-H2A.X at just 24 h posttransfection, which increased significantly in intensity by 48 h posttransfection. This result indicates that double-stranded DNA damage accumulates during the silencing of p60. This accumulation is suggestive of genome fragmentation during apoptosis, which also results in the accumulation of γ-H2A.X (29).
FIG. 3.

Accumulation of fragmented DNA after silencing of p60. (A) Western blot analysis of histone H2A.X phosphorylation (γ-H2A.X). Whole-cell extracts of HeLa cells were prepared at 24 h and 48 h after transfection with nontarget (nt) and p60 siRNAs. Untransfected cell extract was used as an additional control (−). Equal protein amounts were loaded and probed with a monoclonal antibody specific for γ-H2A.X (S139; Upstate). As a loading control, the membrane was reprobed with actin-specific antibodies. (B) TUNEL assay. Cells were grown on glass coverslips and transfected with nontarget and p60 siRNAs as indicated. As a positive control, etoposide was applied at a concentration of 40 μg/ml to untransfected cells 16 h prior to processing. At 48 h posttransfection, DNA breaks were labeled with digoxigenin-dUTP by using terminal transferase (see Materials and Methods). DNA was counterstained with propidium iodide (red signal), and digoxigenin was detected by fluorescein-conjugated antidigoxigenin Fab fragments (green signal). Both signals were detected by confocal fluorescence microscopy, and merged channels are presented. Sites of fragmented DNA appear in yellow. (C) Quantitation of TUNEL assay results. The percentages of intact cells (light grey bars) and apoptotic cells (dark grey bars) were determined from larger microscopic fields than are shown in panel B, containing at least 200 nuclei per reaction.
To corroborate these data independently, we used the TUNEL assay to ask whether the mechanism of cell death at 48 h posttransfection occurs through programmed cell death, or apoptosis (Fig. 3B and C). As a negative control, untreated proliferating human cells did not stain for fragmented nuclear DNA. Transfection with the nontarget siRNA resulted in DNA fragmentation in 11% of the cell population, perhaps due to unspecific cytotoxic insult arising from the RNAi transfection reagent or procedure. In marked contrast, 73% of the cells transfected with the p60-specific siRNA stained strongly for apoptotic genomic DNA fragmentation (Fig. 3B and C). In a positive control, 43% of the positive-staining cells were observed in the sample treated with the DNA topoisomerase poison etoposide (Fig. 3B), which causes severe DNA damage leading to the activation of programmed cell death (2, 12). It is worth noting that approximately 38% of the p60-silenced cells at 48 h posttransfection did not effectively counterstain for total DNA and appeared severely deteriorated (Fig. 3B), suggesting that they had undergone apoptosis prior to this time point. In summary, we conclude that silencing of the p60 subunit of CAF-1 leads to an induction of DNA breaks and the activation of programmed cell death.
Cell cycle effects after silencing of p60.
It has been reported that expression of a dominant-negative mutant of p150 in human cells results in S-phase arrest and activation of a DNA damage checkpoint response (45). Therefore, we tested whether the induction of DNA breaks and cell death caused by p60 silencing is preceded by an arrest of the transfected cells in S phase. An analysis of nuclear DNA content by flow cytometry of nuclei taken at 36 h posttransfection showed no significant enrichment of S-phase nuclei in either of the transfected samples (Fig. 4A). An analysis of nuclei taken at 24 h posttransfection gave virtually identical results (data not shown).
FIG. 4.

After silencing of p60, proliferating cells do not accumulate in S phase before cell death. (A) Cell cycle analysis. HeLa cells were transfected with nontarget (nt) and p60 siRNAs as indicated. Untransfected cells were used as a control. At 36 h posttransfection, nuclear DNA content was analyzed by flow cytometry. Identical results were obtained at 24 h posttransfection. (B) Quantitative analysis of DNA replication. Nuclei from the cells shown in panel A at 24 h posttransfection were incubated in their cognate cytosol in vitro to label active DNA replication foci (see Materials and Methods) (17). The percentages of nuclei with active DNA replication foci were determined by confocal fluorescent microscopy (see below). (C) Representative fields of replicating nuclei. Replication reactions were performed as indicated, and nuclei were analyzed by confocal fluorescence microscopy. Nuclear DNA was counterstained with propidium iodide (red signal) and replicated DNA was counterstained with fluorescein-conjugated antidigoxigenin Fab fragments (green signal). Merged images are presented, showing sites of replicated nuclear DNA in yellow and nonreplicating nuclei in red.
