

Summary
Triosephosphate isomerase (TPI) is a glycolytic enzyme that converts dihydroxyacetone phosphate (DHAP) into glyceraldehyde 3-phosphate (GAP). Glycolytic enzyme dysfunction leads to metabolic diseases collectively known as glycolytic enzymopathies. Of these enzymopathies, TPI deficiency is unique in the severity of neurological symptoms. The Drosophila sugarkill mutant closely models TPI deficiency and encodes a protein prematurely degraded by the proteasome. This led us to question whether enzyme catalytic activity was crucial to the pathogenesis of TPI sugarkill neurological phenotypes. To study TPI deficiency in vivo we developed a genomic engineering system for the TPI locus that enables the efficient generation of novel TPI genetic variants. Using this system we demonstrate that TPI sugarkill can be genetically complemented by TPI encoding a catalytically inactive enzyme. Furthermore, our results demonstrate a non-metabolic function for TPI, the loss of which contributes significantly to the neurological dysfunction in this animal model.
Key words: TPI, Triosephosphate isomerase, Drosophila melanogaster, Glycolysis, Longevity, Locomotor function
Introduction
Triosephosphate isomerase (TPI) is a homodimeric enzyme that functions in a non-linear step of glycolysis, converting dihydroxyacetone phosphate (DHAP) into glyceraldehyde 3-phosphate (GAP). Both DHAP and GAP are produced from the catabolism of fructose 1,6-bisphosphate by the enzyme aldolase; however, only GAP can be utilized by the remaining steps in glycolysis. Therefore, TPI is responsible for the net gain of glycolysis-derived ATP as well as the production of an extra molecule of pyruvate per molecule of glucose. This enhancement of glycolysis is important in the bioenergetics of both aerobic and anaerobic metabolism.
TPI deficiency is a recessive loss-of-function human disease resulting from missense mutations in the TPI gene. TPI deficiency is clinically characterized by symptoms such as hemolytic anemia, cardiomyopathy, neurological dysfunction and degeneration, and premature death (Schneider, 2000; Orosz et al., 2006). Pathogenic TPI deficiency mutations can affect the promoter or coding sequence and all have been reported to dramatically reduced TPI activity owing to changes in catalysis and/or enzyme stability (Daar et al., 1986; Hollán et al., 1993; Arya et al., 1997). TPI deficiency has a very poor genotype-phenotype correlation and studies to elucidate pathogenesis are extremely limited, especially in animal systems.
Drosophila are the only model system identified to date in which mutants have been shown to recapitulate the neurological phenotypes seen in human patients (Celotto et al., 2006; Gnerer et al., 2006). We have previously isolated an animal model of TPI deficiency known as TPIsugarkill (sgk). TPIsgk is characterized by shortened lifespan, neurodegeneration, and conditional behavioral abnormalities (Celotto et al., 2006) resulting from a missense mutation causing a methionine to threonine substitution (M80T). The affected methionine is present near the dimer interface yet does not seem to result in a shift in monomer-dimer populations in vivo (Seigle et al., 2008). However, the TPIsgk mutation has been shown to induce abnormal proteasomal degradation of TPI resulting in reduced total TPI protein (Seigle et al., 2008; Hrizo and Palladino, 2010). Interestingly, we have previously shown that this loss-of-function mutation can be attenuated by overexpressing mutant TPIsgk (Celotto et al., 2006). This result led us to question whether the presence of the enzyme or its catalytic activity was most important to the pathogenesis of sugarkill. Here we demonstrate that the M80T substitution in TPIsgk reduces its isomerase activity and, based on the proximity to one of the TPI catalytic sites, we predict that the reduced catalytic efficiency is a result of the influence of the mutation on dimeric cooperation.
To address whether the presence of the enzyme or its catalysis were most important to the TPIsgk disease phenotypes in vivo, we developed genomic engineering (GE) of the Drosophila TPI locus. This process establishes an attP ΔTPI founder line which can be used to modify the gene locus using highly efficient attP/attB transgenesis. We hypothesized that if the presence of the enzyme was crucial to pathogenesis independent of catalytic activity, we would be able to rescue the disease phenotypes with a catalytically inactive variant of the protein. Lys11 of TPI is a fully conserved catalytic residue known to be required for substrate binding and substitution to Met completely abolishes catalysis (Lodi et al., 1994; Wierenga et al., 2010). We have generated the attP ΔTPI founder line and have used GE to create Drosophila TPIK11M, encoding a catalytically inactive TPI. Here we demonstrate that TPIK11M genetically complements the longevity and behavior of the TPIsgk animal model of TPI deficiency. Furthermore, catalytically inactive TPI complements TPIsgk phenotypes without enhancing its stability, catalysis or reducing the associated metabolic stress. Collectively, these data suggest a function of TPI independent of its catalytic activity which is crucial to behavior and longevity.
Results
Recombinant TPI enzyme activity
Previous studies established that TPIsgk is a recessive loss-of-function mutation characterized by reduced TPI levels (Seigle et al., 2008). Genetic data suggested that TPIsgk retained sufficient function to rescue mutant survival and behavioral phenotypes if overexpressed (Seigle et al., 2008). These data led us to hypothesize that reduced TPI catalysis was crucial to the pathogenesis of TPI deficiency. To investigate this hypothesis further we generated recombinant Drosophila WT (dWT) and TPIsgk (dM80T) and examined the kinetics of isomerase activity for each enzyme (Table 1). These data demonstrate that TPIsgk (dM80T) exhibits a substantial reduction in isomerase activity. The dM80T protein has a 33% decrease in substrate affinity and ∼11-fold reduction in catalytic activity compared to WT enzyme. This ultimately resulted in a ∼15-fold reduction in enzyme efficiency. Both enzymes displayed typical Michaelis–Menten kinetics (supplementary material Fig. S1).
Table 1. Kinetic parameters of Drosophila wild-type (dWT) and sugarkill (dM80T) triosephosphate isomerase enzymes.

dWT, Drosophila wild-type triosephosphate isomerase; dM80T, Drosophila sugarkill triosephosphate isomerase.
Values for kcat and Km are means ± s.e.m.
To assess the role of the M80 position within TPI function, we analyzed the crystal structure of TPI from G. gallus (Zhang et al., 1994) – Drosophila TPI shows 67% identity and 80% functional conservation with G. gallus. This structure was determined in the presence of the transition-state analog phosphoglycolohydroxamic acid (PGH), and the homologous methionine (M81) is located within the third loop of TPI (residues 63 to 86; Fig. 1A). This loop coordinates numerous interactions within the dimerization interface of TPI including important contacts between M81 and M13 and N14 of the neighboring subunit (Fig. 1B). These hydrophobic interactions help position the third loop, and are aided by hydrogen bonding between residues T74 and Q76 (also on the third loop), which interact with H94 and R97, respectively. The sum of these interactions is a coordinated network using conserved residues to mediate both hydrophobic and hydrogen-bonding interactions that ultimately positions both K11 and H94 properly within the active site of the enzyme (Fig. 1B,C). These residues are crucial for enzymatic function in TPI (Lambeir et al., 1987; Norledge et al., 2001) and establish a connection between M80 in Drosophila TPI and reduced catalysis for TPIsgk.
