

Abstract
Accumulating evidence suggests that regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSC) are elevated in cancer patients and tumor-bearing hosts, and that depletion of Tregs and MDSC may enhance the anti-tumor immunity of the host. Sorafenib, a novel multikinase inhibitor, is approved for the treatment of several human cancers including advanced hepatocellular carcinoma (HCC). Sorafenib is believed to inhibit tumor growth via anti-angiogenesis, cell cycle arrest, and inducing apoptosis. However, the impact of Sorafenib on immune cell populations in tumor-bearing hosts is unclear. In this report, we show that Tregs and MDSC are increased in the spleens and bone marrows of the BALB/c mice with liver hepatoma. The increase in Tregs and MDSCs was positively correlated with tumor burden. Treatment of Sorafenib not only inhibited HCC cell growth in the mice, but also significantly decreased the suppressive immune cell populations: Tregs and MDSCs. In conclusion, our study strongly suggests that Sorafenib can enhance antitumor immunity via modulating immunosuppressive cell populations in the murine liver cancer model.
Keywords: Hepatocellular carcinoma, Sorafenib, regulatory T cells, myeloid-derived suppressor cells, kinase inhibitor
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide. Most patients present in late stages of the disease when surgical resection is not feasible, which leads to dismal clinical outcomes. The annual mortality rate is almost equal to the annual incidence, which is approximately 600,000 patients. 1, 2 Radiation therapy and chemotherapy are generally not very effective for HCC. Therefore developing effective new strategies to target liver cancer is urgently needed. Many studies have investigated immune responses in patients with HCC 3-6 and have explore approaches to harness antitumor immunity. 3, 7-10 Immunotherapy for liver cancer has also been actively investigated, and some clinical trials have been reported 11. Attention has mainly focused on the induction of HCC-tumor specific immunity by introducing tumor-associated antigens (TAA). 12-14 Among these TAAs, alpha-fetal protein (AFP) and glypican3 have generated significant interest. 15, 16, 17 Both proteins are expressed during embryogenesis, but silent in most adult tissues. These two proteins are valuable for HCC diagnosis and they have the potential to be target antigens for immunotherapy. Although evidence of immune response to these two antigens in HCC patients has been demonstrated, eliciting antitumor immunity using these two antigens is still not robust. 18-21 One of the potential explanations is that the expression of these two proteins during embryogenesis may have caused immune tolerance in adults. However, it is still unclear how the host immunosurvillence mechanisms interact with HCC to allow tumor to grow. Recently, many studies have suggested the presence of suppressor immune cells that play an important role in immune homeostasis and cancer development/progression. 22-22
Several studies, including ours, have shown that development of HCC is associated with a number of immune suppressive mechanisms. 5, 23-25 Two groups of cells are implicated as main mediators of immune suppression in cancer: regulatory T cells (Tregs) and regulatory myeloid cells - myeloid-derived suppressor cells (MDSCs). MDSCs are a heterogeneous population of cells in tumor-bearing mice and cancer patients that have been shown to inhibit T-cell responses. 26-28 Upregulation of Tregs and MDSC has been demonstrated in patients with HCC. 25, 26, 30 MDSCs (identified as CD11b+Gr-1+cells) have also been shown to accumulate in the spleens, bone marrows and tumor tissues in tumor bearing mice. 28 It has been suggested that MDSCs suppresses specific cellular response to cancer cells. 26 A recent report has also highlighted that MDSCs can inhibit NK cells in HCC patients. 30 It is possible that modulation of these cells may offer a new avenue for immunotherapy against cancer. Interestingly, current chemotherapy may already take advantage of this immunoregulatory mechanism. One recent report has shown that the kinase inhibitor, sunitinib, is capable of reversing the accumulation of the suppressive cell populations: MDSCs and Tregs, which is a potential new mechanism for this chemotherapy. 31 Sorafenib, another multiple kinases inhibitor, has recently been approved as the first agent for advanced liver cancer therapy. Besides its cell growth inhibitory activity, the precise mechanisms by which Sorafenib suppresses HCC in patients remain to be defined.
To understand the immunoregulation in HCC, to define the mechanisms of therapeutic anti-HCC agents, and to develop more effective immunotherapy, it is essential to have a robust and reliable small animal model. Genetically engineered mouse models and xenograft liver cancer models are excellent tools to study the intracellular signaling pathways altered in hepatocarcinogenesis and to evaluate therapeutic efficacy for anticancer molecules. However, models that can be used for studying immunoregulation in HCC are limited. Recently, Gonzalez-Carmona et al have described an immunocompetent murine model using mouse HCC cell line Hepa129 and the C3H/HeNcrl mice for investigating anti-tumor dendritic cells. 32
In this report, we describe a robust mouse liver cancer model in a commonly used immunocompetent BALB/c mouse strain. Immune responses and suppressor cells form a complex network in this HCC cancer bearing mouse model. Moreover, we have shown that Sorafenib is capable of suppressing HCC progression in the mice and down regulating Tregs and MDSCS, which suggests a novel anti-HCC mechanism in this tumor model.
Materials and Methods
Tumor cell line and cell culture
A mouse liver hepatoma cell line BNL 1ME A.7R.1 (referred as 1MEA in this study), was obtained from American Type Culture Collection (ATCC), and maintained in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% heat-inactivated (56°C, 30 min) fetal bovine serum (FBS) (GIBCO) and 100 U/mL penicillin, 100 μg/mL streptomycin (Cellgro), L-glutamin (2 mM) in a humidified atmosphere of 5% CO2 at 37°C. This tumor cell line was developed from BALB/c mouse 33.
