

Abstract
Here we present a workflow to analyze the metabolic profiles for biological samples of interest including; cells, serum, or tissue. The sample is first separated into polar and non-polar fractions by a liquid-liquid phase extraction, and partially purified to facilitate downstream analysis. Both aqueous (polar metabolites) and organic (non-polar metabolites) phases of the initial extraction are processed to survey a broad range of metabolites. Metabolites are separated by different liquid chromatography methods based upon their partition properties. In this method, we present microflow ultra-performance (UP)LC methods, but the protocol is scalable to higher flows and lower pressures. Introduction into the mass spectrometer can be through either general or compound optimized source conditions. Detection of a broad range of ions is carried out in full scan mode in both positive and negative mode over a broad m/z range using high resolution on a recently calibrated instrument. Label-free differential analysis is carried out on bioinformatics platforms. Applications of this approach include metabolic pathway screening, biomarker discovery, and drug development.
Keywords: Biochemistry, Issue 75, Chemistry, Molecular Biology, Cellular Biology, Physiology, Medicine, Pharmacology, Genetics, Genomics, Mass Spectrometry, MS, Metabolism, Metabolomics, untargeted, extraction, lipids, accurate mass, liquid chromatography, ultraperformance liquid chromatography, UPLC, high resolution mass spectrometry, HRMS, spectrometry
Introduction
Due to recent technological advances in the field of HRMS, untargeted, hypothesis-generating metabolomics approaches have become a feasible approach to analysis of complex samples.1 Mass spectrometers capable of 100,000 resolution facilitating routine low part per million (ppm) mass accuracy have become widely available from multiple vendors.2,3 This mass accuracy allows greater specificity and confidence in a preliminary assignment of analyte identity, isotopic pattern recognition, and adduct identification.4 When coupled with an appropriate extraction procedure and high-performance LC or UPLC, complex mixtures can be analyzed with additional specificity derived from retention time data.5 UPLC possesses greater chromatographic efficiency and allows greater sensitivity, resolution and analysis time making a greater coverage of the metabolome possible.6 The resulting large datasets can be integrated into any of multiple differential analysis software and mined for useful patterns or individual analytes of interest.7,8,9,10,11 Putative hits can be initially identified using a combination of peak detection algorithms, accurate mass based chemical formula prediction, fragmentation prediction, and chemical database searching. This approach allows prioritization of targets for time-consuming complete structural identification or for development of more sensitive and more specific stable isotope dilution UPLC/selected or multiple reaction monitoring/MS studies that are the current gold standard methods for quantification.12
The varying nature of biological samples has led to optimization of extraction protocols for urine13, cells14, serum15, or tissue16. This protocol features extractions for cells, serum, and tissue. Where appropriate, comments and additional references have been included for modifications of the procedure to address inclusion of stable isotopes, or for inclusion of especially unstable metabolites.
Protocol
1. Sample Extraction from Cells
For a 10 cm plate of cells: collect 1.5 ml of lifted cell suspension in media into a pre-labeled 10 ml glass centrifuge tube. For adherent lines, cells should be lifted with gentle scraping in 1.5 ml of media kept on ice. Optional: If internal standards are used, add an appropriate aliquot at this step. Comment: Quenching of cellular metabolism is crucial for certain metabolites. For analysis of time-sensitive metabolites, procedures such as cold methanol extraction should be considered.17
Add 6 ml of chloroform (CHCl3)/methanol (CH3OH) (2:1, v/v) to each of the 10 ml glass tubes containing the samples. Set the samples in a shaker or multi-vortexer on low speed for 30 min. After shaking, the samples are centrifuged with a low acceleration/deceleration setting at 1,935 x g for 10 min at 4 °C to completely separate the phases. Optional: To avoid using CHCl3, extractions can be conducted with dichloromethane (CH2Cl2).18,12
Using a long stem Pasteur pipette, transfer the organic (bottom) layer to a new pre-labeled 10 ml glass centrifuge tube. Continue to 4.1.