To visualize actively replicating S-phase nuclei directly, cells were harvested at 24 h posttransfection, before apoptosis became evident, and after the plasma membranes had been disrupted, the cell homogenates were subjected to nuclear run-on DNA replication in vitro (17). About 30% of the untransfected proliferating cells were found to be in S phase, whereas both of the transfected cell populations contained significantly reduced percentages of S-phase cells (Fig. 4B and C). In fact, cells transfected with the p60-specific siRNA showed the lowest proportion, only 12% replicating cell nuclei. Taken together, these data do not support an accumulation of cells in S phase upon silencing of p60. To investigate whether a requirement for CAF-1 for cell viability is specific for proliferating cells, we next analyzed the silencing of p60 in quiescent human cells.
Silencing of p60 and viability in quiescent cells.
Human EJ30 cells were made quiescent by serum deprivation (18) and then subjected to RNAi. We were able to reduce p60 expression in quiescent cells to more than 90% at 48 h posttransfection but only after increasing the concentration of siRNA from 10 nM, as used for proliferating HeLa cells, to 25 nM (Fig. 5A). This higher siRNA concentration led to an increased nonspecific cytopathicity in both of the transfected samples (Fig. 5B). Importantly, and in contrast to findings for proliferating cells, silencing of p60 in quiescent cells did not reduce the percentage of morphologically intact and viable cells beyond the percentage observed in the nontarget siRNA-transfected cells (Fig. 5B and C). This experiment demonstrates that cell death due to p60 silencing is restricted to actively proliferating human cells.
FIG. 5.

Quiescent human cells remain viable after silencing of p60. (A) Transfections of quiescent human EJ30 cells were performed at 10 days after serum deprivation. Western blot analysis of whole-cell extracts prepared at 48 h posttransfection with nontarget (nt) and p60 siRNAs was performed. The untransfected cell extract control is also shown (−). Equal protein amounts were loaded and probed with polyclonal antibodies specific for p60 (22) and replication protein A (RPA) (38); only the RPA-32 band is shown. (B) Cell viability was determined in these quiescent cells at 48 h posttransfection, as detailed in the legend to Fig. 2A. (C) Direct visualization of these cells by phase-contrast light microscopy, as detailed in the legend to Fig. 2B.
Loss of CAF-1 activity in p60-silenced cell extracts.
In the last set of experiments we analyzed whether silencing of p60 removes the cell's ability for rapid chromatin assembly during DNA replication. We prepared nuclear extracts of cells transfected with the negative-control, nontarget siRNA and with the p60-specific siRNA at 36 h posttransfection. Increasing amounts of these extracts were added to cDNA strand synthesis reaction mixtures (Fig. 6), with circular single-stranded DNA templates incubated in a cytosolic extract from human proliferating cells (20). We have shown before that addition of increasing amounts of CAF-1 to this reaction leads to increased supercoiling of the double-stranded DNA reaction products due to a stoichiometric nucleosome assembly activity of CAF-1 (20).
FIG. 6.

Nucleosome assembly during DNA synthesis is impaired in the absence of p60. (A) cDNA strand synthesis reaction mixtures with single-stranded circular DNA templates, incubated in cytosolic extract from untransfected HeLa cells (20), were supplemented with the indicated amounts of a HeLa cell nuclear extract prepared at 36 h after transfection with the nontarget siRNA (nt, lanes 1 to 7). Double-stranded DNA reaction products were visualized by autoradiography after agarose gel electrophoresis. The positions of relaxed circular form II, linear form III, and supercoiled form I DNA are indicated. Nucleosome assembly by CAF-1 induces negative supercoiling in this assay (20). (B) cDNA strand synthesis reaction mixtures were supplemented with the indicated amounts of a HeLa cell nuclear extract prepared at 36 h after transfection with p60 siRNA (lanes 1 to 7). (C) Reconstitution of nucleosome assembly activity of p60-silenced cell nuclear extract upon addition of exogenous p60 protein. Increasing amounts of recombinant p60 protein (0, 0.03, 0.1, and 0.3 μg) prepared from baculovirus-infected Sf9 insect cells were added to reaction mixtures containing 10 μg of nuclear extract from p60-silenced cells.