Mice and tumor injection
BALB/c mice (6–8 wk of age) were obtained from the Jackson Laboratory. Mice were kept in pathogen-free conditions and handled in accordance with the requirements of the Institutional Guideline for Animal Experiments. The right flanks of BALB/c mice were injected subcutaneously (s.c.) with 5 × 106 1MEA cells in 100μL of phosphate-buffered saline (PBS). At the end points of the experiments, mice were sacrificed and spleens were collected. Tumors were separated from mice, measured. Tumor size was indicated as weight (mg). Representative tumor tissues were carefully excised and surface blood was removed by rinsing in cold PBS, then fixed in 10% neutral formalin and processed for histology staining, or prepared into single cells and stained with fluorescence-conjugated antibodies for flow cytometry analysis.
Flow cytometric analysis
Splenic cells were obtained by pressing splenic tissue or lymph node tissue, respectively, through a Nylon Cell Strainer of 40 μm mesh (BD Falcon™). Bone marrow cells were harvested by flushing femurs and tibias with cold 1×PBS in a syringe with a 27-gauge needle. Tumors were prepared as described above to obtain single cell suspensions. Red blood cells were removed using red blood lysing buffer (Sigma). In order to decrease non-specific binding of immunoglobulins, cells were incubated for 30 min with anti-CD16/32 (Fc block). The resulting cells were stained with flueorecence-conjugated antibodies anti-Gr-1, anti-CD11b Abs, anti-CD3, -CD4, -CD25 (BD Pharmingen) and anti-FOXP3 (eBioscience). After washing with PBS twice, at least 200,000 events were collected and analyzed on a FACSCalibur system with CELLQuest software (Becton Dickinson) for each analysis. The data were expressed as a proportion of positive cells (compared with cells stained with an irrelevant control antibody).
CD11b+ cell functional assays
CD11b+ cells with purity of more than 90%, were positively selected from splenocytes derived from tumor-free and tumor-bearing mice by using CD11b microbeads (Miltenyi Biotec), respectively. The CD11b+-depleted splenocytes were used as responders and placed in triplicates into a U-bottom 96-well plate (1 × 105/well) and cocultured with CD11b+ cells (as stimulators) at different ratios in the presence of anti-CD3 ( 5μg/mL ) for 48 hours. Eighteen hours before harvesting, cells were pulsed with [3H]thymidine (1 μCi/well; PerkinElmer). [3H]thymidine uptake was counted using a liquid scintillation counter and expressed as the mean counts per min (cpm).
Sorafenib treatment in vivo
Sorafenib (Nexavar, 200mg / tablets) was dissolved in Cremophor EL / 95% ethanol (termed as solvent) (50:50; Sigma), prepared stock solution at 6 mg/mL and stored in −20°C for in vivo study. After two weeks from tumor cells challenging, mice were divided into two groups ( 10 mice / group) and orally fed Sorafenib at 0.6mg / 100ul/mouse ( 30 mg/kg, once daily) or 100ul Cremophor EL / 95% ethanol (control group), respectively, by gavage for two weeks. At the endpoints of experiments, mice were sacrificed and tumors and spleens were dissociated and weighed, femurs and tibias were collected. Splenocytes, bone marrow cells and single tumor cells were prepared for further analysis.
Immunohistochemistry analysis of Tumor-Infiltrating Lymphocytes
Tumor tissues were immediately removed from the mice following CO2 asphyxiation and fixed in 10% formalin for 4 h. Subsequent tissue processing, including sectioning and staining, was performed using services provided by the University of Florida’s Molecular Pathology and Immunology Core. Briefly, sections through formalin-fixed samples were paraffin-embedded, cut and collected 100 μm apart, then stained with antibodies to CD4, CD8, CD25.
To obtain TIL from tumors, tumor tissues were cut into small pieces and placed into 40 μm Nylon Cell Strainers in 10 mL cold PBS, and prepared single cell suspension by using hub of syringe. After centrifugation, the resulting cell suspension was washed once in HBSS, resuspended in 5 mL Histopaque®-1077 (Sigma) and placed in a sterile 15 mL conical tube, which was centrifuged for 30 minutes at 3300 rpm at room temperature. The grey layer was collected. After washing with DMEM once, the cells were incubated with 1 mL red blood cell lysis buffer (Sigma) for 5 minutes at room temperature; subsequently, 25 mL sterile PBS was added to stop the lysis. The cells were washed, counted and stained with antibodies to CD3, CD4, CD8 and CD25 for flow cytometry analysis.
Statistical analysis
The statistical significance between values was determined by Student’s t test. All data were expressed as the mean ± SD. Probability values of p≤0.05 were considered significant.
Results
Murine tumor cell line grows and initiates immune response in immunocompetent Balb/c mice
The murine cell line 1MEA was a chemically transformed cell line from a Balb/c mouse. To demonstrate the tumorigenic potential, we injected 5 million 1MEA cells per mouse s.c on the right hind flank of the mouse. Tumors could be palpated around 10 days in all the mice injected (total number 25). As demonstrated in Figure 1, the average tumor size was around 1.8 cm in greatest dimension in about two weeks (Figure 1A and 1B). Histologically, the tumor showed features of poorly differentiated hepatocellular carcinoma (Figure 1C). Marked splenomegaly occurred in virtually all the mice that had tumor growth (Figure 1D). On average, the size of the spleen in a tumor-bearing mouse was three times larger than that of the control mouse (Fig. 1E and 1F). The size of the spleens was consistent with the number of splenic cells when total splenic cells were counted. Both CD4 and CD8 cells were present in the spleen, and the ratio of CD4/CD8 was not significantly altered. Histologically, there was no significant morphological difference between spleens from the control mice and the spleens from the tumor-bearing mice (data not shown).
