

Background: Biological functions of mammalian Atat1 and its contribution to α-tubulin acetylation in vivo remain elusive.
Results: Atat1-null mice are viable but possess deficient α-tubulin acetylation and a bulge in the dentate gyrus.
Conclusion: Mouse Atat1 is a predominant α-tubulin acetyltransferase in vivo and fine-tunes hippocampus development.
Significance: Mammalian Atat1 is not required for survival and development but may regulate more advanced functions.
Keywords: Histone Acetylase, Histone Deacetylase, Microtubules, Mouse Genetics, Neural Stem Cell, Post-translational Modification, Tubulin, Lysine Acetylation, Tubulin Acetylation, Tubulin Acetyltransferase
Abstract
α-Tubulin acetylation at Lys-40, located on the luminal side of microtubules, has been widely studied and used as a marker for stable microtubules in the cilia and other subcellular structures, but the functional consequences remain perplexing. Recent studies have shown that Mec-17 and its paralog are responsible for α-tubulin acetylation in Caenorhabditis elegans. There is one such protein known as Atat1 (α-tubulin acetyltransferase 1) per higher organism. Zebrafish Atat1 appears to govern embryo development, raising the intriguing possibility that Atat1 is also critical for development in mammals. In addition to Atat1, three other mammalian acetyltransferases, ARD1-NAT1, ELP3, and GCN5, have been shown to acetylate α-tubulin in vitro, so an important question is how these four enzymes contribute to the acetylation in vivo. We demonstrate here that Atat1 is a major α-tubulin acetyltransferase in mice. It is widely expressed in mouse embryos and tissues. Although Atat1-null animals display no overt phenotypes, α-tubulin acetylation is lost in sperm flagella and the dentate gyrus is slightly deformed. Furthermore, human ATAT1 colocalizes on bundled microtubules with doublecortin. These results thus suggest that mouse Atat1 may regulate advanced functions such as learning and memory, thereby shedding novel light on the physiological roles of α-tubulin acetylation in mammals.
Introduction
Along with the recent proteomic analyses by mass spectrometry, numerous molecular and cell-based studies in the past 17 years have convincingly established lysine acetylation as an important post-translational modification likely to occur in all living species and to rival other well known modifications such as phosphorylation and ubiquitination (1–3). First discovered with histones in the early 1960s (4), the importance of lysine acetylation was initially recognized for its relation to chromatin organization and its impact on chromatin-templated nuclear processes (5–8). However, it is now clear that the spectrum of lysine acetylation is much broader than initially anticipated, comprising thousands of eukaryotic proteins that are not only involved in various nuclear events but also in a diversity of cytoplasmic processes (9–12). In addition, this modification is abundant in prokaryotic cells (13, 14). Unlike N-terminal acetylation, lysine acetylation is an Nϵ-modification and is reversible and highly dynamic, governed by two groups of enzymes called lysine acetyltransferases and deacetylases (15–17). Based on sequence similarity, mammalian lysine acetyltransferases are grouped into three major families (15–17), and the deacetylases are divided into two large families composed of four classes (18–22). Therefore, lysine acetylation is an abundant covalent modification with its level actively maintained by acetyltransferases and deacetylases.
As one of two key components of microtubules (23, 24), α-tubulin is the first Nϵ-acetylated non-histone protein discovered in the mid-1980s (25–28). The acetylated residue was identified as Lys-40 (25). This modification is conserved in a wide variety of species ranging from unicellular organisms, such as the green algae Chlamydomonas and the protist Tetrahymena, to mammals (25–28). Unlike many other tubulin modifications, the acetylation occurs on the luminal side of microtubules (23, 24, 29). Because of the early successful development of monoclonal antibodies (such as the 6-11b-1 clone) specific to Lys-40 acetylation (26), this modification has been used as a routine marker for stable microtubules in cilia, flagella, axons, midbodies, and other subcellular structures (23, 24, 29). However, it still remains debatable whether Lys-40 acetylation plays a causal role in regulating microtubule stability and dynamics (30–33). The acetylation has been linked to microtubule-mediated transport of proteins and organelles (31, 34–38), but it is enigmatic how a luminal modification affects transport processes occurring on the external surface of microtubules (29).
