

Abstract
Purpose
Repeated, intermittent administration of the psychotropic NMDA antagonist phencyclidine (PCP) to laboratory animals causes impairment in cognitive and executive functions, modeling important sequelae of schizophrenia; these effects are thought to be due to a dysregulation of neurotransmission within the prefrontal cortex. Atypical antipsychotic drugs have been reported to have measurable, if incomplete, effects on cognitive dysfunction in this model, and these effects may be due to their ability to normalize a subset of the physiological deficits occurring within the prefrontal cortex. Asenapine is an atypical antipsychotic approved in the US for the treatment of schizophrenia and for the treatment, as monotherapy or adjunctive therapy to lithium or valproate, of acute manic or mixed episodes associated bipolar I disorder. To understand its cognitive and neurochemical actions more fully, we explored the effects of short- and long-term dosing with asenapine on measures of cognitive and motor function in normal monkeys and in those previously exposed for 2 weeks to PCP; we further studied the impact of treatment with asenapine on dopamine and serotonin turnover in discrete brain regions from the same cohort.
Methods
Monkeys were trained to perform reversal learning and object retrieval procedures before twice-daily administration of PCP (0.3 mg/kg intramuscular) or saline for 14 days. Tests confirmed cognitive deficits in PCP-exposed animals before beginning twice-daily administration of saline (control) or asenapine (50, 100, or 150 μg/kg, intramuscular). Dopamine and serotonin turnover were assessed in 15 specific brain regions by high-pressure liquid chromatography measures of the ratio of parent amine to its major metabolite.
Results
On average, PCP-treated monkeys made twice as many errors in the reversal task as did control monkeys. Asenapine facilitated reversal learning performance in PCP-exposed monkeys, with improvements at trend level after 1 week of administration and reaching significance after 2–4 weeks of dosing. In week 4, the improvement with asenapine 150 μg/kg (p=0.01) rendered the performance of PCP-exposed monkeys indistinguishable from that of normal monkeys without compromising fine motor function. Asenapine administration (150 μg/kg twice daily) produced an increase in dopamine and serotonin turnover in most brain regions of control monkeys and asenapine (50–150 μg/kg) increased dopamine and serotonin turnover in several brain regions of subchronic PCP-treated monkeys. No significant changes in the steady-state levels of dopamine or serotonin were observed in any brain region except for the central amygdala, in which a significant depletion of dopamine was observed in PCP-treated control monkeys; asenapine treatment reversed this dopamine depletion. A significant decrease in serotonin utilization was observed in the orbitofrontal cortex and nucleus accumbens in PCP monkeys, which may underlie poor reversal learning. In the same brain regions, dopamine utilization was not affected. Asenapine ameliorated this serotonin deficit in a dose-related manner that matched its efficacy for reversing the cognitive deficit.
Conclusions
In this model of cognitive dysfunction, asenapine produced substantial gains in executive functions that were maintained with long-term administration. The cognition-enhancing effects of asenapine and the neurochemical changes in serotonin and dopamine turnover seen in this study are hypothesized to be primarily related to its potent serotonergic and noradrenergic receptor binding properties, and support the potential for asenapine to reduce cognitive dysfunction in patients with schizophrenia and bipolar disorder.
Keywords: asenapine, cognitive function, phencyclidine, primate, schizophrenia, serotonin
1. Introduction
The cognitive symptoms of schizophrenia remain a target of intensive research, both to uncover the relevant pathophysiological determinants and to highlight potential pharmacotherapeutic strategies. The problems of poor memory and attention, impaired executive and verbal functions and deficits of psychomotor and sensory capabilities combine to dramatically impair psychosocial function in schizophrenia (Kalkstein et al., 2010); however, little is known about how to achieve more than extremely modest gains in these domains. This lack of knowledge is due, in part, to poor insight into the disease processes that ultimately lead to these cognitive problems.
The success of efforts to enhance cognition in schizophrenia depends, to some extent, on the availability of pre-clinical models that sufficiently represent the relevant disease features to permit invasive and/or exploratory research not possible in humans (Castner et al., 2004; Floresco et al., 2005; Green and Braff, 2001; Hagan and Jones, 2005; Jentsch, 2003; Robbins, 2005). Non-human primate models are of particular interest, inasmuch as they can be assessed for cognitive functioning using tasks with both face and construct validity.
An influential model for the pathophysiologic mechanisms in schizophrenia posits that low activity of the N-methyl-D-aspartate/glutamate (NMDA) receptor is a critical component of the disease process. This model is based, in part, upon the fact that in otherwise normal humans, acute administration of NMDA receptor antagonists (such as phencyclidine [PCP] and ketamine) provoke profound cognitive impairments similar to those observed in schizophrenia (Krystal et al., 1994; Malhotra et al., 1997; Malhotra et al., 1996; Newcomer et al., 1999; Umbricht et al., 2000). Importantly, the administration of these agents to monkeys performing complex cognitive tasks results in very similar impairments (Taffe et al., 2002).
Our own work has identified the emergence of deficits of prefrontal cortical cognitive functions after long-term administration of PCP to monkeys. There is relevant evidence that chronic PCP ingestion in humans produces persistent psychotomimetic effects that outlast the direct effects of the drug, and continue to be present after prolonged discontinuation of drug use (Allen and Young, 1978; Rainey and Crowder, 1975). Previous behavioral studies have suggested that following long-term exposure to PCP there are impairments on frontal lobe-dependent task in rats and monkeys, and that these deficits are specific in that basic sensory motivational and motoric processes are unaffected by the drug (Jentsch et al., 1998a; Jentsch et al., 1997b; Jentsch et al., 2000). These impairments appear to depend upon a decline in dopaminergic transmission which is secondary to the low NMDA receptor function (Jentsch et al., 1997b; Jentsch and Roth, 1999; Jentsch et al., 2000; Tsukada et al., 2005). This model has also been pursued using rodents as subjects, with mixed results (Abdul-Monim et al., 2006; Andersen and Pouzet, 2004; Deschenes et al., 2006; Fletcher et al., 2005; Hashimoto et al., 2007; Hashimoto et al., 2005; Jentsch and Taylor, 2001; Laurent and Podhorna, 2004; Marquis et al., 2003; Rodefer et al., 2005; Schwabe et al., 2006; Stefani and Moghaddam, 2002). However, the effects of PCP may depend on testing subjects in procedures that maximize demands upon flexible responding (e.g., reversal learning, continuous alternation tasks). If this hypothesis is correct, the PCP model may represent a powerful approach for dissecting the mechanisms underlying perseverative responding in schizophrenia.
Atypical antipsychotic drugs produce small gains in reversal learning and object retrieval in chronic PCP-exposed monkeys (Jentsch, 2003). Asenapine is an atypical antipsychotic approved in the United States for the treatment of schizophrenia and the acute treatment, as monotherapy or adjunctive therapy to lithium or valproate, of manic or mixed episodes associated with bipolar I disorder (Merck, 2010). In the European Union, asenapine is indicated for the treatment of moderate to severe manic episodes associated with bipolar I disorder in adults (European Medicines Agency, 2010). It has been reported that asenapine improves positive and negative symptoms in patients with schizophrenia at least as effectively as risperidone (Potkin et al., 2007). Asenapine is derived from a chemical template different from that of currently available antipsychotics and has a human receptor signature and functional activity profile different from those of available antipsychotic agents (Figure 1). Asenapine is a multi-target agent with relatively higher affinity for serotonin (5-hydroxytryptamine, 5HT) and noradrenergic receptor subtypes compared to dopamine (DA) and histamine and in particular muscarinic cholinergic receptors, with its most potent interactions at 5HT2A and 5HT2C receptors. In vitro assessments have shown that asenapine acts as an antagonist at monoamine and histamine receptors (Shahid et al., 2009). Although the receptor profile of asenapine is similar in many respects to clozapine, asenapine differs most strikingly from clozapine in its diminished affinity for muscarinic receptors relative to its D2 potency (Shahid et al., 2009).
Figure 1.
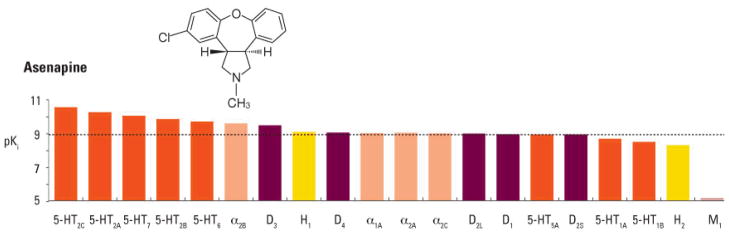
Chemical structure and receptor binding affinity of asenapine. A higher pKi indicates greater binding affinity. The dashed line represents the pKi of asenapine at the D2L receptor. Reprinted with permission from Journal of Psychopharmacology. © Sage Publications.