Figure 1. Tumor growth and enlarged spleens in immune competent BALB/c mice.
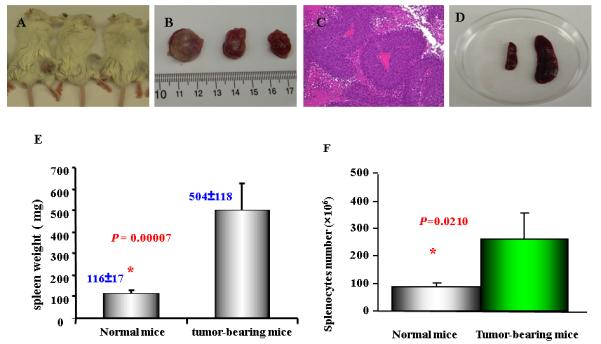
Six to eight weeks old BALB/c mice were injected (s.c.) 1MEA cells ( 5 × 106 / per mouse). After two weeks, the tumors were measurable. As mentioned in Materials and Methods, at the endpoint of experiments (the end of the forth week), spleens and tumor tissues were collected and weighed. Splenocytes were prepared and counted after red cells lysis. A. Tumor-bearing BALB/c mice; B. Tumor tissue separated from the mice. C. Histological features of the liver cancer tissue (H&E stain) showing poorly differentiated hepatocellular carcinoma; D. the size of spleens from Balb/c mice with tumor (right) and normal mice (left); E. the weight of spleens from normal mice and tumor-bearing mice (n=9); F. the splenocytes from normal mice (87± 14 × 106; n=3) and tumor-bearing mice (263 ± 95 × 106; n=5). Data represent the mean ± SD from one of two similar experiments
Liver cancer upregulates immunosuppressive cell population in mice
As we have previously reported, HCC patients have higher percentage of regulatory T cells (CD4+CD25+) 25. To investigate whether the similar phenomenon is also present in the mouse model, we firstly examined the CD4+CD25+ regulatory T cells in the tumor bearing mice. CD3+ T cells were gated for the presence of CD4+ and CD25+ T cells. As shown in Figure 2A and 2B, tumor bearing mice had higher numbers of CD4+CD25+ cells, which is consistent with findings in HCC patients. The data suggests that HCC induces immune suppressive population in this mouse model which is similar to that observed in human disease.
Figure 2. Increased CD4+CD25+ T cells and CD11b+Gr-1+ cells in tumor-bearing mice.
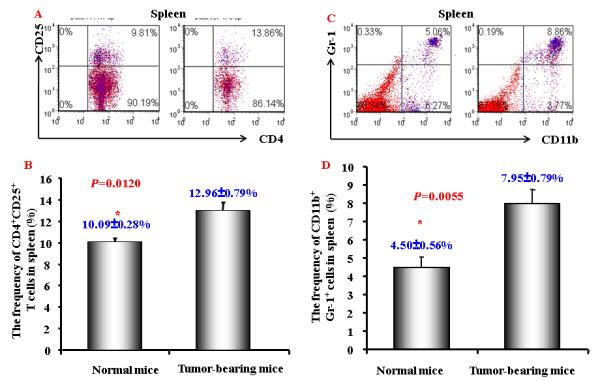
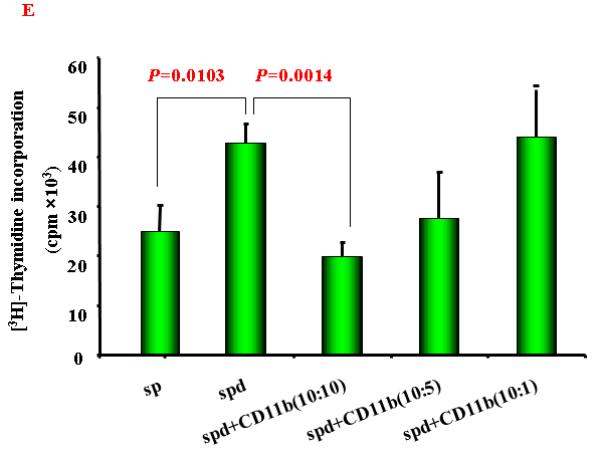
A. A representative flow cytometric data showing the CD4+CD25+ T cells frequency of gated CD4+ T cells in splenocytes from normal and HCC tumor-bearing mice; B. Mean CD4+CD25+ T cells frequency in CD4+ T cells in splenocytes from normal and HCC tumor-bearing mice (n=3) . C and D. A representative (C) and mean ( D) flow cytometry data showing increased CD11b+ Gr-1+cells frequency in splenocytes from normal and HCC tumor-bearing mice (n=3). The gated viable cells were analyzed. Numbers in the figures represent the percentage of fluorescence-positive cells in corresponding areas; E. Functional analysis of CD11b+ cells were evaluated for their ability to suppress autologous CD11b+-depleted splenocytes proliferation by [3H]-thymidine incorporation when stimulated with anti-CD3 and CD28 at various ratios. Values represent mean ± SD (n=3) and are representative examples of three separate experiments.
Another population of suppressive cells, MDSC, has been recently defined as CD11b+Gr-1+. This cell population has been reported to be increased in a number of cancers, including head-neck, pancreatic, HCC, renal and breast cancers. 26, 34, 35 We next examined this population of cells in this animal model. Population of CD11b+Gr-1+MDSC was significantly expanded in tumor-bearing mice (Fig. 2C and 2D). To confirm the immune suppressive activity of these MDSCs, we performed cell proliferation assay. As shown in Figure 2E, MDSCs inhibited T cell proliferation.