Substitution of Lys-40 in lower organisms such as Tetrahymena and Chlamydomonas exerts little impact on their survival (39, 40). Lys-40 of Mec-12 (mechanosensory-defective 12), the sole acetylatable α-tubulin, is not essential for nematodes (41). Moreover, substitution of Lys-40 with arginine exerts little effects on the function of a plant α-tubulin (42). Recent studies indicate that Lys-40 acetylation is important for mammalian neuron migration (36, 43, 44). Further knock-in analyses in genetically amendable mammalian models such as mice should substantiate how important Lys-40 acetylation is in vivo. Insights into the physiological function of α-tubulin acetylation can also be gained from studies of the responsible enzymes.
HDAC6 was discovered to be an α-tubulin deacetylase over 10 years ago (45–47). Global hyperacetylation of α-tubulin was subsequently found in Hdac6−/− mouse tissues and embryonic fibroblasts (48). SIRT2 was reported to be another α-tubulin deacetylase (49), but genetic deletion of mouse Sirt2 does not affect α-tubulin acetylation significantly (48, 50). Thus, HDAC6 is now considered to be the predominant α-tubulin deacetylase in vivo. Despite this unique and crucial function in α-tubulin deacetylation, Hdac6−/− mice are viable and fertile (48), only displaying minor phenotypes, including moderate immune responses (48), anti-depressant-like symptoms (51), and slow platelet activation (52). This unexpected outcome suggests a potential role of HDAC6 and perhaps also α-tubulin acetylation in advanced biological functions.
Multiple acetyltransferases have been shown to acetylate α-tubulin in vitro, including the ARD1-NAT1 Nα-acetyltransferase complex (53), the transcriptional elongator complex subunit ELP3 (43, 54), and the well known acetyltransferase GCN5 (55). The ARD1-NAT1 complex was the first reported to acetylate α-tubulin in vitro (53). ELP3 was then found to acetylate α-tubulin and to regulate the migration of cortical neurons in the developing mouse brain (43). GCN5 was recently described to acetylate α-tubulin upon recruitment to microtubules by Myc-nick, a cytoplasmic isoform of Myc (55). More recent biochemical and genetic studies have demonstrated that a new member of the GNAT family (56), Mec-17 (mechanosensory-defective 17), is an α-tubulin acetyltransferase in Tetrahymena and Caenorhabditis elegans (32, 57). As a result, Mec-17 was renamed Atat1 (α-tubulin N-acetyltransferase 1). Atat1 is conserved from C. elegans to humans, although in C. elegans there are two Atat proteins and only one such ortholog exists in a higher organism (32, 57). Survey of 50 genomes of evolutionarily diverse organisms revealed that the presence of an Atat1 ortholog correlates perfectly with the possession of a cilium or flagellum (32). For example, an Atat1-like protein is encoded in the green algae Chlamydomonas (32), where α-tubulin acetylation was initially discovered (25–28). Moreover, mammalian Atat1 efficiently acetylates α-tubulin both in enzymatic assays with purified proteins and in cultured cells (32, 57). Recent structural analyses have provided elegant insights into how Atat1 targets and acetylates Lys-40 of α-tubulin (58–60). These studies all support that Atat1 is a bona fide α-tubulin acetyltransferase.
Morpholino analysis revealed an essential role of Atat1 in zebrafish development (57), raising the exciting possibility that Atat1 is also important for mammalian development. This is in agreement with a recent study (32) showing that Atat1 regulates assembly of the primary cilium, a unique organelle important for cellular signaling and animal development (61). However, it remains to be formally investigated whether Atat1 indeed plays a critical role in mammalian development. It is also unclear how mammalian Atat1 and the other aforementioned acetyltransferases contribute to α-tubulin acetylation in vivo.
Through analyzing Atat1−/− mutant mice generated by gene targeting, we demonstrate herein that mouse Atat1 is the predominant α-tubulin acetyltransferase in embryonic fibroblasts and various adult tissues. We also show that Atat1 is widely expressed, most highly in the brain, testis, renal pelvis, and gastrointestinal tract. Unexpectedly, Atat1−/− mutant embryos and mice are viable and display no overt phenotypes. Fluorescence microscopy and histological analyses revealed that Atat1 is required for α-tubulin acetylation in sperm flagella and that the dentate gyrus is slightly deformed. These results suggest that although not important for basic functions such as animal survival and development, mouse Atat1 may regulate more advanced functions, thereby shedding novel light on roles of mammalian α-tubulin acetylation in vivo.