Transfer the upper aqueous later into a clean plastic 2 ml Eppendorf tube. Evaporate the sample under nitrogen gas. Continue to 2.6.2 for aqueous phase desalting or continue to 4.1 for direct analysis.
2. Sample Extraction from Serum
Transfer 20 μl of serum into a plastic 2 ml Eppendorf tube. Optional: If internal standards are used, add an appropriate aliquot at this step.
Add 190 μl of CH3OH and vortex for 10 sec.
Add 380 μl of CHCl3 and vortex for 10 sec.
Add 120 μl of H2O to induce phase separation. Vortex the samples for 10 sec and allow to equilibrate at RT for 10 min.
Centrifuge the samples at 8,000 x g for 10 min at 4 °C to separate the phases.
Transfer the lower organic phase into a clean plastic 2 ml Eppendorf tube. Continue to 4.1.
Transfer the upper aqueous later into a clean plastic 2 ml Eppendorf tube. Evaporate the sample under nitrogen gas.
Re-suspend the sample in 200 μl H2O. Acid or base reagents (0.1% Acetic acid, formic acid or 0.1% ammonium hydroxide) may be used to preferentially extract pH sensitive metabolites. Sonicate for 20 min on ice to re-suspend the sample completely.
Activate the C18 spin columns with 500 μl CH3OH.
Equilibrate the spin column with two washes of 500 μl H2O and then load the sample. If acid/base reagents are used maintain this concentration in the wash steps.
Wash with 500 μl H2O to desalt the sample. Again, if acid/base reagents are used maintain this concentration in the wash steps.
Elute with two volumes of 200 μl 80% CH3OH in H2O into a pre-labeled clean 2 ml Eppendorf tube. Again, if acid/base reagents are used maintain this concentration in the wash steps. Continue to 4.1.
3. Sample Extraction from Tissue
Depending on the tissue, use between 10-100 mg of frozen tissue. Add the whole piece of tissue to a pre-labeled 2 ml low retention Eppendorf tube with 1 ml of PBS (1 M, pH 6.8) in an ice bath. Optional: If internal standards are used, add it into the buffer, before adding the tissue.
Homogenize the tissue in ice with an electric tissue grinder for 30 sec in each Eppendorf. Between samples, clean the tissue grinder in two separate tubes containing CH3OH and H2O, respectively.
Transfer the tissue homogenate with a plastic pipette into a pre-labeled 10 ml glass centrifuge tube. After the transfer, rinse each Eppendorf tube 2x with 1 ml of CH3OH and add the washes to the tissue homogenate in the glass tubes.
Add 4 ml of CHCl3 to each of the 10 ml glass tubes containing the tissue homogenate and CH3OH washes. Set the samples in a shaker or multi-vortexer on low speed for 30 min.
After shaking, centrifuge the samples with a low acceleration/deceleration setting at 1,935 x g for 10 min to completely separate the phases. Optional: To avoid using chloroform, extractions have been developed with CH2Cl2.
Using a long stem Pasteur pipette, transfer the organic (bottom) layer to a new pre-labeled 10 ml glass centrifuge tube. Continue to 4.1.
Using a long stem Pasteur pipette, transfer the aqueous layer to a new pre-labeled 10 ml glass centrifuge tube. Go to 2.6.2 for desalting or continue to 4.1.
4. Re-suspension and Filtration of Samples for UPLC
Evaporate the samples under nitrogen gas.
Reconstitute dried down samples in an appropriate volume of the starting solution for the desired UPLC method (see Table 1). 50-100 μl is usually a desirable re-suspension volume. Gently vortex and/or pipette the sample up and down to aid in dissolving the analytes.
Transfer the sample into a 0.22 μm nylon tube filter and spin at up to 14,000 x g until the sample has completely passed through the filter (~5 min). Make sure that there are no visible precipitates remaining in the sample.
Transfer the supernatant of the filtered sample into a pre-labeled UPLC vial with insert. Cap vial, then flick the vial to remove any bubbles from the bottom of the sample vial. Place the sample in the chilled autosampler (4-5 °C).