Sorafenib inhibits HCC growth in the mouse model
Sorafenib has been approved as a systemic therapeutic agent for advanced HCC. Clinical trials indicate that treatment with Sorafenib modestly increases patient survival. 36, 37 To validate the animal model, we tested the effect of Sorafenib on tumor growth. Mice were first injected with 1MEA cells subcutaneously. Two weeks after the injection, when the tumor average size is around 0.4 cm, Sorafenib treatment was initiated with daily dose of 30 mg/kg. After two weeks of treatment, the mice were sacrificed and tumor sizes were measured. As shown in Figure 3, Sorafenib significantly inhibited tumor growth, the average tumor size was three times smaller than the control group.
Figure 3. Sorafnib inhibits HCC growth in BALB/c mice.
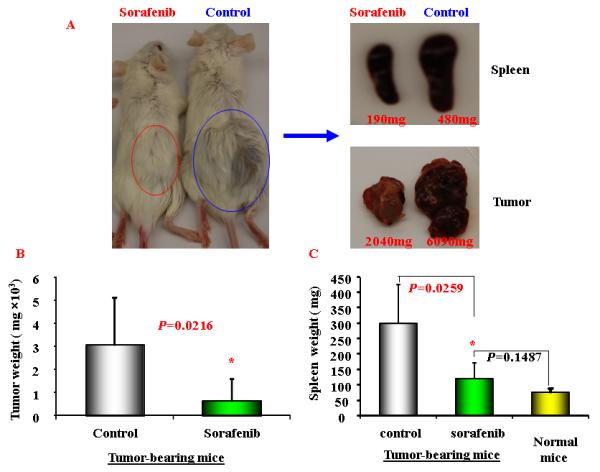
Blab/c mice of 6-8 weeks were subcutaneously injected with 1MEA cells (4 million/mouse) in the right flank. Two weeks later, the mice with measurable tumor were divided into two groups. Each mouse was gavage-fed with Sorafenib (0.6mg/100ul) or solvent (100ul), once daily, five days each week, for two weeks. At endpoint of experiments, mice were sacrificed, tumors and spleens were collected and weighed. A. A representative of HCC tumor-bearing mice after 2 weeks treatment with Soarfenib (left) or solvent (right), spleen (right upper) and tumor (right lower) size. B. the weight of tumor tissues from tumor-bearing mice after 2 weeks treatment with sorafenib or solvent (n=5). C. the weight of spleens from normal mice or tumor-bearing mice after 2 weeks treatment with sorafenib or solvent (n=5). Data represent the mean ± SD of 5 mice per group from one representative experiment of two similar experiments. * P ≤ 0.05
Sorafenib down regulates immunosuppressive cell population
To further evaluate the mechanisms of Sorafenib inhibition of HCC, we tested the hypothesis whether Sorafenib would exert antitumor activity through modulating suppressive populations of the immune cells 38, 399. We first determined the effect of Sorafenib on CD4+, CD8+, CD4+CD25+ and CD11b+Gr-1+ cells in the spleens and bone marrows of normal mice by flow cytometric analysis. As shown in Figure 4A and 4B, Sorafenib had no significant effects on these cells in normal mice. Then we analyzed the effects of Sorafenib on CD4+CD25+FOXP3+ Tregs and CD11b+Gr-1+ MDSCs in tumor environments. As shown in Figure 4C and 4D, tumor bearing mice had significantly elevated Tregs, while the treatment of Sorafenib restored the Tregs population and Foxp3 expression in CD4+CD25+ T cells to the normal level. Sorafenib treatment also significantly reduced the number of CD11b+ Gr-1+cells in both the bone marrows and the spleens. The number of CD11b+ Gr-1+cells were even lower than the normal mice control, suggesting that Sorafenib prevented CD11b+ Gr-1+cell differentiation. To evaluate overall inhibitory effect of Sorafenib to the cellular immune system, we analyzed the absolute cell numbers of CD4+,CD8+, CD4+CD25+ and CD11b+Gr-1+cells in these mice. The data in Table 1 show that splenomegaly in tumor-bearing mice is associated with significantly increased absolute cell number of CD4+CD25+ Tregs and CD11b+ Gr-1+ cells. Sorafenib did not alter the overall CD4+ and CD8+ cells numbers and their ratio (Table 1, Figure 5). By inference, Sorafenib exhibited relative specific activity on Tregs and MDSCs.
Figure 4. Sorafenib treatment significantly decreased the percentage of CD4+CD25+ T cells and CD11b+ cells in tumor-bearing mice.
