

Abstract
Traditional culture-based methods have incompletely defined the etiology of common recalcitrant human fungal skin diseases including athlete’s foot and toenail infections. Skin protects humans from invasion by pathogenic microorganisms, while providing a home for diverse commensal microbiota1. Bacterial genomic sequence data have generated novel hypotheses about species and community structures underlying human disorders2,3,4. However, microbial diversity is not limited to bacteria; microorganisms such as fungi also play major roles in microbial community stability, human health and disease5. Genomic methodologies to identify fungal species and communities have been limited compared with tools available for bacteria6. Fungal evolution can be reconstructed with phylogenetic markers, including ribosomal RNA gene regions and other highly conserved genes7. Here, we sequenced and analyzed fungal communities of 14 skin sites in 10 healthy adults. Eleven core body and arm sites were dominated by Malassezia fungi, with species-level classifications revealing greater topographical resolution between sites. By contrast, three foot sites, plantar heel, toenail, and toeweb, exhibited tremendous fungal diversity. Concurrent analysis of bacterial and fungal communities demonstrated that skin physiologic attributes and topography differentially shape these two microbial communities. These results provide a framework for future investigation of interactions between pathogenic and commensal fungal and bacterial communities in maintaining human health and contributing to disease pathogenesis.
Keywords: fungi, genome, skin, dermatology, microbiome, Malassezia, ITS, 18S rRNA
Since Hippocrates first described oral candidiasis in 400 BC, scientists have sought to explore roles that commensal and pathogenic fungi and microbial communities play in human health and disease8,9. Culture-based studies have reported Malassezia, Rhodotorula, Debaromyces, Cryptococcus, and in some sites, Candida as fungal skin commensals10. Cutaneous fungal infections affect 29 million Americans, yet the role of dermatophytes in common toenail infections are difficult to characterize using culture-based studies11. For other common skin disorders such as seborrheic dermatitis (dandruff), fungal involvement remains incompletely understood12,13. Difficulty in establishing growth conditions14,15 contribute to challenges to rapidly identify and direct treatment against pathogenic fungi.
To compare culture- and DNA sequence-based identification of human skin-associated fungi, we simultaneously sampled four skin sites from adult healthy volunteers (HVs) (Figure S1 and Table S1). We characterized isolates by morphological features and molecular markers. In total, we cultured >130 fungal isolates: 62 Malassezia (species: globosa, restricta, and sympodialis13,16), 25 Penicillium (species: chrysogenum and lanosum), and 19 Aspergillus (species: candidus, terreus, and versicolor) (Table S2). Five or fewer Alternaria, Candida, Chaetomium, Chrysosporium, Cladosporium, Mucor, Rhodotorula, and Trichophyton isolates were cultured.
To explore fungal diversity with culture-independent methods, we prepared DNA directly from clinical swabs, PCR-amplified and sequenced two phylogenetic markers within the rRNA region: 18S rRNA and Intervening Internal Transcribed Spacer (ITS) region7,17,18. We generated a custom ITS database based on sequences deposited in GenBank to classify sequences to genus-level with greater than 97% accuracy (Table S3). 18S rRNA sequences were classified with SILVA database19. We determined the relative abundance of fungal genera of occiput (back of head), nare (nostril), plantar heel, and retroauricular crease (behind the ear). Consistent across 18S rRNA and ITS-characterized samples, the genus Malassezia predominated in the retroauricular crease, nare, and occiput. Plantar heel was the most diverse site with representation of Malassezia, Aspergillus, Cryptococcus, Rhodotorula, Epicoccum, and others (Figure S2). ITS sequencing enabled greater genus-level taxonomic resolution, reflecting the specificity of the genomic region and richness of the molecular database. Based on technical and analytic advantages, we selected ITS1 region for subsequent sequencing and analyses of fungal diversity.