MATERIALS AND METHODS
Animals
Three Atat1tm1(KOMP)Vlcg heterozygous C57BL6 male mice were obtained from the knock-out mouse project (KOMP) repository affiliated with University of California at Davis. Crossing them with wild-type C57BL6 mice produced heterozygous female mutant mice. Interbreeding between heterozygous male and female mice was then carried out to obtain homozygous knock-out embryos and mice. The mice used were housed in barrier cages, with 1–5 animals per cage. The animals were provided with food and water ad libitum. The experimental procedures performed on mice were carried out according to standard operating procedures created by the Veterinary Care Subcommittee, and the use of the mice for this project received approval by the McGill University Animal Care Committee.
Mouse Genotyping
Mice were genotyped by PCR with genomic DNA extracted from tissues from ear punch typically used for mouse identification. After ear punching, each tissue piece (round, roughly 2 mm in the diameter) was carefully collected and immersed in 200 μl of 50 mm NaOH and incubated at 95 °C for 10 min. After addition of 20 μl of 1 m Tris-HCl, pH 6.8, the mixture was vortexed for 7 s and centrifuged in a bench-top centrifuge at ∼20,000 × g for 6 min. 2 μl of the supernatant was used for PCR. The primers used were as follows: Atat-F1 (5′-ACTGA AGGAC ACCTC AGCCC GA-3′), Atat-R1 (5′-TACCT CATTG TGAGC CTCCC GG-3′), LacInF (5′-GGTAA ACTGG CTCGG ATTAG GG-3′), and LacInR (5′-TTGAC TGTAG CGGCT GATGT TG-3′). A 10-μl PCR was set up with 5 μl of 2× GoTaq Green Master Mix (Promega, PRM5122), 2 μl of genomic DNA, 0.25 μl for each of two primers (10 pmol/μl), and 2.5 μl of sterile nuclease-free water. Cycling conditions were as follows: 95 °C for 3 min, 30 PCR cycles (95 °C for 30 s, 50 °C for 30 s, and 72 °C for 30 s per cycle) and 72 °C for 2 min. The reaction was kept at 4 or −20 °C until further analysis by agarose gel electrophoresis.
RT-PCR
Fresh mouse tissues were collected and then rinsed in ice-cold PBS, which was pretreated with 0.1% diethyl pyrocarbonate (DEPC,2 Sigma) to inactivate RNases and then autoclaved to remove residual DEPC. The rinsed mouse tissues were transferred to 1.5-ml microtubes. After addition of 200 μl of PBS pretreated with 0.1% DEPC, tissues were crushed in 1.5-ml Eppendorf tubes and mixed with 1 ml of the TRIzol reagent (Invitrogen, 16520-100). After vortexing, the suspension was then incubated at room temperature for 5 min and centrifuged at ∼20,000 × g and 4 °C for 10 min. The supernatant was carefully transferred to a new microtube and mixed with 200 μl of chloroform. After vigorous vortexing, the suspension was centrifuged at ∼20,000 × g for 2 min. The upper aqueous phase was then pipetted out and transferred to a new microtube for mixing with 500 μl of isopropyl alcohol and subsequent centrifugation at ∼20,000 × g and 4 °C for 15 min. The supernatant was carefully decanted, and the pellet was washed with 750 μl of 75% cold ethanol prepared with sterile DEPC-treated Nano-pure water. The pellet was then air-dried and dissolved in an appropriate amount of sterile DEPC-treated Nano-pure water. After quantification of the RNA concentration with a NanoDrop 2000 spectrophotometer (Thermo Scientific), cDNA was synthesized by use of a QuantiTect reverse transcription kit (Qiagen, 205313) according to the manufacturer's instructions. PCR was performed with the synthesized cDNA as the template. PCR conditions were identical to that used for genotyping, but the reaction volume was adjusted to 25 μl. The primers for Atat1 and LacZ were also the same as the ones used for genotyping. The primers for Gapdh were mGAPDH-RT01 (5′-GCACA GTCAA GGCCG AGAAT-3′) and mGAPDH-RT02 (5′-GCCTT CTCCA TGGTG GTGAA-3′).