Fig. 1.

Positioning of the third loop forms an interaction network connecting the dimerization interface to the catalytic pocket. (A) The crystal structure of TPI dimer from Gallus gallus (Protein Database ID: 1TPH). Monomer A of the dimer is rendered as a surface and colored white, whereas monomer B is shown as a cartoon colored blue, with the third loop highlighted in orange. The position of the transition state analog phosphoglycolohydroxamic acid (PGH, green) is indicated within the catalytic pocket. (B) Local environment of the third loop. Colored as before, with residues making important contributions to the dimerization interface indicated as sticks. Van der Waals interactions between M81 and residues M13 and N14 are indicated as black dotted lines. Hydrogen bonds between residues on the distal end of the third loop and connecting to the catalytic core are shown as green dotted lines. Residues are numbered based on the Gallus gallus sequence and for clarity we are using the TPI numbering standard lacking the initiator methionine. (C) An alignment of TPI protein sequences from H. sapiens, G. gallus, and D. melanogaster. Protein alignment was performed using the ClustalW method, with H. sapiens and G. gallus sharing 63.5% and 67.3% identity with D. melanogaster. Residues that mediate TPI dimerization are indicated by grey circles. Residues within the hydrogen-bonding network crucial for catalysis are indicated by cyan diamonds. The position of the sugarkill mutant (M81, red) and its positions within the third loop are indicated by an orange box.
Establishing that TPIsgk still retained isomerase activity supported a catalytic explanation of our phenotypic attenuation via TPIsgk transgenic overexpression. However, a rigorous assessment of this hypothesis in vivo would require supplementation of TPI levels without changing isomerase activity.
Generation of the founder knockout line by homologous recombination
To create a catalytically inactive allele of TPI we developed a GE approach for the TPI locus. Homologous recombination (HR) in Drosophila had previously been developed (Rong and Golic, 2000; Gong and Golic, 2003) but recent advances in GE technology have made the process considerably more efficient (Huang et al., 2008; Huang et al., 2009). The GE founder line was generated through replacement of the endogenous TPI locus using ends-out homologous recombination with a functional attP element (supplementary material Fig. S2A). TPI is located on the third chromosome between CG31029 (centromeric) and AdoR (telomeric). Homology arms directing the deletion of the entire TPI locus were PCR amplified from the w1118 wild type genomic DNA and cloned into the pGX-attP targeting vector. Traditional P-element transgenesis was used to obtain pGX-attP insertion on the second chromosome. HR was induced as previously outlined (Huang et al., 2009). Approximately four hundred putative positive HR events were identified after screening ∼60,000 animals. Among the putative positives, three positive HR lines were identified and verified using genetic and molecular methods.
Genetic validation included failure to complement a previously identified TPI null allele, TPIJS10 (Celotto et al., 2006), and homozygous lethality. Molecular validation included PCR analyses to confirm proper targeting of left and right homology arms using primers directed outside the homology arms and within construct elements. The confirmed founder line was reduced to remove wmc+ using a CRE recombinase and is designated attP ΔTPI (supplementary material Fig. S2B). The entire attP ΔTPI region was sequenced to confirm the correct placement of the GE elements and the integrity of the homology arms.
GE was performed to create TPI+ (attR), TPI+-CFP (attR), TPIM80T (attR), TPIK11M (attR), and TPIK11M-CFP (attR) animals using standard phiC31 site-directed integration followed by CRE-mediated reduction to remove wmc+ (supplementary material Fig. S2). These animals were molecularly verified using PCR analyses of the attR construct element (supplementary material Fig. S2E) and the locus was sequenced. Importantly, GE TPI+ (attR) animals are homozygous viable and behave normally while animals bearing the M80T substitution are substantially stress sensitive and exhibit reduced longevity (supplementary material Fig. S3). Animals homozygous for TPIK11M (attR) are lethal, indicating a necessity for TPI catalysis during development.
Animal behavior and longevity
To test whether TPI isomerase activity was crucial to disease pathogenesis, TPIsgk/TPI+, TPIsgk/TPIsgk, TPIsgk/TPIJS10, and TPIsgk/TPIK11M animals were collected and aged at 25°C. TPIJS10 is a null allele owing to a 1.6 kb deletion, therefore TPIsgk/TPIJS10 animals represent approximately the amount of TPI produced by one allele of TPIsgk. For the purposes of this study, contrasting TPIsgk/TPIJS10 with TPIsgk/TPIK11M provides the most informative comparison, as each animal population contains one catalytically active TPI allele.
Male and female animals were housed in vials of 10–20 individuals and examined for mechanical- and temperature-dependent locomotor defects on days 3 and 20. Time to recovery and time to paralysis were measured in each assay. While TPIsgk/TPI+ shows no sign of paralysis or seizures upon mechanical stress, TPIsgk/TPIsgk and TPIsgk/TPIJS10 exhibit a clear delay in recovery (Fig. 2A). In contrast, TPIsgk/TPIK11M animals display no signs of stress sensitivity (Fig. 2A). Previously, we have shown that TPIsgk phenotypes are progressive in nature (Celotto et al., 2006; Seigle et al., 2008). We measured these behaviors at two time points, day 3 and day 20, to reveal whether TPIK11M fully complements the TPIsgk phenotypes or simply delays their onset. The catalytically inactive enzyme was shown to complement the mechanical stress sensitivity at both time points indicating that the TPIK11M allele does not simply delay the progression of the disease phenotype (Fig. 2A).
Fig. 2.
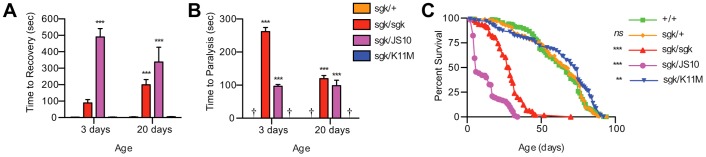
The catalytically inactive TPIK11M rescues behavioral and longevity phenotypes in TPIsgk. (A) TPIK11M complements TPIsgk mechanical stress sensitivity at both day 3 and day 20. (B) TPIK11M expression complements TPIsgk thermal stress sensitivity, again at both day 3 and day 20. † indicates TPIsgk/TPI+ and TPIsgk/TPIK11M animals were not paralyzed. (C) TPIK11M complements the longevity defect of TPIsgk. **P<0.01 and ***P<0.001 relative to TPI+/TPI+; n>15 per genotype for all behavioral assays, and n>150 per genotype for all lifespans. Error bars indicate ± s.e.m.