We generated >5 million ITS1 sequences from 10 HVs each sampled at 14 skin sites (Table S4). Both Ascomycetous and Basidiomycetous fungi were identified as normal skin flora. The genera Malassezia predominated at all eleven core body and arms sites: antecubital crease, back, external auditory canal, glabella, hypothenar palm, inguinal crease, manubrium, nare, occiput, retroauricular crease, and volar forearm (Figure 1). We explored Malassezia species-level resolution with a taxonomic dataset we developed with reference ITS1 sequences and our human-associated Malassezia isolates. Pairwise comparisons of these Malassezia ITS1 sequences demonstrated >91% sequence identity within species and 70–88% identity between species. These Malassezia sequences served as references within the phylogenetic pplacer20 program to classify ~80–90% of Malassezia sequences per skin site to species-level. Species-level identification revealed fungal specificity between body sites (Figure 1). M. restricta predominated in external auditory canal, retroauricular crease, and glabella while M. globosa predominated on back, occiput, and inguinal crease. Sites such as nares, antecubital crease, volar forearm, and hypothenar palm were characterized by multiple species (M. restricta, M. globosa, M. sympodialis). In total, we identified 11 of the 14 known Malassezia species amongst skin sites, suggesting that human skin is colonized with a wide range of Malassezia. Based on species-level resolution, we observed that fungal diversity is more dependent on body site than individual subject. ITS sequences also matched Candida species tropicalis, parapsilosis, and orthopsilosis and Cryptococcus species flavus, dimennae, and diffluens, which are considered part of normal human flora and as possible pathogens in wounds or immunocompromised patients14.
Figure 1. Relative abundance of fungal genera and Malassezia species of human skin sites.

Fungal diversity of individual body sites of healthy volunteers (HV) was taxonomically classified at the genus level with further resolution of Malassezia species. Note: Left side is shown for all body sites except HV7’s right toenail.
Significantly greater diversity was observed on three feet sites (plantar heel, toenail, and toeweb) in both number of genera observed and variation between individuals (Figure S3 and Table S5). The fungal profile of subject HV7 was notably more diverse than other participants (Figure 1). HV7 completed a two-month course of oral antifungals for a toenail infection seven months prior to sampling. Remaining HVs reported no use of either oral or topical antifungals for at least two years prior to sampling. While HV7 is an outlier, the additional genera (e.g.; Aspergillus and Saccharomyces) identified demonstrate that skin is capable of harboring a high degree of fungal diversity. While microbial sequencing is unable to determine causation, these data might suggest that either fungal dysbiosis is associated with recurrent toenail infections, or that antifungals alter fungal communities with alterations persisting beyond seven months. In comparison with culture-based analysis, ITS1 sequencing can provide a more complete range of commensal and potentially pathogenic microbial diversity.
To quantify and compare community similarity and taxonomic richness of skin sites, we assigned fungal sequences to taxonomic units based on genus-level phylogeny rather than percent sequence identity to obviate the high variation noted between species21 with the later metric. Plantar heel was the most complex fungal site (median richness ~80 genera) followed by other foot sites (toeweb and toenail with ~60 and ~40 genera, respectively; Figure 2, Table S6). Arm sites demonstrated intermediate richness ranging from 18–32 genera and core body sites exhibited much lower richness ranging from 2 to ~10 genera. Thus, regional location is a strong determinant of fungal richness. As observed in skin bacterial studies, left-right similarity within an individual was greater than between different individuals at the same body site (Figure S4 and Table S7). To determine temporal stability of the fungal microbiome, six HVs returned 1–3 months after initial sampling. Sites exhibiting Malassezia predominance at initial sampling displayed the same genus and species level predominance with strong community structure stability (Figures S5 and S6 and Table S8). Feet sites continued to exhibit great diversity perhaps reflecting frequent environmental exposure.
Figure 2. Median richness of fungal and bacterial genera.

Median taxonomic richness (or number of observed genera) of fungal and bacterial genera at 14 body sites. Error bars represent the median absolute deviation.
To explore the relationship between skin-associated fungi and bacteria, we sequenced the 16S rRNA gene from the same clinical samples. Consistent with prior studies22,23, bacteria on healthy human skin were predominantly Propionibacterium, Corynebacterium, and Staphylococcus (Figure S7). Similar to other moist skin sites, the toenail (not previously surveyed) was predominantly Corynebacterium and Staphylococcus. Interestingly, although HV7 was an outlier in terms of fungal diversity and membership, HV7’s bacterial profile was normal with respect to taxonomic and ecological measures of diversity (Figure S7). Directed studies may help elucidate how antibacterial and antifungal therapies perturb fungal and bacterial communities.