Addition of nuclear extract from control nontarget siRNA-transfected cells led to increased supercoiling of the reaction products (Fig. 6A). In contrast, a 10-fold-higher amount of nuclear extract from p60 siRNA-transfected cells was required to achieve the same degree of supercoiling as observed with the extract from nontarget siRNA-transfected control cells (Fig. 6, compare lane 4 of panel A with lane 7 of panel B). Therefore, silencing of p60 leads to a reduction of the specific supercoiling activity of nuclear extracts by an order of magnitude. This deficiency in nucleosome assembly could be restored by addition of recombinant p60 from baculovirus-infected insect cells to the reaction (Fig. 6C). Because the nuclear extract from p60-silenced cells still contains the p150 and p48 subunits of CAF-1 (see Fig. 1A), we conclude that addition of recombinant p60 restores the functionality of CAF-1 in these reactions. This observation is consistent with a report of functional reconstitution of CAF-1 from individual recombinant subunits in vitro (10). These experiments therefore demonstrate that CAF-1 is an essential factor for nucleosome assembly during DNA synthesis in human cell extracts. They furthermore suggest that the loss of de novo nucleosome assembly activity of CAF-1 during DNA replication and the resulting perturbations of chromatin structure may be the cause of the induction of cell death in proliferating human cells.
Reduced efficiencies of chromatin assembly and DNA replication in p60-silenced cells.
Finally, we investigated the chromatin structure of nascent DNA in p60-silenced cell nuclei at 24 h posttransfection (Fig. 7). Nuclei were isolated from nontarget siRNA- and p60 siRNA-transfected cells and incubated in a nuclear DNA replication run-on system (17) in the presence of their cognate cytosolic extracts and radiolabeled deoxynucleoside triphosphates. With this experimental system, it was shown before that CAF-1 colocalizes with sites of DNA synthesis in vitro in untreated S-phase nuclei (17). We therefore expected that nucleosome assembly on nascent DNA in these isolated nuclei consists of both passive transfer of parental nucleososmes and active de novo assembly from soluble histones, mediated by CAF-1 (16). Following digestion with micrococcal nuclease, we visualized the nuclease-resistant bulk nuclear DNA after gel electrophoresis by staining with ethidium bromide (Fig. 7A) and the nascent DNA by phosphor imaging (Fig. 7B). We found that undigested nuclear DNA used as a control did not enter the gel (data not shown), indicating that at 24 h posttransfection with either the nontarget or p60 siRNA, apoptotically degraded DNA was not yet detectable. Upon digestion with micrococcal nuclease, bulk chromatin was about 10 times more sensitive to degradation in p60-silenced cells than in the nontarget siRNA-transfected cells (Fig. 7A). However, regular nucleosomal ladders were apparent in both samples, indicating that the bulk of the genomic DNA was still assembled into regularly spaced chromatin upon p60 silencing.
FIG. 7.
