

Background: Increased protein N-glycosylation and aberrant Wnt signaling are features of oral cancer.
Results: Overexpression of pro-migratory protein CTHRC1 is due to hyperglycosylation and transcriptional activation by canonical Wnt.
Conclusion: N-Glycosylation collaborates with canonical Wnt to induce CTHRC1 and drive OSCC cell migration.
Significance: Elucidating how N-glycosylation impacts tumor-promoting proteins is critical to understand cancer development and progression.
Keywords: Cancer, Cell Migration, Glycoprotein, Glycosylation, Wnt Signaling, CTHRC1, DPAGT1, OSCC, Canonical Wnt
Abstract
Oral squamous cell carcinoma (OSCC) is one of the most pernicious malignancies, but the mechanisms underlying its development and progression are poorly understood. One of the key pathways implicated in OSCC is the canonical Wnt/β-catenin signaling pathway. Previously, we reported that canonical Wnt signaling functions in a positive feedback loop with the DPAGT1 gene, a principal regulator of the metabolic pathway of protein N-glycosylation, to hyperglycosylate E-cadherin and reduce intercellular adhesion. Here, we show that in OSCC, DPAGT1 and canonical Wnt signaling converge to up-regulate CTHRC1 (collagen triple helix repeat containing 1), an N-glycoprotein implicated in tumor invasion and metastasis. We found that in human OSCC specimens, amplification of the levels of CTHRC1 was associated with its hyperglycosylation. Partial inhibition of DPAGT1 expression in OSCC CAL27 cells reduced CTHRC1 abundance by increasing protein turnover, indicating that N-glycosylation stabilizes CTHRC1. Additionally, canonical Wnt signaling promoted β-catenin/T-cell factor transcriptional activity at the CTHRC1 promoter to further elevate CTHRC1 levels. We demonstrate that DPAGT1 promotes cell migration and drives the localization of CTHRC1 to cells at the leading edge of a wound front coincident with drastic changes in cell morphology. We propose that in OSCC, dysregulation of canonical Wnt signaling and DPAGT1-dependent N-glycosylation induces CTHRC1, thereby driving OSCC cell migration and tumor spread.
Introduction
Head and neck squamous cell carcinoma ranks as the sixth most common cancer in the world and is associated with high morbidity and poor survival rates (1–3). Oral squamous cell carcinoma (OSCC),2 which involves epithelial neoplasms of the oral cavity and oropharynx, accounts for the majority of head and neck squamous cell carcinomas (1). Although whole-exome sequencing studies have identified components of signaling pathways whose aberrant expression and/or activity has been linked to head and neck squamous cell carcinoma pathogenesis (4, 5), there is a critical need for a better understanding of the molecular events that drive early stages of its development and progression.
The canonical Wnt signaling pathway has acknowledged roles in cell proliferation and cell fate, and its dysregulation is pivotal in the early pathogenesis of many cancers, including head and neck squamous cell carcinoma (4–10, 13). Aberrant activation of canonical Wnt signaling leads to accumulation of β-catenin, which translocates to the nucleus and, through binding to the T-cell factor (TCF)/lymphoid enhancer-binding factor family of transcription factors, regulates the expression of a wide range of genes, including several oncogenic factors, such as cyclin D1 and c-myc.
Canonical Wnt signaling functions within a network of signaling pathways and is intimately linked to cell adhesion receptors and cellular metabolism (11–14). Our previous studies have identified asparagine-linked protein glycosylation (N-glycosylation) as a key metabolic pathway that is integrated with canonical Wnt signaling (15). Evolutionarily conserved and essential for viability, N-glycosylation controls a wide spectrum of activities, including protein folding, sorting, targeting, and secretion, as well as cell adhesion and signaling events (16). We have shown that the first N-glycosylation gene, DPAGT1, which functions at a rate-limiting step in the N-glycosylation pathway, interacts with canonical Wnt signaling via a positive feedback loop (15, 17). DPAGT1 encodes the first enzyme in the dolichol pathway, dolichol-P-dependent N-acetylglucosamine-1-phosphate transferase (GPT), which initiates the synthesis of the lipid-linked oligosaccharide precursor for protein N-glycosylation in the endoplasmic reticulum. Many components of canonical Wnt signaling are N-glycoproteins, and their N-glycosylation status controls Wnt signaling by regulating secretion and membrane localization of a canonical Wnt ligand, Wnt3a, and a co-receptor, LRP6 (17). In turn, canonical Wnt activates DPAGT1 expression via β-catenin at a transcriptional level. Furthermore, N-glycosylation inhibits E-cadherin adhesion and interferes with E-cadherin antagonism of canonical Wnt signaling (17, 18). Aberrant activation of canonical Wnt and N-glycosylation pathways has been associated with tumorigenesis (9, 19–21).
Recently, we reported that OSCC specimens display an aberrantly amplified DPAGT1/canonical Wnt feedback loop (6), where overexpression of DPAGT1 is due to increased occupancy of β-catenin at the DPAGT1 promoter. This leads to hyperglycosylation of E-cadherin and reduced intercellular adhesion, resulting in a continuous activation of N-glycosylation and canonical Wnt signaling.
Inappropriate activation of N-glycosylation and canonical Wnt signaling early in OSCC pathogenesis is likely to trigger downstream events leading to cancer progression. For OSCC tumors to spread, cells must migrate off the primary tumor epithelium. Thus, another consequence of amplified DPAGT1/canonical Wnt signaling could be the activation of key components driving OSCC cell migration. Recently, CTHRC1 (collagen triple helix repeat containing 1), a secreted N-glycoprotein highly conserved in evolution, has emerged as a key player in tumor invasion and metastasis (22). First identified in a screen for novel genes associated with vascular injury and repair, CTHRC1 can be tethered to the cell surface to promote actin polymerization and cell polarity (23). Strikingly, up-regulation of CTHRC1 is a feature of many aggressive human cancers, being linked to tumor cell migration and invasion (24–26).
Here, we report that human OSCC specimens display dramatic up-regulation of CTHRC1, which coincides with its hyperglycosylation. We show that N-glycosylation stabilizes the CTHRC1 protein by decreasing its turnover rate. At the same time, canonical Wnt signaling further induces CTHRC1 expression at a transcriptional level via increased recruitment of β-catenin to the CTHRC1 promoter. In scratch wound assays, N-glycosylation drives the localization of CTHRC1 to cells at the migrating wound front and promotes cell migration, in part, through a non-canonical Wnt pathway. Collectively, our studies show that in OSCC, CTHRC1 is up-regulated by the amplified DPAGT1/canonical Wnt feedback loop. We propose that this aberrant up-regulation of CTHRC1 drives OSCC tumor cell migration and the subsequent invasion and metastasis.