In this study, we explored the effects of short- and long-term asenapine dosing, producing therapeutically relevant plasma concentrations, on reversal learning and object retrieval in normal and chronic PCP-exposed monkeys. By assessing the behavioral and neurochemical impact of asenapine in our PCP primate model, we evaluated the potential mechanisms that might be attributed to the beneficial effects of asenapine in schizophrenia.
To examine this point, we measured the effects of 2 weeks of dosing with PCP (or saline) on performance of a test of discrimination learning and reversal in monkeys. This test contains, within a single session, measures of simple stimulus-reward learning and response flexibility; importantly, patients with schizophrenia exhibit selective deficits when tested on similar reversal learning tasks (Waltz and Gold, 2007). We also assessed performance of a test of simple response inhibition, the object retrieval/detour task, in the same subjects based upon our earlier finding that this task is impacted by subchronic PCP administration (Jentsch et al., 2000). After completion of the behavioral assessment, the impact of this treatment on DA and 5HT turnover in discrete brain regions was measured ex vivo 28 days after initiation of asenapine or saline treatment and 2 hours following the last treatment.
This study used adolescent monkeys for two reasons. First, adolescence appears to be the period when the final neuronal changes that will ultimately underlie schizophrenia occur (Lewis and Levitt, 2002). Additionally, several reports now indicate that developmental exposure to PCP produces more persistent and dramatic behavioral and molecular changes (Schwabe et al., 2006; Stefani and Moghaddam, 2005; Thomsen et al., 2010).
2. Materials and Methods
2.1. Animals
Male and female adolescent vervet monkeys (Chlorocebus aethiops sabeus) at the St. Kitts Biomedical Research Foundation were used in these studies. The monkeys were individually housed in stainless steel primate cages in an open-air, but covered, facility. Individual housing was required because of the need to test animals in their home cages without disruptions from other subjects. Therefore, they experienced a natural day/night cycle. All subjects were maintained with ad libitum water and a diet of monkey chow. The monkeys were fed only after behavioral testing, which allowed a maximal interval until the following day of study. In addition, highly palatable reward items (banana slices and apple bits) were used to motivate performance. Therefore, caloric restriction was not required. Testing was always conducted during the day (between 8:30 am and 4:30 pm) and artificial lights were never turned on after dark. Studies were approved by our Institutional Animal Care and Use Committee. No distressful effects of PCP were observed at the dosage used in this study. The number of animals used was the minimum required to obtain scientifically valid data.
2.2. Habituation and Pre-Training
After a period of approximately 6 weeks of individual housing, the initial training began. Initially, the subjects were shaped to reach out of their cages to retrieve palatable food items (banana slices or apple bits) from the hands of the experimenters.
Once the animals were habituated to the experimenters, a portable Wisconsin General Test apparatus (WGTA) was fitted to the animals’ cage for treat delivery. The WGTA fit flush against the front of the cage and was equipped with an opaque screen that could be lowered and raised by the experimenter. The WGTA was positioned on the cage such that the screen was immediately in front of the cage door, which was opened during testing. The WGTA was also equipped with a tray with 3 equally spaced food wells that were positioned opposite the monkey; therefore, when the screen was open, the animal could reach out and manipulate items placed upon the tray or within its food wells.
The subjects were next trained to reach out and obtain food items placed within the food wells and subsequently to displace novel objects that were partially or completely concealing a food item hidden within the food well. This initial training took between 2 and 4 days of exposure.
2.3. Visual Discrimination Acquisition and Reversal
Subsequently, training on the visual discrimination task (Jentsch et al., 2002) began. In three consecutive daily sessions, the monkeys were tested for the acquisition of a novel discrimination. On the first trial, the screen was raised and the animal was presented with three distinctive objects they had never seen before, each of which concealed a food well. The objects were chosen to be roughly the same size but of different colors, patterns and shapes.
The food well was baited and the objects were arranged only when the opaque screen was lowered. For each animal, 1 of the objects concealed a hidden food reward (termed stimulus A+). The other 2 objects never concealed a food reward (termed B− and C−). On all the trials within a daily session, the monkey was allowed to reach out and displace 1 single object. If it selected A+, it was allowed to retrieve it before the screen was lowered. If the animal selected B− or C−, it was allowed to view the empty well and the screen was quickly lowered. A total of about 10 seconds separated each trial, during which time the experimenter recorded data from the previous trial, the position of the objects was re-arranged and the appropriate well was baited. The relative spatial placements of the three objects were pseudo-randomly varied across all trials to ensure that spatial position could never be used to solve the discrimination task.
Each of these initial test sessions consisted of both a search phase and an acquisition phase. The search phase consisted of all the trials up to and including the first selection of A+. The acquisition phase consisted of all the subsequent trials. The acquisition phase was complete when 1) the monkey successfully chose A+ on at least 7 out of 10 consecutive trials or 2) a total of 40 trials were delivered. The animals were trained on 3 different discriminations in the first three days of training.
Next, the animals were trained on the first within-session acquisition and reversal session. In this condition, the animals were first exposed to 3 novel objects and allowed to first select A+ across consecutive trials (acquisition search phase) and then continue performing under that contingency until they achieved 7 correct responses of that stimulus within 10 consecutive trials (acquisition phase). At this point, the reversal component of the task began; the reward was now placed under 1 of the initially negative stimuli, and stimulus A was now unreinforced (e.g., A−, B+, C−). The subjects were allowed to choose a single object per trial until they first selected the now reinforced stimulus (reversal search phase). After that, the animals were tested with the reversal contingency until 1) they achieved at least 7 correct choices on 10 consecutive trials or 2) 20 trials elapsed. For both the acquisition and reversal search phases, the maximum number of trials was set to 10; if the subject failed to select the reinforced object within 10 trials, it was, on the 10th trial, allowed to displace as many objects as necessary until it found the reward.
Dependent measures collected in each of these sessions included: 1) the number of errors made before first selecting the correct object in the acquisition search phase, 2) the number of trials required to achieve acquisition criterion after the first correct choice, 3) the number of errors made before first selecting the correct object in the reversal search phase, and 4) the number of trials required to achieve reversal criteria after the first correct choice.
After the initial exposure period, the within-session acquisition and reversal design was repeated daily with a novel set of objects until the subjects were regularly achieving both the acquisition and reversal criteria on a daily basis. This required 3 to 9 days of subsequent training.
2.4. Object Retrieval/Detour Task
Next, the animals were exposed to the basic contingencies of the object retrieval/detour task (Jentsch et al., 1997b; Jentsch et al., 2000; Taylor et al., 1990). In this task, a transparent Plexiglas cube with one open face is fitted to the tray. Rewards are inserted into the cube through the open face, which can be directed towards or to the right or left of the monkey. In addition, the position of the cube on the tray (left, right or middle) can be varied. The key to this task is that, when the open face is directed towards the monkey, it simply follows its line of sight in order to retrieve the reward (sensory-guided responding). However, if the open face is directed to the left or right and the reward is placed deep into the cube, the animal sees the reward directly in front of it but must reach around the transparent barrier to retrieve it. Therefore, there is a conflict between its sensory input and its acquired knowledge of the task. Without any training, all monkeys reach directly at the transparent face, rather than negotiating the barrier. However, over time, control monkeys acquire the ability to quickly (in one reach) negotiate the barrier to retrieve their reward, while chronic PCP-treated monkeys have difficulty in acquiring this ability (Jentsch et al., 2000).
In a single baseline session, the animals were exposed to a total of 10 object retrieval/detour trials. Of these 10 trials, 4 involved direct reaches in which the open face of the box was directed towards the animal, 4 involved trials in which the box was directed to the left or right but the reward was placed such that it emerged from the box somewhat and 2 involved trials in which the open face was directed to the left or the right and the reward was placed within the cube. This session was introduced to expose the animals to the basic task contingencies and to the cube.
Three dependent measures were collected: the numbers of successful trials, completed trials and omissions. Because animals were allowed to make as many reaches as necessary to retrieve the reward, all trials were either completed or an omission was scored when no response was made within 2 min. The sum of the number of correct trials and omissions was therefore the number of total available trials per day. Success was scored only when a trial was completed with one single reach (no errors) and therefore reflected the ability to inhibit the pre-potent response (direct reach) and the accurate deployment of the detour reaching strategy.
2.6 Group Assignments
Group assignments were then made. Data for reversal performance on the final 2 days of the baseline testing period were combined, and these results were used to construct the drug treatment groups, which were balanced with respect to pre-drug performance. The dependent measures used to construct balanced groups were reversal search errors and trial to criteria in reversal.