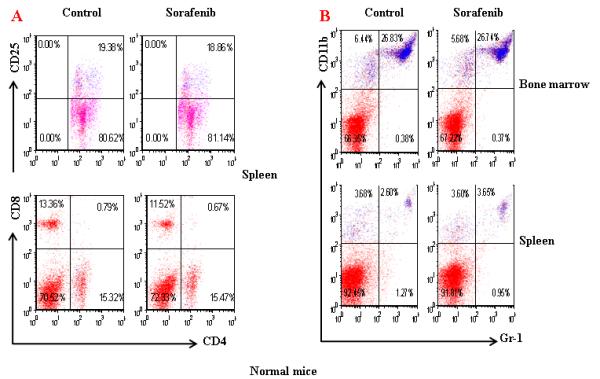
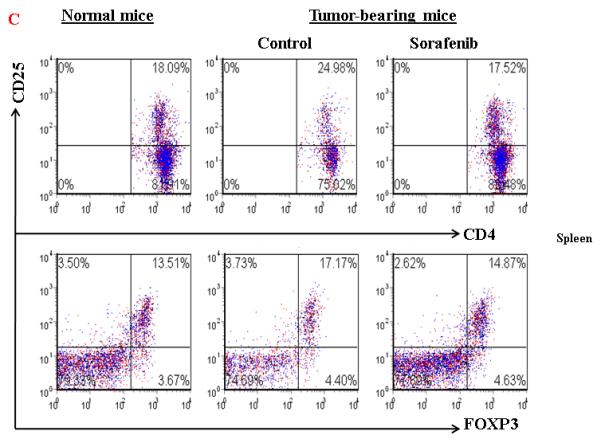
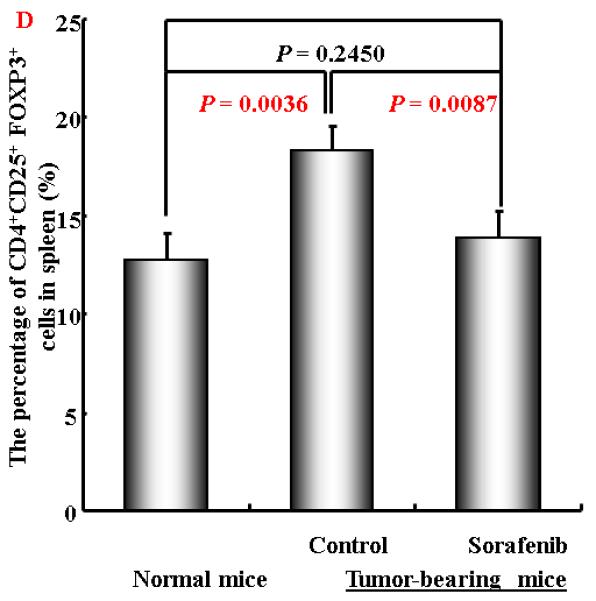
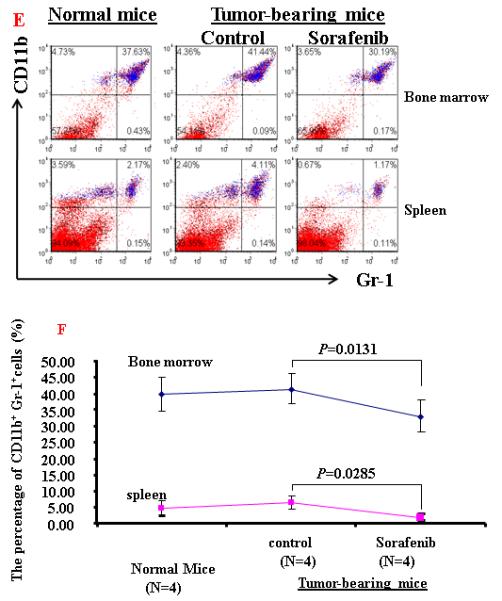
A and B. A representative of flow cytometry data showing the frequency of CD4+CD25+ T cells and CD11b+Gr-1+ cells in spleens and bone marrow from normal mice or tumor-bearing mice treated with solvent or sorafenib; C and D. A representative (C) and collective ( D, n=3) of flow cytometry data showing the expression of FOXP3 in CD4+CD25+ T cells in spleens from normal mice or tumor-bearing mice treated with solvent or sorafenib; E. A representative of flow cytomrtey data showing the frequency of CD11b+ Gr-1+cells in bone marrow (upper panel) and spleen (lower panel) from normal mice or tumor-bearing mice treated with solvent or sorafenib; F. Mean CD11b+ Gr-1+ cells frequency bone marrow and spleen from normal and HCC tumor-bearing mice treated with solvent or sorafenib (n=5) . Data represent the mean ± SD of 3 or 5 mice per group from one representative experiment of two similar experiments.
Table 1.
Increased numbers of CD4+CD25+ regulatory T cells and CD11b+Gr-1+ cells in the spleens of the tumor-bearing mice
| Cell number ( × 106) |
Normal mice | Tumor-bearing mice (Control group) |
Tumor-bearing mice (Sorafenib group) |
|---|---|---|---|
| CD4 | 32.8±4.3 | 32.3±7.4 | 36.8±6.2 |
| CD8 | 20.9±2.8 | 23.7±5.4 | 21.8±3.7 |
| CD4+CD25+ | 3.6±0.5a | 5.3±1.2 | 2.8±0.5a |
| CD11b+Gr-1+ | 7.2±0.9a | 19.2±4.4 | 5.6±0.9a |
Spleens from normal or tumor-bearing mice treated with or without Sorafenib were prepared into single cell suspension. After red cell lysing and wash, cells were suspended, counted, and stained with antibodies for CD3, CD4, CD8, CD25, CD11b and Gr-1, then analyzed by flow cytometry. The absolute numbers of cells were caculated based on the total number of splenocytes and percentages of CD4+, CD8+, CD4+CD25+ and CD11b+Gr-1+ cells in spleens. Values represent mean ± SD from 4 mice per group from one of two independent similar experiments.
vs control group P value <0.05.
Figure 5. Sorafenib treatment maintained the CD4 and CD8 cells in spleen and lymph nodes.