Thermal stress sensitivity of the animals was also evaluated to test the possibility of pathogenic divergence (Fig. 2B). Here, TPIsgk/TPI+ shows no signs of paralysis or seizures upon thermal stress, while TPIsgk/TPIsgk and TPIsgk/TPIJS10 animals show distinct paralytic phenotypes initiated at ∼240 seconds and ∼90 seconds, respectively (Fig. 2B). In contrast, animals expressing the catalytically inactive allele TPIsgk/TPIK11M displayed no signs of paralysis after thermal stress induction (Fig. 2B). Again, we measured these behaviors at two time points, day 3 and day 20, and expression of the catalytically inactive enzyme was shown to complement the thermal stress sensitivity at both time points (Fig. 2B).
To test whether genetic complementation extended to longevity we performed lifespans as previously described (Palladino et al., 2003). TPIsgk animals exhibit a marked reduction in median longevity that was also complemented with the catalytically inactive TPIK11M allele (Fig. 2C; supplementary material Table S1). Finally, all genotypes eclosed at Mendelian rates suggesting the absence of developmental deficits and that the behavior of the adult population is representative of the genotype as a whole. These data show that expression of a catalytically inactive TPI is sufficient to rescue animal behavior and longevity in the TPIsgk model of TPI deficiency.
TPIsgk animal bioenergetics and lysate enzyme activity
Genetic complementation with TPIK11M could indicate a previously unidentified non-isomerase function of the enzyme or it could result from a restoration of isomerase activity by a number of possible mechanisms. Thus, we examined whether TPIK11M provided genetic complementation through a restoration of enzyme isomerase activity. TPI enzyme activity was robust in both TPI+/TPI+ and TPIsgk/TPI+ lysates yet markedly reduced in TPIsgk/TPIsgk and TPIsgk/TPIJS10 animals (Fig. 3A). Most importantly, TPIsgk/TPIK11M animals exhibit levels of isomerase activity similar to TPIsgk/TPIsgk and TPIsgk/TPIJS10 animals demonstrating that the genetic complementation is independent of isomerase activity (Fig. 3A).
Fig. 3.
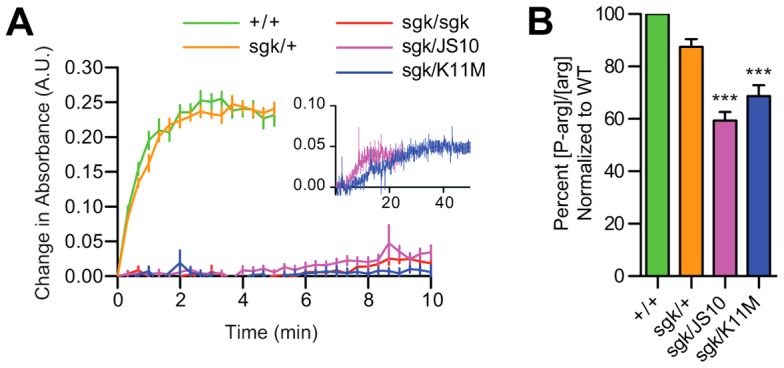
TPIK11M fails to rescue animal bioenergetics and TPI enzyme activity. (A) TPIsgk exhibits little activity in animal lysates. Additionally, complementing TPIsgk with TPIK11M does not rescue its activity in vivo. Inset: a comparison between TPIsgk/TPIJS10 and TPIsgk/TPIK11M lysate kinetics reveals low activity levels in both lysates (note that the units are the same but the scale has been changed). (B) TPIsgk/TPIJS10 animals and TPIsgk/TPIK11M animals both showed reduced ratios of P-arg/arginine indicating bioenergetic stress. ***P<0.001 relative to WT. Error bars indicate ± s.e.m.
The dramatic reduction in lysate isomerase activity suggested that pathogenesis of TPIsgk neurological phenotypes could be unrelated to animal metabolic stress. Previous studies measuring ATP levels in this same mutant have failed to detect a significant change (Gnerer et al., 2006). To further evaluate the relationship between metabolism and TPI deficiency behavioral and longevity phenotypes we measured phosphorylated arginine [P-arg] to arginine [arg] concentration ratios as previously described (Viant et al., 2001), as this is an extremely sensitive assay of Drosophila bioenergetic status (Celotto et al., 2011). [P-arg]/[arg] ratios in TPIsgk mutant animals were depressed compared to wild type, consistent with a strong loss-of-function mutation in glycolysis (Fig. 3B). This represents a reduction in the available pool of [P-arg] normalized to the total [arg] available, and suggests that the animals are utilizing this pool at a higher rate to buffer their ATP levels, similar to how mammals utilize phosphocreatine (Celotto et al., 2011). Importantly, TPIsgk/TPIK11M animals exhibiting low levels of catalytic activity also showed a similar depression in their [P-arg]/[arg] ratios compared to wild type animals (Fig. 3B). These data corroborate the lysate catalysis results and collectively indicate that TPIK11M complements TPIsgk phenotypes by a mechanism independent of the well-established role of TPI in cellular bioenergetics, and may provide an explanation as to the absence of an association between enzyme catalysis and neurological complications in human patients.
TPIsgk and TPIK11M protein levels
The capacity of a catalytically inactive allele of TPI to genetically complement all behavioral and longevity phenotypes independent of isomerase activity and animal metabolism strongly supports the conclusion that TPIsgk phenotypes are due to a change in total TPI protein levels, not a reduction in TPI catalytic capacity. To determine whether the catalytically inactive TPI rescues total TPI levels, we immunoblotted for the presence of TPI with or without a cyan fluorescent protein (CFP) tag; the use of the tag allowed us to discriminate between protein isoforms. The addition of this C-terminal CFP does not influence the capacity of TPIK11M to complement TPIsgk phenotypes (supplementary material Fig. S4). TPIsgk animals display reduced TPI protein levels in vivo (Seigle et al., 2008; Hrizo and Palladino, 2010), and TPI levels were further reduced when TPIsgk was paired with the TPIJS10 null allele (Fig. 4A,C). Interestingly, complementing TPIsgk with the catalytically inactive allele TPIK11M rescues total TPI protein levels to that seen in the aphenotypic TPI+/TPI+ wild type homozygotes. Observing TPIK11M-CFP and TPIsgk independently, it can be seen that TPIK11M-CFP fails to change TPIsgk levels relative to TPIsgk/TPIJS10 or TPIsgk/TPIsgk (Fig. 4A,C) and, conversely, mutant TPIsgk protein does not induce the degradation of TPIK11M-CFP relative to TPI+/TPIK11M-CFP (Fig. 4B,C).