5. UPLC Setup
Using the control software for the liquid chromatograph, create a run for the organic sample based off of the conditions listed in Table 1. Then create a method for the aqueous phase off the conditions listed in Table 1. Optional: If a known set of target analytes are of high interest, optimize the UPLC conditions using the heavy labeled analog of these compounds, and generate a method using these conditions.
Allow the UPLC to adequately equilibrate. This should include a full priming of solvents before attaching a column, and following the manufacturer's directions for equilibration volume of a new column (usually 3-5 column volumes). Maintain a log of starting back pressures to help diagnose future problems.
6. Mass Spectrometer Setup
Using the control software for the high resolution mass spectrometer, create a positive mode method using the conditions listed in Table 2. Then using the same conditions, create a negative mode method. Optional: If a known set of target analytes are of high interest, optimize source and optics conditions using the heavy labeled analog of these compounds, and generate a method using these conditions.
Clean and calibrate the instrument, establish a stable spray into the spectrometer, and allow time for the calibration solution to adequately dissipate from the source and optics. Acceptable stability of spray should give a 3-5% RSD of intensity over at least 100 scans with injection of calibration solution at the same flow rate as the LC method being used.
Set up the sequence for all the samples, set appropriate injection volume, and run a blank before the first sample. If carryover or matrix effects between samples are suspected, run adequate blanks/washes. Samples should be setup in a randomized manner using a random number generator and a key to avoid any batch effects introduced by analysis. Comment: Quality control samples including stable isotope standards or analytical standards of likely targets can be extremely valuable in troubleshooting and assuring reliable, reproducible, and valid results. A technical replicate of a standard only sample followed by a solvent blank can be used to accesses deviation of signal intensity and retention time, as well as carryover into the blank run.
Begin sequence and monitor periodically for problems including pressure fluctuations, or loss of signal intensity across the run. If severe problems are detected, stop the run, and repeat from 6.2.
7. Differential Analysis
Two workflows are presented for this protocol. The first is through SIEVE 2.0, proprietary label-free differential analysis software sold by Thermo Fisher. The second workflow is through XCMS Online, a free platform through Scripps Research Institute.11 Before beginning differential analysis, manually check the TICs or look at filtered chromatograms for any known compound in the sample to ensure reproducibility of runs and injection/UPLC/spray stability throughout all the samples.
- SIEVE
- Transfer the .raw data files from the mass spectrometer to the hard drive of the workstation where SIEVE is installed. (Note: running SIEVE with data stored on a networked or USB port connected drive is not advisable as it can limit the speed of analysis.)
- Open SIEVE, and begin a new experiment.
- Load the appropriate files into SIEVE.
- Assign comparison groups and select a single reference file to allow SIEVE to generate the parameters for analysis.
- Follow the prompts and adjust any parameters per requirements of any individual experiment. It is often advisable to start with stringent parameters and then go back and loosen the parameters as more generous settings can lengthen analysis time.
- Load the correct adduct list for positive or negative mode in the parameters.
- XCMS Online
- Convert files into an acceptable format for XCMS Online. In our case, we convert to mzXML format.19
- Open XCMS Online, and Create Job.
- Upload and name datasets. Only two datasets (e.g. Control vs Treatment) can be uploaded as XCMS stand-alone is limited to head-to-head comparisons.
- Select desired settings from the drop-down list. Pre-populated parameters are available for different instrumentation setups, and these can be further modified for experimental needs. In our case, we use the HPLC-Orbi2 settings.
- Submit and confirm job.
- Data Analysis
- For SIEVE and XCMS a list of frames and features, respectively, will be populated once the analysis is finished. Ensure that the LC alignment overlays any landmark peaks. If any of the unaligned LC runs show large deviation in landmark peaks this may indicate a problem with the sample. Optional: For SIEVE, if internal standards are used, locate the corresponding peak group and normalize to the selected frame. For XCMS On-line, normalization can be done in a spreadsheet after export.