Tissue Extract Preparation and Western Blotting
All these procedures were carried out at 4 °C or on ice. Tissues were weighted and homogenized with a small plastic pestle in 3 volumes of the RIPA buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, and a mixture of protease inhibitors). After additional homogenization by sonication with a Virsonic 100 sonicator (Virtis, Inc.) at setting 5, the suspension was centrifuged at ∼20,000 × g for 10 min at 4 °C, and the supernatant was collected as the soluble extract, snap-frozen on dry ice, and stored at −80 °C. Protein concentration was measured by use of the Bradford assay reagent (Bio-Rad).
Tissue extracts were diluted in a 3× reduced SDS-PAGE sample buffer and denatured by boiling for 5 min. After electrophoresis, proteins were transferred onto nitrocellulose membranes, which were blocked for 1 h in the blocking solution (PBS containing 0.15% Tween 20 (Sigma) and 5% nonfat milk powder (Carnation)). Anti-α-tubulin antibody (Sigma, T5168, diluted at 1:2500) or the 6-11b-1 monoclonal antibody (specific to acetyl-Lys-40 of α-tubulin; Sigma, T7451, diluted at 1:10,000) was then added to the blocking solution for incubation overnight at 4 °C, with agitation. Blots were washed with PBS containing 0.15% Tween 20 five times (8 min each) and incubated with the horseradish peroxidase-conjugated sheep anti-mouse IgG secondary antibody (GE Healthcare, NA931V; diluted at 1:5000) for 1–3 h. After extensive washing, the Supersignal chemiluminescence solution (Pierce) was used to develop the signals.
Whole-mount β-Gal Staining of Embryos and Adult Tissues
Mice were anesthetized and transcardially perfused first with PBS and then with PBS containing 2% PFA. After perfusion was completed, embryos or tissues were collected and fixed at 4 °C for 2–3 h (up to 1 h for embryos) in PBS containing 2% PFA, 0.2% glutaraldehyde, 0.02% Nonidet P-40, and 0.2 mm MgCl2. The fixative solution was changed to the detergent rinse (0.1 m phosphate buffer, pH 7.3, 2 mm MgCl2, 0.01% sodium deoxycholate, and 0.02% Nonidet P-40). After incubation in the detergent rinse (changed 3–5 times) for 2–16 h, tissues or embryos were incubated in the β-galactosidase staining solution composed of 5 mm K3(Fe(CN)6)/K4(Fe(CN)6) and 1 mg/ml X-Gal (62) until the desired intensity of blue color was developed.
Stained embryos were examined and analyzed under a Lumar V12 SteREO dissecting microscope (Zeiss), linked to an Axiocam HRc color camera, and controlled by Axiovision Re 4.8. Images were exported to Adobe Photoshop for processing and then imported to Adobe Illustrator for final presentation.
Frozen Section Preparation
Mice were anesthetized and sacrificed by cervical dislocation. Tissues were fixed overnight in PBS, 4% PFA at 4 °C and equilibrated first with PBS, 15% sucrose at 4 °C for ∼8 h and then overnight with PBS, 30% sucrose. After that, tissues were found to be at the bottom of the tubes and were further equilibrated with the optimal cutting temperature (OCT) compound (Tissue-Tek) for 30 min at 4 °C, with gentle agitation. Tissues were then carefully oriented prior to snap-freezing on dry ice. Tissue blocks were transferred to −80 °C for storage. 30 min prior to sectioning, tissues blocks were transferred to dry ice. The blocks were sectioned on a cryotome electronic cryostat (Thermo Shandon, A77210167), and 10–30-μm sections were collected onto glass slides (Fisher Scientific, 12-550-19). The sections were then air-dried and stored at −80 °C.
β-Gal Staining of Frozen Sections
Sections were post-fixed in a fixative (0.2% PFA in 0.1 m PIPES, pH 6.9, 2 mm MgCl2, and 5 mm EGTA) on ice for 10 min and rinsed with PBS containing 2 mm MgCl2, followed by washing in the same solution for 10 min. Sections were permeabilized by incubation in the detergent rinse on ice for 10 min. The detergent rinse was changed to the β-galactosidase staining solution for incubation at 37 °C in the dark until the desired blue color was reached (1 h to overnight). The slides were then washed three times in PBS containing 2 mm MgCl2 at room temperature for 5 min, followed by washing in distilled water. Counterstaining was carried out in the Nuclear Fast-Red solution (Sigma), with sequential dehydration by incubation with 50, 70, and 100% methanol (5 min each). Sections were cleared twice in Histoclear (Diamed, HS-200) for 5 min and mounted onto glass slides with a mounting medium (American Master Tech, MMC0126).