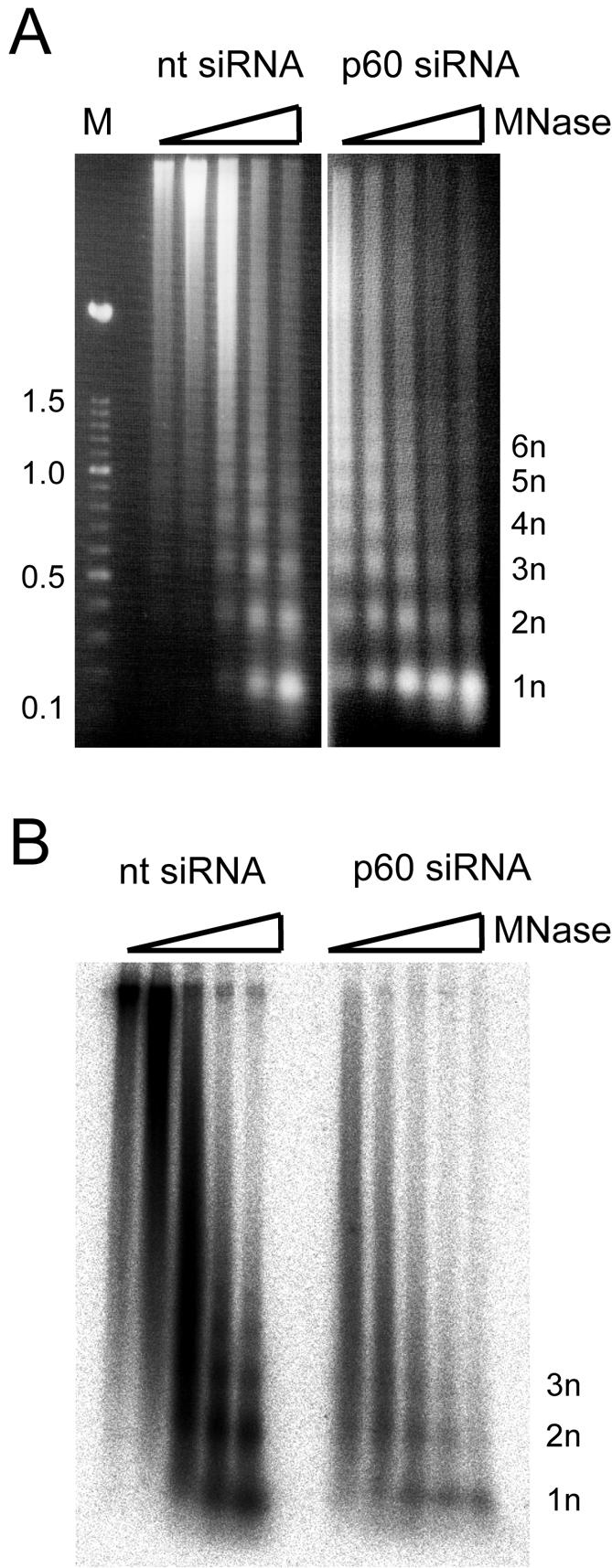
Analysis of chromatin structure in proliferating cells during silencing of p60. HeLa cells were transfected with either nontarget (nt) or p60 siRNAs, and a cell homogenate of each sample was prepared at 24 h posttransfection. Nascent DNA in these nuclei was radiolabeled for 3 h in vitro (see Materials and Methods). Labeled nuclei were treated with 0.1, 0.3, 1, 3, and 10 U of micrococcal nuclease (MNase) per ml (lanes 1 to 5, respectively). Nuclease-resistant DNA from both transfection experiments was isolated and analyzed by agarose gel electrophoresis. Note that DNA prepared from nontarget and p60 siRNA-transfected cell nuclei did not enter the gel after incubation without micrococcal nuclease present and is therefore not shown here. (A) Visualization of digested bulk DNA from transfected cells by staining with ethidium bromide. Lane M, multimeric 100-bp DNA ladder (Roche), with size indicated in kilobase pairs. The positions of nuclease-resistant mono- (1n) and oligonucleosomal DNA fragments are indicated. (B) Visualization of digested replicating, nascent DNA in these nuclei by phosphor imaging of the dried gel.
Nascent DNA in p60-silenced cell nuclei also was about 10 times more sensitive to digestion with micrococcal nuclease compared to the nontarget transfection control (Fig. 7B). Additionally, the p60-silenced cells were less competent in replicating their nuclear DNA. For each DNA replication reaction, the p60-silenced nuclei had incorporated 80% ± 3% less radioactivity than the nontarget-transfected samples (data not shown). The nuclease-resistant DNA in p60-silenced nuclei was predominantly mononucleosomal and contained fewer oligonucleosomal bands than the control (Fig. 7B). These data show that the efficiencies of DNA replication as well as of nucleosome assembly on nascent DNA were both reduced upon p60 silencing in proliferating human cells.