Bacterial and fungal richness were not linearly correlated, but rather grouped into discrete clusters of arm, feet, and core (Figures 2 and S8). Arm sites displayed markedly greater bacterial diversity with lower fungal diversity. In contrast, feet sites displayed markedly greater fungal diversity with lower bacterial diversity. Core body sites clustered together, displaying both lower bacterial and fungal diversity. These data reveal skin microbiome complexity and suggest that different characteristics shape skin bacterial and fungal communities.
Using principal coordinates analysis of community structure; we explored properties that might differentially shape bacterial and fungal communities. Consistent with prior studies5, variation in bacterial communities segregated strongly by skin physiology, grouping into sebaceous, moist and dry sites, with lipophilic bacteria (Corynebacterium, Propionibacterium, Turicella) and staphylococcal species driving variation between communities (Figure 3 and S9). In contrast, fungal communities segregated more strongly by site location, with feet, arm, head, and torso sites forming discrete groups. Different Malassezia species drive variation in arm, torso, and head while a wide range of fungal genera drive variation in feet (Figure 3 and S9). Co-occurrence analysis based on Spearman correlation of fungal and bacterial taxonomic relative abundances of feet sites (Figure S10), provided a preliminary evaluation of major fungal-bacterial associations. For example, a group of primarily Actinobacteria was anti-correlated with resident Ascomycota and Basidiomycota in contrast to a group of primarily Firmicutes and Proteobacteria that was positively correlated with these fungal taxa.
Figure 3. Forces shaping fungal and bacterial communities.
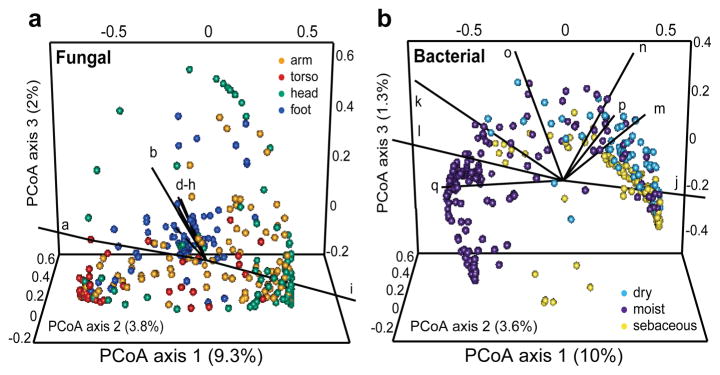
Principal coordinates analysis (PCoA) of degree of fungal and bacterial community similarity at 14 body sites, based on predominant genera and species. Variation in fungal communities segregated strongly along site location, with feet, head, and torso sites forming discrete groups (A). Bacterial community structure was more dependent on site physiology (B). Axes that most significantly contribute to variation and their relative length are defined in Figure S10 legend. For fungi, M. restricta (i; ρ = 0.92) and M. globosa (a; ρ = −0.79) are primary and opposing drivers of variation. For bacteria, Propionibacterium (j; ρ = 0.95) contributes to sebaceous site variation, while Corynebacterium (l; ρ = −0.74), and Turicella (q; ρ = −0.56) contribute most heavily for moist sites.
These datasets can now be used to inform future clinical studies examining fungal shifts from case/control studies or from contralateral uninvolved/involved sites on a patient. Of our study participants, we observed 20% (12/60) had plantar heel scaling, toeweb scaling or toenail changes, consistent with possible fungal infections (Table S1). Of subjects with observed clinical involvement, positive mycological cultures were obtained from toenails (two samples) and plantar heel (one sample) (Trichophyton, Penicillium, and Aspergillus). These percentages are similar to larger studies, which report signs of clinical involvement in up to 60% of feet of healthy individuals with 2–25% culture-positive for fungi. The wide variation in prevalence of clinical involvement and positive fungal cultures is dependent on several factors including population and climate24,25. As an initial inquiry into the etiology of foot fungal disorders, we examined how clinical status of observed involved/uninvolved at plantar heel, toeweb or toenail affected fungal community structure. For uninvolved sites, interpersonal variation in community structure was highly consistent across all feet sites. In contrast, for sites with observed clinical involvement, similarity of community structure was much higher for plantar heel yet much lower for toenail (Figure 4). These data potentially suggest that there might be a common fungal community shared amongst individuals with plantar heel involvement and tremendous fungal diversity underlying toenail infections, but further studies are needed.
