

Summary
The ubiquitously expressed glucocorticoid receptor (GR) is a major drug target for inflammatory disease, but issues of specificity and target tissue sensitivity remain. We now identify high potency, non-steroidal GR ligands, GSK47867A and GSK47869A, which induce a novel conformation of the GR ligand-binding domain (LBD) and augment the efficacy of cellular action. Despite their high potency, GSK47867A and GSK47869A both induce surprisingly slow GR nuclear translocation, followed by prolonged nuclear GR retention, and transcriptional activity following washout. We reveal that GSK47867A and GSK47869A specifically alter the GR LBD structure at the HSP90-binding site. The alteration in the HSP90-binding site was accompanied by resistance to HSP90 antagonism, with persisting transactivation seen after geldanamycin treatment. Taken together, our studies reveal a new mechanism governing GR intracellular trafficking regulated by ligand binding that relies on a specific surface charge patch within the LBD. This conformational change permits extended GR action, probably because of altered GR–HSP90 interaction. This chemical series may offer anti-inflammatory drugs with prolonged duration of action due to altered pharmacodynamics rather than altered pharmacokinetics.
Key words: Nuclear receptor, Glucocorticoid, GR, Heat shock protein 90, Crystal structure, Subcellular trafficking
Introduction
Synthetic glucocorticoids are potent anti-inflammatory drugs used to treat multiple conditions including asthma and rheumatoid arthritis (Schett et al., 2008; Krishnan et al., 2009). Unfortunately glucocorticoid treatment also carries a wide range of serious side effects including hyperglycaemia and osteoporosis (Canalis et al., 2002). In recent years a significant effort has been made to design dissociative ligands with the anti-inflammatory potency of conventional glucocorticoids, but with a reduced spectrum of side-effects (Lin et al., 2002; Bledsoe et al., 2004; Cerasoli, 2006; Wang et al., 2006; McMaster and Ray, 2007; McMaster and Ray, 2008; van Lierop et al., 2012).
Glucocorticoid actions are mediated by the ubiquitously expressed glucocorticoid receptor (GR; NR3C1) a member of the nuclear hormone receptor superfamily with a conserved modular structure consisting of an N-terminal regulatory domain, a DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD) (Hollenberg et al., 1985; Encío and Detera-Wadleigh, 1991). The unliganded GR resides in the cytoplasm in a complex with heat-shock proteins and immunophilins (Grad and Picard, 2007). Ligand binding triggers rapid activation of cytosolic kinase signalling cascades and concomitantly results in exposure of two nuclear localisation signals (NLS1, and NLS2) enabling nuclear import (Picard and Yamamoto, 1987). This is accompanied by replacement of the immunophilin FKBP51 with FKBP52 (Davies et al., 2002) which associates with dynein to drive GR along microtubules (Czar et al., 1994; Harrell et al., 2004). The process of translocation to the nucleus post ligand binding occurs rapidly, with the majority of cellular GR being nuclear 30 minutes after treatment with 100 nM Dex (Nishi et al., 1999). In addition cell cycle phase is able to regulate the subcellular localisation of unliganded GR, but with far slower kinetics of nuclear accumulation (Matthews et al., 2011). In the nucleus GR binds to cis-elements to activate or repress target gene expression, recruiting co-modulator proteins from distinct classes to effect chromatin remodelling, and recruitment of the basal transcriptional machinery (Ford et al., 1997; Jones and Shi, 2003; Ito et al., 2006; Johnson et al., 2008).
GR recruits co-modulator proteins via its transcriptional activation function domains (AF1, and AF2) (Wärnmark et al., 2000; Kumar et al., 2001; Bledsoe et al., 2002). The GR AF1 is the site of various post translational modifications including phosphorylation, both in the presence and absence of ligand. (Wang et al., 2002; Ismaili and Garabedian, 2004; Galliher-Beckley et al., 2008). Phosphorylation directs GR function by impacting protein stability and recruitment of specific co-modulator proteins such as MED14 (Chen et al., 2006; Chen et al., 2008). In addition, co-modulators bind to the GR AF2 domain, within the LBD (Heery et al., 1997). Structural information about bound ligand is transmitted through differential folding of the LBD, which directs GR function by offering differentially attractive signals for co-modulator recruitment. Both GR agonists and antagonists provoke similar rapid kinetics of nuclear translocation, but differ in the profile of co-modulator proteins recruitment, providing a mechanism for their different modes of action (Bledsoe et al., 2002; Kauppi et al., 2003; Stevens et al., 2003).
Here we identify a novel switch mechanism that regulates GR trafficking in response to ligand binding, distinct from an effect attributable to ligand potency. We identify two novel, non-steroidal GR ligands that regulate the GR surface to greatly reduce rates of nuclear translocation and reduce reliance on heat-shock protein for continuing activity. The difference in GR conformation induced by the novel GR ligands reveals a patch of positive charge on the surface of the LBD. We propose that this prevents efficient engagement with the active nuclear translocation mechanism, subsequent export, and protein degradation mechanisms for the GR. The result is generation of ligands with greatly prolonged duration of action as a consequence of altered pharmacodynamics rather than pharmacokinetics.
Results
GSK47867A and GSK47869A are highly potent GR agonists
There is wide interest in understanding how variation in ligand structure (Fig. 1A) affects the function of GR. Here, we use novel, non-steroidal glucocorticoid receptor ligands (NSG) with very high potency, and specificity for GR to determine how ligand structure impacts receptor function (Fig. 1B,C; supplementary material Fig. S1). Transient GR transactivation and transrepression models in HeLa cells were used initially to compare the NSGs to conventional synthetic glucocorticoid ligands. We find that both GSK47867A and GSK47869A were ∼30 times more potent that Dexamethasone (Dex, Fig. 1B,C; Table 1). Similar results were also obtained using A549 cells with stably integrated GRE-Luc or NFκB-Luc templates (supplementary material Fig. S2A,B). The steroidal glucocorticoid Fluticasone Propionate (FP) had similar potency to GSK47867A and GSK47869A. Hydrocortisone was significantly less potent than all the synthetic ligands tested (Fig. 1).
Fig. 1.
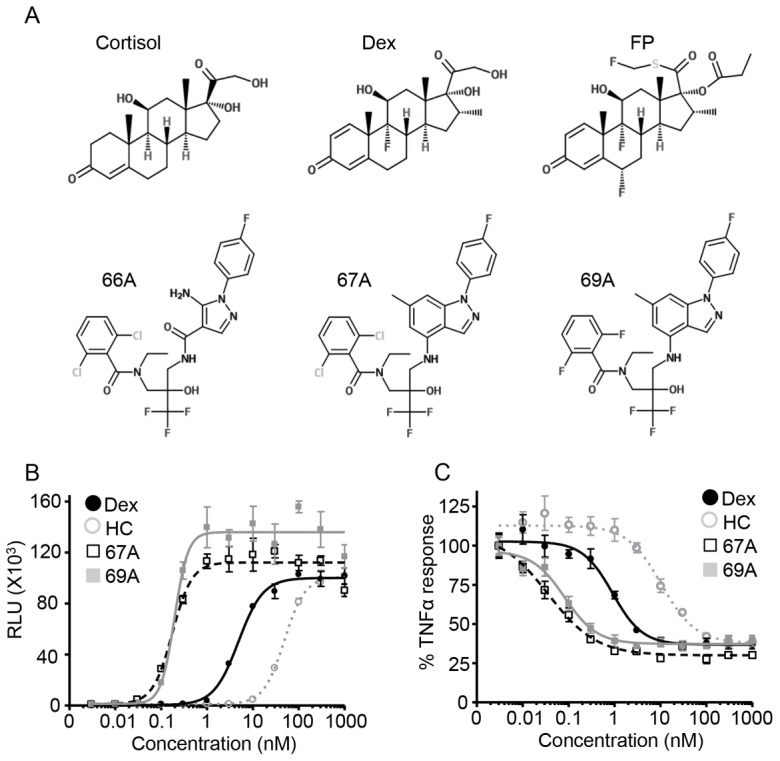
GSK47867A and GSK47869A are highly potent GR agonists. Structure of steroidal and non-steroidal glucocorticoids (A). HeLa cells were transfected with a positive GR reporter gene (TAT3-luc) (B) or with a glucocorticoid-repressed NFκB reporter gene (NRE-luc) (C). At 24 hours post-transfection, NRE-Luc transfected cells were pre-treated with TNF-α (0.5 ng/ml) for 30 minutes. Subsequently all transfected cells were treated with 0.01–1000 nM Dex, hydrocortisone (HC), GSK47867A (67A) or GSK47869A (69A) for 18 hours, and were then lysed and subjected to analysis using a luciferase assay. The graphs (mean±s.d.) show the relative light units (RLU) (B) or percentage inhibition (C) from one of three representative experiments performed in triplicate.