DISCUSSION
The main conclusion from our data is that CAF-1 is an essential protein for the viability of human proliferating cells, as its disruption by RNAi leads to inhibition of chromatin assembly during DNA synthesis, accumulation of DNA damage, and induction of programmed cell death. These experiments provide the strongest phenotype for a knockout of CAF-1 in eukaryotic cells described in the literature to date. We postulate that redundant factors exist in lower eukaryotes that may substitute for CAF-1 function in vivo but that CAF-1 has assumed an essential and unique regulatory role in proliferating higher eukaryotic somatic cells.
Genetic knockout studies in S. cerevisiae have demonstrated that deletion of CAF-1 yielded viable cells showing only a relatively mild phenotype of altered patterns of gene expression and an increased sensitivity to DNA damage (5, 6, 8, 11, 24). A similar situation has been found in Arabidopsis thaliana, in which deletion of CAF-1 results in viable plants which show alterations in gene expression, suggesting that CAF-1 function is required for facilitating stable maintenance of gene expression states (13). These alterations are linked to a phenotypic defect in maintaining the cellular architecture of postembryonic apical meristems (13). Taken together, these observations indicate the existence of redundant chromatin assembly factors which can bypass the requirement for CAF-1 and assemble chromatin that is epigenetically altered yet sufficiently functional for cell viability in S phase. Candidate factors are the evolutionarily conserved Asf1 and Hir proteins, which functionally interact with CAF-1, bind to histones, and can mediate an assembly of nucleosomes that is not dependent on DNA synthesis (15, 21, 27, 31, 32, 41, 42).
In vertebrates, experiments to disrupt CAF-1 function have so far targeted only the p150 subunit of the complex and have led to different and sometimes conflicting conclusions about the physiological role of CAF-1 in vivo. A dominant-negative approach to targeting the p150 subunit of CAF-1 in Xenopus laevis embryos led to an inhibition of the nucleosome assembly activity of CAF-1 during DNA repair (26). Ectopic expression of a dominant-negative p150 in vivo resulted in severe developmental defects during early embryogenesis that were indicative of major cell proliferation defects. However, when applying this dominant-negative approach to a somatic cell line from X. laevis, neither a loss of cell viability nor major defects were reported (26). Therefore, it is possible that CAF-1 function in X. laevis is required only for the early very rapid embryonic cell cycles of alternating S phase and mitosis but that other assembly factors can compensate for a loss of function in the slower-cycling adult somatic cells, as in S. cerevisiae and plants.
In human somatic cells, introduction of a dominant-negative p150 subunit of CAF-1 resulted in reversal of epigenetic gene silencing in otherwise viable cells (39), indicating that CAF-1 might play a role that is at least similar to that in S. cerevisiae or plants. However, a substantially different phenotype was recently reported by Ye and coworkers (45). Ectopic expression of dominant-negative p150 in human U2OS cells to disrupt CAF-1 activity resulted in S-phase arrest accompanied by DNA damage. Furthermore, an S-phase checkpoint was activated through the ataxia telangiectasia mutated (ATM) and/or ATM-related (ATR) kinase pathways, leading to the conclusion that CAF-1 is required for the completion of S phase and that a defect in chromatin assembly during DNA replication can lead to DNA damage (45).
Our observations of induction of severe DNA damage and rapid programmed cell death as a result of silencing of the expression of the p60 subunit of CAF-1 confirm and further extend the physiological relevance of CAF-1 in proliferating human cells. We have shown here that CAF-1 is an essential factor for proliferating cell viability. In contrast to the dominant-negative approaches against p150 outlined above, our experimental strategy targeted the middle subunit of CAF-1 and used RNAi to remove the protein from the cell. Ectopic expression of dominant-negative p150 mutants does not remove the endogenous p150 but interferes with the function of the trimeric CAF-1 complex, most likely through the formation of dysfunctional p150 dimers (26) and/or through interference with the PCNA interaction (45). In contrast to a physical depletion of the protein by RNAi (Fig. 1), these dominant-negative approaches might only partially inactivate the CAF-1 complex by undefined substoichiometric mutant protein levels or by transient interactions in the transfected cells, leading to a range of relatively milder phenotypes and viable cells (26, 39, 45).