Paraffin Section Preparation
Mice were anesthetized for transcardial perfusion first with PBS and then with PBS containing 4% PFA. After perfusion was completed, major tissues were collected and fixed in PBS containing 4% PFA. In addition, Bouin's solution (saturated picric acid (Sigma), 40% formaldehyde, and 5% glacial acetic acid mixed at the 15:5:1 ratio) was also used for fixation of testes. The tissues were kept in PBS containing 4% PFA or in Bouin's solution overnight at 4 °C for subsequent paraffin embedding. The resulting paraffin blocks were sectioned on a Leica Rotary Microtome (Leica RM2125 RTS), and 5-μm sections were collected onto glass slides (Fisher Scientific, 12-550-19). The tissue section-containing slides were baked overnight in a 37 °C incubator and stored at 4 °C.
Nissl Staining of Brain Sections
Paraffin sections were deparaffinized in xylene twice for 5 min each, then rehydrated in 100% alcohol twice (5 min each), followed by 95 and 70% alcohol (3 min each), and subsequently rinsed in tap water and distilled water. The sections were then placed in 0.1% cresyl violet (Sigma) containing 0.3% glacial acetic acid for 7 min and rinsed quickly in distilled water. The sections were subsequently dehydrated in 70 and 95% alcohol for 3 min each, 100% alcohol twice for 5 min each, and cleared in xylene twice (5 min each). The sections were finally mounted with glass coverslips (Fisher Scientific) in the ClearMount mounting solution (Invitrogen). The slides were either examined under a regular light microscope or scanned with a digital slide scanner (ScanScope XT, Aperio ePathology). For the latter, the Spectrum software (Aperio ePathology) was then used to analyze the digitized images, and the ruler tool was employed to measure the size and dimensions of various structures in the sections.
Indirect Immunofluorescence Microscopy
Paraffin-embedded testis sections were fixed in Bouin's solution and prepared as above. The sections were deparaffinized and rehydrated the same as the brain sections mentioned above. Then heat-induced epitope retrieval was performed with the sections by boiling the slides in 10 mm citrate, pH 6.0, for 20 min. After cooling down for 20 min, the sections were circled with a PAP pen (Invitrogen, 00-8877) and rinsed with PBS three times for 5 min each. The remaining fluorescence microscopic analysis was similar to that used for cultured cells (63). After permeabilization with a few drops of the immunofluorescence (IF) buffer (PBS, 0.2% Triton X-100, and 0.05% Tween 20) for 30 min, the sections were incubated with drops of the IF blocking solution (IF buffer containing 2% bovine serum albumin) for 1 h. Then the anti-α-tubulin antibody (diluted at 1:500) or the anti-acetylated α-tubulin antibody (the 6-11b-1 clone, diluted to 1:1000) was added, and the incubation was carried out at room temperature for 3 h or overnight at 4 °C, with gentle agitation. After washing with the IF buffer three times (5 min each), the sections were incubated with Alexa Fluor-labeled anti-mouse IgG (Invitrogen, A11031) for 1 h, followed by three washes with the IF buffer. After incubation with PBS containing DAPI (0.5 ng/ml) for 5 min and subsequently washing twice with water (5 min each), the sections were mounted with coverslips in the ImmuMount mounting medium (Thermo Shandon, 9990402).
Preparation of Mouse Embryonic Fibroblasts (MEFs)
MEFs were derived from E13.5 embryos as described previously (64, 65).
Fluorescence-activated Cell Sorting (FACS) Analysis of MEFs
Atat1+/− and Atat1−/− MEFs were seeded in 10-cm dishes and cultured in MEF medium, composed of DMEM, 10% FBS, nonessential amino acids (1×, diluted from 100× stocks, Invitrogen), sodium pyruvate (1×, diluted from 100× stocks, Invitrogen) and were incubated in a low oxygen incubator (3% O2) for 4 days. Cells were then trypsinized before reaching confluency and washed with PBS. Collected cells were fixed in 75% ethanol at −20 °C for 2–3 h. Cells were incubated with the staining solution (300 μl of PBS, 1 μl of DAPI, and 7 μl of RNase A (10 μg/ml)) for 15 min. Then the stained cells were analyzed on a FACS analyzer.