Table 1. Saturating concentration of ligands calculated from EC50.

To rationalise subsequent matched analyses, saturating concentrations of the ligands were selected, calculated as 10 times the measured EC50 for transactivation (Table 1). At these concentrations all ligands showed similar repression of IL6 and IL8 secretion (supplementary material Fig. S2C,D), and inhibition of cell proliferation (supplementary material Fig. S2E,F).
GR crystal structure reveals ligand-specific altered surface charge
To identify conformational differences in the GR ligand-binding domain (LBD), we first compared the structures of GR-Dex (1M2Z) and GR-GSK47866A (3E7C) a non-steroidal GR ligand similar in structure to GSK47867A (Fig. 1A; Fig. 2). An active site model derived from the coordinates of deposited structure 3E7C was used to dock GSK47867A and GSK47869A. Both GSK47867A and GSK47869A are similar to GSK47866A and gave very high scoring fits in the binding pocket formed by GSK47866A bound to the GR LBD (supplementary material Fig. S3). Inspection of the poses showed sensible, well fitting conformers, indicating that structure 3E7C was a suitable surrogate to compare with 1M2Z.
Fig. 2.
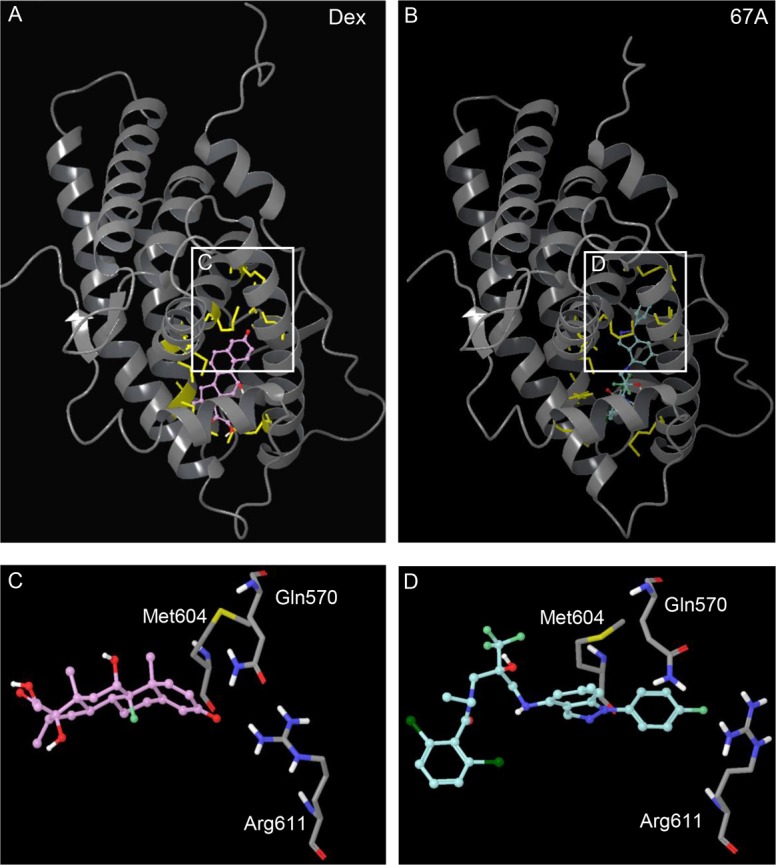
Dex and GSK47867A binding induces different GR LBD structures. (A,B) Comparison of the crystal structures of the GR LBD bound to Dex (A, purple) and GSK47867A (67A) (B, blue). The residues in the binding pocket that show significant movement upon 67A binding are highlighted in yellow. When 67A binds to the GR LBD the head region causes movement of residues Gln570, Met604 and Arg611 (D) when compared with Dex binding (C).
Observation of the ligand-binding pocket in each crystal structure revealed that amino acids in closest proximity to each ligand demonstrated significant movement compared to Dex at the head (A ring, Fig. 2C,D) and tail (D ring, supplementary material Fig. S4C,D). The greatest displacement was seen in amino acids Gln570 and Arg611 (Fig. 2C,D). Less displacement was seen at the opposite end of the ligand; most noticeable here was the movement of Gln642 (supplementary material Fig. S4C,D). The effect of residue movements in the GR LBD upon binding of GSK47866A was examined by visualisation of the molecular surface (Fig. 3; supplementary material Figs S5, S6). This revealed a distinct surface electrostatic potential difference, highlighting a patch of positive charge in the GR-GSK47866A structure resulting from displacement of Arg611 (Fig. 3B,D). This demonstrates that the structural difference between Dex and the NSGs results in a different GR surface charge upon binding, with potential for altered for protein–protein interactions.
Fig. 3.

GR LBD surface charge is altered by GSK47867A binding. (A,C) The region of the GR LBD surface where residues Gln570, Met604 and Arg611 are exposed is highlighted [A, with Dex in purple and C, GSK47867A (67A) in blue]. (B,D) A close-up of this region is shown with an electrostatic charge map that reveals the creation of a patch of positive surface charge due to the movement of Arg611 upon 67A binding.
NSG induce different kinetics of endogenous glucocorticoid target gene regulation
To determine whether the alteration in GR surface charge upon binding NSG had any functional consequence, transcript levels of endogenous glucocorticoid induced (GILZ and FKBP5) and glucocorticoid repressed (IL6 and IL8) target genes were quantified at multiple time points (Fig. 4A,B; supplementary material Fig. S7A,B). Both the steroidal and NSG ligands displayed equivalent kinetics of FKBP5 induction (Fig. 4A). Although NSG treatment did not induce GILZ transcript at 1 hour, similar induction was observed at later time points (Fig. 4B). Similarly NSG treatment did not repress IL6 or IL8 transcripts at 1 hour but comparable repression was observed at later time points (supplementary material Fig. S7A,B).
Fig. 4.

GSK47867A and GSK47869A induce slow kinetics of GR activation. HeLa cells were treated with DMSO vehicle, 100 nM Dex, 3 nM GSK47867A (67A) or 3 nM GSK47869A (69A) for 1, 4 or 24 hours. Cells were then lysed and RNA was extracted using an RNeasy kit. RNA was reverse transcribed and subjected to qPCR for FKBP5 (A) and GILZ (B) using Sybr Green detection in an ABI q-PCR machine and with data analysed by the ΔΔCT method. Graphs (mean±s.e.m.) combine data from three separate experiments and display fold change over that in vehicle-treated control. (C) Following transfection with HaloTag-GR, HeLa cells were incubated with 100 nM Dex, 3 nM FP, 3 nM 67A or 69A. Cells were imaged in real-time at 37°C to determine the subcellular localisation of the GR (white) at the times indicated. Scale bars: 25 µm. Images are representative of three independent experiments. (D) HeLa cells transfected with a TAT3-Luc reporter plasmid were treated with 100 nM Dex, 3 nM FP, 3 nM 67A or 69A for up to 24 hours. The production of luciferase was tracked by measuring the relative light units (RLU) emitted from each sample; D tracks RLU production over the first 5 hours following addition of treatment and is representative of three separate experiments. The time taken to reach half the maximal light output was measured for all treatments (E). Statistical significance was evaluated by one-way ANOVA followed by Tukey post-test. *P<0.005 compared with control; **P<0.001 compared with Dex.