2.7. Chronic PCP Exposure Period
After all pre-training was completed, the monkeys entered the subchronic PCP treatment period (Jentsch et al., 1997b). Of the monkeys reported here, 16 received twice-daily intramuscular injections with sterile, isotonic saline (0.1 ml/kg). The remaining monkeys were injected twice-daily with phencyclidine hydrochloride (Sigma, St Louis MO; 0.3 mg/kg); the drug was dissolved in sterile saline and delivered intra-muscularly. The injections were delivered for 14 consecutive days at ~7:30 am and ~4:30 pm. As expected, no distressful effects of PCP treatment were observed at this dose.
2.8. Asenapine Treatment
All the subjects described here received twice-daily intramuscular injections of saline or asenapine (50, 100, or 150 μg/kg) for the 4 weeks following the PCP treatment period. No adverse effects of asenapine treatment were observed and the final dose of saline or asenapine was administered 1 hour before tissue collection.
2.9. Post-PCP Testing
Animals were tested 5 days per week for up to 26 days after the last PCP treatment. On Mondays and Thursdays, they were assessed for the acquisition and reversal of a novel discrimination. On Tuesdays and Fridays, they were tested on the object retrieval/detour task (data not shown).
2.10. Tissue Collection and Analyses
Euthanasia was carried out after injection of an overdose of pentobarbital. Brains were perfused with cold saline, and the following regions dissected, then frozen, from 4 mm sections: dorsolateral prefrontal cortex (DLPFC; area 46), medial prefrontal cortex (area 9), orbital frontal cortex (area 12), prelimbic cortex (area 32), supplementary motor area (area 6M), infralimbic cortex (area 25), premotor cortex (area 6V), frontal eye (area 8A), primary motor cortex (area 4), entorhinal cortex (olfactory), caudate nucleus, putamen, nucleus accumbens, amygdala, hippocampus. Tissue samples were sonicated and extracted on an alumina column prior to high performance liquid chromatography (HPLC) electrochemical detection analysis for DA. A portion of the supernatant was used for direct injection (no alumina separation) on HPLC column for separation of 5HT, 5-hydroxyindoleacetic acid (5HIAA) and homovanillic acid (HVA) (Elsworth et al., 1996; Elsworth et al., 1989).
2.11. Statistical Analyses
Group differences between animals treated with either saline-only or PCP-only were examined with independent samples t-tests (Statview, SAS Institute Inc.); unless otherwise noted, these were two-tailed tests. Analysis of the effects of asenapine on group means of behavioral data were performed with 1-factor or 2-factor ANOVA, as appropriate, followed by post-hoc tests, using p< 0.05 as significant.
The statistical approach for analysis of the neurochemical effects of asenapine was based on planned comparisons between mean values in specific groups. The comparisons were based on the behavioral data. Firstly, as a relevant neurochemical change that contributed to the PCP-induced cognitive deficit would be reflected in a difference (increase or decrease) between the group treated with only saline (SAL-SAL) and the group treated only with PCP (PCP-SAL), mean values from these 2 groups were initially compared. Secondly, as a neurochemical change that was involved in restoration of cognitive performance would be shown by a reversal of the change induced by PCP (PCP-SAL) by the group treated with 150 μg/kg asenapine (PCP-ASE150), mean values from these 2 groups were compared. PCP-treated animals given lower doses of asenapine (50 or 100 μg/kg) did not exhibit significant behavioral improvement, so PCP-ASE150 was the only group that displayed significant behavioral improvement. Thus, planned comparisons of these 2 pairs of group means (df =1) were performed by contrast analysis following an overall ANOVA, using a general linear modeling program (“SuperAnova”, Abacus Concepts, Berkeley, CA).
3. Results
3.1. Behavioral deficits in the PCP model
3.1.1. Reversal Learning
We first examined whether PCP administration successfully produced cognitive deficits, as expected, by comparing performance of control and PCP-exposed monkeys (n=8/group) on the discrimination acquisition and reversal task across the 7 post-PCP assessments. Repeated measures ANOVA (between subjects factor: treatment group; within subjects factor: time) demonstrated main effects of treatment group and a treatment group x time interaction for reversal search errors (treatment group: F(1,14)=105.8, p<0.0001; group x time: F(6,14)=3.0, p=0.01) and trials to criteria in reversal (treatment group: F(1,14)=6.3, p=0.03; group x time: F(6,14)=3.4, p=0.005). Neither acquisition search errors, nor trials to criteria in acquisition, demonstrated similar effects or interactions (all Fs<1.8, p>0.05). Based upon these results, we further examined reversal learning performance during each of the post-PCP administration weeks of testing.
One week after the last dose of PCP or saline, PCP-exposed monkeys made more errors before first selecting the newly-reinforced object in reversal (Figure 2; reversal search errors; t14=−2.91, p=0.01) and required more trials overall to reach criteria in reversal (t14=−2.8, p=0.040. The same was true at 2 weeks post-PCP, the drug-treated subjects again made some reversal search errors (Figure 2; t14=−7.78, p<0.001) and needed more trials to reach criteria in reversal (t14=−3.27, p=0.006) than the saline subjects. By 3 weeks after the last dose of PCP, impairments were still measurable; PCP-treated monkeys made more reversal search errors (Figure 2; t14=−4.23, p<0.001), though they no longer required more trials, overall, to reach criteria after selection of the positive object. In the fourth week, PCP-exposed monkeys still made more reversal search errors than did saline controls (Figure 2; t=−3.07, p=0.008); trials to criteria in reversal were not affected by chronic dosing with PCP at this time point (t14=1.59, p=0.15).
Figure 2.

Prolonged deficit in reversal learning induced by PCP. Withdrawal from subchronic PCP treatment led to an increased number of search errors in the reversal learning paradigm, which persisted for 4 weeks. Saline-Saline group was treated with saline twice daily for 6 weeks. PCP-Saline group was treated with PCP twice daily for 2 weeks, followed by saline twice daily for 1, 2, 3, or 4 weeks. *Significant increase compared with respective saline controls (n=8 per group).
3.1.2 Object Retrieval/Detour (data not illustrated)
Object retrieval/detour is an acquisition task which tests the ability to learn a cognitively, as opposed to visually-guided, reach strategy. Consistent with this, damage to the frontal cortex or chronic PCP exposure produced transient acquisition deficits in the object retrieval/detour task (Jentsch et al., 2000; Wallis et al., 2001). In this study, the main effect of treatment group on the measure of success narrowly missed statistical significance (F(1,14)=3.9, p=0.06) and there was no treatment x time interaction (F(6,14)=0.9, p=0.47). In the first week after the last dose of PCP, the PCP-treated subjects completed fewer trials in 1 reach than their saline-treated counterpart (t14=2.6, p=0.02). There were no group differences at later time points of assessment. Numbers of omissions were not modulated by chronic treatment at any time point, suggesting that both groups were equally motivated to perform the task.
3.2. Effects of asenapine on behavior in the PCP model
A number of animals that received daily dosing with asenapine did not complete 1 or more reversal learning session (6 of 8 animals dosed with asenapine 150 μg/kg after chronic saline; 1 of 8 animals dosed with asenapine 50 μg/kg after chronic PCP; 3 of 8 animals dosed with asenapine 100 μg/kg after chronic PCP; and 3 of 8 animals dosed with asenapine 150 μg/kg after chronic PCP); a full within subject analysis was impossible because of the missing cells. Because most animals completed at least one reversal learning assessment each week, we averaged the dependent variables for the two test sessions in each week for each subject. Using this strategy, we still had a few animals with empty cells. For this reason, ANOVA results relate to the remaining subjects for which there were data available in each week.
Because reversal search errors and trials to criteria in reversal were the measures modulated by chronic PCP treatment, the primary analyses focused on these measures. For reversal search errors, ANOVA with treatment group as the between subjects factor and week as the within subjects factor showed a main effect of group (F(5,39)=8.3, p<0.0001) and a significant group x week interaction (F(15,117)=1.9, p=0.03). Post hoc Tukey HSD tests revealed that the chronic PCP group not treated with asenapine differed from all other groups (mean differences of 2.4–3.2, all ps<0.002); none of the remaining groups (the saline-only controls, the saline controls treated with asenapine 150 μg/kg or any of the PCP-exposed monkeys subsequently treated with any dose of asenapine) differed from one another (all mean differences < 1, all ps>0.05) for reversal search errors. For total trials to criteria in reversal, once again, ANOVA revealed a main effect of treatment group (F(5,36)=3.0, p=0.02) and a significant group x week interaction (F(15,108)=2.3, p=0.007). Tukey HSD did not identify reliable group differences for this dependent variable.
Our exploratory analyses focused on potential group differences in performance at the end of treatment, in week 4. As shown in Figure 3, PCP-exposed monkeys that were not treated with asenapine made more reversal search errors than did saline-only controls (t14=−3.1, p=0.01). Treatment with asenapine 150 μg/kg after chronic PCP fully reversed this deficit (t14=4.0, p=0.001). The lower doses produced intermediate effects, consistent with dose responsivity of the modulation of reversal learning.