Figure 4. Clinical involvement alters shared fungal community structure.

Community structure measures the type and relative abundance of each genus. A value of 1 implies identical community structure and 0 implies dissimilar structures. Amongst uninvolved feet sites, community structure is fairly consistent at plantar heel, toenail and toeweb. For involved sites, plantar heel has much greater shared community structure and toenails have much lower shared community structure. Error bars represent the standard error of the mean.
This systematic study clearly demonstrates that human skin surfaces are complex ecosystems, providing diverse environments for microorganisms that inhabit our bodies. Different factors determine bacterial and fungal communities depending on skin physiological properties. Malassezia species predominate on all core body and arm sites. In contrast, feet sites display tremendous fungal diversity and dramatically lower stability over time. Microbial community instability may provide an opportunity for potentially pathogenic microbes to establish disease. Plantar heel, toeweb and toenail are common sites of recurrent human fungal disease, which can be recalcitrant to treatment. This study also investigated fungal diversity at sites of predilection for other skin disorders including seborrheic dermatitis, tinea cruris, and forms of atopic dermatitis. With genomic advances, such as shotgun metagenomic sequencing, we can begin to address interactions between microbes (bacteria-fungal, bacteria-bacteria, fungal-fungal) residing in these complex environments. The role of fungal commensals in educating the human immune system is gaining new appreciation in intestinal disease26. Further studies in healthy skin and dermatologic disorders are needed to explore these host-microbial interactions. Additionally, antifungal medications including azoles, echinocandins, and amphotericin B have potentially serious side effects such as liver or kidney damage27. Therefore, new treatment strategies are required to strategically target microbial dysbiosis and to combat the increasingly observed resistance against our current arsenal of antimicrobials.
Subject recruitment and sampling
Healthy adult male and female volunteers (HVs) 18–40 years of age were recruited from the Washington, DC metropolitan region from September 2009 to September 2011. This natural history study was approved by the Institutional Review Board of the National Human Genome Research Institute (clinicaltrials.gov/NCT00605878) and all subjects provided written informed consent prior to participation. Subjects provided medical and medication history and underwent a physical examination. Exclusion criteria included history of chronic medical conditions, including chronic dermatologic diseases, and use of antimicrobials (antibiotics or antifungals) 6 months prior to sampling (see Table S1 for HV information). Bathing/showering with only non-antibacterial soap/cleansers was allowed during the 7 days prior to sample collection. No bathing, shampooing, or moisturizing was permitted for 24 hours prior to sample collection. Some HVs returned 1–3 months after their initial visit for follow-up sampling.
Fourteen skin sites representing a range of physiological characteristics and sites of predilection for fungal-associated dermatologic diseases were selected including core/proximal body sites: middle upper back, external auditory canal (inside the ear), retroauricular crease (behind the ear), occiput (back of scalp), glabella (central forehead between eyebrows), inguinal crease (skin fold midway between hip and groin area), manubrium (upper central chest), and nare (inside the nostril); and distal body sites: antecubital fossa (inner elbow), volar forearm (mid-forearm), hypothenar palm (palm of hand closer to little finger), plantar heel (bottom of heel), toenail, and toe web (webspace between 3rd and 4th toes) (Figure S1). All clinical findings observed at sampling sites were documented, including any scaling on the feet and any changes of the toenails. Body sites with left-right symmetry (10 of the 14 body sites) were sampled bilaterally to calculate intrapersonal variation (see Figure S1 for sites sampled).