EXPERIMENTAL PROCEDURES
Reagents
Anti-CTHRC1 monoclonal antibody was used for immunoblotting and immunoprecipitation analyses. A polyclonal antibody to the last 11 amino acids of the C terminus of GPT was prepared commercially (Covance). For immunofluorescence studies, anti-CTHRC1 antibody was purchased from Abcam, as was anti-Wnt5a polyclonal antibody. Monoclonal antibodies to β-catenin, Rac1, and the IgG isotype control were obtained from BD Biosciences. Polyclonal antibodies to TCF3/4 and Fzd6 were from Exalpha Biologicals and R&D Systems, respectively. Anti-RhoA antibody was from Santa Cruz Biotechnology, and antibody to active GTP-bound RhoA was from NewEast Biosciences. Anti-Dvl2 polyclonal antibody was from Cell Signaling. Antibodies to GAPDH and FLAG were from Sigma and anti-pan-actin antibody was from NeoMarkers.
Cell Culture and Transfections
Cultures of OSCC CAL27 cells were grown to 70% confluence in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, penicillin, and streptomycin as described (6). Total cell lysates were prepared by extraction with Triton X-100/β-octyl glucoside buffer. When indicated, CAL27 cells were treated with 50% Wnt3a-containing conditioned medium for 24 h as described (18). CAL27 cells expressing recombinant DPAGT1 were obtained by transfection of passage 2 CAL27 cells with full-length DPAGT1 (RefSeq NM_001382) or transcript variant (Refseq NM_203316) cDNA clones (OriGene) at 80–90% confluence using Lipofectamine 2000. Controls included untransfected cells and cells transfected with a control pCMV6-Entry vector. After 14 h, the media were changed, and cells were divided into several plates and grown in the presence of G418. Media were changed every 2–3 days and supplemented with G418. After 2 weeks, cells were processed for RNA isolation and preparation of total cell lysates. For immunofluorescence analyses, stable transfectants were plated in chamber slides at a density of 5–6 × 103/cm2 and processed as described (32).
RNA Interference and Quantitative Real-time PCR
SMARTpool siRNAs targeting CTHRC1 and DPAGT1 (referred to as S siRNA) were obtained from Dharmacon. The non-silencing negative control siRNA (referred to as NS siRNA) was from Qiagen. CAL27 cells were transfected at 60% confluence with either NS or S siRNA (150 nm) using Lipofectamine 2000 (Invitrogen) and cultured for 48 h. Total RNAs isolated from CAL27 cells transfected with either NS or S siRNA were used for cDNA synthesis to assess CTHRC1 and DPAGT1 expression by real-time PCR. The gene expression profiles were generated by normalizing the Ct (threshold cycle numbers) of CTHRC1 and DPAGT1 with a housekeeping gene (18 S rRNA) and comparing the gene expression of cells treated with NS or S siRNA.
Cell Migration and Scratch Wound Assay
For cell migration assays, serum-free medium containing 1 × 105 cells was placed into the upper compartment of Transwell inserts (Corning), and the lower compartment was filled with medium containing 10% FBS. Cells in Transwells were then incubated for 20 h in 5% CO2 at 37 °C. Cell migration was quantified by counting crystal violet-positive cells (Fisher). For wounding studies, CAL27 cells transfected with either NS or S siRNAs, as well as CAL27 cells transfected with DPAGT1 cDNA, were grown to confluence in P60 plates and wounded with a sterile 200- or 1000-μl pipette tip, washed three times with growth medium, and returned to the incubator. At the indicated times, wound edges were photographed using a phase-contrast Nikon Eclipse TE300 microscope and 10× objective. For immunofluorescence analyses of wounded cells, confluent cultures of CAL27 cells transfected with non-silencing RNAs or siRNAs to DPAGT1 were grown in chamber slides. At 18 h post-wounding, cells were fixed and processed for immunofluorescence localization of CTHRC1 and for F-actin organization by counterstaining with rhodamine-phalloidin. Cells were then examined on a Zeiss LSM 510 META confocal microscope.
Tissue Specimens
All studies with surgical OSCC specimens were approved by the Institutional Review Board at the Boston University Medical Campus. Fresh tissues were obtained from patients with moderately differentiated to poorly differentiated OSCC of the tongue, maxillary gingiva, and floor of mouth. Regions of OSCC and adjacent epithelia (AE), defined by an on-site pathology analysis, were snap-frozen at −80 °C. Tissues were divided for H&E analyses, biochemistry, and immunofluorescence staining. OCT-embedded fresh tumor tissues were used for preparation of frozen sections (5 μm). One frozen section was set aside for H&E staining, whereas the remaining sections were processed for immunofluorescence analyses as described below. For biochemical analyses, total tissue lysates (TTLs) from AE and OSCC were prepared by extraction with Triton X-100/β-octyl-glucoside buffer as described previously (24). Protein concentrations were determined using the BCA assay (Pierce).
Immunoblotting and Immunoprecipitation
Cell and tissue lysates were fractionated on 7.5 or 10% SDS-polyacrylamide gel, transferred onto polyvinylidene difluoride membranes, blocked with 10% nonfat dry milk, and incubated with primary antibodies to selected proteins. Protein-specific detection was carried out with horseradish peroxidase-labeled secondary antibodies and an ECL Plus kit (Amersham Biosciences). For co-immunoprecipitation studies, equal amounts of protein (500 μg) were precleared with antibody isotype controls and protein G beads (Sigma). The resulting supernatants were incubated with anti-CTHRC1 antibody (2 μg) and 50 μl of protein G beads for 2 h at 4 °C. The beads were washed with lysis buffer, and samples were analyzed by immunoblotting. Due to the similar migration of CTHRC1 and the 25-kDa IgG chain, CTHRC1 immunoprecipitates were fractionated on 10% nonreducing gels. Immunoblots were quantified using ImageJ software.
Peptide N-Glycosidase F Digestion
Tissue lysates were digested with 100 units of peptide N-glycosidase F (PNGase F; New England Biolabs) for 1 h at 37 °C and analyzed by immunoblotting. Control samples were incubated without the enzyme.