Figure 3.

One month of asenapine (ASE) treatment reverses PCP deficit in reversal learning paradigm. The number following ASE is the dose (micrograms/kg). *Significant increase in search errors compared with saline controls (n=8 per group).
For completeness, we also examined acquisition search errors and trials to criterion in acquisition across this period. Neither measure exhibited a main effect of group, nor a group x week interaction (all Fs<2.2, all ps>0.05).
The effect of asenapine on the ORD task was not evaluated, as the PCP-induced impairment on this task was only evident for 1 week following PCP treatment.
3.3. Effects on asenapine on neurochemistry in controls
In animals not treated with PCP, chronic twice-daily treatment with asenapine (150 μg/kg) significantly elevated DA and 5HT turnover in a number of cortical and subcortical brain regions (Table 1); no significant changes were found in other examined brain regions (not shown).
Table 1.
Effect of asenapine on dopamine and serotonin turnover in brain
| DOPAMINE TURNOVER | ||
|---|---|---|
| REGION | SALINE-SALINE | SALINE-ASENAPINE 150 |
| caudate nucleus | 0.87 ± 0.04 | 1.33 ± 0.08* |
| putamen | 1.15 ± 0.12 | 1.72 ± 0.04* |
| prelimbic cortex | 2.79 ± 0.17 | 4.97 ± 0.33* |
| premotor cortex | 2.93 ± 0.18 | 4.23 ± 0.38* |
| frontal eye field | 7.00 ± 0.43 | 10.9 ± 1.47* |
| SEROTONIN TURNOVER | ||
|---|---|---|
| REGION | SALINE-SALINE | SALINE-ASENAPINE 150 |
| caudate nucleus | 1.50 ± 0.10 | 2.37 ± 0.19* |
| putamen | 2.28 ± 0.24 | 3.28 ± 0.44* |
| prelimbic cortex | 0.43 ± 0.03 | 0.69 ± 0.08* |
| premotor cortex | 0.37 ± 0.02 | 0.56 ± 0.05* |
| frontal eye field | 0.46 ± 0.07 | 0.70 ± 0.06* |
| medial prefrontal cortex | 0.37 ± 0.03 | 0.59 ± 0.06* |
| supplementary motor cortex | 0.34 ± 0.02 | 0.84 ± 0.21* |
| primary motor cortex | 0.49 ± 0.05 | 0.73 ± 0.07* |
| amygdala | 0.66 ± 0.06 | 1.00 ± 0.08* |
| hippocampus | 0.89 ± 0.13 | 2.37 ± 0.18* |
Saline-Saline group was treated with saline twice daily for 6 weeks.
Saline-asenapine group was treated with saline twice daily for 2 weeks, followed by asenapine twice daily (150 μg/kg) for 4 weeks.
significantly different from saline-saline group (2-way unpaired t-test, p<0.05) 8 monkeys in each group
3.4. Neurochemical effects in the PCP model
Our prior studies have demonstrated a selective deficit in DA turnover in DLPFC and prelimbic cortex at 1–2 weeks after PCP treatment in our non-human primate PCP model (Jentsch et al., 1997b; Jentsch et al., 1999). Even after 28 days, a lower but significant deficit in DA utilization was still present in the DLPFC (t14=1.98, p<0.05), yet in the prelimbic cortex the decrease was no longer evident (Figure 4). In the orbital frontal cortex and nucleus accumbens DA utilization was not affected (Figure 4). No significant changes were observed in other examined cortical or subcortical regions at either time after PCP treatment.
Figure 4.

Alteration in regional brain DA turnover after withdrawal from PCP treatment. * denotes significant decrease compared with saline controls in a one-tailed t-test (n=8 per group). Treatments conditions for Saline-saline and PCP-saline groups, as in Figure 2. Abbreviations: DA=dopamine; dlpfc, dorsolateral prefrontal cortex; HVA=homovanillic acid; plimb, prelimbic cortex; lorb, lateral orbital cortex; nac, nucleus accumbens.
Surprisingly, we found a significant lowering of 5HT utilization in PCP-treated monkeys in the orbitofrontal cortex (t14=2.2, p<0.05) and nucleus accumbens (t14=2.8, p<0.05) (Figure 5), and an increase in the amygdala (t14=2.8, p<0.05). In our published studies in adult monkeys, no significant changes in 5HT turnover were noted in any brain region examined (Jentsch et al., 1997a; Jentsch et al., 1999). Serotonin turnover was not affected by PCP in other examined cortical or subcortical brains regions.
Figure 5.

Alteration in regional brain 5HT turnover after PCP treatment. *Significant decrease compared with saline controls (n=8 per group). Treatment codes, as in Figure 2. 5-HIAA=5-hydroxyindoleacetic acid; amyg=amygdala; other abbreviations, as in Figure 4.
No significant PCP-induced changes in the steady-state levels of DA or 5HT were observed in any brain region, except for the central amygdala in which DA level was reduced (see below).
3.5. Effects of asenapine on neurochemistry in the PCP model
Asenapine elicited an increase in DA turnover in many brain regions of subchronic PCP-treated monkeys (not shown), but data are shown only for the region (DLPFC) in which there was a significant reversal by asenapine of a PCP-induced decrease in DA turnover. Figure 6 illustrates the impact of asenapine on DA turnover in the DLPFC of control and PCP-treated monkeys. While no difference in DA turnover was revealed by contrast analysis between mean values for the PCP-SAL and SAL-SAL groups, an unpaired t-test between these group means, to test whether the PCP-induced decrease found at 1-week was maintained at 4-weeks after PCP, was significant [t(14)=1.9, p<0.05]. Contrast analysis showed that the mean of the PCP-ASE150 group was higher than for PCP-SAL [F=3.2, p<0.05], illustrating that a behaviorally relevant dose of asenapine increased DA turnover in DLPFC. In central amygdala, contrast analysis showed that the DA level (not turnover) was reduced in the PCP-SAL group compared with the SAL-SAL group [F=6.7, p<0.05], and DA level was increased above the mean value for the PCP-SAL group by PCP-ASE150 [F=5.5, p<0.05] (Figure 7).
Figure 6.

One month of asenapine treatment attenuates PCP-induced decrease in DA turnover in DLPFC. DA=dopamine; HVA= homovanillic acid; other abbreviations and treatment codes, as in Figure 3. # Significant decrease compared with Sal-Sal (see text), † significant increase compared with PCP-Sal (n=8 per group).
Figure 7.

PCP-induced decrease in DA level in the amygdala is reversed by asenapine. Eight animals were in each group. DA=dopamine; other abbreviations and treatment codes, as in Figure 3. *Significant decrease compared with Sal-Sal, † significant increase compared with PCP-Sal (n=8 per group).
Asenapine elicited a change in 5HT turnover in many brain regions of subchronic PCP-treated monkeys (not shown), but data are shown only for the regions (orbital frontal cortex], nucleus accumbens, and amygdala) in which there was a significant reversal by asenapine of a PCP-induced change in 5HT turnover. In lateral orbital cortex [Figure 8] contrast analysis revealed that 5HT turnover was reduced in the PCP-SAL group compared with the SAL-SAL group [F=4.3, p<0.05], and 5HT turnover was increased above the mean value for the PCP-SAL group by PCP-ASE150 [F=5.2, p<0.05]. In addition, contrast analysis demonstrated that in nucleus accumbens [Figure 9] 5HT turnover was reduced in the PCP-SAL group compared with the SAL-SAL group [F=4.0, p=0.05], and that 5HT turnover was increased above the mean PCP-SAL value by PCP-ASE150 [F=7.4, p<0.05]. It is noteworthy that in the amygdala [Figure 10], 5HT turnover was increased in the PCP-SAL group compared with the SAL-SAL group [F=17.0, p<0.0005], and 5HT turnover was reduced below the mean PCP-SAL value by PCP-ASE150 [F=7.1, p<0.05].
Figure 8.

One month of asenapine treatment attenuates PCP-induced decrease in 5HT turnover in orbital frontal cortex. Abbreviations and treatment codes, as in Figure 3. *Significant decrease compared with Sal-Sal, † significant increase compared with PCP-Sal (n=8 per group).
Figure 9.

One month of asenapine treatment attenuates PCP-induced decrease in 5HT turnover in nucleus accumbens. Abbreviations and treatment codes, as in Figure 3. *Significant decrease compared with Sal-Sal, † significant increase compared with PCP-Sal (n=8 per group).
Figure 10.

PCP increased 5HT turnover in central amygdala; this effect was partially reversed by asenapine. Animals were treated for 2 weeks twice daily with saline (Sal) or PCP, followed by 4 weeks of twice daily treatment with saline or asenapine (ASE). The number following ASE is the dose (micrograms/kg). 5-HIAA=5-hydroxyindoleacetic acid. *Significant increase compared with Sal-Sal, † significant decrease compared with PCP-Sal (n=8 per group).