Fungal culturing and characterization
For fungal cultures, superficial skin scrapes were collected from a 4-cm2 area with a sterile surgical blade and placed directly in media. Skin scrapings were spread on fungal culturing plates (under laminar flow hood to minimize contamination) to isolate pathogenic and non-pathogenic fungi, including fastidious yeasts. Selective media containing antibiotics to selectively suppress bacterial growth included: Inhibitory Mold Agar with Gentamicin; Brain Heart Infusion Agar with Chloramphenicol and Gentamicin (Remel, Lanexa, KS); and Sabouraud with Chloramphenicol and Cycloheximide (Remel, Lanexa, KS) augmented with olive oil to promote Malassezia growth. Plates were incubated at 30°C, checked daily for the first week and 2–3 days thereafter. Isolates that flourished in culture were re-streaked for single colonies followed by sub-culturing to ensure purity and characterized by morphological features and molecular markers. DNA was extracted with MasterPure™ Yeast DNA Purification Kit (Epicentre, Madison, WI) according to manufacturer’s instruction with the addition of 5mm steel beads to mechanically disrupt fungal cell wells (Qiagen, Valencia, CA). The Internal Transcribed Spacer (ITS) 1 and 2 regions were amplified from purified genomic fungal DNA using primers 18S-F (5′-GTAAAAGTCGTAACAAGGTTTC-3′) and 5.8S-1R (5′-GTTCAAAGAYTCGATGATTCAC-3′) for ITS1 and 5.8S-F (5′-GTGAATCATCGARTCTTTGAAC-3′) and 28S1-R (5′-TATGCTTAAGTTCAGCGGGTA-3′). PCR products were purified and sent to ACGT, Inc. (Wheeling, IL) for sequencing and BLAST was performed on the resulting amplicon sequence to identity each isolate1.
Clinical sample collection, DNA extraction, PCR amplification and sequencing of 18S rRNA gene and ITS1
For DNA analyses, samples were collected, including negative controls, as previously described2. Catch-All™ Sample Collection Swabs (Epicentre, Madison, WI) were used for skin swab sample collection across all sites with the exception of the toenail (toenail clippings were collected)3, and swabs were stored in lysis solution provided with the MasterPure™ Yeast DNA Purification Kit (Epicentre, Madision, WI). To pre-digest the toenail clippings, Proteinase K (Invitrogen, Carlsbad, CA) was added to the sample and incubated overnight with shaking at 55°C. Skin samples were incubated in yeast lysis buffer and lysozyme (20 mg/mL) for 1 hour with shaking at 37°C. Then, 5 mm steel beads were added to mechanically disrupt fungal cell walls using a Tissuelyser (Qiagen, Valencia, CA) for 2 minutes at 30 Hz. The Invitrogen PureLink Genomic DNA Kit (Invitrogen, Carlsbad, CA) was utilized for all subsequent steps.
For 18S rRNA amplicon sequencing, each DNA was amplified with SR6 (5′-TACCTGGTTGATTCTGC-3′) and SR1R (5′-TGTTACGACTTTTACTT-3′) primers. The following PCR conditions were used: 2.5 μl 10X AccuPrime Buffer II, 0.2 μl Accuprime Taq, 0.5 μl primer SR6 (20 μM), 0.5 μl primer SR1R (20 μM), and 4 μl of isolated microbial genomic DNA. PCR was performed in duplicate if possible and a portion of the reaction was run on an agarose gel to verify the presence of the 18S PCR product. Cycle number was determined such that amplification was still in the linear range of the reaction and yielded sufficient PCR product for cloning (maximum of 32 cycles). Negative controls on both the mock swab and no template DNA were performed with each set of amplifications. The PCR product was ligated into the PCR4 TOPO vector (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol. 384 resulting bacterial colonies per ligation were picked, plasmid DNA purified and inserts sequenced at NISC on an ABI 3730xl sequencer (Applied Biosystems Inc., Foster City, CA) using M13 primers flanking the insert.