Protein Stability
CAL27 cells were stimulated with conditioned medium overexpressing Wnt3a for 24 h and then transfected with either NS siRNA or siRNA to DPAGT1 and grown for 48 h. Next, cycloheximide (20 μg/ml) was added to NS and S siRNA-transfected cells, and cells were harvested at 0, 4, 8, and 12 h. CTHRC1 protein levels were examined by immunoblotting and quantified with ImageJ. The half-life of CTHRC1 in NS and S siRNA-transfected cells was determined by calculating the time when 50% of CTHRC1 remained using a line of best fit in Excel.
Immunofluorescence
For indirect immunofluorescence analyses, CAL27 cells were grown to 80% confluence, fixed in 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 10% goat serum, and incubated with primary antibodies. Cells were then incubated with FITC-tagged secondary antibodies (Invitrogen), counterstained for F-actin with rhodamine-phalloidin, mounted in VECTASHIELD, and analyzed with a Zeiss LSM 510 META confocal microscope. For indirect immunofluorescence analyses of human AE and OSCC specimens, tissue sections were blocked with 10% goat serum and incubated with antibodies against selected proteins, followed by secondary antibodies conjugated with FITC. Negative controls lacked primary antibodies. The slides were mounted in VECTASHIELD, and optical sections were analyzed with either a Nikon Eclipse TE300 microscope a Zeiss LSM 510 META confocal microscope.
Chromatin Immunoprecipitation
ChIP assays were carried out with chromatin cross-linked with 1% formaldehyde from CAL27 cells. For tissue ChIP analyses, AE and OSCC were incubated in DMEM containing 1% formaldehyde, homogenized, and sonicated at 4 °C. The sonicated extract was immunoprecipitated, and genomic DNA was isolated from the complex after reverse cross-linking. The primers and the TaqMan probe surrounding the Wnt-responsive element of human CTHRC1 were designed using the Primer Express program (Applied Biosystems). They were as follows: forward primer, 5′-TGC AGT GAT CCC TTT ACC ATT ATA TAA-3′; reverse primer, 5′-TGC AAT CCT AGT CTC TGA TAA AAC AGA-3′; and TaqMan probe, 6Fam-TCCTTCTTTGTCTTTTCTG-MGB.
Statistical Analyses
Data are presented as the mean ± S.E. Statistical significance was determined using two-tailed Student's t tests. A p value <0.05 was considered significant.
RESULTS
Augmented DPAGT1 Levels in Human OSCC Specimens Are Associated with Up-regulation of CTHRC1 Expression
Previously, we reported that aberrantly amplified DPAGT1 leads to hyperglycosylation of E-cadherin and consequently reduced intercellular adhesion (6, 20). Recent studies have identified an N-glycoprotein, CTHRC1, as a key regulator of cell motility in cancer (25, 26). Thus, we hypothesized that increased DPAGT1-mediated N-glycosylation may promote additional molecular events that influence OSCC progression, such as cell migration. We therefore examined the expression and localization of the protein encoded by the DPAGT1 gene (GPT), a canonical Wnt effector (β-catenin), and a pro-migratory N-glycoprotein (CTHRC1) in nine moderately to poorly differentiated human OSCC tongue tumor specimens and corresponding cytologically normal AE. A representative H&E image of AE and OSCC used in this study is shown in Fig. 1A. As reported previously (6), immunoblots of TTLs showed that β-catenin was up-regulated by ∼2-fold in OSCC compared with AE (Fig. 1B). Immunofluorescence analyses indicated that in AE, β-catenin was highly expressed in the proliferating basal layer of the epithelium, with diminished levels in the cytodifferentiating spinous layer. A similar localization pattern was observed for GPT (Fig. 1C). In contrast, in OSCC, β-catenin and GPT exhibited robust expression throughout tumor regions (Fig. 1C). Furthermore, immunoblot analysis confirmed that similar to β-catenin, GPT was more abundant in OSCC compared with AE (Fig. 1E). Although GPT overexpression in these OSCC tumors averaged 4-fold, even a modest increase in DPAGT1 expression has been shown to result in a robust activation of canonical Wnt pathway activity and hyperglycosylation of proteins (17). Immunofluorescence analyses revealed low levels of CTHRC1 in AE, but prominent expression in OSCC (Fig. 1D), with the majority of CTHRC1 localizing to cells at the invasive tumor front (data not shown). Immunoblots confirmed that AE had very low levels of CTHRC1 compared with OSCC, which exhibited increases in CTHRC1 abundance of >50-fold (n = 9) (Fig. 1F).
FIGURE 1.

Expression of β-catenin, GPT, and CTHRC1 in OSCC. A, H&E staining comparing cytologically normal AE and OSCC. B, left, immunoblot (IB) of β-catenin expression levels in AE and OSCC. Right, -fold change in GPT levels after normalization to actin. **, p < 0.01. C, immunofluorescence localization of GPT (green) and β-catenin (red) in AE and OSCC. D, immunofluorescence localization of CTHRC1 (green) and β-catenin (red) in AE and OSCC. E, left, immunoblot of GPT protein levels in AE and OSCC. Right, -fold change in GPT levels after normalization to actin. *, p < 0.05. F, left, immunoblot of CTHRC1 expression in AE and OSCC. Right, -fold change in CTHRC1 levels after normalization to actin. **, p < 0.01. G, immunoblot of control (C) or PNGase F (F)-treated CTHRC1 from AE and OSCC.
Next, we examined the N-glycosylation status of CTHRC1 in OSCC. Immunoblot analyses revealed that in TTLs from AE, CTHRC1 migrated mostly as a 30-kDa species, whereas in TTLs from OSCC, a higher molecular mass isoform was observed in addition to the 30-kDa species (Fig. 1G, arrows). Treatment of TTLs with PNGase F, an N-glycanase that cleaves most N-glycans at asparagine residues, converted both the high molecular mass and 30-kDa isoforms to deglycosylated species of 27 kDa (Fig. 2C), indicating that both are modified with N-glycans. Furthermore, the ratios between CTHRC1 in AE and OSCC before and after PNGase F treatment were similar, confirming that changes in mobilities were due to differences in N-glycosylation. We conclude that a hyperglycosylated CTHRC1 glycoform exists in OSCC. Because CTHRC1 has only one N-glycan consensus sequence, and activation of the N-glycosylation pathway is typically accompanied by greater modification with complex and more highly branched N-glycans (15, 17), our results indicate enhanced N-glycan heterogeneity on CTHRC1 in OSCC.