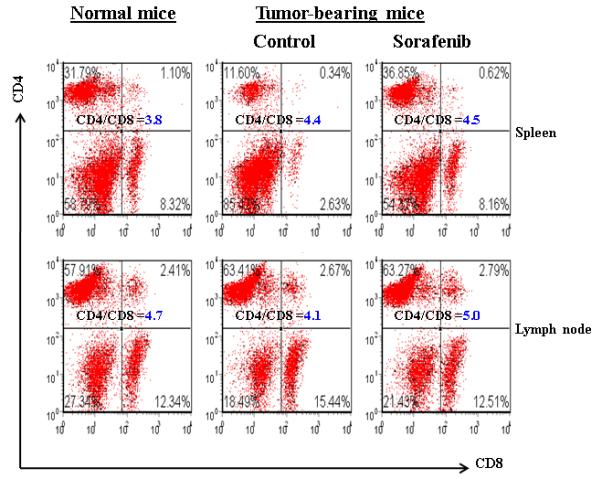
A representative flow cytometric data showing the CD4+and CD8+ T cells frequency of gated lymphocytes in spleen and lymph nodes from normal and HCC tumor-bearing mice treated with solvent or soarfenib. Numbers in the figures represent the percentage of fluorescence-positive cells in corresponding areas, representative data of three independent experiments.
Lastly, we evaluated for the presence of tumor-infiltrating lymphocytes by using immunohistochemical staining and flow cytometry. Unexpectedly, no significant numbers of CD3, CD4 or CD8 T cells were detected in the tumor tissues (data not shown).
Discussion
There are a number of animal models being utilized for the study of liver cancer. However, most of the models rely on transgenic and knockout mice. The major disadvantage of these mouse models is the limitation of studying immune regulation, because of the immune tolerance developed in these mice. Recently, an immunocompentent mouse model has been described for the study of dendritic cells. 32 There is emerging evidence that immune responses play a significant role in the development and progression of HCC. Thus, a robust mouse model that possesses intact immunity would be a valuable system for the study of HCC.
A murine HCC cell line, 1MEA, was first established two decades ago through chemical carcinogenesis of the immortalized cell line BNL derived from a BALB/c mouse. 33 The 1MEA cells can readily grow into tumor in BALB/c mice, indicating that there is no immune rejection to this tumor in these mice. This observation prompted us to ask the following questions: 1) Can the 1MEA tumor cells initiate immune response in the mice? 2) What is the nature of these immune responses that allow the tumor growth in an immune competent mouse? And 3) Can the immune responses be modulated and tumor growth altered? Through a series experiments, we have begun to answer these questions.
Murine HCC cell line initiates robust immune response in BALB/c tumor. Our experiments clearly demonstrated that 1MEA cell line can grow in BALB/c mice with a high rate of efficiency. With 5 million cells in the initial injection, a one-centimeter tumor is visible within two weeks. While we focused on tumor ectopic growth for the purpose of conveniently monitoring tumor size, we have also implanted the tumor cells in the mouse livers and achieved the similar tumor growth. The tumors are locally invasive to the surrounding tissue, but metastasis is not observed, even in mice with primary tumor size is as large as 4.5 cm.
One notable gross finding of the tumor bearing mice is the enlargement of spleens, which has been noticed in several other cancer murine models 40, 41. The enlarged spleens suggest the initiation of immune responses to the tumors. The sizes of the spleens positively correlated with the tumor burdens as shown in Figure 1. Extensive evaluation of splenocytes suggests that the splenic enlargement is associated with increased numbers of immune cells, mainly Tregs and MDSC cells. The overall CD4 and CD8 cells and their ratios are not significantly altered. Histologically, there was no distinctive morphological difference between the spleens from the normal mice and the tumor-bearing mice. The observation of the splenomegaly in the mice is interesting considering that such phenomenon does not exist in human liver cancer patients.
As our experiments demonstrated, HCC could readily grow in the mice in spite of the robust immune responses. This observation would suggest that specific antitumor activity must have been inhibited. We have reported before that human HCC induces an immunosuppressive phenotype as reflected by the increased circulating Tregs 25. Recently, another suppressive population of cells, MDSCs, have been shown to be elevated in HCC patients 26. Therefore, we decided to examine the Tregs and MDSCs in the tumor bearing mice. Our data show that both Tregs and MDSCs were significantly upregulated in these mice, and they were positively correlated with tumor burden. Moreover, the MDSC cells also exhibited typical suppressive activity when co-cultured with effector T cells. We have previously reported that supernatants from human HCC cell lines can promote Tregs in vitro, indicating that tumor cells can secrete factors that modulate host immune system by promoting immunosuppressive populations. 23 Identification of these factors would have a significant impact on the understanding of the tumor immunity and immune modulation for antitumor activity. One potential limitation of our current model is the fact that we have injected tumor in an ectopic site rather than the natural location of the liver. Further experiments will validate these finding using an orthotopic model.
The precise mechanisms by which Sorefanib influences HCC progression and patient survival is unclear. Since it is a kinase inhibitor, one possible mechanism is that it directly induces cancer cell apoptosis or inhibits cancer cell proliferation. Such activity has been reported previously. 42 We tested the in-vitro activity of Sorafenib in 1MEA cells and no significant tumor cell death was seen, but there was a significant impact on 1MEA cell proliferation (data not shown). We then tested the effect of Sorafenib in the mouse model. As our data shown in Figure 3, Sorafenib exhibited overt antitumor activity, which is unlikely due to anti-proliferation alone. We then examined the possibility of the immune modulatory effect of Sorafenib in the HCC model. Indeed, the animals treated with the drug exhibited significantly reduced numbers of both Tregs and MSDCs. This data suggests an immunomodulatory mechanism of Sorafenib on HCC in vivo. The marked decrease in MDSC from the bone marrow also suggests a direct effect on progenitor cell production, which requires further investigation. One note of caution is that we cannot exclude the possibility of the secondary effect on the suppressive populations, i.e, the slow growth of HCC caused by Sorafenib would have less capacity to induce Tregs and MDSCs. We believe that both direct and indirect effects may exist in the Sorafenib-treated mice.