For ITS1 amplicon sequencing, each DNA was amplified with adapter+18SF (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGGTAAAAGTCGTAACAAGGTTTC) and 5.8S-1R+ barcode (5′-GTTCAAAGAYTCGATGATTCAC) primers4. The following PCR conditions were used: 2.5 μl 10X AccuPrime Buffer II, 0.2 μl Accuprime Taq (Invitrogen, Carlsbad, CA), 0.1 μl primer B adapter+18SF (100 μM), 2 μl primer 5.8S-1R+barcode (5 μM), and 4 μl of isolated microbial genomic DNA. The PCR was performed in duplicate for 32 cycles. Duplicate amplicons were combined, purified (Agencourt AMPure XP-PCR Purification Kit (Beckman Coulter, Inc., Brea, CA)), and quantified (QuantIT dsDNA High-Sensitivity Assay Kit (Invitrogen, Carlsbad, CA)). An average of ~8 ng DNA of 94 amplicons were pooled together, purified (MinElute PCR Purification Kit (Qiagen, Valencia, CA)) and sequenced on a Roche 454 GS20/FLX platform with Titanium chemistry (Roche, Branford, Connecticut). Flow-grams were processed with the 454 Basecalling pipeline (v2.5.3).
For 16S rRNA amplicon sequencing, each DNA was amplified with adapter+V1_27F (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGAGAGTTTGATCCTGGCTCAG) and V3_534R+barcode primers (5′-CAGCACGCATTACCGCGGCTGCTGG)4. The following PCR conditions were used: 2 μl 10X AccuPrime Buffer II, 0.15 μl Accuprime Taq (Invitrogen, Carlsbad, CA), 0.04 μl adapter+V1_27F (100 μM), 2 μl primer V3_354R+barcode (2 μM), and 2 μl of isolated microbial genomic DNA. PCR was performed in duplicate for 30 cycles and then PCR-clean up, amplicon pooling of ~10 ng DNA, purification, and sequencing were performed as described above for ITS.
Custom generated fungal ITS1 reference database
ITS sequences were extracted from GenBank using the query: (ITS1[All Fields] OR ITS2[All Fields] OR 5.8S[All Fields]) AND Fungi[All Fields]) NOT “uncultured”[All Fields]. Taxonomy classifications associated with sequences were recorded as strings in the following order: kingdom, phylum, class, order, family and genus, and recorded as unclassified if the levels were not clearly defined. Sequence classification was manually curated and any discrepancies in taxonomy strings were resolved using the Taxonomy database in Pubmed. When both anamorphic (asexual) and teleomorphic (sexual) names were represented for a species within GenBank, the strings were manually curated and the anamorphic taxonomic nomenclature was selected. The sequences were then clustered to 95% sequence identity using CD-HIT5. Representative sequences were chosen by CD-HIT and a consensus taxonomy string was generated for the sequence, starting from the highest level (kingdom) and moving to the lowest level (genus). If the most highly represented classification was twice as frequent as the next one, this classification was chosen for the level. If no classification satisfied this criterion, this and all lower levels were set as unclassified. Sequences clearly misclassified as fungi were removed from the curated database.
Sequence classification and analyses
Sequences were pre-processed to remove primers and barcodes. Possible chimeras created during PCR amplification were identified with UCHIME in mothur6,7. Reference was set to self and included in the names file to check for chimeras using more abundant sequences as references7. With the ITS database described above as the reference, these chimera checked sequences were classified to the genus level with the BLAST option and the k-Nearest Neighbor (knn) algorithm in mothur7. 16S rRNA sequences were classified to the genus or species level using the RDP classifier with training set (v6) and as previously described8. Sequences were assigned to taxonomic units based on their genus level phylogenetic classification. R statistical software was implemented to generate plots representing the relative abundance of fungal genera.
Community richness (Chao1), diversity (Shannon Index), membership (Jaccard Index) and structure (Theta Index) were calculated within mothur as previously described after using a subsampling cutoff of 1000 sequences/sample2,9,10. Diversity indices for left and right symmetric sites were averaged for body sites with bilateral symmetry. All statistical analyses are represented as the standard error of the mean unless otherwise indicated.
18S rRNA sequence classification
To pre-process the 18S rRNA sequences, traces were base called using Phred (v 0.990722.g), trimmed with Crossmatch, and each clone assembled using Phrap (v 0.990329). The default parameters were used except force level was 9 and the mismatch penalty was −111,12. For approximately 15% of read pairs, the overlap was not sufficient for de novo assembly and a scaffolded assembly was attempted. Scaffolded assembly was done using the AmosCmp16Spipeline (available from http://microbiomeutil.sourceforge.net) and non-redundant reference sequences from the SILVA small subunit rRNA database13. 18S rRNA sequences were classified using the SILVA v108 database. R was implemented to generate plots representing the relative abundance of fungal genera.