FIGURE 2.
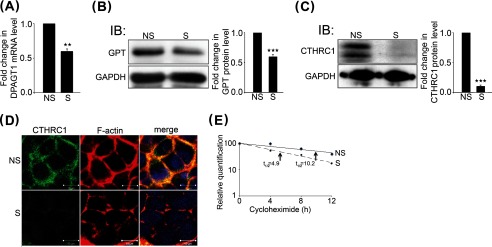
N-Glycosylation increases CTHRC1 abundance by protein stabilization. A, quantitative PCR of DPAGT1 transcript levels in NS (non-silenced) and S (DPAGT1-silenced) siRNA-transfected CAL27 cells. **, p < 0.01. B, left, immunoblot (IB) of GPT expression in NS and S siRNA-transfected CAL27 cells. Right, -fold change in GPT levels after normalization to GAPDH, ***, p < 0.001. C, left, immunoblot of CTHRC1 in NS and S siRNA-transfected CAL27 cells. Right, -fold change in CTHRC1 levels after normalization to GAPDH, ***, p < 0.001. D, immunofluorescence localization of CTHRC1 (green) and F-actin (red) in NS and S siRNA-transfected cells. E, comparison of CTHRC1 protein decay rates in NS and S (DPAGT1) siRNA-transfected cells. CAL27 cells were stimulated with conditioned medium overexpressing Wnt3a for 24 h, and total cell lysates were collected from cells grown in the presences of 20 μg/ml cycloheximide at 0, 4, 8, and 12 h.
Increased N-Glycosylation Drives Overexpression of CTHRC1 in OSCC
Because CTHRC1 is linked to the pathogenesis of several human cancers (25, 26), we next examined the mechanisms responsible for its up-regulation in OSCC. CTHRC1 is an N-glycoprotein with one N-glycan consensus addition sequence in the C-terminal region at Asn-188, and N-glycosylation was shown to promote its tethering to the membrane (23). Thus, it was possible that N-glycosylation contributed to up-regulation of CTHRC1 in OSCC.
To determine whether N-glycosylation impacts CTHRC1 abundance, we examined the effects of knocking down DPAGT1 levels on CTHRC1 in human OSCC CAL27 cells. We note that because DPAGT1 is essential for viability (27), we used conditions to achieve only partial knockdown of its expression (28). As shown in Fig. 2A, partial inhibition of DPAGT1 with S siRNA reduced its mRNA levels by 40% compared with NS control siRNA. This corresponded to a 40% reduction in GPT abundance (Fig. 2B). Because DPAGT1 functions at a rate-limiting step in the N-glycosylation pathway, even modest changes in its expression have pronounced effects on the N-glycosylation and function of proteins. Indeed, immunoblot analyses demonstrated that inhibition of GPT in S siRNA-transfected cells led to reduced levels of CTHRC1 compared with NS siRNA-transfected cells (Fig. 2C). Immunofluorescence staining of CTHRC1 in NS siRNA-transfected CAL27 cells showed that it localized primarily to the cell membrane, and this staining was greatly diminished in S siRNA-transfected cells (Fig. 2D).
Our observations that down-regulation of DPAGT1 reduced CTHRC1 levels in CAL27 cells suggested that N-glycosylation plays a role in CTHRC1 stability. To examine this possibility, we examined the turnover rate of CTHRC1 in CAL27 cells following siRNA-mediated DPAGT1 knockdown and subsequent inhibition of protein synthesis with cycloheximide. We note that following DPAGT1 knockdown, CAL27 cells express very modest levels of CTHRC1. Therefore, to efficiently detect CTHRC1 at different time points, CAL27 cells were first treated with Wnt3a-conditioned medium for 24 h to enhance CTHRC1 abundance (Fig. 3). The results show that in NS siRNA-transfected cells, the half-life of CTHRC1 was ∼10 h (Fig. 2E). In contrast, in S siRNA-transfected cells, the half-life of CTHRC1 was reduced by 50% to ∼5 h (Fig. 2E). Therefore, down-regulation of DPAGT1 increased the turnover of CTHRC1 in CAL27 cells, indicating that N-glycosylation regulates CTHRC1 stability.
FIGURE 3.

Wnt/β-catenin signaling increases CTHRC1 transcription. A, TCF-binding sites in the human CTHRC1 promoter. B, quantitative PCR of CTHRC1 transcript levels in CAL27 cells grown either in conditioned medium (CM) or in conditioned medium containing Wnt3a. **, p < 0.01. C, left, immunoblot (IB) of CTHRC1 and actin expression in CAL27 cells stimulated with either conditioned medium alone or from cells overexpressing Wnt3a. Right, -fold change in CTHRC1 levels after normalization to GAPDH. **, p < 0.01. D, immunofluorescence localization of CTHRC1 (green) and F-actin (red) in conditioned medium- or Wnt3a-stimulated cells. E, ChIP analyses of β-catenin and TCF at the CTHRC1 promoter in CAL27 cells compared with the IgG control. ***, p < 0.001. F, ChIP analyses of β-catenin and TCF at the CTHRC1 promoter after normalization to the IgG control in AE and OSCC. *, p < 0.05; **, p < 0.01.
Canonical Wnt/β-Catenin Signaling Induces CTHRC1 Transcription in OSCC
We previously reported that overexpression of DPAGT1 in OSCC promotes canonical Wnt/β-catenin signaling (6). Thus, we examined whether increased levels of CTHRC1 in OSCC may also be a consequence of its activation at a transcriptional level by Wnt-induced β-catenin-TCF complexes. The human CTHRC1 promoter has two TCF-binding sites spanning sequence−1946 to −1952 and sequence−1963 to −1969 (Fig. 3A). To determine whether these sites are Wnt-responsive, we examined the consequences of treating CAL27 cells with the canonical Wnt ligand, Wnt3a. The results show that there was a modest but significant 1.5-fold increase in the CTHRC1 transcript level (Fig. 3B). Accordingly, we detected a 2-fold up-regulation of CTHRC1 protein abundance by immunoblotting (Fig. 3C). Up-regulation of CTHRC1 by Wnt3a was further confirmed by immunofluorescence staining, which showed a pronounced increase in CTHRC1 membrane localization (Fig. 3D). This was associated with increased immunofluorescence staining for F-actin (Fig. 3D), as reported previously (29, 30).