4. Discussion
This study found that following 2 weeks of PCP treatment, significant deficits in reversal learning occurred and were sustained for up to 4 weeks in monkeys. These deficits in executive function were reversed by asenapine treatment initiated after PCP exposure. The reversal of PCP-induced cognitive dysfunction by asenapine was paralleled by changes in serotonin and dopamine turnover in lateral orbital cortex, dorsolateral prefrontal cortex, nucleus accumbens and amygdala.
4.1 PCP-induced behavioral deficits
Two weeks of dosing with the psychotomimetic drug PCP produces a fairly persistent defect in the ability to respond flexibly in a reversal learning task in monkeys. The impairments in reversal learning were indicated by the increase in the number of reversal search errors; these are the errors the animal makes immediately after the switch. Generally, the total number of reversal search errors is a function of the number of perseverative responses the animal makes to the initially reinforced stimulus. All the errors described in the analysis are “choice errors”, meaning that a response was made. Therefore, a change in motivation level could not have accounted for the differences in reversal search errors. Throughout the first 2 weeks of testing post-PCP, drug-exposed animals also required more trials to reach criteria after the first correct choice; this effect was likely due to a re-emergence of perseverative responses even after making a selection of the now-reinforced object. Collectively, these data suggest that repeated exposure to PCP produces a deficit in the ability to change or update behavior in this task similar to that reported in patients with schizophrenia (Waltz and Gold, 2007).
These impairments are consistent with observations indicating reversal learning deficits in rodents after subchronic exposure to PCP (Abdul-Monim et al., 2006; Jentsch and Taylor, 2001; Laurent and Podhorna, 2004); see also (Fletcher et al., 2005; Rodefer et al., 2005). Taken with the learning deficits in the object retrieval/detour task, with no obvious motor or motivational impairments, these data indicate that, under a certain set of dosing conditions, long-term exposure to PCP erodes the ability of subjects to adaptively alter their behavioral strategies in order to obtain reward; rather, they are more likely to perseveratively emit innate or well-learned, conditioned responses despite the fact that those responses no longer gain them desired outcomes (e.g., reward). These impairments are qualitatively similar to those exhibited by patients with schizophrenia (Crider, 1997; Klingner et al., 1972; Kopp and Rist, 1994; Oades, 1997) and patients with inferior frontal or orbitofrontal damage (Berlin et al., 2004; Fellows and Farah, 2003; Frank and Claus, 2006; Rolls, 2004).
4.2. Behavioral Basis of Poor Reversal Learning
Several different forms of behavioral dysfunction could theoretically contribute to the poor reversal learning in PCP-exposed monkeys. First, PCP could affect the ability of the subjects to adequately process the complex visual features of the cues (a perceptual deficit); this is not, however, an adequate explanation of our results since such an effect would be predicted to lead to generally poor discrimination learning and performance, affecting acquisition and reversal equally, and this outcome was not found. Second, a decline in motivational function could lead to reversal learning deficits, but none of the measures of index motivation (trials completed/omissions committed in the discrimination and object retrieval tasks, total number of rewards retrieved in the fine motor task) differentiated PCP and control subjects, eliminating this as a possibility. Third, PCP-exposed animals could exhibit a general defect in their ability to learn about the associative relationships between the discriminanda and reward; however, this is also not a parsimonious explanation of our results because there were no group differences in the ability to learn a novel visual discrimination (i.e., no impairments in acquisition), even though this phase of the task equally relies upon a subject’s ability to acquire Pavlovian relationships.
Finally, the deficits in reversal learning could be attributed to an inability to inhibit the previously learned response when the rules change in the task. This possibility is supported by a significant amount of data. Generally, the increased errors in reversal in PCP-exposed monkeys were limited to the reversal search phase, when the interference from the previously learned rule is strongest. Additionally, the deficit was specific to reversal, as opposed to acquisition, and the need to inhibit a previously learned response is one of the few factors that differentiate these two conditions. Finally, the learning defect in the object retrieval/detour task, where subjects need to inhibit the innate tendency to reach directly for the reward in order to learn the detour reaching strategy, also supports a failure of response inhibition in these subjects. Taken together, we propose that subchronic PCP exposure specifically impairs tasks that require response inhibition and may therefore have greater effects on any task that requires response alternation or switching. This proposal clearly requires additional study in rodent and non-human primate models.
4.3. Neuroanatomical and Neurochemical Considerations
Observational studies in humans and experimental studies in non-human primates have implicated the inferior frontal/orbitofrontal cortex as a critical substrate underlying reversal learning (Dias et al., 1996; Fellows and Farah, 2003; Rolls, 2004; Schoenbaum et al., 2002) or other tasks involving response inhibition (Aron et al., 2004; Iversen and Mishkin, 1970; Wallis et al., 2001). The inferior frontal cortical regions involved in reversal learning and response inhibition may exert their actions through corticostriatal projections (Rogers et al., 2000; Schwartzbaum and Donovick, 1968) or through the so-called hyper-direct pathway (Aron and Poldrack, 2006; Winstanley et al., 2005); the latter circuit has been identified through studies that emphasize rapidly stopping responses. Recent work suggests that the hyper-direct pathway is functionally associated with goal-directed interruption of global behavioral output, rather than selective inhibition of particular responses (Majid et al., 2011). In the case of reversal learning, the task requires the selective withholding of only one response, with other behaviors still being required to gain access to the desired outcome, possibly relying to a greater degree on cortical innervation of the striatum.
Although the exact role of DA within frontal cortex on reversal learning is unclear, behavioral pharmacological data do clearly implicate DA D2-like receptor mediated signaling in reversal learning (Lee et al., 2007). Of course, these effects need not be mediated through DA mechanisms within the frontal cortex. Long-term exposure to PCP dysregulates both cortical and subcortical DA transmission (Balla et al., 2001; Jentsch et al., 1997b; Jentsch et al., 1999; Jentsch et al., 1997c; Noda et al., 2000). In that sense, there are multiple potential mechanisms by which reversal learning impairments may arise in PCP-exposed monkeys (Jentsch and Roth, 1999).
Recent experiments have demonstrated that serotonergic, rather than dopaminergic, denervation of monkey frontal cortex impairs the ability to respond flexibly in a reversal learning task (Clarke et al., 2004; Clarke et al., 2005; Clarke et al., 2007) or in an object retrieval/detour task (Walker et al., 2006). Serotonin is strongly implicated in the ability to shift behavior in response to changing stimulus-reward contingencies. Converging evidence from a number of studies has implicated 5HT innervation of the orbitofrontal cortex in the behavioral flexibility necessary for the reversal learning task (Boulougouris et al., 2007; Chudasama and Robbins, 2003; Rolls, 2004). Remarkably, in the current study we found a significant lowering of 5HT utilization in the orbitofrontal cortex in PCP monkeys, which may be a mechanism underlying poor reversal learning (Clarke et al., 2005; Robbins and Roberts, 2007). In the same brain region, DA utilization was not affected.
4.4 Asenapine reversal of PCP-induced behavioral and neurochemical deficits
Daily treatment with asenapine (150 μg/kg) for 4 weeks following PCP administration fully reversed the search errors in the reversal learning task. The lower doses of asenapine (50 and 100 μg/kg) produced intermediate effects that are consistent with dose responsivity in the modulation of reversal learning.
Asenapine ameliorated the PCP-induced 5HT deficit in orbitofrontal cortex in a dose-related manner that paralleled the reversal of the behavioral impairment. A deficit in 5HT turnover was also found in the nucleus accumbens, which was attenuated by chronic asenapine treatment. As the nucleus accumbens appears to play a role in reversal learning (Cardinal et al., 2001; Masaki et al., 2006; Stern and Passingham, 1995), restoration of the 5HT deficit in this brain region could also be important in for the behavioral effects of asenapine.
Although the mechanism by which asenapine reverses the 5HT turnover deficit in the orbitofrontal cortex is unclear, recent data support the view that 5HT2C receptors play a role in cognitive flexibility and response inhibition (Boulougouris et al., 2008). The improved performance in reversal learning following 5HT2C antagonism suggests the possibility that these receptors may be involved in the ability of asenapine to ameliorate the reversal learning deficits observed in this study. However, the relatively high occupancy of 5-HT2A compared with the D2 receptor may be critically important for some atypical antipsychotic drug actions, such as low extrapyramidal symptoms and efficacy in treatment-resistant cognitive deficits of schizophrenia (Boulougouris et al., 2008; Meltzer, 1989; Meltzer and Huang, 2008; Meltzer et al., 2003). This selective action on 5HT subtypes may in part explain the attenuation of the reversal learning deficit and restoration of 5HT turnover in the orbital frontal cortex and nucleus accumbens in the monkey PCP model. 5-HT2A receptor blockade has, for example, been reported to be a key mechanism for alleviating, in part, negative symptoms in schizophrenia (Lane et al., 2005; Marquis et al., 2003), while other evidence also lends support to a role for the 5-HT2C receptor in this regard (Reynolds et al., 2005). The combined potent 5-HT2A- and 5-HT2C-receptor blockade of asenapine may therefore provide a concerted and efficient means of addressing negative symptoms compared with antagonism at either receptor alone (Meltzer et al., 2003). The subnanomolar affinity of asenapine for these two receptors, combined with a higher selectivity relative to D2 receptors, offers an opportunity to achieve high occupancy of 5-HT2A and 5-HT2C receptors while maintaining adequate blockade of D2 receptors.