- Plot the data by coefficient of variation (CV) within a like sample population (e.g. control samples). Any large CV values may indicate a problem with one or more sample. Sort the list based on desired traits (e.g. p-value<0.05, Fold Change>1.5, low standard deviation within a group, etc).
- Examine each peak for appropriate peak alignment across groups, peak integration, Gaussian peak shape, signal intensity, isotopes, and adduct assignment.
- Export data as desired for further analysis or to generate targeted mass lists for confirmation and MSn experiments.
Representative Results
The results presented show selected data from a 6-hr treatment of SH-SY5Y glioblastoma cells with the pesticide and mitochondrial complex I inhibitor rotenone. For brevity, only the organic phase positive mode data is presented. The samples were processed and analyzed as described above (Figure 1, Table 1, Table 2) and loaded onto two differential analysis platforms for label-free quantification, SIEVE and XCMS online. Although a large number of hits (Figure 2, Figure 3) are identified by the two programs used for differential analysis these features include likely artifacts, poorly integrated peaks, and other features of questionable analytical value. This can be judged by screening the hits for appropriate signal intensity, low variation between samples of the same group, background levels, and good chromatographic peak shapes (Figure 3).
To demonstrate the downstream confirmation of initial hits, we isolate a feature with a mass corresponding to our treatment compound rotenone within 3 ppm (Figure 3, Figure 4). We confirm the identity of this analyte with UPLC-HR/tandem mass spectrometry (MS/MS) of our sample and the pure compound (Figure 5). We also identify a differentially abundant feature reduced in the rotenone treated group, tentatively identified as D-pantethine by accurate mass database searching (Figure 3).
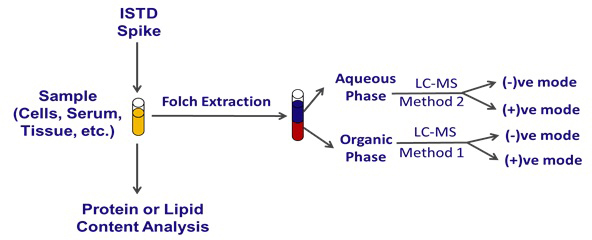 Figure 1. General Experimental Outline for Sample Preparation and Analysis by Microspray UPLC-HRMS. A general outline of sample preparation, including the optional introduction of internal standards and protein or lipid/protein content analysis. Phase separation, liquid chromatography, and analysis by different ion modes in the mass spectrometer are outlined.
Figure 1. General Experimental Outline for Sample Preparation and Analysis by Microspray UPLC-HRMS. A general outline of sample preparation, including the optional introduction of internal standards and protein or lipid/protein content analysis. Phase separation, liquid chromatography, and analysis by different ion modes in the mass spectrometer are outlined.
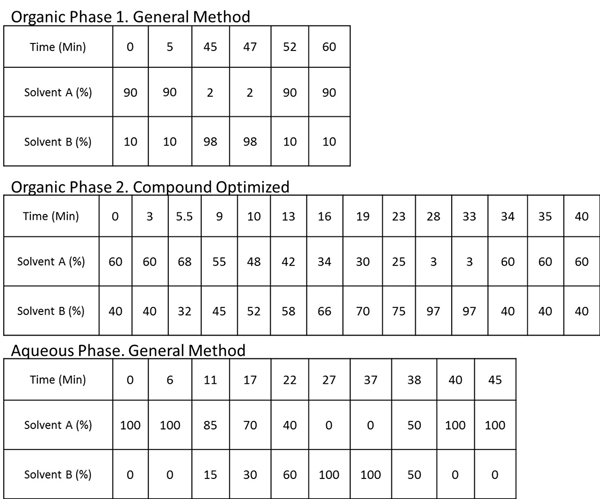 Table 1. UPLC Conditions. General and compound optimized UPLC conditions. For organic phase 1, constant flow rate of 1.8 μl/min through a Waters nanoACQUITY C18 BEH130 column kept in a heater at 35 °C was used. Solvent A: 99.5% water/0.5% acetonitrile with 0.1% formic acid, Solvent B: 98% acetonitrile/2% water with 0.1% formic acid For organic phase 2, constant flow rate of 3.4 μl/min through an Acentis Express C8 column kept in a heater at 35 °C was used. Solvent A: 40% water/20% acetonitrile with 0.1% formic acid and 10 mM ammonium formate, Solvent B: 90% isopropanol/10% acetonitrile with 0.1% formic acid and 10 mM ammonium formate For the aqueous phase, constant flow rate of 1.2 μl/min through a Waters nanoACQUITY C18 BEH130 column kept in a heater at 35 °C was used. Solvent A: 95% water/5% methanol with 0.1% ammonium hydroxide, Solvent B: 95% methanol/5% water with 0.1% ammonium hydroxide.