To confirm that β-catenin was present at the CTHRC1 promoter in CAL27 cells in vivo, we carried out ChIP assays with antibodies to TCF and β-catenin. The results show that compared with the IgG control, there was a >2-fold increase in β-catenin bound to the CTHRC1 promoter (Fig. 3E). We also examined the recruitment of β-catenin to the CTHRC1 promoter by ChIP in fresh human AE and OSCC specimens. We found a 4-fold increase in β-catenin at the TCF sites at the CTHRC1 promoter in OSCC tumor specimens compared with AE (Fig. 3F). Therefore, in addition to increased N-glycosylation, overactive canonical Wnt signaling in OSCC contributes to increased expression of CTHRC1 at a transcriptional level.
DPAGT1 Promotes Cell Migration and Localization of CTHRC1 to the Wound Front
Given that the knockdown of DPAGT1 strongly impacted CTHRC1 levels, we wanted to examine whether increasing DPAGT1 expression would have a similar effect. Therefore, we transfected CAL27 cells with Myc-tagged DPAGT1 cDNA. Following selection of stable DPAGT1 transfectants, CTHRC1 expression was compared in cells overexpressing DPAGT1 with empty vector-transfected cells by immunoblotting. Because CAL27 cells are already along a tumorigenic pathway and have increased DPAGT1/canonical Wnt signaling, transfection of DPAGT1 cDNA resulted in only a 25% increased DPAGT1 transcript abundance (Fig. 4A). These increased DPAGT1 transcript levels corresponded to a robust expression of the DPAGT1 protein product, GPT, as determined by immunoblotting using anti-Myc antibody (Fig. 4B). We note that the immunoblot analyses detected two GPT isoforms, previously reported to be the products of the DPAGT1 gene (31). This augmented DPAGT1 expression caused a >8-fold increase in CTHRC1 abundance (Fig. 4C). Accordingly, knockdown of DPAGT1 expression resulted in diminished CTHRC1 abundance (Fig. 4D). CTHRC1 from the DPAGT1 transfectants migrated with a higher molecular mass than CTHRC1 from the empty vector-transfected cells (Fig. 4E, arrows). Moreover, treatment with PNGase F produced smaller molecular mass CTHRC1 species from both DPAGT1- and empty vector-transfected cells (Fig. 4, E and F), indicating that differences in their molecular size were due to N-glycosylation. We note that to detect CTHRC1 in empty vector-transfected cells, the amount of total cellular protein loaded onto the SDS-polyacrylamide gel was three times higher than in DPAGT1-transfected cells. Thus, up-regulation of DPAGT1 expression in CAL27 cells leads to increases in the abundance and N-glycosylation of CTHRC1.
FIGURE 4.

Overexpression of DPAGT1 is associated with increased cell migration and augmented levels and localization of CTHRC1 to the wound front. A, quantitative PCR of DPAGT1 transcript levels in control empty vector (E)- and DPAGT1 cDNA-transfected CAL27 cells. **, p < 0.01. B, immunoblot (IB) of recombinant GPT isoforms (Myc tag) from empty vector and DPAGT1 transfectants. C, left, immunoblot of CTHRC1 from empty vector and DPAGT1 transfectants. Right, -fold change in CTHRC1 levels after normalization to GAPDH. **, p < 0.01. D, left, immunoblot of CTHRC1 from DPAGT1 transfectants treated with either NS or S (DPAGT1) siRNA. Right, -fold change in DPAGT1 levels after normalization to GAPDH. ***, p < 0.001. E, immunoblot of control (C) or PNGase F (F)-treated CTHRC1 from empty vector and DPAGT1 transfectants. F, scratch wound assay of empty vector- and DPAGT1-transfected CAL27 cells at 0, 6, and 12 h (×20 magnification). G, scratch wound assay of DPAGT1-transfected CAL27 cells in the absence or presence of a ROCK inhibitor, Y-27632 (30 μm), at 0, 25, and 43 h (×4 magnification). H, immunofluorescence localization of CTHRC1 (green) and F-actin (red) at the edge of a wound in empty vector- and DPAGT1-transfected cells at 18 h.
Furthermore, increased expression of DPAGT1 in CAL27 cells promoted motility compared with empty vector-transfected cells in a scratch wound closure assay (Fig. 4F). Because cell migration is regulated by RhoA activity, an effector of ROCK (Rho-associated coiled coil-forming protein serine/threonine kinase), we next inhibited ROCK using Y-27632 in DPAGT1-transfected cells. As shown in Fig. 4G, inhibition of ROCK delayed wound closure, suggesting that DPAGT1 promotes oral cancer cell migration through a RhoA-dependent mechanism.
Next, to determine how DPAGT1-dependent overexpression of CTHRC1 influenced its function, we examined the localization of CTHRC1 in DPAGT1-expressing CAL27 transfectants in a scratch wound healing assay. Immunofluorescence staining revealed that DPAGT1 transfectants exhibited increased localization of CTHRC1 to cells at the wound front (Fig. 4H). Moreover, whereas CAL27 cells transfected with an empty vector displayed multicellular outgrowths with leader cells at the front, cells overexpressing DPAGT1 formed an evenly aligned migrating edge with fewer leader cells. This was associated with dramatic changes in cell morphology. In particular, DPAGT1-transfected cells at the wound edge formed increased cytoskeletal projections facing the wound and marginal actin bundles (Fig. 4H). Thus, up-regulation of DPAGT1 in CAL27 cells results in increased expression of CTHRC1, which localizes predominantly to the leading edge of migrating cells.
CTHRC1 Drives OSCC Cell Migration
Our collective observations that CTHRC1 was induced by DPAGT1, coincident with its increased localization to cells at the leading edge of wounds, suggested that CTHRC1 promotes OSCC cell migration. Thus, we carried out Transwell migration assays with CAL27 cells in which CTHRC1 expression was inhibited by treatment with S siRNA or cells treated with scrambled NS control siRNA. Inhibition of CTHRC1 expression by 85% (Fig. 5A) reduced S siRNA-transfected cell migration by 90% compared with NS siRNA-transfected cells (Fig. 5B). Furthermore, knockdown of CTHRC1 reduced the motile behavior of S siRNA-transfected cells compared with NS siRNA-transfected cells in a scratch wound closure assay (Fig. 5C). These results indicate that in cultured CAL27 cells, CTHRC1 promotes cell migration.