Fig. 4.
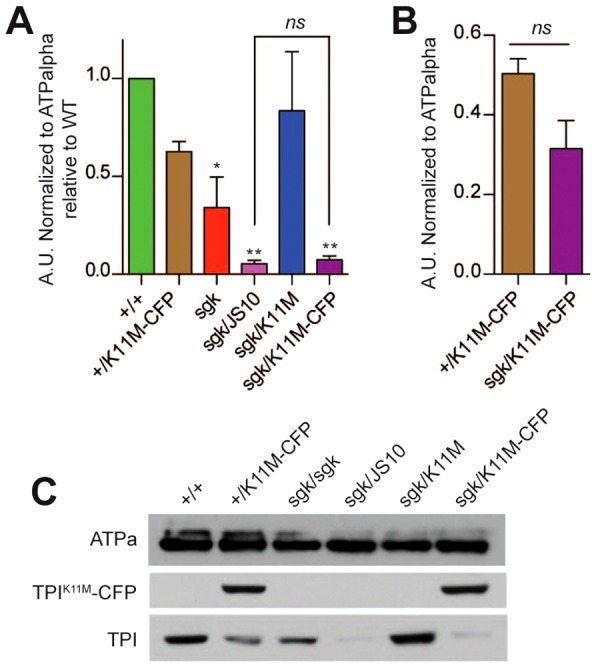
TPIK11M expression does not prevent the degradation of TPIsgk. (A) Quantification of the levels of untagged TPI. TPIsgk complemented with TPIK11M shows elevated levels of total TPI similar to WT. Additionally, TPIsgk/TPIJS10 has similar levels of TPI to TPIsgk/TPIK11M-CFP. **P<0.01 and ***P<0.001 compared to WT; ns indicates no significant change compared with TPIsgk/TPIJS10. n = 4. (B) TPIsgk does not induce the degradation of TPIK11M-CFP. Quantification and comparison of the levels of TPIK11M-CFP (shown in C). Student's t-test was used to compare the two groups and found no significant difference (ns). n = 4. Error bars indicate ± s.e.m. (C) A representative blot from which the data in A,B were obtained. The loading control used was ATPalpha (Na,K-ATPase).
The inability of TPIK11M-CFP to modulate TPIsgk levels suggests that any phenotypic rescue is likely being performed by the catalytically inactive isoform, and not merely increasing the presence of a still catalytically active TPIsgk. Secondly, it is of particular interest that TPIsgk does not significantly influence the levels of TPIK11M-CFP. These results suggest three possible interpretations: (1) TPIsgk is degraded prior to but stable upon heterodimerization; (2) TPIsgk monomers are selectively degraded from heterodimer complexes; or (3) that TPIsgk and TPIK11M never exist as a heterodimer. Taken together, these results confirm that TPIK11M complements the TPIsgk phenotypes independent of isomerase activity demonstrating a non-catalytic function of TPI crucial to the pathogenesis and severity of TPI deficiency.
Discussion
The sugarkill model of TPI deficiency used in these experiments is characterized by reduced catalysis and protein stability, characteristics found in several of the more toxic human TPI alleles (Hollán et al., 1993; Watanabe et al., 1996; Arya et al., 1997). Similarly, the M80T mutation recapitulates the neurological dysfunction and toxicity exhibited in many of the human patients. Using Drosophila we have genomically engineered (GE) the TPI locus to make targeted in vivo modifications to the endogenous gene. GE allows us to study TPI manipulations under endogenous regulatory control, allowing rigorous experiments that would not be feasible with traditional transgenic approaches (Huang et al., 2011; Pollarolo et al., 2011; Venken et al., 2011). The power of GE is born in its capacity to study a gene's function in vivo while reducing artifacts due to transgenic overexpression and miss expression. Previous studies analyzing TPI deficiency have primarily utilized human patient erythrocytes, yeast and mammalian cellular expression systems. Although these experimental paradigms are valuable, they do not provide the elegant endogenous genetic control of GE and preclude the examination of in vivo phenotypes such as behavior and longevity.
We initiated this study asking whether the presence of the enzyme or its catalytic activity was most important to the pathogenesis of our mutant. Our experiments assessing enzyme activity identified that the M80T substitution in TPIsgk resulted in a ∼11-fold reduction in enzyme catalysis with modest changes in substrate affinity (Table 1). This reduction in catalytic activity was more severe than those reported in two human mutations (Orosz et al., 2001; Rodríguez-Almazán et al., 2008), but was not nearly as severe as those modifying crucial catalytic components (Raines et al., 1986; Nickbarg et al., 1988; Casteleijn et al., 2006; Go et al., 2010). An analysis of the TPI enzyme structure taken from G. gallus suggested that this mutation might affect isomerase activity through its proximity to several catalytic residues (Fig. 1). These results initially supported a catalytic mechanism of disease pathogenesis, but further experimentation revealed the capacity for an isomerase-inactive isoform of TPI to genetically complement all behavioral and longevity phenotypes of TPIsgk. The capacity for a catalytically inactive TPI to rescue a severe variant of TPI deficiency without a change in isomerase activity was striking. Most research on TPI deficiency focuses on two core principles: reduced TPI activity slows glycolysis, and inhibition of TPI leads to a buildup of excess DHAP and henceforth toxic advanced glycation end-products (AGEs) (Orosz et al., 2009). Here we have shown the capacity to rescue animal behavior and longevity without restoring TPI catalysis, making it unlikely that AGEs are playing a significant role in the genesis of our behavioral or longevity phenotypes. Further, the metabolic change detected in our mutant was also not rescued upon attenuation of behavior. These data suggest a new model of TPI function and we propose that this non-catalytic function is crucial to the neurological complications that contribute to TPI deficiency pathogenesis (Fig. 5).
Fig. 5.
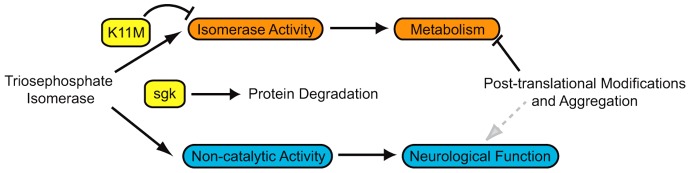
Model of TPI function and pathogenesis. The results of this study suggest that TPI has catalytic (orange) and non-catalytic (blue) functions. Impairment of TPI function (yellow) was studied in vivo using K11M (abolishes catalytic activity) and sgk (leads to protein degradation). The data demonstrate the presence of a non-isomerase TPI function required for normal neural function and suggests that loss of this function underlies neuropathogenesis in TPI deficiency.