Clinical studies have indicated that α2 adrenergic antagonists may be beneficial not only in the pharmacological treatment of depression (Grossman et al., 1999) and dementia (Coull et al., 1996; Sahakian et al., 1994) but also in schizophrenia (Litman et al., 1996). It has been hypothesized that α2 adrenergic antagonist actions of certain antipsychotic drugs, such as clozapine, may contribute to their actions on DA systems. It has been appreciated for some time that α2 adrenergic receptors can modulate the function of the mesoprefrontal cortical DA system (Gresch et al., 1995; Jentsch et al., 2008; Jentsch et al., 1998b). The potent α2 adrenergic receptor antagonist, idazoxan, as well as clozapine, preferentially increase DA output in the prefrontal cortex by acting at the nerve terminal level (Devoto et al., 2003; Hertel et al., 1999a; Hertel et al., 1999b). Similar to clozapine, asenapine has high affinity for α2 adrenergic receptors, and increases DA and noradrenaline release from prefrontal cortex (Franberg et al., 2009; Marcus et al., 2010). This interaction of asenapine with both 5-HT2A/2C and α2 adrenergic receptors may be important with regard to its ability to attenuate PCP-induced cognitive dysfunction and alter DA turnover in DLPFC and 5HT turnover in lateral orbital cortex and nucleus accumbens in this monkey model. This action may also have bearing on its clinical efficacy in the treatment of schizophrenia and bipolar disorder.
4.5. Conclusions
In this model of cognitive dysfunction, asenapine produced substantial gains in executive functions that were maintained with long-term dosing. The reversal of both PCP-induced cognitive impairment and neurochemical changes in 5HT and DA turnover by asenapine seen in this study may be related to the unique receptor signature of asenapine, and support the potential for asenapine to reduce cognitive impairment in patients with schizophrenia.
Research Highlights.
In a nonhuman primate PCP model, cognitive dysfunction persists for 1 month.
Asenapine reversed cognitive dysfunction in this PCP model.
The cognitive effects of asenapine were maintained with long-term administration.
The cognitive effects of asenapine were linked with normalized monoamine turnover.
Acknowledgments
We gratefully acknowledge the staff of the St. Kitts Biomedical Research Foundation for expert assistance in the care and treatment of the animals. These studies were funded, in part, by National Institutes of Health grants MH-57483, MH-57483S (to RHR), and RR-20750, MH-77248, DA-22539 (to JDJ), and Organon Research Laboratories, Ltd. (formerly Schering-Plough, now Merck, Whitehouse Station, NJ, USA), and Pfizer Central Research. Editorial support was provided by Complete Healthcare Communications Inc and was funded by Schering-Plough Corporation, now Merck (Whitehouse Station, NJ, USA. We thank the sponsors for help in formatting and copy-editing this manuscript and preparing it for submission to the special issue on schizophrenia in Neuropharmacology.
Footnotes
Conflicts of Interest
JDJ and RHR were recipients of grant funding from Pfizer Central Research and Organon Research Laboratories (formerly Schering-Plough, now Merck, Whitehouse Station, NJ, USA). JDJ is a consultant to Pfizer. MS and HM are employees of MSD, Lanarkshire, UK. EW was an employee of Pfizer, Ann Arbor,MI, USA. Merck/MSD currently distributes and has a commercial interest in asenapine. Co-authors who were employees at sponsoring organizations participated in and approved the experimental design of these experiments and the final version of this manuscript.
References
- Abdul-Monim Z, Reynolds GP, Neill JC. The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav Brain Res. 2006;169:263–273. doi: 10.1016/j.bbr.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Allen RM, Young SJ. Phencyclidine-induced psychosis. Am J Psychiatry. 1978;135:1081–1084. doi: 10.1176/ajp.135.9.1081. [DOI] [PubMed] [Google Scholar]
- Andersen JD, Pouzet B. Spatial memory deficits induced by perinatal treatment of rats with PCP and reversal effect of D-serine. Neuropsychopharmacology. 2004;29:1080–1090. doi: 10.1038/sj.npp.1300394. [DOI] [PubMed] [Google Scholar]
- Aron AR, Poldrack RA. Cortical and subcortical contributions to Stop signal response inhibition: role of the subthalamic nucleus. J Neurosci. 2006;26:2424–2433. doi: 10.1523/JNEUROSCI.4682-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron AR, Robbins TW, Poldrack RA. Inhibition and the right inferior frontal cortex. Trends Cogn Sci. 2004;8:170–177. doi: 10.1016/j.tics.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Balla A, Koneru R, Smiley J, Sershen H, Javitt DC. Continuous phencyclidine treatment induces schizophrenia-like hyperreactivity of striatal dopamine release. Neuropsychopharmacology. 2001;25:157–164. doi: 10.1016/S0893-133X(01)00230-5. [DOI] [PubMed] [Google Scholar]
- Berlin HA, Rolls ET, Kischka U. Impulsivity, time perception, emotion and reinforcement sensitivity in patients with orbitofrontal cortex lesions. Brain. 2004;127:1108–1126. doi: 10.1093/brain/awh135. [DOI] [PubMed] [Google Scholar]
- Boulougouris V, Dalley JW, Robbins TW. Effects of orbitofrontal, infralimbic and prelimbic cortical lesions on serial spatial reversal learning in the rat. Behav Brain Res. 2007;179:219–228. doi: 10.1016/j.bbr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Boulougouris V, Glennon JC, Robbins TW. Dissociable effects of selective 5-HT2A and 5-HT2C receptor antagonists on serial spatial reversal learning in rats. Neuropsychopharmacology. 2008;33:2007–2019. doi: 10.1038/sj.npp.1301584. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Pennicott DR, Sugathapala CL, Robbins TW, Everitt BJ. Impulsive choice induced in rats by lesions of the nucleus accumbens core. Science. 2001;292:2499–2501. doi: 10.1126/science.1060818. [DOI] [PubMed] [Google Scholar]
- Castner SA, Goldman-Rakic PS, Williams GV. Animal models of working memory: insights for targeting cognitive dysfunction in schizophrenia. Psychopharmacology (Berl) 2004;174:111–125. doi: 10.1007/s00213-003-1710-9. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. Dissociable contributions of the orbitofrontal and infralimbic cortex to pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex. J Neurosci. 2003;23:8771–8780. doi: 10.1523/JNEUROSCI.23-25-08771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke HF, Dalley JW, Crofts HS, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion. Science. 2004;304:878–880. doi: 10.1126/science.1094987. [DOI] [PubMed] [Google Scholar]
- Clarke HF, Walker SC, Crofts HS, Dalley JW, Robbins TW, Roberts AC. Prefrontal serotonin depletion affects reversal learning but not attentional set shifting. J Neurosci. 2005;25:532–538. doi: 10.1523/JNEUROSCI.3690-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke HF, Walker SC, Dalley JW, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion is behaviorally and neurochemically specific. Cereb Cortex. 2007;17:18–27. doi: 10.1093/cercor/bhj120. [DOI] [PubMed] [Google Scholar]
- Coull JT, Sahakian BJ, Hodges JR. The alpha(2) antagonist idazoxan remediates certain attentional and executive dysfunction in patients with dementia of frontal type. Psychopharmacology (Berl) 1996;123:239–249. doi: 10.1007/BF02246578. [DOI] [PubMed] [Google Scholar]
- Crider A. Perseveration in schizophrenia. Schizophr Bull. 1997;23:63–74. doi: 10.1093/schbul/23.1.63. [DOI] [PubMed] [Google Scholar]
- Deschenes A, Goulet S, Dore FY. Rule shift under long-term PCP challenge in rats. Behav Brain Res. 2006;167:134–140. doi: 10.1016/j.bbr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Devoto P, Flore G, Longu G, Pira L, Gessa GL. Origin of extracellular dopamine from dopamine and noradrenaline neurons in the medial prefrontal and occipital cortex. Synapse. 2003;50:200–205. doi: 10.1002/syn.10264. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- Elsworth JD, Brittan MS, Taylor JR, Sladek JR, Jr, al-Tikriti MS, Zea-Ponce Y, Innis RB, Redmond DE, Jr, Roth RH. Restoration of dopamine transporter density in the striatum of fetal ventral mesencephalon-grafted, but not sham-grafted, MPTP-treated parkinsonian monkeys. Cell Transplant. 1996;5:315–325. doi: 10.1177/096368979600500220. [DOI] [PubMed] [Google Scholar]
- Elsworth JD, Deutch AY, Redmond DE, Jr, Taylor JR, Sladek JR, Jr, Roth RH. Symptomatic and asymptomatic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates: biochemical changes in striatal regions. Neuroscience. 1989;33:323–331. doi: 10.1016/0306-4522(89)90212-1. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Sycrest (asenapine) 2010 http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/001177/human_med_001379.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001d124&jsenabled=true.