Furthermore, it is possible that the p60 and p150 subunits subtly fulfill distinct functions in linking nucleosome assembly during DNA synthesis to the cell cycle control machinery. Inactivation of p150 by binding to a dominant-negative version of itself could elicit a signal leading to S-phase arrest. Activation of the DNA damage-signaling protein kinases ATR and ATM has been shown under these conditions (45). A similar response of S-phase arrest and ATR signaling in viable cells was recently found when p150 was silenced by RNAi (9), indicating that disruption of CAF-1 activity via the p150 subunit results in S-phase arrest. In contrast, the absence of p60 could signal through a different and as yet unknown pathway to elicit its response, i.e., programmed cell death. In this context it is worth noting that p60 interacts directly with the cell cycle control machinery (14, 22). The p60 subunit is reversibly phosphorylated by the S-phase-specific protein kinase complexes cyclin A/Cdk2 and cyclin E/Cdk2, and the nucleosome assembly activity of CAF-1 itself depends on ongoing reversible phosphorylation by these kinases (14). Interference with this control pathway by depleting p60 could rapidly result in the physiological cell response of programmed cell death, as reported here. Further experimentation is required to resolve this issue.
However, the most widely supported scenario, which is not mutually exclusive with either of the possibilities discussed above, is that any inactivation of CAF-1 leads to the inhibition of de novo nucleosome assembly during DNA replication (Fig. 6) (45), resulting in incompletely assembled chromatin on nascent DNA in vivo (Fig. 7). The extent of this defect would be influenced by the depletion protocol used and elicit physiological cell responses with effects ranging from (i) loss of a repressive chromatin structure and activated expression of a silenced transgene (39) to (ii) checkpoint activation and S-phase arrest (9, 45) to (iii) rapid programmed cell death (this paper). The observation that extracts from p60-silenced cells exhibited a 10-fold-reduced level of nucleosome assembly and thus did not completely lose all assembly activity (Fig. 6) suggests that redundant pathways can still mediate de novo nucleosome assembly on replicating DNA. Even in the complete absence of the de novo nucleosome assembly activity of CAF-1, each nascent DNA strand would still receive half the complement of parental nucleosomes that are passively transferred past the replication fork during S phase (16). However, these additional assembly pathways would not suffice for a fast genome-wide and complete assembly of nascent DNA into functional chromatin.
Importantly, the response of induced rapid cell death upon p60 depletion depends on the physiological state of proliferating cells, because quiescent cells remain viable after depletion (Fig. 5). This lends strong independent support for a vital role of CAF-1 in S phase, which is already supported by the observation of S-phase arrest in the absence of functional CAF-1 (9, 45). Together, these findings have identified CAF-1 as a potential target for anticancer drugs with cytostatic or, indeed, cytotoxic effects for proliferating cells.
Acknowledgments
We thank Dávid Szüts for critical reading of the manuscript and Bruce Stillman and Keiishi Shibahara for reagents.
This work was funded by a research grant (RGP0375/2001) from the Human Frontier Science Program Organization. Arman Nabatiyan is supported by a fellowship from the Boehringer Ingelheim Fonds.