NSG treatment results in delayed kinetics of GR 211 phosphorylation
Transactivation of IGFBP1 is reliant on Ser211 phosphorylation of the GR, a signal to recruit the co-activator protein MED14. Dex treatment resulted in significant induction of IGFBP1 transcript by 1 hour (supplementary material Fig. S8A), but the NSG ligands failed to induce transcript at this early time point. This lack of transcript regulation at an early time point was similarly seen with GILZ, IL6 and IL8. Ligand induction of GR Ser211 phosphorylation was compared. Treatment with Dex resulted in rapid phosphorylation of GR at both serine residues 203 and 211 (supplementary material Fig. S8B). The NSG ligands induced slower onset of phosphorylation of both serine residues 203 and 211 (supplementary material Fig. S8B).
NSG treatment results in slow rate of GR nuclear translocation
The delay in endogenous gene transactivation and receptor phosphorylation seen with NSG treatment suggested that nuclear translocation may also be delayed. Use of a halo tagged GR clearly demonstrated a slower rate of nuclear translocation with both GSK47867A and GSK47869A (supplementary material Movies 1 and 2; Fig. 4C). Ligand-bound nuclear GR has a signature FRAP signal, with reduced intranuclear mobility resulting in delayed recovery from photobleaching. FRAP studies revealed that at 1 hour following NSG treatment nuclear GR displayed characteristics of an unliganded receptor (supplementary material Fig. S9A,B). However with 4 hour NSG treatment nuclear GR displayed the typical signature of liganded receptor, indicative of a delay in adoption of the activated GR conformation (supplementary material Fig. S9C,D).
Altered kinetics of GR phosphorylation may explain the observed differences in nuclear translocation rate and transactivation of endogenous glucocorticoid target genes. Therefore, we made GR mutants Ser211Ala (phosphodeficient) and Ser211Asp (phosphomimetic) to assess the importance of this phosphorylation site (supplementary material Fig. S10A). However, the phosphomimetic GR did not significantly increase the rate of GR translocation with either GSK47867A or GSK47869A treatment (supplementary material Fig. S10C,D). Likewise the phosphodeficient GR had no significant impact on the rate of translocation seen with Dex treatment (supplementary material Fig. S10B,D).
NSG treatment results in slower onset of GR transactivation
Treatment with NSG results in slowed GR nuclear translocation and delayed transactivation of endogenous glucocorticoid target genes. To measure the kinetics of GR transactivation more precisely, real-time luciferase analysis was used (Meng et al., 2008; McMaster et al., 2008) (Fig. 4D). This revealed that the NSG ligands consistently took longer to reach half-maximal transactivation compared to either Dex, or the higher potency FP (Fig. 4E). Interestingly all three high potency ligands resulted in greater maximal transactivation (Fig. 4D).
Delayed action of NSG ligands cannot be explained by impaired cellular uptake
One possible explanation for these observations is altered ligand access to the intracellular GR. Initially mass spectroscopy analysis of cell lysates was performed after 10 minutes ligand exposure (Fig. 5A). A 10 µM concentration of each ligand was compared, to permit detection of the ligand by mass spectrometry in cell lysates. Strikingly, the NSG ligands showed greater than 10-fold increased concentrations within the cells compared to Dex, effectively ruling out delayed ligand penetration.
Fig. 5.

GSK47867A and GSK47869A rapidly accumulate in cells. A549 cells were treated with 10 µM Dex, FP, GSK47867A (67A) or GSK47869A (69A) for 10 minutes and subsequently washed and lysed. The cell samples were analysed for ligand uptake by mass spectrometry (A). HeLa cells were treated with DMSO vehicle (not shown), 100 nM Dex, 3 nM FP, 3 nM 67A or 3 nM 69A either for 4 hours or for 10 minutes followed by washout (WO) and culture in ligand-free medium for 4 hours. Subsequently cells were lysed and RNA extracted using an RNeasy kit. RNA was reverse transcribed and subjected to qPCR for GILZ (B), FKBP5 (C) and IGFBP1 (D) using Sybr Green detection in an ABI q-PCR machine and data were analysed by the ΔΔCT method. Graphs (mean±s.e.m.) combine data from three separate experiments and display percentage induction compared with the equivalent 4 hours of constant treatment. (E,F) Following transfection with HaloTag-GR HeLa cells were placed on ice for 10 minutes and subsequently incubated with 100 nM Dex, 3 nM FP, 3 nM 67A or 69A for 1 hour on ice. Following treatment, cell images were captured in real-time at 37°C to determine the subcellular localisation of the GR (white, E). Scale bars: 25 µm. F displays the average time (mean±s.e.m) to exclusively nuclear GR following 1 hour with ligand on ice, calculated from three separate experiments. Statistical significance was evaluated by one-way ANOVA followed by Tukey post-test. *P<0.001 compared with Dex.
To further evaluate cell pharmacokinetics, cells were incubated with 100 nM Dex or 3 nM FP, GSK47867A or GSK47869A for 10 minutes, washed and then incubated in ligand-free medium for 4 hours. These samples were compared to cells treated with ligand continuously for 4 hours (Fig. 5B–D). Short exposure to both NSG ligands resulted in greater induction of GILZ and FKBP5 although not IGFBP1 compared to Dex, again demonstrating rapid cellular accumulation of ligand. Furthermore cells incubated with NSG on ice for 1 hour to permit ligand access in the absence of GR activation still showed delayed nuclear translocation (supplementary material Movies 3 and 4; Fig. 5E,F), implicating a post receptor mechanism of action. The observed differences could not be attributed to Dex activation of mineralocorticoid receptor, as the mineralocorticoid receptor antagonist Spironolactone did not affect the Dex induction (supplementary material Fig. S11A,B).
NSG bound GR shows prolonged nuclear retention
As treatment with both NSG ligands results in delayed nuclear translocation, we investigated whether nuclear export of GR may also be slower. To measure GR export HeLa cells were treated with 100 nM Dex or 3 nM NSGs for 1 hour then washed and placed in serum free media and imaged over 24 hours (supplementary material Fig. S12A). In cells treated with NSG the GR-GFP was not exported from the nucleus during the 24-hour wash-out period, but Dex treated cells exported GR from the nucleus within 6 hours (supplementary material Movies 5 and 6; Fig. S12B).
Structural modelling suggests that NSGs modify the HSP90 interaction surface
Our data clearly demonstrates that when bound to NSG there is altered interaction of GR with the translocation machinery resulting in delayed nuclear import, delayed transcriptional activity and receptor export. The chaperone heat shock protein 90 (HSP90) is known to play key roles in this aspect of GR biology, including maintaining GR structure, ligand-binding activity, and trafficking of GR between nucleus and cytoplasm (Segnitz and Gehring, 1997; Tago et al., 2004; Kakar et al., 2006; Grad and Picard, 2007; Echeverría et al., 2009). GR residues identified by Ricketson and co-workers (Ricketson et al., 2007) as important for HSP90 interaction were mapped onto the crystal structure of GR bound to Dex (Fig. 6A). Surface map analysis of GR following replacement of Met604 with Thr604, which has been shown to inhibit HSP90 recruitment, was in the same part of the GR structure that was differentially affected by NSG binding (Fig. 6B,C).
Fig. 6.

Disruption of the microtubule network increases the rate of GR translocation in a ligand-specific manner. (A) The ribbon structure of the GR LBD bound to Dex. The residues highlighted in yellow were identified by Ricketson et al. (Ricketson et al., 2007) as important for interaction between GR and HSP90. (B) The region of the GR LBD surface where the NSGs cause an alteration in surface charge. (C) The region of the GR LBD surface where Met604 is exposed is highlighted in yellow. This area overlaps the region identified as having altered surface charge upon binding NSG, supporting the lack of HSP90 engagement with NSG treatment. (D) Untreated HeLa cells with GFP-labelled microtubules. Incubation for 1 hour with 2 µM colcemid disrupts the microtubule network (E). Following transfection with a halo-tagged GR, HeLa cells were incubated with 2 µM Colcemid for 1 hour then subsequently co-treated with 100 nM Dex, 3 nM FP, 3 nM GSK47867A (67A) or 3 nM GSK47869A (69A) (F). Cells were imaged in real-time and analysed for subcellular localisation of the GR (white). Scale bars: 25 µm. (G) The average time taken (mean±s.e.m) for the GR to be exclusively nuclear. Statistical significance was evaluated by one-way ANOVA followed by Tukey post-test. *P<0.005 compared with treatment without colcemid.