- Fellows LK, Farah MJ. Ventromedial frontal cortex mediates affective shifting in humans: evidence from a reversal learning paradigm. Brain. 2003;126:1830–1837. doi: 10.1093/brain/awg180. [DOI] [PubMed] [Google Scholar]
- Fletcher PJ, Tenn CC, Rizos Z, Lovic V, Kapur S. Sensitization to amphetamine, but not PCP, impairs attentional set shifting: reversal by a D1 receptor agonist injected into the medial prefrontal cortex. Psychopharmacology (Berl) 2005;183:190–200. doi: 10.1007/s00213-005-0157-6. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Geyer MA, Gold LH, Grace AA. Developing predictive animal models and establishing a preclinical trials network for assessing treatment effects on cognition in schizophrenia. Schizophr Bull. 2005;31:888–894. doi: 10.1093/schbul/sbi041. [DOI] [PubMed] [Google Scholar]
- Franberg O, Marcus MM, Ivanov V, Schilstrom B, Shahid M, Svensson TH. Asenapine elevates cortical dopamine, noradrenaline and serotonin release. Evidence for activation of cortical and subcortical dopamine systems by different mechanisms. Psychopharmacology (Berl) 2009;204:251–264. doi: 10.1007/s00213-008-1456-5. [DOI] [PubMed] [Google Scholar]
- Frank MJ, Claus ED. Anatomy of a decision: striato-orbitofrontal interactions in reinforcement learning, decision making, and reversal. Psychol Rev. 2006;113:300–326. doi: 10.1037/0033-295X.113.2.300. [DOI] [PubMed] [Google Scholar]
- Green MF, Braff DL. Translating the basic and clinical cognitive neuroscience of schizophrenia to drug development and clinical trials of antipsychotic medications. Biol Psychiatry. 2001;49:374–384. doi: 10.1016/s0006-3223(00)01027-1. [DOI] [PubMed] [Google Scholar]
- Gresch PJ, Sved AF, Zigmond MJ, Finlay JM. Local influence of endogenous norepinephrine on extracellular dopamine in rat medial prefrontal cortex. J Neurochem. 1995;65:111–116. doi: 10.1046/j.1471-4159.1995.65010111.x. [DOI] [PubMed] [Google Scholar]
- Grossman F, Potter WZ, Brown EA, Maislin G. A double-blind study comparing idazoxan and bupropion in bipolar depressed patients. J Affect Disord. 1999;56:237–243. doi: 10.1016/s0165-0327(99)00041-5. [DOI] [PubMed] [Google Scholar]
- Hagan JJ, Jones DN. Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull. 2005;31:830–853. doi: 10.1093/schbul/sbi058. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Fujita Y, Iyo M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of fluvoxamine: role of sigma-1 receptors. Neuropsychopharmacology. 2007;32:514–521. doi: 10.1038/sj.npp.1301047. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Fujita Y, Shimizu E, Iyo M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of clozapine, but not haloperidol. Eur J Pharmacol. 2005;519:114–117. doi: 10.1016/j.ejphar.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Hertel P, Fagerquist MV, Svensson TH. Enhanced cortical dopamine output and antipsychotic-like effects of raclopride by alpha2 adrenoceptor blockade. Science. 1999a;286:105–107. doi: 10.1126/science.286.5437.105. [DOI] [PubMed] [Google Scholar]
- Hertel P, Nomikos GG, Svensson TH. Idazoxan preferentially increases dopamine output in the rat medial prefrontal cortex at the nerve terminal level. Eur J Pharmacol. 1999b;371:153–158. doi: 10.1016/s0014-2999(99)00175-2. [DOI] [PubMed] [Google Scholar]
- Iversen SD, Mishkin M. Perseverative interference in monkeys following selective lesions of the inferior prefrontal convexity. Exp Brain Res. 1970;11:376–386. doi: 10.1007/BF00237911. [DOI] [PubMed] [Google Scholar]
- Jentsch JD. Pre-clinical models of cognitive dysfunction in schizophrenia: new avenues to addressing unmet needs. Clin Neurosci Res. 2003;3:303–315. [Google Scholar]
- Jentsch JD, Elsworth JD, Redmond DE, Jr, Roth RH. Phencyclidine increases forebrain monoamine metabolism in rats and monkeys: modulation by the isomers of HA966. J Neurosci. 1997a;17:1769–1775. doi: 10.1523/JNEUROSCI.17-05-01769.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Elsworth JD, Taylor JR, Redmond DE, Jr, Roth RH. Dysregulation of mesoprefrontal dopamine neurons induced by acute and repeated phencyclidine administration in the nonhuman primate: implications for schizophrenia. Adv Pharmacol. 1998a;42:810–814. doi: 10.1016/s1054-3589(08)60870-4. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Olausson P, De La Garza R, Taylor JR. Impairments of reversal learning and response perseveration after repeated, intermittent cocaine administrations to monkeys. Neuropsychopharmacology. 2002;26:183–190. doi: 10.1016/S0893-133X(01)00355-4. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Redmond DE, Jr, Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science. 1997b;277:953–955. doi: 10.1126/science.277.5328.953. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH, Taylor JR. Object retrieval/detour deficits in monkeys produced by prior subchronic phencyclidine administration: evidence for cognitive impulsivity. Biol Psychiatry. 2000;48:415–424. doi: 10.1016/s0006-3223(00)00926-4. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Sanchez D, Elsworth JD, Roth RH. Clonidine and guanfacine attenuate phencyclidine-induced dopamine overflow in rat prefrontal cortex: mediating influence of the alpha-2A adrenoceptor subtype. Brain Res. 2008;1246:41–46. doi: 10.1016/j.brainres.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR. Impaired inhibition of conditioned responses produced by subchronic administration of phencyclidine to rats. Neuropsychopharmacology. 2001;24:66–74. doi: 10.1016/S0893-133X(00)00174-3. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR, Elsworth JD, Redmond DE, Jr, Roth RH. Altered frontal cortical dopaminergic transmission in monkeys after subchronic phencyclidine exposure: involvement in frontostriatal cognitive deficits. Neuroscience. 1999;90:823–832. doi: 10.1016/s0306-4522(98)00481-3. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Tran A, Le D, Youngren KD, Roth RH. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology. 1997c;17:92–99. doi: 10.1016/S0893-133X(97)00034-1. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Wise A, Katz Z, Roth RH. Alpha-noradrenergic receptor modulation of the phencyclidine- and delta9-tetrahydrocannabinol-induced increases in dopamine utilization in rat prefrontal cortex. Synapse. 1998b;28:21–26. doi: 10.1002/(SICI)1098-2396(199801)28:1<21::AID-SYN3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Kalkstein S, Hurford I, Gur RC. Neurocognition in schizophrenia. Curr Top Behav Neurosci. 2010;4:373–390. doi: 10.1007/7854_2010_42. [DOI] [PubMed] [Google Scholar]
- Klingner A, Ban TA, Lehmann HE. Transference, discrimination and reversal. A comparison between normals and pathological groups. Cond Reflex. 1972;7:216–225. [PubMed] [Google Scholar]
- Kopp B, Rist F. Error-correcting behavior in schizophrenic patients. Schizophr Res. 1994;13:11–22. doi: 10.1016/0920-9964(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lane HY, Lee CC, Liu YC, Chang WH. Pharmacogenetic studies of response to risperidone and other newer atypical antipsychotics. Pharmacogenomics. 2005;6:139–149. doi: 10.1517/14622416.6.2.139. [DOI] [PubMed] [Google Scholar]
- Laurent V, Podhorna J. Subchronic phencyclidine treatment impairs performance of C57BL/6 mice in the attentional set-shifting task. Behav Pharmacol. 2004;15:141–148. doi: 10.1097/00008877-200403000-00006. [DOI] [PubMed] [Google Scholar]
- Lee B, Groman SM, London ED, Jentsch JD. Dopamine D2/D3 receptors play a specific role in the reversal of a learned visual discrimination in monkeys. Neuropsychopharmacology. 2007 doi: 10.1038/sj.npp.1301337. in press. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Litman RE, Su TP, Potter WZ, Hong WW, Pickar D. Idazoxan and response to typical neuroleptics in treatment-resistant schizophrenia. Comparison with the atypical neuroleptic, clozapine. Br J Psychiatry. 1996;168:571–579. doi: 10.1192/bjp.168.5.571. [DOI] [PubMed] [Google Scholar]