REFERENCES
- 1.Akey, C. W., and K. Luger. 2003. Histone chaperones and nucleosome assembly. Curr. Opin. Struct. Biol. 13:6-14. [DOI] [PubMed] [Google Scholar]
- 2.Burden, D. A., and N. Osheroff. 1998. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim. Biophys. Acta 1400:139-154. [DOI] [PubMed] [Google Scholar]
- 3.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 4.Elbashir, S. M., J. Harborth, K. Weber, and T. Tuschl. 2002. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 26:199-213. [DOI] [PubMed] [Google Scholar]
- 5.Enomoto, S., and J. Berman. 1998. Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev. 12:219-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enomoto, S., P. D. McCune-Zierath, M. Gerami-Nejad, M. A. Sanders, and J. Berman. 1997. RLF2, a subunit of yeast chromatin assembly factor-I, is required for telomeric chromatin function in vivo. Genes Dev. 11:358-370. [DOI] [PubMed] [Google Scholar]
- 7.Gaillard, P. H., E. M. Martini, P. D. Kaufman, B. Stillman, E. Moustacchi, and G. Almouzni. 1996. Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell 86:887-896. [DOI] [PubMed] [Google Scholar]
- 8.Game, J. C., and P. D. Kaufman. 1999. Role of Saccharomyces cerevisiae chromatin assembly factor-I in repair of ultraviolet radiation damage in vivo. Genetics 151:485-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoek, M., and B. Stillman. 2003. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. USA 100:12183-12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufman, P. D., R. Kobayashi, N. Kessler, and B. Stillman. 1995. The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell 81:1105-1114. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman, P. D., R. Kobayashi, and B. Stillman. 1997. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 11:345-357. [DOI] [PubMed] [Google Scholar]
- 12.Kaufmann, S. H. 1998. Cell death induced by topoisomerase-targeted drugs: more questions than answers. Biochim. Biophys. Acta 1400:195-211. [DOI] [PubMed] [Google Scholar]
- 13.Kaya, H., K. Shibahara, K. Taoka, M. Iwabuchi, B. Stillman, and T. Araki. 2001. FASCIATA genes for chromatin assembly factor-1 in Arabidopsis maintain the cellular organization of apical meristems. Cell 104:131-142. [DOI] [PubMed] [Google Scholar]
- 14.Keller, C., and T. Krude. 2000. Requirement of cyclin/Cdk2 and protein phosphatase 1 activity for CAF-1 dependent chromatin assembly during DNA synthesis. J. Biol. Chem. 275:35512-35521. [DOI] [PubMed] [Google Scholar]
- 15.Krawitz, D. C., T. Kama, and P. D. Kaufman. 2002. Chromatin assembly factor I mutants defective for PCNA binding require Asf1/Hir proteins for silencing. Mol. Cell. Biol. 22:614-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krude, T. 1999. Chromatin assembly during DNA replication in somatic cells. Eur. J. Biochem. 263:1-5. [DOI] [PubMed] [Google Scholar]
- 17.Krude, T. 1995. Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in HeLa cell nuclei. Exp. Cell Res. 220:304-311. [DOI] [PubMed] [Google Scholar]
- 18.Krude, T. 1999. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res. 247:148-159. [DOI] [PubMed] [Google Scholar]
- 19.Krude, T., and C. Keller. 2001. Chromatin assembly during S phase: contributions from histone deposition, DNA replication and the cell division cycle. Cell. Mol. Life Sci. 58:665-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krude, T., and R. Knippers. 1993. Nucleosome assembly during complementary DNA strand synthesis in extracts from mammalian cells. J. Biol. Chem. 268:14432-14442. [PubMed] [Google Scholar]
- 21.Lorain, S., J. P. Quivy, F. Monier-Gavelle, C. Scamps, Y. Lecluse, G. Almouzni, and M. Lipinski. 1998. Core histones and HIRIP3, a novel histone-binding protein, directly interact with WD repeat protein HIRA. Mol. Cell. Biol. 18:5546-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marheineke, K., and T. Krude. 1998. Nucleosome assembly activity and intracellular localization of human CAF-1 changes during the cell division cycle. J. Biol. Chem. 273:15279-15286. [DOI] [PubMed] [Google Scholar]