Live Green/Red Fluorescence Microscopy
The microscopic analysis was performed as described previously (66). Briefly, HEK293 cells were plated onto 12-well plates at 4 × 104 per well. 16–24 h later, the cells were transfected with expression plasmids for eGFP-ATAT1, eGFP-α-tubulin, and RFP-DCX as indicated. The eGFP-ATAT1 construct was prepared from a human cDNA clone from Open Biosystems, whereas the ones for eGFP-α-tubulin and RFP-DCX were obtained from Addgene (plasmids 30387 and 32851, respectively). The transfections were performed with 1.5 μl of Lipofectamine 2000 (Invitrogen). 16–18 h post-transfection, green and red fluorescence signals in the cells were analyzed, without any further processing, under an Axiovert 200 inverted microscope (Zeiss), linked to an Axiocam HRm camera and controlled by Axiovision Re 4.8. Fluorescence images were taken and further processed with CS5 Adobe Photoshop and Illustrator.
RESULTS
Targeted Deletion of Mouse Atat1
To determine the necessity of Atat1 in mice and to investigate what roles Atat1 plays at the physiological level, we analyzed mutant mice in which Atat1 is inactivated by homologous recombination. These mice were generated by gene targeting through a BacVec-derived targeting vector (KOMP affiliated with the University of California at Davis). In the resulting mutant allele (Fig. 1A), a 9.2-kb genomic fragment spanning all coding exons of Atat1 was replaced with a promoterless lacZ knock-in cassette and a selection marker flanked by a splice acceptor and a polyadenylation site. The lacZ coding sequence is under the control of the 5′ promoter of Atat1 and thereby provides a convenient and sensitive reporter system for assessment of Atat1 transcription by detecting β-galactosidase activities in whole-mount embryos and tissues or in thin sections at the single cell level.
FIGURE 1.

Analysis of mouse Atat1 by gene targeting. A, strategy for generation of the Atat1-null allele. Pink boxes denote the 5′- and 3′-untranslated sequences, and solid boxes indicate exons of the Atat1 gene located at the mouse chromosome 17qB1 (UCSC Genome Browser). A Bac targeting vector was utilized to replace the 9.2-kb genomic sequence of Atat1, spanning the entire coding region, with the promoterless lacZ coding sequence and a ubiquitin C (UBC) promoter-neomycin selection cassette flanked by two LoxP sites. SA, splicing acceptor; pA, polyadenylation signal. Rough positions of the PCR genotyping primers are indicated with tiny bars; F1 and R1 are for amplifying a 321-bp fragment from wild-type allele, and InF and InR are used to amplify a 210-bp fragment from the knock-out allele. B, PCR genotyping with the indicated primer pairs. The asterisk denotes nonspecific bands. M indicates the 100-bp DNA ladder as the molecular size marker (lane 1). C, adult Atat1−/− mice are indistinguishable from wild-type littermates by gross examination. D, RT-PCR analysis of Atat1 expression in tissues from the wild-type and homozygous knock-out mice. Atat1 mRNA was detected in the indicated tissues from the wild-type but not Atat1−/− mouse. Gapdh, internal control. E, RT-PCR analysis of lacZ mRNA in various tissues from the wild-type and knock-out mice. The cDNA templates were the same as those used in D, with PCR primers Lac-InF/-InR (A). M, 100-bp DNA ladder (lane 1).
Three male heterozygous mice were obtained from the KOMP repository and mated with wild-type female mice to obtain male and female heterozygous knock-out mice. Interbreeding was then carried out to yield wild-type, heterozygous, and homozygous littermates for subsequent experimental analyses. Genotyping by PCR with two specific primer pairs confirmed the expected genotypes (Fig. 1B). Mating between Atat1+/− mice yielded offspring with the three genotypes at the expected Mendelian ratio.
Atat1−/− mice were viable and did not yield obvious phenotypes (Fig. 1C and data not shown). Both male and female Atat1−/− mice were indistinguishable from their wild-type or Atat1+/− littermates by gross examination. Genetic deletion of Atat1 did not affect the size and the overall growth (data not shown), and both Atat1+/− and Atat1−/− mice were fertile. In addition, compared with the wild-type counterparts, the mutant mice showed no obvious differences in pup retrieval and nest building (data not shown). Overall, no overt phenotypes were observed.