There are recent reports about the effect of sunitinib, another kinase inhibitor, on the inhibition of MDSC. 31 Ozao-Choy et al. hypothesized that malate, a receptor tyrosine kinase inhibitor, could reverse MDSC-mediated immune suppression and modulate the tumor microenvironment, thereby improving the efficacy of immune-based therapies. Treatment with sunitinib decreased the number of MDSC and Treg in advanced tumor-bearing animals. Furthermore, it not only reduced the suppressive function of MDSCs but also prevented tumor-specific T-cell anergy and Treg development. Interestingly, sunitinib treatment resulted in reduced expression of interleukin (IL)-10, transforming growth factor-beta, and Foxp3 but enhanced expression of Th1 cytokine IFN-gamma and increased CTL responses in isolated tumor-infiltrating leukocytes. 38
In a variety of human solid tumors, tumor-infiltrating lymphocytes (TIL) are considered to play an important role in anticancer immune surveillance. Among the mature human T-cell population, three subtypes seem to play a major role in immunosurveillance against tumor growth: CD8+, CD4+, and T Treg cells. Curiel et al reported that tumor infiltration by Treg cells predicted reduced survival, 43 but others have failed to reproduce this finding. 44 In many rodent tumor models, the mobilization of T cell responses is also sufficient to eradicate tumors early during their establishment. 45, 46 However, such mobilization is clearly less effective when it occurs late during tumor progression, in both rodent tumor models and human cancer patients. 47, 48 Studies suggested an inability of functionally normal T cells to infiltrate or persist in late stage solid tumors because of physical barriers and/or emigration from the tumor site. 48, 49 In our study, we did not detect significant infiltration of T cells in the tumor tissues. The reason may be partially due to late stage of tumor progression or the ectopic nature of the tumor.
In summary, we have established a novel, immune competent HCC mouse model that demonstrates the relevance to human HCC. Using this model, we have studied the immune response to HCC and examined the treatment effect of kinase inhibitor, Sorafenib. Our results show that HCC can upregulates immunosuppressive populations of cells: Tregs and MDSCs. We also found that Sorefanib modulated immune responses by suppressing Tregs and MDSCs. We believe that this validated model can be used for understanding the immunoregulations in HCC, immunotherapy, and evaluating novel therapeutic agents.
Acknowledgement
The work was supported in part by DK02958 and RR023976 from NIH to Chen Liu, and AI061158 and UL1RR029890 to David Nelson.
Footnotes
Note: Drs. Liu and Nelson share the senior authorship.
References
- 1.Bosch FX, Ribes J, Diaz M, et al. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127:S27–34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Matar P, Alaniz L, Rozados V, et al. Immunotherapy for liver tumors: present status and future prospects. J Biomed Sci. 2009;16:30. doi: 10.1186/1423-0127-16-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Unitt E, Marshall A, Gelson W, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol. 2006;45:246–53. doi: 10.1016/j.jhep.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 5.Cabrera R, Ararat M, Cao M, et al. Hepatocellular Carcinoma Immunopathogenesis: Clinical Evidence for Global T Cell Defects and an Immunomodulatory Role for Soluble CD25 (sCD25) Dig Dis Sci. 2009 doi: 10.1007/s10620-009-0955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Zhou Y, Qiu S, et al. Autoantibodies to tumor-associated antigens combined with abnormal alpha-fetoprotein enhance immunodiagnosis of hepatocellular carcinoma. Cancer Lett. 2009 doi: 10.1016/j.canlet.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stift A, Friedl J, Dubsky P, et al. Dendritic cell-based vaccination in solid cancer. J Clin Oncol. 2003;21:135–42. doi: 10.1200/JCO.2003.02.135. [DOI] [PubMed] [Google Scholar]
- 8.Llovet JM. Updated treatment approach to hepatocellular carcinoma. J Gastroenterol. 2005;40:225–35. doi: 10.1007/s00535-005-1566-3. [DOI] [PubMed] [Google Scholar]
- 9.Wang XG, Revskaya E, Bryan RA, et al. Treating cancer as an infectious disease--viral antigens as novel targets for treatment and potential prevention of tumors of viral etiology. PLoS One. 2007;2:e1114. doi: 10.1371/journal.pone.0001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibolet O, Alper R, Zlotogarov L, et al. Suppression of hepatocellular carcinoma growth via oral immune regulation towards tumor-associated antigens is associated with increased NKT and CD8+ lymphocytes. Oncology. 2004;66:323–30. doi: 10.1159/000078334. [DOI] [PubMed] [Google Scholar]
- 11.Greten TF, Manns MP, Korangy F. Immunotherapy of HCC. Rev Recent Clin Trials. 2008;3:31–9. doi: 10.2174/157488708783330549. [DOI] [PubMed] [Google Scholar]
- 12.Butterfield LH. Immunotherapeutic strategies for hepatocellular carcinoma. Gastroenterology. 2004;127:S232–41. doi: 10.1053/j.gastro.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 13.Sung YK, Hwang SY, Park MK, et al. Glypican-3 is overexpressed in human hepatocellular carcinoma. Cancer Sci. 2003;94:259–62. doi: 10.1111/j.1349-7006.2003.tb01430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuen MF, Lai CL. Serological markers of liver cancer. Best Pract Res Clin Gastroenterol. 2005;19:91–9. doi: 10.1016/j.bpg.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Carmona MA, Marten A, Hoffmann P, et al. Patient-derived dendritic cells transduced with an a-fetoprotein-encoding adenovirus and co-cultured with autologous cytokine-induced lymphocytes induce a specific and strong immune response against hepatocellular carcinoma cells. Liver Int. 2006;26:369–79. doi: 10.1111/j.1478-3231.2005.01235.x. [DOI] [PubMed] [Google Scholar]