FIGURE 5.

CTHRC1 promotes OSCC cell migration. A, quantitative PCR of CTHRC1 transcript levels in NS and S (CTHRC1) siRNA-transfected CAL27 cells. ***, p < 0.001. B, Transwell migration of NS and S siRNA-transfected CAL27 cells. ***, p < 0.001. C, scratch wound assay of NS and S siRNA-transfected CAL27 cells at 0, 18, and 24 h. D, immunoblot of a co-immunoprecipitate (IP) of CTHRC1 with Wnt5a and Fzd6 from CAL27 cells. E, immunoblot of a co-immunoprecipitate of CTHRC1 with Wnt5a and Fzd3 from empty vector (E)- and DPAGT1-transfected CAL27 cells. F, left, immunoblot (IB) of active RhoA and total RhoA expression in empty vector- and DPAGT1-transfected CAL27 cells. Right, -fold change in active RhoA/total RhoA levels after normalization to GAPDH. ***, p < 0.001. TCL, total cell lysate. G, left, immunoblot of Wnt5a expression in AE and OSCC. Right, -fold change in Wnt5a levels after normalization to actin. *, p < 0.05. H, left, immunoblot of Dvl2 protein levels in AE and OSCC. Right, -fold change in Dvl2 levels after normalization to actin. ***, p < 0.001. I, left, immunoblot of Rac1 and RhoA in AE and OSCC. Right, -fold change in Rac1 and RhoA levels after normalization to actin. *, p < 0.05. J, left, immunoblot of phospho-JNK and total JNK in AE and OSCC. Right, -fold change in phospho-JNK (p-JNK) and total JNK levels after normalization to actin. *, p < 0.05.
CTHRC1 has been shown to activate the Wnt/planar cell polarity (PCP) pathway downstream of non-canonical Wnt ligands and receptors (23). The increased levels and localization of CTHRC1 to the leading edge of DPAGT1-expressing CAL27 transfectants (Fig. 4H) suggested that CTHRC1 may promote non-canonical Wnt signaling in this context. Indeed, immunoprecipitation of CTHRC1 followed by immunoblotting for components of non-canonical Wnt/PCP pathways demonstrated that CTHRC1 interacted with a non-canonical Wnt ligand (Wnt5a) and receptor (Fzd6) in CAL27 cells (Fig. 5D). Moreover, in DPAGT1-transfected CAL27 cells, the interaction between Wnt5a and CTHRC1 was more prominent (Fig. 5E). We note that because anti-Fzd6 antibodies gave very high background levels in DPAGT1-transfected CAL27 cells, we used an antibody to a different non-canonical Wnt receptor, Fzd3, as both function in the non-canonical Wnt/PCP pathway (Fig. 5E). Consistent with activation of a non-canonical Wnt/PCP pathway, the abundance of active RhoA was also higher in DPAGT1-expressing CAL27 transfectants (Fig. 5F). Accordingly, we found that the levels of Wnt5a were >5-fold higher in human OSCC specimens compared with AE (n = 4) (Fig. 5G). Likewise, expression of Dvl2, a downstream effector of the non-canonical Wnt/PCP pathway, was up-regulated in OSCC (n = 3) (Fig. 5H). Similarly, the abundance of total Rac1 and RhoA was increased in OSCC (n = 4) (Fig. 5I). Because deregulation of Rho protein signaling in cancer has been shown to involve both activation and expression levels (32), these results suggest that in OSCC, CTHRC1 promotes cell migration via the non-canonical Wnt/PCP pathway. Accordingly, the levels of a downstream Rac1 effector, phospho-JNK, were increased in OSCC compared with AE, whereas the total levels of JNK were reduced (Fig. 5J).
DISCUSSION
Canonical Wnt/β-catenin signaling plays key roles in cancer initiation and progression (7, 33). Likewise, increased protein N-glycosylation is linked to neoplasia (34, 35). Previously, we showed that human OSCC specimens display aberrant amplification of the DPAGT1/canonical Wnt signaling positive feedback loop that leads to hyperglycosylation of E-cadherin and diminished intercellular adhesion (6). In this study, we have provided evidence that increased activity of this loop leads to overexpression of CTHRC1, an N-glycoprotein shown to drive tumor progression and metastasis (26). We have demonstrated that in OSCC, DPAGT1 and canonical Wnt synergize to up-regulate CTHRC1 through hyperglycosylation/protein stabilization and transcriptional activation, respectively. Because changes in CTHRC1 in OSCC CAL27 cells are associated with corresponding changes in cell migration, our studies suggest that increased levels of CTHRC1 in human OSCC tumors drive cell migration and promote oral cancer spread. Several lines of evidence support this notion. First, overexpression of DPAGT1 promotes cell migration. Second, increased expression of CTHRC1 via up-regulation of DPAGT1 results in a more prominent localization of CTHRC1 to cells at the migrating wound edge. Moreover, DPAGT1 expression results in distinct changes in the morphology of cells at the migrating wound front. Third, inhibition of CTHRC1 in OSCC CAL27 cells reduces migration and wound closure.
In mouse cochlear sensory hair cells, CTHRC1 stabilizes the Wnt-Fzd complex to activate non-canonical Wnt/PCP signaling (30). Consistent with these observations, we found that in CAL27 cells, CTHRC1 forms a complex with Fzd6/Fzd3 and Wnt5a. Furthermore, increased expression of CTHRC1 in DPAGT1-expressing CAL27 transfectants correlates with augmented levels of active RhoA, a downstream effector in the Wnt/PCP pathway (23). Also, overexpression of DPAGT1 promotes cell migration in a RhoA-dependent manner. These data suggest that in CAL27 cells, CTHRC1 activates a non-canonical Wnt/PCP pathway to drive cytoskeletal reorganization and migration. Activation of a non-canonical Wnt/PCP pathway in OSCC is further supported by increased levels of this pathway's effectors in fresh tumor specimens, including Wnt5a, RhoA, Rac1, and active JNK, all known to be up-regulated in human cancers (36). Moreover, Wnt/PCP signaling has been shown to drive breast cancer cell migration (37). We note that CTHRC1 can also activate other oncogenic signaling pathways, including Src/focal adhesion kinase and MEK/ERK (26). Further studies are needed to determine the details of signaling events activated by CTHRC1 in OSCC.