Next, we sought to validate the knock-out strategy by analyzing the expected mRNA transcripts. For this, RNA from different tissues in wild-type and Atat1−/− mice was extracted for RT-PCR, to compare the levels of mRNA species transcribed from endogenous Atat1 and the exogenously inserted lacZ reporter gene. As shown in Fig. 1D, the Atat1 transcript was widely expressed in the wild-type mice but was absent in the Atat1−/− mice. Conversely, the exogenous lacZ reporter was expressed in the Atat1−/− but not wild-type mice (Fig. 1E). These results confirm the knock-out accuracy and efficiency.
Atat1 as the Predominant α-Tubulin Acetyltransferase in Mice
To substantiate genetic ablation of Atat1 at the functional level, we extracted soluble proteins from wild-type and Atat1−/− mice and performed Western blotting using antibodies specific to regular and acetylated α-tubulin. For acetylated α-tubulin, we employed the widely used monoclonal antibody 6-11b-1. As shown in Fig. 2A, α-tubulin acetylation was totally ablated in major organs such as the testis (lanes 1 and 2), kidney (lanes 3 and 4), liver (lanes 5 and 6), and brain (lanes 13 and 14) of the Atat1−/− mice. It is interesting to note that the acetylation was the highest in the brain (Fig. 2A). Although α-tubulin acetylation was still detectable in the lung (Fig. 2A, lanes 7 and 8) and spleen (lanes 9 and 10), the levels dramatically or significantly decreased. In the white fat tissue, the acetylation was only minimally affected (Fig. 2A, lanes 11 and 12). These results demonstrate that Atat1−/− mice exhibit complete or dramatic loss of α-tubulin acetylation in major tissues, including the testis, kidney, liver, lung, and brain, indicating that Atat1 is the predominant α-tubulin acetyltransferase in vivo. The retained acetylation in the mutant lung, spleen, and fat tissues may be due to other α-tubulin acetyltransferases. The candidates include Ard1-Nat1, ELP3, and GCN5, which are known to acetylate α-tubulin in vitro (43, 53, 55). Further genetic analyses are needed to investigate whether any of them are responsible for the remaining α-tubulin acetylation detected in the mutant lung, spleen, and fat.
FIGURE 2.

Mouse Atat1 is the predominant α-tubulin acetyltransferase in vivo. A, protein extracts were prepared from various tissues of wild-type and Atat1−/− young male mice for Western blotting (WB) using the monoclonal antibody recognizing α-tubulin acetylated at Lys-40 (top panel) or the anti-α-tubulin antibody (bottom panel). B, same as A except that protein extracts were prepared from fibroblasts prepared from E13.5 embryos with the indicated genotypes. Yolk sacs were initially used to determine the genotypes as shown in Fig. 1B, and the genotypes were subsequently confirmed by PCR with genomic DNA directly isolated from the expanded MEFs. To illustrate the residual acetylation signal detected in Atat1−/− MEF extracts, two exposures for the same blot are shown (top two blots).
In addition to tissue analysis, we also examined extracts prepared from MEFs, which were isolated from heterozygous and homozygous mutant embryos dissected out at gestation day E13.5. Embryonic days were calculated according to the convention that E0.5 corresponds to the noon after the morning when the copulation plug was found if the mating was assumed to occur at the preceding midnight. Compared with that in the heterozygous fibroblasts, α-tubulin acetylation in the homozygous MEFs decreased dramatically to a minimal level (Fig. 2B), supporting that Atat1 is also a major α-tubulin acetyltransferase in mouse embryos. As discussed above for adult tissues, the residual level of acetylation (Fig. 2B, lane 4, middle panel) could be due to the existence of additional α-tubulin acetyltransferases.
Broad Expression of Atat1 in Mouse Embryos
Taking advantage of the lacZ knock-in cassette in the mutant allele (Fig. 1A), we then determined the expression pattern of mouse Atat1 during embryonic development. For this, embryos at E10.75 and E13.5 were used for whole-mount β-galactosidase staining. Gross morphological examination of the resulting wild-type and homozygous knock-out embryos did not reveal any obvious defects during the embryonic period (Fig. 3). This is expected in light of grossly normal Atat1−/− mice (Fig. 1C and data not shown). At both E10.75 and E13.5, Atat1 was highly expressed throughout the embryos (Fig. 3), with the spinal cord being the most intensely stained in the E13.5 mutant embryo (data not shown). These results indicate that mouse Atat1 is not required for embryonic development and is widely expressed. Therefore, Atat1 does not appear to play a major role in either embryonic or postnatal development.