Although glycolytic enzymes are not often thought of as important players in non-metabolic cellular mechanisms, work spanning the past two decades has clearly outlined vital non-metabolic roles for some of these ancient proteins. Glycolytic enzymes have been described in a multitude of different non-catalytic roles, including assistance in assembly and function of the vacuolar-type proton-ATPase by aldolase (Lu et al., 2004; Lu et al., 2007), inhibition of apoptosis through the modulation of Bax, Bak and Bad by hexokinase (Pastorino et al., 2002; Majewski et al., 2004), transcriptional regulation of the histone H2B gene during cell cycle progression by glyceraldehyde 3-phosphate dehydrogenase (Zheng et al., 2003), and the induction of cell motility and invasion through the secretion and binding of glucose 6-phosphate isomerase to the autocrine motility factor receptor gp78 (Niinaka et al., 1998). The potential for glycolytic enzymes to perform crucial functions dramatically independent from their roles in glycolysis is evident (reviewed by Kim and Dang, 2005). Our findings are particularly relevant as TPI is one of the primary targets of nitrotyrosination in Alzheimer's disease and is sequestered in the neurofibrillary tangles in patient brains (Coma et al., 2005; Guix et al., 2009). Sequestration such as this could inhibit a crucial non-catalytic function of TPI, further contributing to the neurological dysfunction known to be associated with tau aggregation and toxic amyloid beta.
Recently TPI has been identified as a target of cyclin dependent kinase 2 (cdk2) (Lee et al., 2010) and arginine methyltransferase 5 (PRMT5) (Kim et al., 2010). Together, these studies conclude that TPI is more highly regulated than previously appreciated. Furthermore, it was noted that TPI protein levels could be modulated via methylation (Kim et al., 2010). These results portend the enticing possibility that TPI protein levels are regulated in a cell-cycle-dependent manner, and that its relationship to the cell cycle could be perturbed in TPI-deficient patients experiencing neurological symptoms. Indeed, a relationship has already been well established between cell cycle misregulation and neurodegeneration (reviewed by Khurana and Feany, 2007), and inappropriate regulation of the cell cycle has been described in Alzheimer's disease (Yang et al., 2001). It is clear that further work is needed to define the biological role of post-translational modifications of TPI, and the role of TPI as either a direct or indirect component of the cell cycle.
In conclusion, our data demonstrate the capacity of a catalytically inactive TPI enzyme to genetically complement TPI deficiency behavioral phenotypes independent of changes in bioenergetics or enzyme catalysis. The identification of an isomerase-independent function for this crucial protein opens up new avenues of investigation that will prove crucial to understanding the role of TPI in maintaining normal neural function and TPI deficiency pathogenesis.
Materials and Methods
Animal strains
The Drosophila w*; P{GawB} 477w-; TM2/TM6B, Tb1 and y,w/Y, hs-hid; hs-FLP, hs-I-SceI/CyO, hs-hid; stocks were obtained from Dr Yang Hong. TPIsgk is a missense mutation resulting in a Met-to-Thr change at position 80, while TPIJS10 is a null allele owing to a 1.6 kb deletion of two of the three constitutively expressed exons of the TPI locus (Celotto et al., 2006). This numbering uses the established nomenclature for TPI mutations, assuming the start methionine is removed following translation (Orosz et al., 2006); and for consistency all residue numbering in this study uses the same convention. The Cre recombinase stock was used to reduce the locus following homologous recombination and phiC31 integration. The strain used, y1 w67c23 P{y[+mDint2] = Crey}1b; D*/TM3, Sb1, was obtained from the Bloomington Drosophila Stock Center (Bloomington, IN, USA). Care was taken to ensure all animal populations assessed were approximately equivalent mixtures of males and females, with the exception of the TPIsgk/TPIJS10 day 20 behavioral tests – in this genotype males died noticeably faster than females and as a result this small population of survivors consisted mostly of female animals.
Genomic engineering
We performed the TPI GE similar to previously published methods (Huang et al., 2008; Huang et al., 2009). Briefly, ∼2.6 kb homology arms were generated by PCR amplification to areas 5′ and 3′ of the TPI locus. These homology arms were inserted into the pGX-attB vector (Huang et al., 2009) and transferred onto the second chromosome of w1118 animals using traditional P-element transgenesis. Ends-out homologous recombination was performed and founder lines were molecularly verified. The wild type TPI locus was cloned into the pGE-attB vector via PCR and verified by sequencing. Founder lines were mated to vasa-phiC31ZH-2A animals expressing the integrase on the X chromosome and their progeny injected with pGE-attBTPI+, pGE-attBTPI+-CFP, pGE-attBTPIM80T, pGE-attBTPIK11M, or pGE-attBTPIK11M-CFP constructs. Integration events between attP and attB elements produce an attL and attR and reconstitute the target locus. Such events were identified based on the presence of the w+ phenotype and verified molecularly.
Mutagenesis
Site directed mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Mutagenesis primers were generated (Integrated DNA Technologies, Coralville, IA, USA) to introduce a Lys-to-Met codon change affecting position 11 and a Met-to-Thr codon change affecting position 80 of the protein. Mutagenesis was performed on the pGE-attBTPI+ plasmid and confirmed by sequencing.
Drosophila TPI purification
The coding sequence for Drosophila TPI was cloned into the bacterial expression vector pLC3 using standard techniques. The resulting plasmid directs expression of TPI containing N-terminal His6- and MBP tags, both of which can be removed with TEV protease. TPI protein was expressed in BL21(DE3) Codon-Plus (RILP) E. coli (Agilent Technologies) grown in ZY auto-induction media (Studier, 2005) at room temperature for 24–30 hours. Cells were harvested by centrifugation, lysed via homogenization in 25 mM Tris pH 8.0, 500 mM NaCl, 10% glycerol, 5 mM imidazole, 1 mM β-mercaptoethanol and cleared by centrifugation at 30,000 g. TPI was purified by nickel affinity chromatography followed by overnight TEV protease treatment to cleave the His6-MBP tag from TPI. A second round of nickel affinity purification was performed to separate the His6-MBP and TEV protease. TPI protein was further purified using cation-exchange chromatography (HiTrap-QP) followed by gel filtration (Sephacryl S-200, GE Healthcare, Little Chalfont, England, UK). Peak fractions were concentrated to 4–8 mg/ml in 20 mM Tris pH 8.8, 25 mM NaCl, 2.0% glycerol and 1 mM β-mercaptoethanol using a Vivaspin concentrator (GE Healthcare). The purity was >99% as verified by SDS-PAGE. Expression and purification of the Drosophila TPI M80T sgk mutant was performed as described above.