- 23.Moggs, J. G., P. Grandi, J. P. Quivy, Z. O. Jonsson, U. Hubscher, P. B. Becker, and G. Almouzni. 2000. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 20:1206-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monson, E. K., D. de Bruin, and V. A. Zakian. 1997. The yeast Cac1 protein is required for the stable inheritance of transcriptionally repressed chromatin at telomeres. Proc. Natl. Acad. Sci. USA 94:13081-13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian, Y. W., and E. Y. Lee. 1995. Dual retinoblastoma-binding proteins with properties related to a negative regulator of ras in yeast. J. Biol. Chem. 270:25507-25513. [DOI] [PubMed] [Google Scholar]
- 26.Quivy, J. P., P. Grandi, and G. Almouzni. 2001. Dimerization of the largest subunit of chromatin assembly factor 1: importance in vitro and during Xenopus early development. EMBO J. 20:2015-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray-Gallet, D., J. P. Quivy, C. Scamps, E. M. Martini, M. Lipinski, and G. Almouzni. 2002. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell 9:1091-1100. [DOI] [PubMed] [Google Scholar]
- 28.Rogakou, E. P., C. Boon, C. Redon, and W. M. Bonner. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogakou, E. P., W. Nieves-Neira, C. Boon, Y. Pommier, and W. M. Bonner. 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275:9390-9395. [DOI] [PubMed] [Google Scholar]
- 30.Rogakou, E. P., D. R. Pilch, A. H. Orr, V. S. Ivanova, and W. M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858-5868. [DOI] [PubMed] [Google Scholar]
- 31.Sharp, J. A., E. T. Fouts, D. C. Krawitz, and P. D. Kaufman. 2001. Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr. Biol. 11:463-473. [DOI] [PubMed] [Google Scholar]
- 32.Sharp, J. A., A. A. Franco, M. A. Osley, and P. D. Kaufman. 2002. Chromatin assembly factor I and Hir proteins contribute to building functional kinetochores in S. cerevisiae. Genes Dev. 16:85-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibahara, K., and B. Stillman. 1999. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96:575-585. [DOI] [PubMed] [Google Scholar]
- 34.Shimamura, A., D. Tremethick, and A. Worcel. 1988. Characterization of the repressed 5S DNA minichromosomes assembled in vitro with a high-speed supernatant of Xenopus laevis oocytes. Mol. Cell. Biol. 8:4257-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith, S., and B. Stillman. 1991. Immunological characterization of chromatin assembly factor I, a human cell factor required for chromatin assembly during DNA replication in vitro. J. Biol. Chem. 266:12041-12047. [PubMed] [Google Scholar]
- 36.Smith, S., and B. Stillman. 1989. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 58:15-25. [DOI] [PubMed] [Google Scholar]
- 37.Smith, S., and B. Stillman. 1991. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 10:971-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szüts, D., L. Kitching, C. Christov, A. Budd, S. Peak-Chew, and T. Krude. 2003. RPA is an initiation factor for human chromosomal DNA replication. Nucleic Acids Res. 31:1725-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tchénio, T., J. F. Casella, and T. Heidmann. 2001. A truncated form of the human CAF-1 p150 subunit impairs the maintenance of transcriptional gene silencing in mammalian cells. Mol. Cell. Biol. 21:1953-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyler, J. K. 2002. Chromatin assembly. Cooperation between histone chaperones and ATP-dependent nucleosome remodeling machines. Eur. J. Biochem. 269:2268-2274. [DOI] [PubMed] [Google Scholar]
- 41.Tyler, J. K., C. R. Adams, S. R. Chen, R. Kobayashi, R. T. Kamakaka, and J. T. Kadonaga. 1999. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402:555-560. [DOI] [PubMed] [Google Scholar]
- 42.Tyler, J. K., K. A. Collins, J. Prasad-Sinha, E. Amiott, M. Bulger, P. J. Harte, R. Kobayashi, and J. T. Kadonaga. 2001. Interaction between the Drosophila CAF-1 and ASF1 chromatin assembly factors. Mol. Cell. Biol. 21:6574-6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verreault, A. 2003. Histone deposition at the replication fork: a matter of urgency. Mol. Cell 11:283-284. [DOI] [PubMed] [Google Scholar]
- 44.Verreault, A., P. D. Kaufman, R. Kobayashi, and B. Stillman. 1996. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 87:95-104. [DOI] [PubMed] [Google Scholar]
- 45.Ye, X., A. A. Franco, H. Santos, D. M. Nelson, P. D. Kaufman, and P. D. Adams. 2003. Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol. Cell 11:341-351. [DOI] [PubMed] [Google Scholar]