Microtubule disruption improves nuclear translocation rate
HSP90 anchors the GR to the microtubule network, so permitting rapid, energy-dependent nuclear translocation. HSP90 antagonism slows the rate of nuclear translocation (Galigniana et al., 1998). However, in addition, GR can translocate using a diffusion mechanism (Nishi et al., 1999). Disruption of the microtubule network using colcemid restores rapid GR translocation even in the presence HSP90 inhibitor geldanamycin (Segnitz and Gehring, 1997; Galigniana et al., 1998). Therefore, we used colcemid to determine if the microtubule architecture was slowing NSG mediated nuclear translocation. Colcemid significantly increased the rate of NSG-driven nuclear translocation, but had no effect on that promoted by Dex (supplementary material Movies 7 and 8; Fig. 6D–G), suggesting a diffusion mechanism for translocation.
NSGs mediate prolonged duration of action
The duration of ligand-dependent activity depends on continuing presence of ligand, and maintaining GR in a ligand-binding compatible conformation. To investigate these phenomena we initially undertook washout studies, using real time reporter gene luciferase analysis. These revealed a striking prolongation of transactivation following NSG ligand withdrawal compared to either Dex or FP, which was not explained by increased ligand potency (Fig. 7A).
Fig. 7.

Antagonism of HSP90 has less impact on the activity of NSG ligands. (A,D) HeLa cells transfected with a TAT3-Luc reporter plasmid were treated with 100 nM Dex, 3 nM GSK47867A (67A) or 3 nM GSK47869A (69A) for 24 hours. Subsequently cells were either co-treated with 10 mM geldanamycin (GA) (D) or washed (WO) and placed in serum-free recording medium (A) for a further 24 hours. The production of luciferase was tracked by measuring the relative light units (RLU) emitted from each sample. Graphs tracks RLU production for 24 hours following GA addition or ligand removal. Graphs are representative of three separate experiments. (B,C) HeLa cells were treated with DMSO vehicle (not shown), 100 nM Dex, 3 nM FP, 3 nM 67A or 3 nM 69A for 24 hours or 1 hour followed by washes (WO) and then cultured in ligand-free medium for 24 hours. Subsequently cells were lysed and RNA was extracted using an RNeasy kit. RNA was reverse transcribed and subjected to qPCR of GILZ (B) and FKBP5 (C) using Sybr Green detection in an ABI q-PCR machine and data analysed by the ΔΔCT method. Graphs (mean±s.e.m.) combine data from three separate experiments and display percentage induction compared with the equivalent constant treatment for 24 hours. HeLa cells were treated with 100 nM Dex, 3 nM 67A or 69A for 2 hours and then co-treated with 10 mM GA for a further 2 hours (E) or 22 hours (F), and a constant 4-hour or 24-hour treatment was used as a comparison. Following treatment, cells were lysed in RIPA buffer containing phosphatase and protease inhibitors and analysed by immunoblotting for GR abundance and GR Ser211 phosphorylation. α-Tubulin was used as a loading control. Statistical significance was evaluated by one-way ANOVA followed by Tukey post-test. *P<0.01 compared with both Dex and FP. (G) Mechanism of GR action. Upon binding glucocorticoids (Gc) (1) the GR interacts with the translocation machinery enabling nuclear import (2). In the nucleus, GR binds to cis-elements to activate or repress target gene expression (3). The GR undergoes dynamic cycles of dissociation, and re-binding of ligand, which occurs in an HSP90-dependent manner (4). Interaction with PP5 facilitates nuclear export of the GR (5) enabling it to be recycled or targeted for degradation by the proteasome (6).
To corroborate these observations with endogenous genes a 2-hour ligand exposure was chased with a 24-hour washout before measurement of GILZ and FKBP5 transcripts (Fig. 7B,C). There was significantly enhanced preservation of transactivation seen with both the NSGs compared to the potency matched control steroid FP.
To determine the role of HSP90 in mediating prolonged GR transactivation, geldanamycin was used. As HSP90 activity is required for initial GR ligand binding, these studies were performed sequentially, adding geldanamycin after ligand activation. The geldanamycin was added to cells at the time of maximal transactivation, in the presence of continuing ligand exposure (Fig. 7D). Both FP and Dex showed exponential decay of transactivation, as predicted. However, the NSG ligands showed a striking biphasic response, with initial potentiation, followed by decay (Fig. 7D).
As HSP90 is also essential for maintaining GR protein stability investigation of receptor abundance and phosphorylation was undertaken. Inhibition of HSP90 preserves GR protein levels following Dex treatment for 4 hours (Fig. 7E), but not at a later time point (Fig. 7F). Strikingly, the NSG ligands did not show such a ligand-dependent loss of GR protein (Fig. 7E,F), again identifying differences in HSP90 interaction with the novel NSGs. Additionally treatment of cells with Dex in the presence of geldanamycin results marked dephosphorylation of GR at serine 211 (Fig. 7E). However treatment with the NSG was protective for serine 211 phosphorylation (Fig. 7E). Collectively, these studies suggest that GR-HSP90 interactions can be modulated by ligand structure, to influence the properties of the glucocorticoid response.
Discussion
Understanding how the GR interprets its ligands to permit appropriate cellular responses is of vital interest in both physiology and pharmacology, as the GR remains an important drug target in inflammation and malignancy (Barnes, 2011; De Iudicibus et al., 2011). The advent of drug design based on the crystal structure predicted pharmacophore has permitted new generations of ligands to be synthesised, including those studied here (Kauppi et al., 2003; Bledsoe et al., 2004). Our initial findings identified that although highly potent, the NSG ligands surprisingly result in slowed kinetics of GR phosphorylation, nuclear import and delayed onset of GR-dependent gene transactivation. Our data suggests that the NSG ligands fundamentally alter the mechanism of GR activation.
A possible explanation for the delayed kinetics of cellular response to GSK47867A and GSK47869A is reduced efficiency of cellular uptake of ligand. Although the NSGs retain the highly lipophilic characteristics of steroidal ligands, they may interact differentially with membrane components. However our mass spec studies in fact showed an accelerated ligand accumulation with the NSGs compared to Dex. We also undertook a functional assay, washing off ligand after a short incubation, and tracking response of glucocorticoid target genes. Again, the NSGs produced enhanced target gene transactivation compared with Dex, indicating rapid ligand accumulation. Furthermore treatment of cells with ligand for 1 hour on ice allowed for saturation of the receptor without translocation. When the cells were returned to 37°C the GR rapidly translocated with both Dex and FP but translocation was slower for both the NSG ligands, supporting defective interaction with the nuclear translocation machinery post ligand binding.
To explain these observations we interrogated the crystal structure of GR LBD bound to GSK47867A and GSK47869A. This revealed a very similar conformation to that seen with Dex, but there was a single difference, namely the addition of a patch of positive charge on the external surface of the LBD. Ricketson and co-workers were able to demonstrate, through amino acid substitution, that this surface is required for HSP90 interaction (Ricketson et al., 2007). HSP90 recognises the GR LBD through two, defined hydrophobic sites and binds to a solvent accessible major groove maintaining GR stability and permitting high-affinity ligand binding (Fang et al., 2006), as depicted in Fig. 7G. Following ligand binding, HSP90 undergoes a conformation change to bind to the same region of the GR LBD, but with a different motif. This is required to couple the GR to the dynein active transport mechanism through the bridging effect of immunophilins (Harrell et al., 2004) (Fig. 7G). HSP90 remains associated with the GR in the nucleus, where binding to the major groove of the GR LBD competes with recruitment of co-activators (Caamaño et al., 1998; Kang et al., 1999; Fang et al., 2006), and also promotes nuclear retention (Tago et al., 2004; Kakar et al., 2006). Binding of NSGs to the GR LBD forces the movement of Arg611, leading to the creation of a novel interaction surface which could be the mechanism by which interaction with HSP90 is altered. Therefore, we measured the impact of HSP90 manipulation on GR function with both the steroidal ligands, and NSGs.