Enzyme assays
Isomerase activity was determined using an NADH-linked assay as previously detailed (Williams et al., 1999). TPI activity was measured with three different enzyme concentrations of both purified Drosophila WT and M80T enzyme. Initial velocity of the enzyme was calculated over a GAP (Sigma-Aldrich, St. Louis, MO, USA) range of 0.94–5.64 mM. All kinetic measurements were performed three times in triplicate by monitoring the absorbance of NADH at 340 nm in a SpectraMax Plus 384 microplate reader (Molecular Devices, Sunnyvale, CA, USA). The assay was performed using 80 µl mixtures containing varied GAP and enzyme concentrations, 0.42 mM NADH (Sigma-Aldrich), and 1 unit glycerol-3-phosphate dehydrogenase (Sigma-Aldrich) in 100 mM triethanolamine (TEA), pH 7.6. Enzyme activity curves were normalized to reactions performed without GAP. Enzyme kinetics were determined by assessing initial velocities taken during the linear phase of each reaction, and the data were fit to the Michaelis–Menten equation using nonlinear regression in Graphpad Prism 5.0b (GraphPad Software, La Jolla, CA, USA).
For lysate assays, animals were collected and aged to day 3 at 25°C and frozen in liquid nitrogen. Bodies lacking the head and appendages (abdomen and thorax) were isolated after vigorous mechanical shaking of the frozen animals. Tissues lacking eye pigments were used to reduce background absorbance. The bodies were homogenized in 0.75 ml of 1× PBS (2.7 mM KCl, 137 mM NaCl, 2 mM NaH2PO4, 10 mM Na2HPO4 pH 7.4) with protease inhibitors leupeptin 1∶1000 (Acros Organics, Geel, Belgium), pepstatin 1∶1000 (Sigma-Aldrich), PMSF 1∶100 (Pierce, Rockford, IL, USA). The homogenates were ice bath-sonicated for 10 minutes then centrifuged at 4°C for 5 minute at 5,000 g to remove exoskeletal debris. Lysates were diluted to 1 µg/µl in 100 mM TEA, pH 7.6 + inhibitors and TPI enzyme activity was assessed using a linked-enzyme assay, similar to that outlined above. The assay was performed using 80 µl mixtures containing 0.47 mM GAP, 0.42 mM NADH, 1 unit glycerol-3-phosphate dehydrogenase and 30 µg of lysate protein in 100 mM TEA, pH 7.6. Enzyme activity curves were performed three times in triplicate and normalized to reactions performed without GAP. A one-way ANOVA was performed to assess variance and data sets were compared using Tukey's post-hoc analysis.
Behavioral testing and lifespan analysis
Mechanical stress sensitivity was determined by vortexing the animals in a standard media vial for 20 seconds and measuring time to recovery, similar to (Ganetzky and Wu, 1982). Thermal stress sensitivity was assessed by acutely shifting animals to 38°C and measuring time to paralysis, as previously described (Palladino et al., 2002; Palladino et al., 2003). All behavioral responses were capped at 600 seconds. Testing days were selected based on previous experience with TPI deficiency progression in Drosophila (Celotto et al., 2006). Animal lifespans were performed at 25°C as previously described (Palladino et al., 2003). Two-way ANOVAs were performed with Bonferroni's post test to compare genotype behavior over time, and lifespans were compared with Log-rank (Mantel–Cox) survival tests.
HPLC phosphoarginine assay
Three sets of 25 animals per genotype were collected and aged to day 3 at 25°C and frozen in liquid nitrogen. The animals were homogenized in 200 µl 0.6 M perchloric acid and neutralized with 25 µl of 2 M potassium carbonate. The lysates were then centrifuged at 12,000 g for 10 minutes at 4°C and the supernatant filtered through a PVDF 0.45 µm spin column (National Scientific, Rockwood, TN, USA) at 12,000 g for 5 minutes at 4°C as previously described (Celotto et al., 2011). These extracts were injected onto a Phenomenex Luna 5 µm NH2 250×4.6 mm column (Torrance, CA, USA) using a Shimadzu high performance liquid chromatography (HPLC) system (Kyoto, Japan) and separated with a 95∶5 20 mM KH2PO4 pH 2.6: acetonitrile linear mobile phase at a flow rate of 0.6 ml/minute. Arginine (Acros Organics) and phospho-arginine (P-arg) standards were detected at 205 nm, and all samples were measured in triplicate (Celotto et al., 2011). Phospho-arginine standards were generated as previously described (Morrison et al., 1957). Peak analysis was performed using EZStart 7.3 software (Shimadzu), and P-arg levels were normalized to total arginine and compared across genotypes. Retention times for arginine and phospho-arginine were 3.7 minutes and 5.4 minutes, respectively. A one-way ANOVA was performed to assess variance and data sets were compared using Tukey's post-hoc analysis.
Immunoblots
Animals were collected and aged at 29°C. After 24 hours, ten fly heads were collected in triplicate from each genotype and processed as outlined previously (Hrizo and Palladino, 2010). Briefly, the fly heads were ground by pestle in 80 µl 2× SDS–PAGE sample buffer (4% SDS, 4% β-mercaptoethanol, 130 mM Tris–HCl pH 6.8, 20% glycerol) and centrifuged for 5 minutes at 5000 g to pellet the exoskeleton. Proteins were resolved by SDS–PAGE and transferred onto PVDF membrane. Following treatment in 1% milk PBST, the blots were incubated with anti-TPI (1∶5000; rabbit polyclonal FL-249; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-ATPalpha (1∶10,000; mouse monoclonal alpha5; Developmental Studies Hybridoma Bank, Iowa City, IA, USA). The blots were washed in PBST, incubated in the appropriate HRP-conjugated secondary antibody, and developed using ECL (Pierce). Densitometric analyses of the scanned films were performed digitally using ImageJ software available from the National Institutes of Health. A one-way ANOVA was performed to assess variance of TPI levels and data sets were compared using Tukey's post-hoc analysis.
Supplementary Material
Acknowledgments
We thank Dr Yang Hong for fly strains and clones to perform the genomic engineering; the Bloomington Stock center for fly strains; Samula Mula, Henry Liu, Mallory Hensel, Matt Baumgartner, Brian Kosik, Krupa Patel and Alicia Olsen for assistance with the genomic engineering.
Footnotes
Author contributions
B.P.R., K.A.S., A.M.C., and M.J.P. designed the research; B.P.R., K.A.S., S.B.L., and A.M.C. performed the research; C.G.A. and A.P.V. contributed new reagents, B.P.R., C.G.A., A.P.V. and M.J.P. analyzed the data, and B.P.R. and M.J.P. wrote the paper.