Malassezia sequence classification to the species level
To classify Malassezia ITS1 sequences from the genus to species level, we used the get.lineage command in mothur to retrieve only Malassezia sequences. As an internal check, these skin-associated Malassezia ITS1 sequences were aligned with the Malassezia reference package in mothur, and all discrepancies resolved. Next, we curated and aligned a reference library of Malassezia type-strain ITS1 sequences retrieved from GenBank augmented by those from the fungal cultures described above. Two to ten representatives were included in the database for Malassezia species (M. globosa, M. restricta, M. sympodialis, M. slooffiae, M. furfur, M. pachydermatis, M. dermatis, M. yamatoensis, M. obtusa, M. japonica, and M. nana) for a total of 52 ITS1 sequences. Sequences were aligned with MUSCLE to generate the reference alignment14.
This curated library was used as the reference to phylogenetically place and classify novel skin-associated Malassezia ITS1 sequences to the species level with the software package pplacer15. Sequence placement on the reference tree along with confidence scores were visualized in Archaeopteryx using the guppy command15,16. guppy classify and a light-weight SQL database were employed to make and store taxonomic classifications. A likelihood score of at least 0.65 was used for classifications produced by the guppy classify command. Finally, classifications were converted into mothur compatible taxonomic strings to create the tax.summary file for community-based analyses as above. Similarly, species-level designations for bacterial Staphylococcus sequences were generated using pplacer with a 16S rRNA reference database built from rRNA records extracted from RefSeq genomes (as of April 2012) and RDP type species sequences (Release 10, Update 24)8.
ITS1 and 16S rRNA comparisons
Taxonomic units were defined from genus- and, where available, species-level ITS and 16S rRNA phylotypes. Groups were each subsampled to 1800 sequences and the Yue-Clayton theta index generated to compare the similarity between communities. Principal coordinate analysis of the theta index was performed and the Spearman correlation of the relative abundance of each taxonomic unit versus the top three axes was calculated to assess how each taxonomic unit contributed to variation along the axes.
Co-occurrence between bacteria and fungi was assessed by calculating the partial Spearman correlation of the relative abundances of the different taxa, adjusted for multiple within-patient measurements. Calculations were performed on Fisher-transformed r values. Comparisons were limited to those taxa which occurred in >25% of samples for either ITS or 16S rRNA, and for ITS, if mean abundance across all samples exceeded 0.25%. Because of the relatively higher fungal diversity, only feet sites (Ph, Tn, and Tw) were used.
Supplementary Material
Acknowledgments
We thank Joseph Heitman, Anthony Amend, Yvonne Shea, Maria Turner, Isaac Brownell, and Mark Udey for helpful discussions. We thank Julia Fekecs for assistance with the figures. This work was supported by NIH NHGRI and NCI Intramural Research Programs and in part by 1K99AR059222 (HHK). Sequencing was funded by grants from the National Institutes of Health (1UH2AR057504-01 and 4UH3AR057504-02).
Footnotes
Sequence data from this study have been submitted to GenBank (KC669797 -KC675175) and SRA and can be accessed through BioProject ID 46333. Patient and sample metadata have been deposited in the controlled access database dbGaP under study accession phs000266.v1.p1.
Author Contributions
K.F., H.H.K and J.A.S. designed the outline of the study. D.S. and E.N. recruited and consented human subjects. K.F. and J.Y. assembled and curated the fungal database. J.A.M and C.D. prepared the clinical samples for sequencing. NISC performed sequencing. K.F., J.O, S.C. and M.P. analyzed sequence data. K.F., H.H.K and J.A.S. drafted the manuscript with specific contributions from J.O, J.Y. and S.C. All authors read and approved the final version. H.H.K. and J.A.S. contributed equally to this study.
The authors declare no competing financial interests.