GR is anchored to the microtubule network through interaction with HSP90 to facilitate nuclear translocation. Antagonism of HSP90 therefore reduces the rate of GR nuclear translocation and can be overcome by disrupting the microtubule network (Galigniana et al., 1998; Nishi et al., 1999). Here we show that the absence of an intact microtubule network significantly increases the rate of GR translocation in response to the NSGs but not Dex, which suggests an impaired interaction of GR-NSG with HSP90. Evidence has emerged that persisting glucocorticoid action requires cycles of dissociation, and re-binding of ligand to the GR, which occurs in a HSP90-dependent manner (Stavreva et al., 2004; Conway-Campbell et al., 2011) (Fig. 7G). To test the role of HSP90 we used the inhibitor geldanamycin (Segnitz and Gehring, 1997). As predicted, geldanamycin curtailed the glucocorticoid transcriptional response rapidly, irrespective of ligand potency, for the two steroid agonists. However, in keeping with the hypothesis that HSP90 binding was disrupted by the final conformation adopted by the NSG bound GR there was greatly prolonged transactivation observed, with a gradual decay likely due to degradation of GR protein. It was, however, striking that the pattern of response for both NSGs included an initial augmentation of response, which is compatible with displacement of HSP90 from the major groove, and subsequent promotion of co-activator recruitment. It is also possible that disruption of the HSP90 interaction surface also affects interaction between GR, and co-modulator protein partners (Caamaño et al., 1998; Kang et al., 1999; Fang et al., 2006).
Altered NSG-driven nuclear translocation, and interaction with HSP90 may also affect GR nuclear export, and the duration of cellular response. Indeed, our washout studies showed a dramatic difference between the steroidal and NSG ligands, with marked reduction in GR export rate and prolongation of action seen with the NSGs, observed with both transfected reporter genes, and endogenous gene transcripts. A similar prolongation of action was seen in cells treated with geldanamycin which may result from stabilised GR-ligand interaction, due to altered engagement with HSP90, and its associated protein complex, including enzymes such as protein phosphatase 5 (PP5). PP5 is responsible for removing phosphate modification from GR Ser211, and promoting GR nuclear export (DeFranco et al., 1991; Silverstein et al., 1997; Galigniana et al., 2002; Hinds and Sánchez, 2008) (Fig. 7G).
Geldanamycin treatment resulted in loss of the Dex ligand-dependent GR Ser211 phosphorylation. However NSG-liganded GR was not dephosphorylated under the same conditions, implying altered recruitment of PP5. PP5 also associates with HSP90 as part of the chaperone complex (Silverstein et al., 1997; Hinds and Sánchez, 2008) (Fig. 7G), and contains a peptidylprolyl isomerase domain that is capable of dynein interaction and therefore forming a bridge between the GR and the nuclear export machinery (DeFranco et al., 1991; Galigniana et al., 2002) (Fig. 7G). Therefore, as PP5 has been implicated in the nuclear export of the GR, the lack of dephosphorylation seen with NSG treatment is compatible with a broader change in protein recruitment with the NSG ligands. Interestingly, it was also observed that NSG treatment preserved GR protein expression compared with Dex treatment. This would further suggest that the conformation adopted by GR following NSG binding decouples protein recruitment required for terminating the GR transcriptional signal (Nawaz and O'Malley, 2004) (Fig. 7G).
In conclusion we have identified two NSGs that bind to GR with high specificity but paradoxically result in profoundly slowed kinetics of cellular response. Analysis of the structural effects of these NSGs bound to GR suggests a change to the GR surface, through the movement of Arg611 in the ligand-binding pocket of the GR, resulting in an alteration in the GR surface charge. The change in electrostatic charge is close to the known binding site for HSP90, and co-modulator proteins. This alteration carries with it the consequence of delayed GR phosphorylation and nuclear translocation, which in turn results in delayed early glucocorticoid target gene regulation. The ability to manipulate the kinetics of GR activation by designing novel NSGs has implications for therapy, by targeting cellular pharmacodynamics rather than organismal pharmacokinetics.
Materials and Methods
Anti-hGR (clone 41, BD Biosciences, Oxford, UK); anti-phospho-(Ser211)-GR, anti α-tubulin (Cell Signalling Technology, MA, USA); Horseradish peroxidase conjugated anti-mouse and anti-rabbit (GE Healthcare, Buckinghamshire, UK); dexamethasone, hydrocortisone and Fluticasone Propionate (Sigma, Dorset, UK). TAT3-Luciferase, and NRE-luciferase have been previously described (Matthews et al., 2008; Matthews et al., 2009).
Cell culture and maintenance
HeLa cells and A549 cells (ATCC, Teddington, UK) were cultured in low glucose (1 g/l) Dulbecco's modified Eagle's medium (DMEM; PAA, Yeovil, UK) supplemented with stable 2 mM glutamine (PAA) and 10% heat inactivated fetal bovine serum (FBS; Invitrogen, Paisley, UK) or 10% charcoal dextran stripped fetal calf serum (sFCS; Invitrogen). A549's stably transfected with GRE-Luc and NRE-Luc were also supplemented with 1% Non-essential amino acid (NEAA; Invitrogen) and 1% Geneticin (Invitrogen). All cells were grown in a humidified atmosphere of 5% carbon dioxide at 37°C.
Immunoblot analysis
Following treatment cells were lysed in RIPA buffer [50 mM Tris-HCl pH 7.4, 1% NP40 (Igepal), 0.25% Na-deoxycholate 150 mM NaCl, 1 mM EDTA] containing protease and phosphatase inhibitors (Sigma), and 10 µg protein was electrophoresed on Tris/Glycine 4–12% gels (Invitrogen) and transferred to 0.2 micron nitrocellulose membranes (BioRad Laboratories, Hertfordshire, UK) overnight at 4°C. Membranes were blocked for 2 hours (NaCl 0.15 M, 2% dried milk, 0.1% Tween 20) and incubated with primary antibodies (diluted in blocking buffer) overnight at 4°C. After three 10-minute washes (88 mM Tris pH 7.8, 0.25% dried milk, 0.1% Tween 20), membranes were incubated with a species-specific horseradish peroxidase-conjugated secondary antibody (diluted in wash buffer) for 1 hour at room temperature, and washed a further three times, each for 10 minutes. Immunoreactive proteins were visualised using enhanced chemiluminescence (ECL Advance, GE Healthcare).
Reporter gene assays
HeLa cells seeded in DMEM containing sFCS were co-transfected with 2 µg reporter gene and 0.5 µg CMV-renilla luciferase (to correct for transfection efficiency) using Fugene 6 (Roche Diagnostics, West Sussex, UK) at a ratio of 3:1 (v/w). 24 hours post transfection, cells were treated as specified in results prior to lysis, then assayed for luciferase activity using a dual-luciferase reporter assay system following the manufacturer's instructions (Promega, Southampton, UK).
Stable A549 GRE-Luc or NRE-Luc cells were seeded in DMEM containing sFCS into 96-well plates and incubated overnight. Cells were treated as specified in results and 18 hours later each well washed twice with PBS (first without Mg2+, Ca2+, then with Mg2+, Ca2+). Renilla Glo (Promega, E2720) or Bright Glo (Promega E2620) lysis buffer was added to the GRE cells or the NRE cells respectively according to the manufacturer's instructions. Cell lysates were read using a luminometer (Wallac 1450 MicroBeta Trilux Liquid Scintillation counter and luminometer). Ten 1-second reads were taken per well and the average RLU determined.