Funding
This work was supported by a fellowship from Achievement Rewards for College Scientists: Pittsburgh Chapter [to B.P.R.]; and the National Institutes of Health [grant numbers R01AG027453, R01AG025046, R01GM103369 to M.J.P., R01GM097204 to A.P.V. and T32GM8424-17 to B.P.R.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.124586/-/DC1
References
- Arya R., Lalloz M. R., Bellingham A. J., Layton D. M. (1997). Evidence for founder effect of the Glu104Asp substitution and identification of new mutations in triosephosphate isomerase deficiency. Hum. Mutat. 10, 290–294 [DOI] [PubMed] [Google Scholar]
- Casteleijn M. G., Alahuhta M., Groebel K., El-Sayed I., Augustyns K., Lambeir A. M., Neubauer P., Wierenga R. K. (2006). Functional role of the conserved active site proline of triosephosphate isomerase. Biochemistry 45, 15483–15494 10.1021/bi061683j [DOI] [PubMed] [Google Scholar]
- Celotto A. M., Frank A. C., Seigle J. L., Palladino M. J. (2006). Drosophila model of human inherited triosephosphate isomerase deficiency glycolytic enzymopathy. Genetics 174, 1237–1246 10.1534/genetics.106.063206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotto A. M., Chiu W. K., Van Voorhies W., Palladino M. J. (2011). Modes of metabolic compensation during mitochondrial disease using the Drosophila model of ATP6 dysfunction. PLoS ONE 6, e25823 10.1371/journal.pone.0025823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coma M., Guix F. X., Uribesalgo I., Espuña G., Solé M., Andreu D., Muñoz F. J. (2005). Lack of oestrogen protection in amyloid-mediated endothelial damage due to protein nitrotyrosination. Brain 128, 1613–1621 10.1093/brain/awh492 [DOI] [PubMed] [Google Scholar]
- Daar I. O., Artymiuk P. J., Phillips D. C., Maquat L. E. (1986). Human triose-phosphate isomerase deficiency: a single amino acid substitution results in a thermolabile enzyme. Proc. Natl. Acad. Sci. USA 83, 7903–7907 10.1073/pnas.83.20.7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B., Wu C. F. (1982). Indirect suppression involving behavioral mutants with altered nerve excitability in Drosophila melanogaster. Genetics 100, 597–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerer J. P., Kreber R. A., Ganetzky B. (2006). wasted away, a Drosophila mutation in triosephosphate isomerase, causes paralysis, neurodegeneration, and early death. Proc. Natl. Acad. Sci. USA 103, 14987–14993 10.1073/pnas.0606887103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K., Koudelka A., Amyes T. L., Richard J. P. (2010). Role of Lys-12 in catalysis by triosephosphate isomerase: a two-part substrate approach. Biochemistry 49, 5377–5389 10.1021/bi100538b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W. J., Golic K. G. (2003). Ends-out, or replacement, gene targeting in Drosophila. Proc. Natl. Acad. Sci. USA 100, 2556–2561 10.1073/pnas.0535280100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guix F. X., Ill-Raga G., Bravo R., Nakaya T., de Fabritiis G., Coma M., Miscione G. P., Villà-Freixa J., Suzuki T., Fernàndez-Busquets X. et al. (2009). Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 132, 1335–1345 10.1093/brain/awp023 [DOI] [PubMed] [Google Scholar]
- Hollán S., Fujii H., Hirono A., Hirono K., Karro H., Miwa S., Harsányi V., Gyódi E., Inselt-Kovács M. (1993). Hereditary triosephosphate isomerase (TPI) deficiency: two severely affected brothers one with and one without neurological symptoms. Hum. Genet. 92, 486–490 10.1007/BF00216456 [DOI] [PubMed] [Google Scholar]
- Hrizo S. L., Palladino M. J. (2010). Hsp70- and Hsp90-mediated proteasomal degradation underlies TPI sugarkill pathogenesis in Drosophila. Neurobiol. Dis. 40, 676–683 10.1016/j.nbd.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Zhou W., Watson A. M., Jan Y. N., Hong Y. (2008). Efficient ends-out gene targeting in Drosophila. Genetics 180, 703–707 10.1534/genetics.108.090563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Zhou W., Dong W., Watson A. M., Hong Y. (2009). From the Cover: Directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc. Natl. Acad. Sci. USA 106, 8284–8289 10.1073/pnas.0900641106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Huang L., Chen Y. J., Austin E., Devor C. E., Roegiers F., Hong Y. (2011). Differential regulation of adherens junction dynamics during apical-basal polarization. J. Cell Sci. 124, 4001–4013 10.1242/jcs.086694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V., Feany M. B. (2007). Connecting cell-cycle activation to neurodegeneration in Drosophila. Biochim. Biophys. Acta 1772, 446–456 10.1016/j.bbadis.2006.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. W., Dang C. V. (2005). Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142–150 10.1016/j.tibs.2005.01.005 [DOI] [PubMed] [Google Scholar]
- Kim C., Lim Y., Yoo B. C., Won N. H., Kim S., Kim G. (2010). Regulation of post-translational protein arginine methylation during HeLa cell cycle. Biochim. Biophys. Acta 1800, 977–985 10.1016/j.bbagen.2010.06.004 [DOI] [PubMed] [Google Scholar]
- Lambeir A. M., Opperdoes F. R., Wierenga R. K. (1987). Kinetic properties of triose-phosphate isomerase from Trypanosoma brucei brucei. A comparison with the rabbit muscle and yeast enzymes. Eur. J. Biochem. 168, 69–74 10.1111/j.1432-1033.1987.tb13388.x [DOI] [PubMed] [Google Scholar]
- Lee W. H., Choi J. S., Byun M. R., Koo K. T., Shin S., Lee S. K., Surh Y. J. (2010). Functional inactivation of triosephosphate isomerase through phosphorylation during etoposide-induced apoptosis in HeLa cells: potential role of Cdk2. Toxicology 278, 224–228 10.1016/j.tox.2010.02.005 [DOI] [PubMed] [Google Scholar]
- Lodi P. J., Chang L. C., Knowles J. R., Komives E. A. (1994). Triosephosphate isomerase requires a positively charged active site: the role of lysine-12. Biochemistry 33, 2809–2814 10.1021/bi00176a009 [DOI] [PubMed] [Google Scholar]
- Lu M., Sautin Y. Y., Holliday L. S., Gluck S. L. (2004). The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar H+-ATPase. J. Biol. Chem. 279, 8732–8739 10.1074/jbc.M303871200 [DOI] [PubMed] [Google Scholar]