Table 1. UPLC Conditions. General and compound optimized UPLC conditions. For organic phase 1, constant flow rate of 1.8 μl/min through a Waters nanoACQUITY C18 BEH130 column kept in a heater at 35 °C was used. Solvent A: 99.5% water/0.5% acetonitrile with 0.1% formic acid, Solvent B: 98% acetonitrile/2% water with 0.1% formic acid For organic phase 2, constant flow rate of 3.4 μl/min through an Acentis Express C8 column kept in a heater at 35 °C was used. Solvent A: 40% water/20% acetonitrile with 0.1% formic acid and 10 mM ammonium formate, Solvent B: 90% isopropanol/10% acetonitrile with 0.1% formic acid and 10 mM ammonium formate For the aqueous phase, constant flow rate of 1.2 μl/min through a Waters nanoACQUITY C18 BEH130 column kept in a heater at 35 °C was used. Solvent A: 95% water/5% methanol with 0.1% ammonium hydroxide, Solvent B: 95% methanol/5% water with 0.1% ammonium hydroxide.
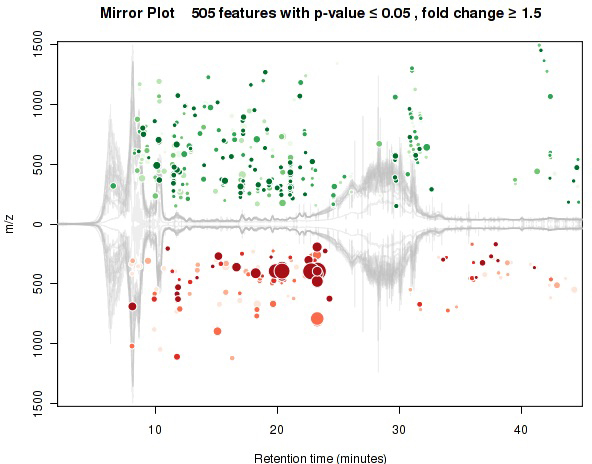 Figure 2. Mirror Plot Generated with XCMS. A mirror plot showing differentially abundant features as detected and quantified through XCMS online from the organic phase positive mode UPLC-MS analysis. Each dot represents a distinct "feature." Green dots are features more abundant (larger integrated area) in the control, whereas red dots are more abundant in the treatment. Increasing size of the dot indicates increasing magnitude of fold change between the groups, and the intensity of color indicates a decreasing p-value with increasing saturation of color.
Figure 2. Mirror Plot Generated with XCMS. A mirror plot showing differentially abundant features as detected and quantified through XCMS online from the organic phase positive mode UPLC-MS analysis. Each dot represents a distinct "feature." Green dots are features more abundant (larger integrated area) in the control, whereas red dots are more abundant in the treatment. Increasing size of the dot indicates increasing magnitude of fold change between the groups, and the intensity of color indicates a decreasing p-value with increasing saturation of color.