Immunofluorescence
Fixed cells: Following 24 hours in DMEM containing sFCS, HeLa cells were transfected (Fugene 6) with hGR-GFP and treated as specified in results. Cells were fixed with 4% paraformaldehyde for 30 minutes at 4°C, and subsequently stained with Hoeschst (Sigma) in PBS (2 µg/ml) for 20 minutes at 4°C. Following three 5 minute washes in PBS, coverslips were mounted using Vectamount AQ (Vector Laboratories, Peterborough, UK). Images were acquired on a Delta Vision RT (Applied Precision, GE Healthcare) restoration microscope using a 40×/0.85 Uplan Apo objective and the Sedat Quad filter set (Chroma 86000v2, VT, USA). The images were collected using a Coolsnap HQ (Photometrics, AZ, USA) camera with a Z optical spacing of 0.5 µm. Raw images were then deconvolved using the Softworx software (GE Healthcare) and average intensity projections of these deconvolved images processed using Image J (Rasband, 1997).
Live cells: Following 24 hours in DMEM containing sFCS, HeLa cells were transfected (Fugene 6) with 5 µg GR-GFP and transferred to glass-bottomed 24-well plates. Alternatively HeLa cells were plated into a glass-bottomed 24-well plate in DMEM containing sFCS. Each well was transfected (Fugene 6) with 0.5 µg HaloTag-GR (Catalog number FHC10483, Promega) and incubated for 16 hours with 0.25 µl Halo ligand (HaloTag TMRDirect, Catalog number G2991, Promega) to enable visualisation. Subcellular GR trafficking was tracked in real-time at 37°C with 5% CO2. Images were acquired on a Nikon TE2000 PFS microscope using a 60×/1.40 Plan Apo or 40×/1.25 Plan Apl objective and the Sedat filter set (Chroma 89,000). The images were collected using a Cascade II EMCCD camera (Photometrics). Raw images were then processed using Image J.
Fluorescent recovery after photobleaching (FRAP)
HeLa cells were transfected (Fugene 6) with 5 µg hGR-GFP then seeded into a glass bottomed 24-well plate. Cells were maintained at 37°C and 5% CO2 and images collected on a Leica TCS SP5 AOBS inverted confocal (Leica, Milton Keynes, UK) using a 63×/0.50 Plan Fluotar objective and 7×confocal zoom. The confocal settings were as follows, pinhole 1 airy unit, scan speed 1000 Hz unidirectional, format 1024×1024. Images were collected using the following detection mirror settings; FITC 494–530 nm using the 488 nm (13%).
MTS Assay
Cells were seeded into a 96-well plate were treated as described in the results. Upon completion of the treatment 10 µl of MTS reagent (Promega) was added to each well. Cells were incubated for 4 hours, reading at 490 nm every hour.
Q-RTPCR
Cells were treated as required, then lysed and RNA extracted using an RNeasy kit (Qiagen). 10 ng RNA was reverse transcribed, and subjected to qPCR using Sybr Green detection in an ABI q-PCR machine (Applied biosystems, CA, USA) and data analysed by δδCT method (Livak and Schmittgen, 2001).
Bioluminescence real-time recording
HeLa cells transfected (Fugene 6) with 2 µg TAT3-luc plasmid were grown to 80% confluency in 35-mm tissue culture dishes in phenol red free DMEM with 10% FCS and 1% glutamine. Prior to the experiment, cells were supplemented with 0.1 mM Luciferin substrate (Izumo et al., 2003; Yamazaki and Takahashi, 2005). Each dish lid was replaced with a glass cover then sealed with vacuum grease before being placed in a light-tight and temperature-controlled (37°C) environment. Light emission (bioluminescence) was measured continuously using a Photomultiplier tube (PMT, H6240 MOD1, Hamamatsu Photonics, Hertfordshire, UK). Baseline measurements (photon counts per minute) were taken for each PMT prior to treatment and then deducted from the experimental values attained.
Measurement of ligand uptake using mass spectroscopy
A549 cells were grown to 90% confluency in 6-well plates. Following treatment the media was removed from the cells and retained for analysis. The cells were washed three times with PBS and lysed in 300 µl of M-Per mammalian protein extraction reagent (#78503, Thermoscientific, Essex, UK) on the shaker at 750 rpm at room temperature for 5 minutes. The whole cell lysate was collected, then centrifuged at 10,000 rpm for 10 minutes, then the supernatant collected and analysed by mass spectrometry.
Measurement of cytokine production
A549 cells were seeded into a 96-well plate into DMEM with 10% FCS and incubated overnight. In order to slow cell proliferation and prevent any interference from steroid present in FCS the media was changed to DMEM with 1% sFCS prior to ligand treatment. Following treatment supernatents were collected and assayed for IL6 and IL8 concentration using a Luminex 100 (Merck Millipore, MA, USA) with StarStation software according to the manufacturer's instructions.
Computational modelling of GR crystal structure
Crystal structures of GR bound to Dex (1M2Z) and GSK47866A (3E7C) (Madauss et al., 2008) were downloaded from the RCSB Protein Data Bank (PDB) (Berman et al., 2007). The structures were imported into Maestro (Schrodinger, 2012) and prepared using the Protein Preparation module. Each Ligand was extracted and scrambled conformationally before docking back into the native active site models to verify that the docking program (GLIDE) (Schrodinger, 2009) was competent at reproducing the X-ray pose for each complex.
Models of compounds GSK47866A, GSK47867A and GSK47869A (S-isomers) were prepared using the Ligprep module and a set of 272 conformers generated using the confgen module of Maestro. This set of conformers was docked in the 3E7C active site model yielding 62 successful poses. Again, as found in the bootstrapping exercise, GSK47866A best scoring pose was extremely close in conformation and position within the active site pocket (RMSD ∼0.2), indicative of a robust model.
Crystal structures 1M2Z and 3E7C were superposed and conformations of residues within 6 Angstrom of the Dex ligand in 1M2Z were compared visually. Any differing substantially were coloured differently (Fig. 2A,B), and these atom colours projected onto a molecular surface to reveal regions of the protein surface impacted by the residue movements induced by binding of GSK47866A (Fig. 3A,C). The regions of surface modification thus highlighted guided where to look for differences in electrostatic potential, projected onto the same molecular surface (Fig. 3B,D).
Modelling of GR mutant with impaired HSP90 interaction
The original 1M2Z X-ray coordinates, already optimised for use with the OPLS forcefield in Maestro, were used to mutate M604 to Threonine. The built-in residue mutation building tool was employed for this. The mutated structure was optimised using the Protein Preparation Wizard option to perform a restrained, all-atom minimisation. Surface and electrostatic potential colouring was calculated as for all other examples, ensuring a consistent range of electrostatic potential values of −0.2 to 0.2 for the blue-white-red colour ramp.
Supplementary Material
Acknowledgments
We thank Bill Leavens and Midori Kayahara for technical assistance. Special thanks to Peter March and Roger Meadows for their help with the microscopy. The Bioimaging Facility microscopes used in this study were purchased with grants from BBSRC, Wellcome and the University of Manchester Strategic Fund.
Footnotes
Author contributions
D.R., L.M. and S.F. conceived the idea; P.T., D.R. and L.M. wrote the paper; P.T. and L.M. performed the experiments; K.A.S. and K.D.S. provided technical assistance for the cytokine production and reporter gene assays. J.W. performed the computational modeling.