- Lu M., Ammar D., Ives H., Albrecht F., Gluck S. L. (2007). Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 282, 24495–24503 10.1074/jbc.M702598200 [DOI] [PubMed] [Google Scholar]
- Majewski N., Nogueira V., Bhaskar P., Coy P. E., Skeen J. E., Gottlob K., Chandel N. S., Thompson C. B., Robey R. B., Hay N. (2004). Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell 16, 819–830 10.1016/j.molcel.2004.11.014 [DOI] [PubMed] [Google Scholar]
- Morrison J. F., Griffiths D. E., Ennor A. H. (1957). The purification and properties of arginine phosphokinase. Biochem. J. 65, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickbarg E. B., Davenport R. C., Petsko G. A., Knowles J. R. (1988). Triosephosphate isomerase: removal of a putatively electrophilic histidine residue results in a subtle change in catalytic mechanism. Biochemistry 27, 5948–5960 10.1021/bi00416a019 [DOI] [PubMed] [Google Scholar]
- Niinaka Y., Paku S., Haga A., Watanabe H., Raz A. (1998). Expression and secretion of neuroleukin/phosphohexose isomerase/maturation factor as autocrine motility factor by tumor cells. Cancer Res. 58, 2667–2674 [PubMed] [Google Scholar]
- Norledge B. V., Lambeir A. M., Abagyan R. A., Rottmann A., Fernandez A. M., Filimonov V. V., Peter M. G., Wierenga R. K. (2001). Modeling, mutagenesis, and structural studies on the fully conserved phosphate-binding loop (loop 8) of triosephosphate isomerase: toward a new substrate specificity. Proteins 42, 383–389 [DOI] [PubMed] [Google Scholar]
- Orosz F., Oláh J., Alvarez M., Keseru G. M., Szabó B., Wágner G., Kovári Z., Horányi M., Baróti K., Martial J. A. et al. (2001). Distinct behavior of mutant triosephosphate isomerase in hemolysate and in isolated form: molecular basis of enzyme deficiency. Blood 98, 3106–3112 10.1182/blood.V98.10.3106 [DOI] [PubMed] [Google Scholar]
- Orosz F., Oláh J., Ovádi J. (2006). Triosephosphate isomerase deficiency: facts and doubts. IUBMB Life 58, 703–715 10.1080/15216540601115960 [DOI] [PubMed] [Google Scholar]
- Orosz F., Oláh J., Ovádi J. (2009). Triosephosphate isomerase deficiency: new insights into an enigmatic disease. Biochim. Biophys. Acta 1792, 1168–1174 10.1016/j.bbadis.2009.09.012 [DOI] [PubMed] [Google Scholar]
- Palladino M. J., Hadley T. J., Ganetzky B. (2002). Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics 161, 1197–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino M. J., Bower J. E., Kreber R., Ganetzky B. (2003). Neural dysfunction and neurodegeneration in Drosophila Na+/K+ ATPase alpha subunit mutants. J. Neurosci. 23, 1276–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino J. G., Shulga N., Hoek J. B. (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618 10.1074/jbc.M109950200 [DOI] [PubMed] [Google Scholar]
- Pollarolo G., Schulz J. G., Munck S., Dotti C. G. (2011). Cytokinesis remnants define first neuronal asymmetry in vivo. Nat. Neurosci. 14, 1525–1533 10.1038/nn.2976 [DOI] [PubMed] [Google Scholar]
- Raines R. T., Sutton E. L., Straus D. R., Gilbert W., Knowles J. R. (1986). Reaction energetics of a mutant triosephosphate isomerase in which the active-site glutamate has been changed to aspartate. Biochemistry 25, 7142–7154 10.1021/bi00370a057 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Almazán C., Arreola R., Rodríguez-Larrea D., Aguirre-López B., de Gómez-Puyou M. T., Pérez-Montfort R., Costas M., Gómez-Puyou A., Torres-Larios A. (2008). Structural basis of human triosephosphate isomerase deficiency: mutation E104D is related to alterations of a conserved water network at the dimer interface. J. Biol. Chem. 283, 23254–23263 10.1074/jbc.M802145200 [DOI] [PubMed] [Google Scholar]
- Rong Y. S., Golic K. G. (2000). Gene targeting by homologous recombination in Drosophila. Science 288, 2013–2018 10.1126/science.288.5473.2013 [DOI] [PubMed] [Google Scholar]
- Schneider A. S. (2000). Triosephosphate isomerase deficiency: historical perspectives and molecular aspects. Baillieres Best Pract. Res. Clin. Haematol. 13, 119–140 10.1053/beha.2000.0061 [DOI] [PubMed] [Google Scholar]
- Seigle J. L., Celotto A. M., Palladino M. J. (2008). Degradation of functional triose phosphate isomerase protein underlies sugarkill pathology. Genetics 179, 855–862 10.1534/genetics.108.087551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- Venken K. J., Simpson J. H., Bellen H. J. (2011). Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron 72, 202–230 10.1016/j.neuron.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant M. R., Rosenblum E. S., Tjeerdema R. S. (2001). Optimized method for the determination of phosphoarginine in abalone tissue by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 765, 107–111 10.1016/S0378-4347(01)00428-5 [DOI] [PubMed] [Google Scholar]
- Watanabe M., Zingg B. C., Mohrenweiser H. W. (1996). Molecular analysis of a series of alleles in humans with reduced activity at the triosephosphate isomerase locus. Am. J. Hum. Genet. 58, 308–316 [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K., Kapetaniou E. G., Venkatesan R. (2010). Triosephosphate isomerase: a highly evolved biocatalyst. Cell. Mol. Life Sci. 67, 3961–3982 10.1007/s00018-010-0473-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. C., Zeelen J. P., Neubauer G., Vriend G., Backmann J., Michels P. A., Lambeir A. M., Wierenga R. K. (1999). Structural and mutagenesis studies of leishmania triosephosphate isomerase: a point mutation can convert a mesophilic enzyme into a superstable enzyme without losing catalytic power. Protein Eng. 12, 243–250 10.1093/protein/12.3.243 [DOI] [PubMed] [Google Scholar]
- Yang Y., Geldmacher D. S., Herrup K. (2001). DNA replication precedes neuronal cell death in Alzheimer's disease. J. Neurosci. 21, 2661–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Sugio S., Komives E. A., Liu K. D., Knowles J. R., Petsko G. A., Ringe D. (1994). Crystal structure of recombinant chicken triosephosphate isomerase-phosphoglycolohydroxamate complex at 1.8-A resolution. Biochemistry 33, 2830–2837 10.1021/bi00176a012 [DOI] [PubMed] [Google Scholar]
- Zheng L., Roeder R. G., Luo Y. (2003). S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114, 255–266 10.1016/S0092-8674(03)00552-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.