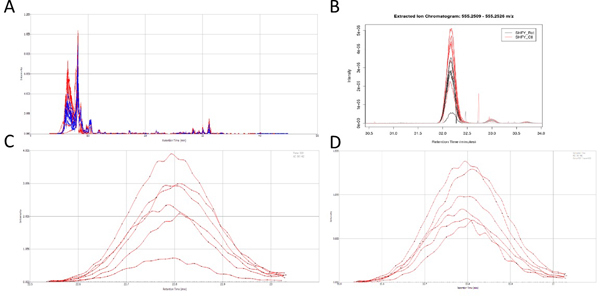 Figure 3. Analyte Peaks. Chromatograms showing the organic phase positive ion mode UPLC-MS analysis. A) Corrected (aligned) Total ion current (TIC) from SIEVE 2.0. B) Extracted Ion Chromatogram for the feature tentatively identified as D-pantethine through XCMS. C) Extracted Ion Chromatogram for the feature later identified as rotenone from SIEVE. D) XIC for the feature later identified as rotenone from SIEVE normalized to TIC. Click here to view larger figure.
Figure 3. Analyte Peaks. Chromatograms showing the organic phase positive ion mode UPLC-MS analysis. A) Corrected (aligned) Total ion current (TIC) from SIEVE 2.0. B) Extracted Ion Chromatogram for the feature tentatively identified as D-pantethine through XCMS. C) Extracted Ion Chromatogram for the feature later identified as rotenone from SIEVE. D) XIC for the feature later identified as rotenone from SIEVE normalized to TIC. Click here to view larger figure.
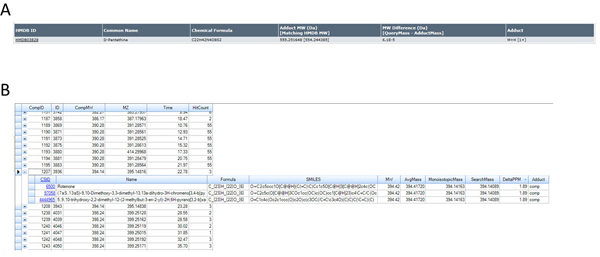 Figure 4. Database Search Results. Database hits for A) Human Metabolome Database (http://www.hmdb.ca) and B) ChemSpider/KEGG (via SIEVE) (ChemSpider alone can also be employed - http://www.chemspider.com) using accurate mass determinations for the features shown in Figures 3B, 3C, and 3D corresponding to the ions of m/z 555.2516 and 395.1481 based on the identification of these ions as [MH]+ species. Click here to view larger figure.
Figure 4. Database Search Results. Database hits for A) Human Metabolome Database (http://www.hmdb.ca) and B) ChemSpider/KEGG (via SIEVE) (ChemSpider alone can also be employed - http://www.chemspider.com) using accurate mass determinations for the features shown in Figures 3B, 3C, and 3D corresponding to the ions of m/z 555.2516 and 395.1481 based on the identification of these ions as [MH]+ species. Click here to view larger figure.
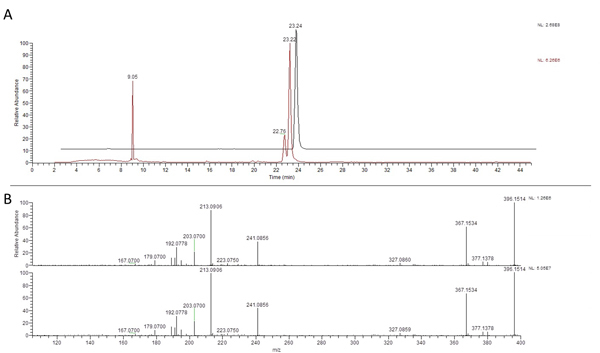 Figure 5. HR/MS/MS Structural Confirmation. Confirmation of identification of the feature corresponding to the ion at 395.1481 m/z with a retention time of 23.22 min as rotenone based on UPLC-MS/MS comparison to a commercial standard with A) similar retention time and B) the same m/z (395.1481, ~0 ppm error) with nearly identical fragmentation. Click here to view larger figure.