- 16.Abelev GI, Eraiser TL. Cellular aspects of alpha-fetoprotein reexpression in tumors. Semin Cancer Biol. 1999;9:95–107. doi: 10.1006/scbi.1998.0084. [DOI] [PubMed] [Google Scholar]
- 17.Kandil DH, Cooper K. Glypican-3: a novel diagnostic marker for hepatocellular carcinoma and more. Adv Anat Pathol. 2009;16:125–9. doi: 10.1097/PAP.0b013e3181992455. [DOI] [PubMed] [Google Scholar]
- 18.Zhang HM, Zhang LW, Ren J, et al. Induction of alpha-fetoprotein-specific CD4- and CD8-mediated T-cell response using RNA-transfected dendritic cells. Cell Immunol. 2006;239:144–50. doi: 10.1016/j.cellimm.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Mizejewski GJ. Alpha-fetoprotein (AFP)-derived peptides as epitopes for hepatoma immunotherapy: a commentary. Cancer Immunol Immunother. 2009;58:159–70. doi: 10.1007/s00262-008-0548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alisa A, Ives A, Pathan AA, et al. Analysis of CD4+ T-Cell responses to a novel alpha-fetoprotein-derived epitope in hepatocellular carcinoma patients. Clin Cancer Res. 2005;11:6686–94. doi: 10.1158/1078-0432.CCR-05-0382. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi E, Motomura Y, Shirakawa H, et al. Detection of glypican-3-specific CTLs in chronic hepatitis and liver cirrhosis. Oncol Rep. 2009;22:149–54. doi: 10.3892/or_00000418. [DOI] [PubMed] [Google Scholar]
- 22.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 23.Cao M, Cabrera R, Xu Y, et al. Hepatocellular carcinoma cell supernatants increase expansion and function of CD4(+)CD25(+) regulatory T cells. Lab Invest. 2007;87:582–90. doi: 10.1038/labinvest.3700540. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi N, Hiraoka N, Yamagami W, et al. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. 2007;13:902–11. doi: 10.1158/1078-0432.CCR-06-2363. [DOI] [PubMed] [Google Scholar]
- 25.Cabrera R, Tu Z, Xu Y, et al. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology. 2004;40:1062–71. doi: 10.1002/hep.20454. [DOI] [PubMed] [Google Scholar]
- 26.Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135:234–43. doi: 10.1053/j.gastro.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–23. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 28.Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marigo I, Dolcetti L, Serafini P, et al. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 30.Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807. doi: 10.1002/hep.23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Carmona MA, Lukacs-Kornek V, Timmerman A, et al. CD40ligand-expressing dendritic cells induce regression of hepatocellular carcinoma by activating innate and acquired immunity in vivo. Hepatology. 2008;48:157–68. doi: 10.1002/hep.22296. [DOI] [PubMed] [Google Scholar]
- 33.Patek PQ, Collins JL, Cohn M. Transformed cell lines susceptible or resistant to in vivo surveillance against tumorigenesis. Nature. 1978;276:510–1. doi: 10.1038/276510a0. [DOI] [PubMed] [Google Scholar]
- 34.Ochoa AC, Zea AH, Hernandez C, et al. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–726s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]
- 35.Youn JI, Nagaraj S, Collazo M, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24:4293–300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- 37.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–27. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xin H, Zhang C, Herrmann A, et al. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DuPre SA, Hunter KW., Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol. 2007;82:12–24. doi: 10.1016/j.yexmp.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Sato N, Michaelides MC, Wallack MK. Characterization of tumorigenicity, mortality, metastasis, and splenomegaly of two cultured murine colon lines. Cancer Res. 1981;41:2267–72. [PubMed] [Google Scholar]
- 42.Peng CL, Guo W, Ji T, et al. Sorafenib induces growth inhibition and apoptosis in human synovial sarcoma cells via inhibiting the RAF/MEK/ERK signaling pathway. Cancer Biol Ther. 2009;8:1729–36. doi: 10.4161/cbt.8.18.9208. [DOI] [PubMed] [Google Scholar]
- 43.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 44.Mori M, Ohtani H, Naito Y, et al. Infiltration of CD8+ T cells in non-small cell lung cancer is associated with dedifferentiation of cancer cells, but not with prognosis. Tohoku J Exp Med. 2000;191:113–8. doi: 10.1620/tjem.191.113. [DOI] [PubMed] [Google Scholar]
- 45.Sampson JH, Archer GE, Ashley DM, et al. Subcutaneous vaccination with irradiated, cytokine-producing tumor cells stimulates CD8+ cell-mediated immunity against tumors located in the “immunologically privileged” central nervous system. Proc Natl Acad Sci U S A. 1996;93:10399–404. doi: 10.1073/pnas.93.19.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zitvogel L, Mayordomo JI, Tjandrawan T, et al. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T helper cell 1-associated cytokines. J Exp Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson HL, Donermeyer DL, Ikeda H, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–76. doi: 10.1016/s1074-7613(00)00026-1. [DOI] [PubMed] [Google Scholar]
- 48.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–4. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 49.Shrikant P, Mescher MF. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. 1999;162:2858–66. [PubMed] [Google Scholar]