Funding
P.T. is supported by a Biotechnology and Biological Sciences Research Council CASE studentship. L.M. is supported by a Faculty of Medical and Human Sciences (FMHS) Stepping Stones Fellowship. D.R. is supported by the Wellcome Trust. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.124784/-/DC1
References
- Barnes P. J. (2011). Glucocorticosteroids: current and future directions. Br. J. Pharmacol. 163, 29–43 10.1111/j.1476-5381.2010.01199.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H., Henrick K., Nakamura H., Markley J. L. (2007). The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 35 Database issue, D301–D303 10.1093/nar/gkl971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledsoe R. K., Montana V. G., Stanley T. B., Delves C. J., Apolito C. J., McKee D. D., Consler T. G., Parks D. J., Stewart E. L., Willson T. M. et al. (2002). Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110, 93–105 10.1016/S0092-8674(02)00817-6 [DOI] [PubMed] [Google Scholar]
- Bledsoe R. K., Stewart E. L., Pearce K. H. (2004). Structure and function of the glucocorticoid receptor ligand binding domain. Vitam. Horm. 68, 49–91 10.1016/S0083-6729(04)68002-2 [DOI] [PubMed] [Google Scholar]
- Caamaño C. A., Morano M. I., Dalman F. C., Pratt W. B., Akil H. (1998). A conserved proline in the hsp90 binding region of the glucocorticoid receptor is required for hsp90 heterocomplex stabilization and receptor signaling. J. Biol. Chem. 273, 20473–20480 10.1074/jbc.273.32.20473 [DOI] [PubMed] [Google Scholar]
- Canalis E., Pereira R. C., Delany A. M. (2002). Effects of glucocorticoids on the skeleton. J. Pediatr. Endocrinol. Metab. 15, Suppl. 51341–1345 [PubMed] [Google Scholar]
- Cerasoli F., Jr (2006). Developing the ideal inhaled corticosteroid. Chest 130 Suppl, 54S–64S 10.1378/chest.130.1_suppl.54S [DOI] [PubMed] [Google Scholar]
- Chen W., Rogatsky I., Garabedian M. J. (2006). MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol. Endocrinol. 20, 560–572 10.1210/me.2005-0318 [DOI] [PubMed] [Google Scholar]
- Chen W., Dang T., Blind R. D., Wang Z., Cavasotto C. N., Hittelman A. B., Rogatsky I., Logan S. K., Garabedian M. J. (2008). Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 22, 1754–1766 10.1210/me.2007-0219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway-Campbell B. L., George C. L., Pooley J. R., Knight D. M., Norman M. R., Hager G. L., Lightman S. L. (2011). The HSP90 molecular chaperone cycle regulates cyclical transcriptional dynamics of the glucocorticoid receptor and its coregulatory molecules CBP/p300 during ultradian ligand treatment. Mol. Endocrinol. 25, 944–954 10.1210/me.2010-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czar M. J., Owens-Grillo J. K., Yem A. W., Leach K. L., Deibel M. R., Jr, Welsh M. J., Pratt W. B. (1994). The hsp56 immunophilin component of untransformed steroid receptor complexes is localized both to microtubules in the cytoplasm and to the same nonrandom regions within the nucleus as the steroid receptor. Mol. Endocrinol. 8, 1731–1741 10.1210/me.8.12.1731 [DOI] [PubMed] [Google Scholar]
- Davies T. H., Ning Y. M., Sánchez E. R. (2002). A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 277, 4597–4600 10.1074/jbc.C100531200 [DOI] [PubMed] [Google Scholar]
- De Iudicibus S., Franca R., Martelossi S., Ventura A., Decorti G. (2011). Molecular mechanism of glucocorticoid resistance in inflammatory bowel disease. World J. Gastroenterol. 17, 1095–1108 10.3748/wjg.v17.i9.1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFranco D. B., Qi M., Borror K. C., Garabedian M. J., Brautigan D. L. (1991). Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol. Endocrinol. 5, 1215–1228 10.1210/mend-5-9-1215 [DOI] [PubMed] [Google Scholar]
- Echeverría P. C., Mazaira G., Erlejman A., Gomez-Sanchez C., Piwien Pilipuk G., Galigniana M. D. (2009). Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol. Cell. Biol. 29, 4788–4797 10.1128/MCB.00649-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encío I. J., Detera-Wadleigh S. D. (1991). The genomic structure of the human glucocorticoid receptor. J. Biol. Chem. 266, 7182–7188 [PubMed] [Google Scholar]
- Fang L., Ricketson D., Getubig L., Darimont B. (2006). Unliganded and hormone-bound glucocorticoid receptors interact with distinct hydrophobic sites in the Hsp90 C-terminal domain. Proc. Natl. Acad. Sci. USA 103, 18487–18492 10.1073/pnas.0609163103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford J., McEwan I. J., Wright A. P., Gustafsson J. A. (1997). Involvement of the transcription factor IID protein complex in gene activation by the N-terminal transactivation domain of the glucocorticoid receptor in vitro. Mol. Endocrinol. 11, 1467–1475 10.1210/me.11.10.1467 [DOI] [PubMed] [Google Scholar]
- Galigniana M. D., Scruggs J. L., Herrington J., Welsh M. J., Carter-Su C., Housley P. R., Pratt W. B. (1998). Heat shock protein 90-dependent (geldanamycin-inhibited) movement of the glucocorticoid receptor through the cytoplasm to the nucleus requires intact cytoskeleton. Mol. Endocrinol. 12, 1903–1913 10.1210/me.12.12.1903 [DOI] [PubMed] [Google Scholar]
- Galigniana M. D., Harrell J. M., Murphy P. J., Chinkers M., Radanyi C., Renoir J. M., Zhang M., Pratt W. B. (2002). Binding of hsp90-associated immunophilins to cytoplasmic dynein: direct binding and in vivo evidence that the peptidylprolyl isomerase domain is a dynein interaction domain. Biochemistry 41, 13602–13610 10.1021/bi020399z [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley A. J., Williams J. G., Collins J. B., Cidlowski J. A. (2008). Glycogen synthase kinase 3beta-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol. Cell. Biol. 28, 7309–7322 10.1128/MCB.00808-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad I., Picard D. (2007). The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 275, 2–12 10.1016/j.mce.2007.05.018 [DOI] [PubMed] [Google Scholar]
- Harrell J. M., Murphy P. J., Morishima Y., Chen H., Mansfield J. F., Galigniana M. D., Pratt W. B. (2004). Evidence for glucocorticoid receptor transport on microtubules by dynein. J. Biol. Chem. 279, 54647–54654 10.1074/jbc.M406863200 [DOI] [PubMed] [Google Scholar]
- Heery D. M., Kalkhoven E., Hoare S., Parker M. G. (1997). A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387, 733–736 10.1038/42750 [DOI] [PubMed] [Google Scholar]
- Hinds T. D., Jr, Sánchez E. R. (2008). Protein phosphatase 5. Int. J. Biochem. Cell Biol. 40, 2358–2362 10.1016/j.biocel.2007.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg S. M., Weinberger C., Ong E. S., Cerelli G., Oro A., Lebo R., Thompson E. B., Rosenfeld M. G., Evans R. M. (1985). Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318, 635–641 10.1038/318635a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismaili N., Garabedian M. J. (2004). Modulation of glucocorticoid receptor function via phosphorylation. Ann. N. Y. Acad. Sci. 1024, 86–101 10.1196/annals.1321.007 [DOI] [PubMed] [Google Scholar]
- Ito K., Yamamura S., Essilfie-Quaye S., Cosio B., Ito M., Barnes P. J., Adcock I. M. (2006). Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 203, 7–13 10.1084/jem.20050466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumo M., Johnson C. H., Yamazaki S. (2003). Circadian gene expression in mammalian fibroblasts revealed by real-time luminescence reporting: temperature compensation and damping. Proc. Natl. Acad. Sci. USA 100, 16089–16094 10.1073/pnas.2536313100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. A., Elbi C., Parekh B. S., Hager G. L., John S. (2008). Chromatin remodeling complexes interact dynamically with a glucocorticoid receptor-regulated promoter. Mol. Biol. Cell 19, 3308–3322 10.1091/mbc.E08-02-0123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. L., Shi Y. B. (2003). N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr. Top. Microbiol. Immunol. 274, 237–268 10.1007/978-3-642-55747-7_9 [DOI] [PubMed] [Google Scholar]