Figure 5. HR/MS/MS Structural Confirmation. Confirmation of identification of the feature corresponding to the ion at 395.1481 m/z with a retention time of 23.22 min as rotenone based on UPLC-MS/MS comparison to a commercial standard with A) similar retention time and B) the same m/z (395.1481, ~0 ppm error) with nearly identical fragmentation. Click here to view larger figure.
| Mass Spectrometry Parameter | Setting | Comment |
| Source | ESI | Michrom CaptiveSpray |
| Ion Model | Positive or Negative | |
| Method Length | Variable (~40-60 min) | |
| Resolution | 60,000-100,000 | |
| Spray (kV) | 1.75 | |
| Tube Lens (V) | 130 | Target Compound Dependent |
| Capillary Temperature (°C) | 250 | May shorten life of spray tip |
| Capillary Voltage (V) | 50 | Target Compound Dependent |
| Data Type | Centroid | (for Profile data, see Discussion) |
Table 2. MS Settings. General and compound optimized mass spectrometer settings used for the LTQ XL-Orbitrap with a Michrom Thermo Advance captive spray ESI source.
Discussion
Untargeted metabolomics offers a powerful tool for investigating endogenous or xenobiotic biotransformations, or capturing a metabolic profile from a sample of interest. The output of the technique scales with the resolution and sensitivity of the technology used to separate and analyze the sample, the ability to deal with the large datasets generated, and the ability to mine the dataset for useful information (e.g. accurate mass database searching). Recently, this has been facilitated by advances in high resolution mass spectrometers, and high- or ultra-performance liquid chromatography. Differential analysis software has addressed the analysis bottleneck, and can accomplish peak detection adjustment for retention time shifts, filtering, and statistical analysis with high-throughput.20,21,22 Selection of the proper informatics pipeline should include considerations as to data centroiding algorithms, peak detection, peak integration, alignment, ability to integrate MS/MS data, and ability to deal with isotopes or adducts.23 Selection of appropriate cheminformatics databases should also be considered.24,25,26,27 The current inadequacy of any particular database to comprehensively identify compounds and integrate accurate mass data, MS/MS data, or LC data remains a major problem in the field.
The workflow presented here integrates liquid-liquid extraction, micro-flow liquid chromatography, and high resolution mass spectrometry with two different differential analysis software platforms demonstrated in a cell culture treatment model. Additionally, extraction protocols are listed for serum and tissue, as these may serve as useful samples for similar analysis.
Although any method used for untargeted metabolomics should be optimized for repeatability, stable UPLC conditions, and source stability, some variation is inevitable. Both SIEVE and XCMS allow correction for retention time shift, but special attention should be paid to ensure that the parameters set in the experiment are adequate to correct variation. Also, stable isotope labeled internal standard(s) can be easily integrated into this procedure to reduce artifacts and inter-sample variation caused by differences in sample extractions or LC-MS analysis.12 As with all sensitive LC-MS methodology, it is crucial to use high purity reagents, and ensure that the sample preparation removes undesirable particulate matter or aggregate. Artifacts can be generated by the normal variation in contaminants, and it may be desirable to confirm that putative hits are not in fact ubiquitous contaminants of the extraction or analysis process.
Lastly, although the method is labeled as "untargeted" or "unbiased" metabolomics, this is a partial misnomer. The nature of the extraction, separation, and analysis will favor metabolites with certain characteristics including stability during extraction, interaction with stationary and mobile phases during separation, and ionization at the source of the mass spectrometer.28,29,30 Depending on the available instrumentation, this procedure can be adapted to different extractions, flow rates, LC pressures, ion sources, or mass spectrometers. Therefore, we have included notes in the procedure where certain conditions can be optimized based on general knowledge or internal standards representative of a class of molecules of interest.
Disclosures
Authors have nothing to disclose.
Acknowledgments
We acknowledge support of NIH grants P30ES013508 and 5T32GM008076. We also thank Thermo Scientific for access to SIEVE 2.0 and Drs. Eugene Ciccimaro and Mark Sanders of Thermo Scientific for useful discussions.