References
- 1.Marples M, editor. The Ecology of the Human Skin. Bannerstone House; Springfield, Illinois: 1965. [Google Scholar]
- 2.Grice EA, Segre JA. The Human Microbiome: Our Second Genome. Annu Rev Genomics Hum Genet. 2012 doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Human Microbiome Project Consortium. Structure function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu Rev Pathol. 2012;7:99–122. doi: 10.1146/annurev-pathol-011811-132421. [DOI] [PubMed] [Google Scholar]
- 5.Peleg AY, Hogan DA, Mylonakis E. Medically important bacterial-fungal interactions. Nat Rev Microbiol. 2010;8:340–9. doi: 10.1038/nrmicro2313. [DOI] [PubMed] [Google Scholar]
- 6.Dollive S, et al. A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol. 2012;13:R60. doi: 10.1186/gb-2012-13-7-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James TY, et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature. 2006;443:818–822. doi: 10.1038/nature05110. [DOI] [PubMed] [Google Scholar]
- 8.Ghannoum MA, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713. doi: 10.1371/journal.ppat.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulino LC, Tseng CH, Strober BE, Blaser MJ. Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. Journal of clinical microbiology. 2006;44:2933–41. doi: 10.1128/JCM.00785-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roth RR, James WD. Microbial ecology of the skin. Annu Rev Microbiol. 1988;42:441–64. doi: 10.1146/annurev.mi.42.100188.002301. [DOI] [PubMed] [Google Scholar]
- 11.Bickers DR, et al. The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55:490–500. doi: 10.1016/j.jaad.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 12.Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. The Malassezia genus in skin and systemic diseases. Clinical microbiology reviews. 2012;25:106–41. doi: 10.1128/CMR.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saunders CW, Scheynius A, Heitman J. Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS pathogens. 2012;8:e1002701. doi: 10.1371/journal.ppat.1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larone DH. Medically Important Fungi: A guide to identification. ASM Press; Washington DC: 2002. [Google Scholar]
- 15.St-Germain G, Summerbell R. Identifying Fungi: A Clinical Laboratory Handbook. Star Publishing Company; Belmont, CA: 2011. [Google Scholar]
- 16.Gioti A, et al. Genomic Insights into the Atopic Eczema-Associated Skin Commensal Yeast Malassezia sympodialis. mBio. 2013;4 doi: 10.1128/mBio.00572-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruns TD, et al. Evolutionary Relationships within the Fungi: Analyses of Nuclear Small Subunit rRNA Sequences. Molecular Phylogenetics and Evolution. 1992;1:231–241. doi: 10.1016/1055-7903(92)90020-h. [DOI] [PubMed] [Google Scholar]
- 18.Schoch CL, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. 2012;109:6241–6. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quast C, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research. 2013;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen AD, Wolak A, Alkan M, Shalev R, Vardy DA. Prevalence and risk factors for tinea pedis in Israeli soldiers. Int J Dermatol. 2005;44:1002–5. doi: 10.1111/j.1365-4632.2005.02281.x. [DOI] [PubMed] [Google Scholar]
- 25.Perea S, et al. Prevalence and risk factors of tinea unguium and tinea pedis in the general population in Spain. J Clin Microbiol. 2000;38:3226–30. doi: 10.1128/jcm.38.9.3226-3230.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–7. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perfect JR, Lindsay MH, Drew RH. Adverse drug reactions to systemic antifungals. Prevention and management. Drug Saf. 1992;7:323–63. doi: 10.2165/00002018-199207050-00003. [DOI] [PubMed] [Google Scholar]
- 28.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grice EA, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043–50. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lennon NJ, et al. A scalable, fully automated process for construction of sequence-ready barcoded libraries for 454. Genome Biol. 2010;11:R15. doi: 10.1186/gb-2010-11-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 6.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conlan S, Kong HH, Segre JA. Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PloS one. 2012;7:e47075. doi: 10.1371/journal.pone.0047075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong HH, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–9. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue JC, Clayton MK. A similarity measure based on species proportions. Communications in Statistics-Theory and Methods. 2005;34:2123–2131. [Google Scholar]
- 11.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–94. [PubMed] [Google Scholar]
- 12.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–85. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 13.Pruesse E, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han MV, Zmasek CM. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics. 2009;10:356. doi: 10.1186/1471-2105-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.