- Kakar M., Kanwal C., Davis J. R., Li H., Lim C. S. (2006). Geldanamycin, an inhibitor of Hsp90, blocks cytoplasmic retention of progesterone receptors and glucocorticoid receptors via their respective ligand binding domains. AAPS J. 8, E718–E728 10.1208/aapsj080481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K. I., Meng X., Devin-Leclerc J., Bouhouche I., Chadli A., Cadepond F., Baulieu E. E., Catelli M. G. (1999). The molecular chaperone Hsp90 can negatively regulate the activity of a glucocorticosteroid-dependent promoter. Proc. Natl. Acad. Sci. USA 96, 1439–1444 10.1073/pnas.96.4.1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi B., Jakob C., Färnegårdh M., Yang J., Ahola H., Alarcon M., Calles K., Engström O., Harlan J., Muchmore S. et al. (2003). The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J. Biol. Chem. 278, 22748–22754 10.1074/jbc.M212711200 [DOI] [PubMed] [Google Scholar]
- Krishnan J. A., Davis S. Q., Naureckas E. T., Gibson P., Rowe B. H. (2009). An umbrella review: corticosteroid therapy for adults with acute asthma. Am. J. Med. 122, 977–991 10.1016/j.amjmed.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Lee J. C., Bolen D. W., Thompson E. B. (2001). The conformation of the glucocorticoid receptor af1/tau1 domain induced by osmolyte binds co-regulatory proteins. J. Biol. Chem. 276, 18146–18152 10.1074/jbc.M100825200 [DOI] [PubMed] [Google Scholar]
- Lin C. W., Nakane M., Stashko M., Falls D., Kuk J., Miller L., Huang R., Tyree C., Miner J. N., Rosen J. et al. (2002). trans-Activation and repression properties of the novel nonsteroid glucocorticoid receptor ligand 2,5-dihydro-9-hydroxy-10-methoxy-2,2,4-trimethyl-5-(1-methylcyclohexen-3-y1)-1H-[1]benzopyrano[3,4-f]quinoline (A276575) and its four stereoisomers. Mol. Pharmacol. 62, 297–303 10.1124/mol.62.2.297 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Madauss K. P., Bledsoe R. K., Mclay I., Stewart E. L., Uings I. J., Weingarten G., Williams S. P. (2008). The first X-ray crystal structure of the glucocorticoid receptor bound to a non-steroidal agonist. Bioorg. Med. Chem. Lett. 18, 6097–6099 10.1016/j.bmcl.2008.10.021 [DOI] [PubMed] [Google Scholar]
- Matthews L., Berry A., Ohanian V., Ohanian J., Garside H., Ray D. (2008). Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol. Endocrinol. 22, 1320–1330 10.1210/me.2007-0154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L., Berry A., Tersigni M., D'Acquisto F., Ianaro A., Ray D. (2009). Thiazolidinediones are partial agonists for the glucocorticoid receptor. Endocrinology 150, 75–86 10.1210/en.2008-0196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L., Johnson J., Berry A., Trebble P., Cookson A., Spiller D., Rivers C., Norman M., White M., Ray D. (2011). Cell cycle phase regulates glucocorticoid receptor function. PLoS ONE 6, e22289 10.1371/journal.pone.0022289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMaster A., Ray D. W. (2007). Modelling the glucocorticoid receptor and producing therapeutic agents with anti-inflammatory effects but reduced side-effects. Exp. Physiol. 92, 299–309 10.1113/expphysiol.2006.036194 [DOI] [PubMed] [Google Scholar]
- McMaster A., Ray D. W. (2008). Drug insight: selective agonists and antagonists of the glucocorticoid receptor. Nat. Clin Pract. Endocrinol. Metab. 4, 91–101 10.1038/ncpendmet0745 [DOI] [PubMed] [Google Scholar]
- McMaster A., Chambers T., Meng Q. J., Grundy S., Loudon A. S., Donn R., Ray D. W. (2008). Real-time analysis of gene regulation by glucocorticoid hormones. J. Endocrinol. 197, 205–211 10.1677/JOE-07-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q. J., McMaster A., Beesley S., Lu W. Q., Gibbs J., Parks D., Collins J., Farrow S., Donn R., Ray D. et al. (2008). Ligand modulation of REV-ERBalpha function resets the peripheral circadian clock in a phasic manner. J. Cell Sci. 121, 3629–3635 10.1242/jcs.035048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz Z., O'Malley B. W. (2004). Urban renewal in the nucleus: is protein turnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol. Endocrinol. 18, 493–499 10.1210/me.2003-0388 [DOI] [PubMed] [Google Scholar]
- Nishi M., Takenaka N., Morita N., Ito T., Ozawa H., Kawata M. (1999). Real-time imaging of glucocorticoid receptor dynamics in living neurons and glial cells in comparison with non-neural cells. Eur. J. Neurosci. 11, 1927–1936 10.1046/j.1460-9568.1999.00616.x [DOI] [PubMed] [Google Scholar]
- Picard D., Yamamoto K. R. (1987). Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J. 6, 3333–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband W. S., Image J. (1997). National Institutes of Health, Bethesda, Maryland, USA. 1997. Ref Type: Computer Program [Google Scholar]
- Ricketson D., Hostick U., Fang L., Yamamoto K. R., Darimont B. D. (2007). A conformational switch in the ligand-binding domain regulates the dependence of the glucocorticoid receptor on Hsp90. J. Mol. Biol. 368, 729–741 10.1016/j.jmb.2007.02.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett G., Stach C., Zwerina J., Voll R., Manger B. (2008). How antirheumatic drugs protect joints from damage in rheumatoid arthritis. Arthritis Rheum. 58, 2936–2948 10.1002/art.23951 [DOI] [PubMed] [Google Scholar]
- Schrodinger(2009). Glide version 5.5 New York, NY: Schrodinger LLC [Google Scholar]
- Schrodinger(2012). Maestro version 9.3 New York, NY: Schrodinger LLC [Google Scholar]
- Segnitz B., Gehring U. (1997). The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J. Biol. Chem. 272, 18694–18701 10.1074/jbc.272.30.18694 [DOI] [PubMed] [Google Scholar]
- Silverstein A. M., Galigniana M. D., Chen M. S., Owens-Grillo J. K., Chinkers M., Pratt W. B. (1997). Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J. Biol. Chem. 272, 16224–16230 10.1074/jbc.272.26.16224 [DOI] [PubMed] [Google Scholar]
- Stavreva D. A., Müller W. G., Hager G. L., Smith C. L., McNally J. G. (2004). Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 24, 2682–2697 10.1128/MCB.24.7.2682-2697.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A., Garside H., Berry A., Waters C., White A., Ray D. (2003). Dissociation of steroid receptor coactivator 1 and nuclear receptor corepressor recruitment to the human glucocorticoid receptor by modification of the ligand-receptor interface: the role of tyrosine 735. Mol. Endocrinol. 17, 845–859 10.1210/me.2002-0320 [DOI] [PubMed] [Google Scholar]
- Tago K., Tsukahara F., Naruse M., Yoshioka T., Takano K. (2004). Regulation of nuclear retention of glucocorticoid receptor by nuclear Hsp90. Mol. Cell. Endocrinol. 213, 131–138 10.1016/j.mce.2003.10.057 [DOI] [PubMed] [Google Scholar]
- van Lierop M. J., Alkema W., Laskewitz A. J., Dijkema R., van der Maaden H. M., Smit M. J., Plate R., Conti P. G., Jans C. G., Timmers C. M. et al. (2012). Org 214007-0: a novel non-steroidal selective glucocorticoid receptor modulator with full anti-inflammatory properties and improved therapeutic index. PLoS ONE 7, e48385 10.1371/journal.pone.0048385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Frederick J., Garabedian M. J. (2002). Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J. Biol. Chem. 277, 26573–26580 10.1074/jbc.M110530200 [DOI] [PubMed] [Google Scholar]
- Wang J. C., Shah N., Pantoja C., Meijsing S. H., Ho J. D., Scanlan T. S., Yamamoto K. R. (2006). Novel arylpyrazole compounds selectively modulate glucocorticoid receptor regulatory activity. Genes Dev. 20, 689–699 10.1101/gad.1400506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wärnmark A., Gustafsson J. A., Wright A. P. (2000). Architectural principles for the structure and function of the glucocorticoid receptor tau 1 core activation domain. J. Biol. Chem. 275, 15014–15018 10.1074/jbc.M001007200 [DOI] [PubMed] [Google Scholar]
- Yamazaki S., Takahashi J. S. (2005). Real-time luminescence reporting of circadian gene expression in mammals. Methods Enzymol. 393, 288–301 10.1016/S0076-6879(05)93012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.