References
- Pluskal T, Nakamura T, Villar-Briones A, Yanagida M. Metabolic profiling of the fission yeast S. pombe: quantification of compounds under different temperatures and genetic perturbation. Mol. Biosyst. 2010;6(1):182–198. doi: 10.1039/b908784b. [DOI] [PubMed] [Google Scholar]
- Makarov A, Denisov E, et al. Performance Evaluation of a Hybrid Linear Ion Trap/Orbitrap Mass Spectrometer. Analytical Chemistry. 2006;78(7):2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- Timischl B, Dettmer K, Kaspar H, Thieme M, Oefner PJ. Development of a quantitative, validated capillary electrophoresis-time of flight-mass spectrometry method with integrated high-confidence analyte identification for metabolomics. Electrophoresis. 2008;29(10):2203–2214. doi: 10.1002/elps.200700517. [DOI] [PubMed] [Google Scholar]
- Katajamaa M, Oresic M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A. 2007;1158(1-2):318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Katajamaa M, Oresic M. Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics. 2005;6:179. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ID, Nicholson JK, et al. High resolution ultra performance liquid chromatography coupled to q-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. Journal of Proteome Research. 2005;4(2):591–598. doi: 10.1021/pr049769r. [DOI] [PubMed] [Google Scholar]
- Benton HP, Wong DM, Trauger SA, Siuzdak G. XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem. 2008;80(16):6382–6389. doi: 10.1021/ac800795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajamaa M, Miettinen J, Oresic M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22(5):634–636. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11(1):395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Analytical Chemistry. 2006;78(3):779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Analytical Chemistry. 2012;84(11):5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelhaus SL, Mesaros AC, Blair IA. Cellular Lipid Extraction for Targeted Stable Isotope Dilution Liquid Chromatography-Mass Spectrometry Analysis. J. Vis. Exp. 2011. p. e3399. [DOI] [PMC free article] [PubMed]
- Want EJ, Wilson ID, et al. Global metabolic profiling procedures for urine using UPLCGÇôMS. Nature Protocols. 2010;5(6):1005–1018. doi: 10.1038/nprot.2010.50. [DOI] [PubMed] [Google Scholar]
- Sellick CA, Hansen R, Stephens GM, Goodacre R, Dickson AJ. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nature Protocols. 2011;6(8):1241–1249. doi: 10.1038/nprot.2011.366. [DOI] [PubMed] [Google Scholar]
- Dunn WB, Broadhurst D, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature protocols. 2011;6(7):1060–1083. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- Masson P, Alves AC, Ebbels TMD, Nicholson JK, Want EJ. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Analytical Chemistry. 2010;82(18):7779–7786. doi: 10.1021/ac101722e. [DOI] [PubMed] [Google Scholar]
- Shaham O, Slate NG, et al. A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proceedings of the National Academy of Sciences. 2010;107(4):1571–1575. doi: 10.1073/pnas.0906039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cequier-Saünchez E, Rodriüguez C, Ravelo AG, Zaürate R. Dichloromethane as a Solvent for Lipid Extraction and Assessment of Lipid Classes and Fatty Acids from Samples of Different Natures. Journal of Agricultural and Food Chemistry. 2008;56(12):4297–4303. doi: 10.1021/jf073471e. [DOI] [PubMed] [Google Scholar]
- Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005;1 doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland WS, Devlin SJ. Locally weighted regression - an approach to regression-analysis by local fitting. J. Am. Stat. Assoc. 1988;83(403):596–610. [Google Scholar]
- Lange E, Tautenhahn R, Neumann S, Gropl C. Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics. 2008;9(1):375. doi: 10.1186/1471-2105-9-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R, Bottcher C, Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 2008;9(1):504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C, Tautenhahn R, Bottcher C, Larson TR, Neumann S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Analytical Chemistry. 2012;84(1):283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, O'Maille G, et al. METLIN: a metabolite mass spectral database. Ther. Drug Monit. 2005;27(6):747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37 Web Server:W623–W633. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37 Database:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian Journal of Biochemistry and Physiology. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Folch J. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Avery MJ. Quantitative characterization of differential ion suppression on liquid chromatography/atmospheric pressure ionization mass spectrometric bioanalytical methods. Rapid Communications in Mass Spectrometry. 2003;17(3):197–201. doi: 10.1002/rcm.895. [DOI] [PubMed] [Google Scholar]