N-Glycosylation has been shown to be required for effective secretion of CTHRC1 and for its anchorage at the cell surface (23). Here, we show for the first time that N-glycosylation stabilizes the CTHRC1 protein. Comparison of CTHRC1 abundance in DPAGT1 siRNA-treated CAL27 cells in the presence of cycloheximide revealed that the half-life of CTHRC1 was diminished in cells with attenuated DPAGT1 expression. Moreover, forced expression of DPAGT1 in CAL27 cells led to increases in the levels of CTHRC1 and its N-glycosylation status. Because overexpression of DPAGT1 in OSCC cells is associated with increased abundance and hyperglycosylation of CTHRC1, our results indicate that N-glycosylation promotes the accumulation of CTHRC1 in OSCC by enhancing protein stability. Furthermore, increased N-glycosylation was also associated with preferential localization of CTHRC1 to cells at the migrating wound front. Because inhibition of CTHRC1 interferes with CAL27 cell migration, these data suggest that N-glycosylation facilitates the pro-migratory function of CTHRC1. A model representing the cross-talk between DPAGT1 and canonical Wnt signaling and their downstream effects on CTHRC1 expression and tumor cell migration is shown in Fig. 6.
FIGURE 6.

CTHRC1 is up-regulated by DPAGT1 and canonical Wnt signaling in OSCC. The schematic shows the interactions between DPAGT1 and canonical Wnt signaling and their downstream effects on CTHRC1 expression and non-canonical Wnt pathway-dependent tumor spread.
N-Glycosylation and canonical Wnt signaling represent highly conserved core pathways whose dysregulation is likely to drive early events in OSCC pathogenesis. Both pathways play pivotal roles in embryogenesis and are overactive in many malignancies. The initial aberrant activation of the DPAGT1/canonical Wnt feedback loop may involve a mutation in any of the components of the canonical Wnt signaling pathway. Likewise, up-regulation of DPAGT1 either by activating mutations or by increased availability of the GPT substrate UDP-GlcNAc from glycolysis and the hexosamine pathway will promote activation of canonical Wnt signaling (21). Once either DPAGT1 or canonical Wnt signaling is increased, it sets off a sequence of events that involve hyperglycosylation and transcriptional activation of downstream targets with key roles in tumorigenesis, such as CTHRC1. We note that CTHRC1 has been shown to be up-regulated by promoter demethylation and in response to TGFβ signaling in gastric cancer (38). Thus, we cannot exclude that additional mechanisms drive CTHRC1 expression in OSCC, although TGFβ receptors have also been shown to be regulated by N-glycosylation (39).
In addition to E-cadherin and CTHRC1, other important targets of the dysregulated DPAGT1/canonical Wnt signaling loop are receptor tyrosine kinases (RTKs), such as EGF receptor, ErbB2, ErbB3 and insulin-like growth factor-1 receptor, which play pivotal roles in cell proliferation, survival, and migration. These RTKs are N-glycoproteins that require N-glycosylation for activity, and some RTKs are also transcriptional targets of canonical Wnt (40). Because aberrant activation of RTKs promotes tumor cell proliferation, survival, and migration, RTKs have served as therapeutic targets for cancer treatment. Indeed, tunicamycin, a specific inhibitor of the DPAGT1 protein product, GPT, disrupts RTK signaling in tumor cells (41). However, because tumor cells develop resistance to chemotherapy by the activation of parallel signaling pathways, RTKs have not proven effective for sustained anticancer therapy. Here, our studies have identified CTHRC1 as a key downstream target of the DPAGT1/canonical Wnt feedback loop involved in OSCC cell migration and provide novel insights into the mechanisms driving its overexpression. The network formed between DPAGT1/canonical Wnt signaling and CTHRC1 highlights important upstream signaling events in OSCC pathogenesis. We propose that targeting this network may represent an effective strategy for therapeutic intervention for OSCC. Moreover, because CTHRC1 influences tumor invasion and metastasis of many aggressive neoplasms, our findings may reveal relevant mechanisms for the pathogenesis of other cancers.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DE15304.
- OSCC
- oral squamous cell carcinoma
- TCF
- T-cell factor
- GPT
- dolichol-P-dependent N-acetylglucosamine-1-phosphate transferase
- AE
- adjacent epithelia
- TTL
- total tissue lysate
- PNGase F
- peptide N-glycosidase F
- PCP
- planar cell polarity
- RTK
- receptor tyrosine kinase.
REFERENCES
- 1. Choi S., Myers J. N. (2008) Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J. Dent. Res. 87, 14–32 [DOI] [PubMed] [Google Scholar]
- 2. Rothenberg S. M., Ellisen L. W. (2012) The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Invest. 122, 1951–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perez-Ordoñez B., Beauchemin M., Jordan R. C. (2006) Molecular biology of squamous cell carcinoma of the head and neck. J. Clin. Pathol. 59, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pérez-Sayáns M., Suárez-Peñaranda J. M., Herranz-Carnero M., Gayoso-Diz P., Barros-Angueira F., Gándara-Rey J. M., García-García A. (2012) The role of the adenomatous polyposis coli (APC) in oral squamous cell carcinoma. Oral Oncol. 48, 56–60 [DOI] [PubMed] [Google Scholar]
- 5. Priyadarsini V. R., Murugan S. R., Nagini S. (2012) Aberrant activation of Wnt/β-catenin signaling pathway contributes to the sequential progression of DMBA-induced HBP carcinomas. Oral Oncol. 48, 33–39 [DOI] [PubMed] [Google Scholar]
- 6. Jamal B., Sengupta P. K., Gao Z. N., Nita-Lazar M., Amin B., Jalisi S., Bouchie M. P., Kukuruzinska M. A. (2012) Aberrant amplification of the crosstalk between canonical Wnt signaling and N-glycosylation gene DPAGT1 promotes oral cancer. Oral Oncol. 48, 523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. MacDonald B. T., Tamai K., He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 9. Wend P., Holland J. D., Ziebold U., Birchmeier W. (2010) Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 21, 855–863 [DOI] [PubMed] [Google Scholar]
- 10. Yang F., Zeng Q., Yu G., Li S., Wang C. Y. (2006) Wnt/β-catenin signaling inhibits death receptor-mediated apoptosis and promotes invasive growth of HNSCC. Cell. Signal. 18, 679–687 [DOI] [PubMed] [Google Scholar]