- Majid DS, Cai W, George JS, Verbruggen F, Aron AR. Transcranial Magnetic Stimulation Reveals Dissociable Mechanisms for Global Versus Selective Corticomotor Suppression Underlying the Stopping of Action. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–150. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- Marcus MM, Wiker C, Franberg O, Konradsson-Geuken A, Langlois X, Jardemark K, Svensson TH. Adjunctive alpha2-adrenoceptor blockade enhances the antipsychotic-like effect of risperidone and facilitates cortical dopaminergic and glutamatergic, NMDA receptor-mediated transmission. Int J Neuropsychopharmacol. 2010;13:891–903. doi: 10.1017/S1461145709990794. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Schizophrenia-like syndrome inducing agent phencyclidine failed to impair memory for temporal order in rats. Neurobiol Learn Mem. 2003;80:158–167. doi: 10.1016/s1074-7427(03)00067-4. [DOI] [PubMed] [Google Scholar]
- Masaki D, Yokoyama C, Kinoshita S, Tsuchida H, Nakatomi Y, Yoshimoto K, Fukui K. Relationship between limbic and cortical 5-HT neurotransmission and acquisition and reversal learning in a go/no-go task in rats. Psychopharmacology (Berl) 2006;189:249–258. doi: 10.1007/s00213-006-0559-0. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. Clinical studies on the mechanism of action of clozapine: the dopamine-serotonin hypothesis of schizophrenia. Psychopharmacology (Berl) 1989;99(Suppl):S18–27. doi: 10.1007/BF00442554. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Huang M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog Brain Res. 2008;172:177–197. doi: 10.1016/S0079-6123(08)00909-6. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Serotonin receptors: their key role in drugs to treat schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1159–1172. doi: 10.1016/j.pnpbp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Merck. Saphris®. 2010 http://www.saphris.com/
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Noda Y, Kamei H, Mamiya T, Furukawa H, Nabeshima T. Repeated phencyclidine treatment induces negative symptom-like behavior in forced swimming test in mice: imbalance of prefrontal serotonergic and dopaminergic functions. Neuropsychopharmacology. 2000;23:375–387. doi: 10.1016/S0893-133X(00)00138-X. [DOI] [PubMed] [Google Scholar]
- Oades RD. Stimulus dimension shifts in patients with schizophrenia, with and without paranoid hallucinatory symptoms, or obsessive compulsive disorder: strategies, blocking and monoamine status. Behav Brain Res. 1997;88:115–131. doi: 10.1016/s0166-4328(97)02304-8. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Cohen M, Panagides J. Efficacy and tolerability of asenapine in acute schizophrenia: a placebo- and risperidone-controlled trial. J Clin Psychiatry. 2007;68:1492–1500. doi: 10.4088/jcp.v68n1004. [DOI] [PubMed] [Google Scholar]
- Rainey JM, Jr, Crowder MK. Prolonged psychosis attributed to phencyclidine: report of three cases. Am J Psychiatry. 1975;132:1076–1078. doi: 10.1176/ajp.132.10.1076. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Yao Z, Zhang X, Sun J, Zhang Z. Pharmacogenetics of treatment in first-episode schizophrenia: D3 and 5-HT2C receptor polymorphisms separately associate with positive and negative symptom response. Eur Neuropsychopharmacol. 2005;15:143–151. doi: 10.1016/j.euroneuro.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Robbins TW. Synthesizing schizophrenia: a bottom-up, symptomatic approach. Schizophr Bull. 2005;31:854–864. doi: 10.1093/schbul/sbi044. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Roberts AC. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cereb Cortex. 2007;17(Suppl 1):i151–160. doi: 10.1093/cercor/bhm066. [DOI] [PubMed] [Google Scholar]
- Rodefer JS, Murphy ER, Baxter MG. PDE10A inhibition reverses subchronic PCP-induced deficits in attentional set-shifting in rats. Eur J Neurosci. 2005;21:1070–1076. doi: 10.1111/j.1460-9568.2005.03937.x. [DOI] [PubMed] [Google Scholar]
- Rogers RD, Andrews TC, Grasby PM, Brooks DJ, Robbins TW. Contrasting cortical and subcortical activations produced by attentional-set shifting and reversal learning in humans. J Cogn Neurosci. 2000;12:142–162. doi: 10.1162/089892900561931. [DOI] [PubMed] [Google Scholar]
- Rolls ET. The functions of the orbitofrontal cortex. Brain Cogn. 2004;55:11–29. doi: 10.1016/S0278-2626(03)00277-X. [DOI] [PubMed] [Google Scholar]
- Sahakian BJ, Coull JJ, Hodges JR. Selective enhancement of executive function by idazoxan in a patient with dementia of the frontal lobe type. J Neurol Neurosurg Psychiatry. 1994;57:120–121. doi: 10.1136/jnnp.57.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Nugent SL, Saddoris MP, Setlow B. Orbitofrontal lesions in rats impair reversal but not acquisition of go, no-go odor discriminations. Neuroreport. 2002;13:885–890. doi: 10.1097/00001756-200205070-00030. [DOI] [PubMed] [Google Scholar]
- Schwabe K, Klein S, Koch M. Behavioural effects of neonatal lesions of the medial prefrontal cortex and subchronic pubertal treatment with phencyclidine of adult rats. Behav Brain Res. 2006;168:150–160. doi: 10.1016/j.bbr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Schwartzbaum JS, Donovick PJ. Discrimination reversal and spatial alternation associated with septal and caudate dysfunction in rats. J Comp Physiol Psychol. 1968;65:83–92. doi: 10.1037/h0025394. [DOI] [PubMed] [Google Scholar]
- Shahid M, Walker G, Zorn S, Wong E. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23:65–73. doi: 10.1177/0269881107082944. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Effects of repeated treatment with amphetamine or phencyclidine on working memory in the rat. Behav Brain Res. 2002;134:267–274. doi: 10.1016/s0166-4328(02)00040-2. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-D-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Stern CE, Passingham RE. The nucleus accumbens in monkeys (Macaca fascicularis). III. Reversal learning. Exp Brain Res. 1995;106:239–247. doi: 10.1007/BF00241119. [DOI] [PubMed] [Google Scholar]
- Taffe MA, Davis SA, Gutierrez T, Gold LH. Ketamine impairs multiple cognitive domains in rhesus monkeys. Drug Alcohol Depend. 2002;68:175–187. doi: 10.1016/s0376-8716(02)00194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Elsworth JD, Roth RH, Sladek JR, Jr, Redmond DE., Jr Cognitive and motor deficits in the acquisition of an object retrieval/detour task in MPTP-treated monkeys. Brain. 1990;113:617–637. doi: 10.1093/brain/113.3.617. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Hansen HH, Mikkelsen JD. Opposite effect of phencyclidine on activity-regulated cytoskeleton-associated protein (Arc) in juvenile and adult limbic rat brain regions. Neurochem Int. 2010;56:270–275. doi: 10.1016/j.neuint.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Tsukada H, Nishiyama S, Fukumoto D, Sato K, Kakiuchi T, Domino EF. Chronic NMDA antagonism impairs working memory, decreases extracellular dopamine, and increases D1 receptor binding in prefrontal cortex of conscious monkeys. Neuropsychopharmacology. 2005;30:1861–1869. doi: 10.1038/sj.npp.1300732. [DOI] [PubMed] [Google Scholar]
- Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- Walker SC, Mikheenko YP, Argyle LD, Robbins TW, Roberts AC. Selective prefrontal serotonin depletion impairs acquisition of a detour-reaching task. Eur J Neurosci. 2006;23:3119–3123. doi: 10.1111/j.1460-9568.2006.04826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis JD, Dias R, Robbins TW, Roberts AC. Dissociable contributions of the orbitofrontal and lateral prefrontal cortex of the marmoset to performance on a detour reaching task. Eur J Neurosci. 2001;13:1797–1808. doi: 10.1046/j.0953-816x.2001.01546.x. [DOI] [PubMed] [Google Scholar]
- Waltz JA, Gold JM. Probabilistic reversal learning impairments in schizophrenia: further evidence of orbitofrontal dysfunction. Schizophr Res. 2007;93:296–303. doi: 10.1016/j.schres.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Baunez C, Theobald DE, Robbins TW. Lesions to the subthalamic nucleus decrease impulsive choice but impair autoshaping in rats: the importance of the basal ganglia in Pavlovian conditioning and impulse control. Eur J Neurosci. 2005;21:3107–3116. doi: 10.1111/j.1460-9568.2005.04143.x. [DOI] [PubMed] [Google Scholar]