- 11. Gordon M. D., Nusse R. (2006) Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429–22433 [DOI] [PubMed] [Google Scholar]
- 12. van Amerongen R., Nusse R. (2009) Towards an integrated view of Wnt signaling in development. Development 136, 3205–3214 [DOI] [PubMed] [Google Scholar]
- 13. Sethi J. K., Vidal-Puig A. (2010) Wnt signalling and the control of cellular metabolism. Biochem. J. 427, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heuberger J., Birchmeier W. (2010) Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2, a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sengupta P. K., Bouchie M. P., Kukuruzinska M. A. (2010) N-Glycosylation gene DPAGT1 is a target of the Wnt/β-catenin signaling pathway. J. Biol. Chem. 285, 31164–31173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Helenius A., Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 17. Sengupta P. K., Bouchie M. P., Nita-Lazar M., Yang H. Y., Kukuruzinska M. A. (2013) Coordinate regulation of N-glycosylation gene DPAGT1, canonical Wnt signaling and E-cadherin adhesion. J. Cell Sci. 126, 484–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liwosz A., Lei T., Kukuruzinska M. A. (2006) N-Glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 281, 23138–23149 [DOI] [PubMed] [Google Scholar]
- 19. Dennis J. W., Granovsky M., Warren C.E. (1999) Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta 1473, 21–34 [DOI] [PubMed] [Google Scholar]
- 20. Nita-Lazar M., Noonan V., Rebustini I., Walker J., Menko A. S., Kukuruzinska M. A. (2009) Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res. 69, 5673–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dennis J. W., Nabi I. R., Demetriou M. (2009) Metabolism, cell surface organization, and disease. Cell 139, 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pyagay P., Heroult M., Wang Q., Lehnert W., Belden J., Liaw L., Friesel R. E., Lindner V. (2005) Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ. Res. 96, 261–268 [DOI] [PubMed] [Google Scholar]
- 23. Yamamoto S., Nishimura O., Misaki K., Nishita M., Minami Y., Yonemura S., Tarui H., Sasaki H. (2008) Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev. Cell 15, 23–36 [DOI] [PubMed] [Google Scholar]
- 24. Turashvili G., Bouchal J., Baumforth K., Wei W., Dziechciarkova M., Ehrmann J., Klein J., Fridman E., Skarda J., Srovnal J., Hajduch M., Murray P., Kolar Z. (2007) Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang L., Dai D. L., Su M., Martinka M., Li G., Zhou Y. (2006) Aberrant expression of collagen triple helix repeat containing 1 in human solid cancers. Clin. Cancer Res. 12, 3716–3722 [DOI] [PubMed] [Google Scholar]
- 26. Park E. H., Kim S., Jo J. Y., Kim S. J., Hwang Y., Kim J. M., Song S. Y., Lee D. K., Koh S. S. (2013) Collagen triple helix repeat containing-1 promotes pancreatic cancer progression by regulating migration and adhesion of tumor cells. Carcinogenesis 34, 694–702 [DOI] [PubMed] [Google Scholar]
- 27. Goto M., Mitra R. S., Liu M., Lee J., Henson B. S., Carey T., Bradford C., Prince M., Wang C. Y., Fearon E. R., D'Silva N. J. (2010) Rap1 stabilizes β-catenin and enhances β-catenin-dependent transcription and invasion in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 16, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nita-Lazar M., Rebustini I., Walker J., Kukuruzinska M. A. (2010) Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp. Cell Res. 316, 1871–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carthy J. M., Garmaroudi F. S., Luo Z., McManus B. M. (2011) Wnt3a induces myofibroblast differentiation by up-regulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS One 6, e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shibamoto S., Higano K., Takada R., Ito F., Takeichi M., Takada S. (1998) Cytoskeletal reorganization by soluble Wnt-3a protein signalling. Genes Cells 3, 659–670 [DOI] [PubMed] [Google Scholar]
- 31. Huang G. T., Lennon K., Kukuruzinska M. A. (1998) Characterization of multiple transcripts of the hamster dolichol-P-dependent N-acetylglucosamine-1-P transferase suggests functionally complex expression. Mol. Cell. Biochem. 181, 97–106 [DOI] [PubMed] [Google Scholar]
- 32. Parri M., Chiarugi P. (2010) Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polakis P. (2000) Wnt signaling and cancer. Genes Dev. 14, 1837–1851 [PubMed] [Google Scholar]
- 34. Lau K. S., Partridge E. A., Grigorian A., Silvescu C. I., Reinhold V. N., Demetriou M., Dennis J. W. (2007) Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 129, 123–134 [DOI] [PubMed] [Google Scholar]
- 35. Mendelsohn R., Cheung P., Berger L., Partridge E., Lau K., Datti A., Pawling J., Dennis J. W. (2007) Complex N-glycan and metabolic control in tumor cells. Cancer Res. 67, 9771–9780 [DOI] [PubMed] [Google Scholar]
- 36. Katoh M. (2005) WNT/PCP signaling pathway and human cancer (review). Oncol. Rep. 14, 1583–1588 [PubMed] [Google Scholar]
- 37. Luga V., Zhang L., Viloria-Petit A. M., Ogunjimi A. A., Inanlou M. R., Chiu E., Buchanan M., Hosein A. N., Basik M., Wrana J. L. (2012) Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 151, 1542–1556 [DOI] [PubMed] [Google Scholar]
- 38. Wang P., Wang Y. C., Chen X. Y., Shen Z. Y., Cao H., Zhang Y. J., Yu J., Zhu J. D., Lu Y. Y., Fang J. Y. (2012) CTHRC1 is up-regulated by promoter demethylation and transforming growth factor-β1 and may be associated with metastasis in human gastric cancer. Cancer Sci. 103, 1327–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watanabe S., Misawa M., Matsuzaki T., Sakurai T., Muramatsu T., Sato M. (2011) A novel glycosylation signal regulates transforming growth factor β receptors as evidenced by endo-β-galactosidase C expression in rodent cells. Glycobiology 21, 482–492 [DOI] [PubMed] [Google Scholar]
- 40. Guturi K. K., Mandal T., Chatterjee A., Sarkar M., Bhattacharya S., Chatterjee U., Ghosh M. K. (2012) Mechanism of β-catenin-mediated transcriptional regulation of epidermal growth factor receptor expression in glycogen synthase kinase 3β-inactivated prostate cancer cells. J. Biol. Chem. 287, 18287–18296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Contessa J. N., Bhojani M. S., Freeze H. H., Rehemtulla A., Lawrence T. S. (2008) Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 68, 3803–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]