FIGURE 3.

Whole-mount β-galactosidase staining of wild-type and Atat1−/− embryos. At both E10.75 (A) and E13.5 (B), LacZ expression was wide in the Atat1−/− but not wild-type embryo (data not shown for the wild-type E10.75 embryo, which displayed no staining). No morphological defects were found in the mutant embryos at these two and other embryonic stages (data not shown), indicating that Atat1 is not required for normal embryonic development. Yolk sacs were used to determine the genotypes as shown in Fig. 1B.
Wide Atat1 Expression in Adult Mouse Tissues
We next set out to determine the global expression pattern of Atat1 in adult mice. For this, we collected a variety of tissues from wild-type and Atat1−/− mice for whole-mount β-galactosidase staining. By gross examination of the appearance, no signs of obvious pathogenicity were found in the tissues from the Atat1−/− mice, and the size of the tissues was also similar between the wild-type and Atat1−/− mice. As shown in Fig. 4, the expression of Atat1 varied among different tissues, with the testis, the renal pelvis, the gastrointestinal tract (stomach, small intestine and rectum), and the brain having the most intense β-galactosidase staining (Fig. 4, A, B, H–J, and L–N, respectively). In addition, high β-galactosidase activities were detected in the habenula, as well as in islands of Calleja located within the olfactory tubercle, of the mutant mouse brain (data not shown).
FIGURE 4.

Whole-mount β-galactosidase staining of adult mouse organs. A–N, strong expression was observed in the testis, renal pelvis, gastrointestinal tract, and brain of the Atat1−/− but not wild-type mouse. For the testis (A) and brain (M and N), two boxed regions are shown at higher magnifications to illustrate β-galactosidase activities detected in seminiferous tubules and the hippocampus of Atat1−/− mice. Although not clear from L, M, and N, high β-galactosidase activities were detected in the habenula as well as in islands of Calleja located within the olfactory tubercle. Different structures in the brain were labeled according to published atlases (71, 72). Abbreviations for regions marked in L, M, and N are as follows: CA1 and CA3, cornu ammonis areas 1 and 3 of the hippocampus, respectively; DG, dentate gyrus; Cc, cerebral cortex; Cb, cerebellum; Mb, midbrain; Mo, medulla oblongata; Hp, hippocampus; Ht, hypothalamus; Ob, olfactory bulb; Sc, spinal cord; Th, thalamus.
However, no strong signals were observed in the spleen and heart (Fig. 4, E and F, respectively). These results suggest that Atat1 is highly expressed in many but not all tissues. Robust Atat1 expressions in the testis and brain (Fig. 4, A and L–N) are consistent with high levels of α-tubulin acetylation detected in these tissue extracts (Fig. 2A). Related to this, it has been reported that α-tubulin acetylation is associated with stable, long-lived microtubules found in the basal bodies and axonemes of sperm flagella (26, 67) and in axons of neurons (68–70). In contrast to the robust expression of Atat1 in the testis, renal pelvis, gastrointestinal tract, and brain, gross examination revealed no overt defects in these tissues of Atat1−/− embryos and mice (Fig. 4 and data not shown). To determine whether there were minor defects, we carried out more thorough and careful analyses of the testis and brain, two tissues that have high α-tubulin acetylation and Atat1 expression (see below).
Requirement of Atat1 for α-Tubulin Acetylation on Sperm Flagella
To gain further insights about Atat1 functions at the histological level, we examined β-galactosidase staining in thin tissue sections prepared from the mouse testes in light of the high expression in this organ (Fig. 4A). For this, frozen tissue blocks were prepared from testes and sectioned to 10–30 μm on a cryostat. Hematoxylin and eosin (H&E) and β-galactosidase stainings were then performed to gain information on tissue morphology and Atat1 expression, as well as on the potential impact of Atat1 deletion.
Specific Atat1 expression was observed in internal cell layers of the mutant seminiferous tubules, where spermatids and spermatocytes reside (Fig. 5A). By indirect immunofluorescence microscopy, we detected α-tubulin in all cells within wild-type and mutant seminiferous tubules, with the signals localized exclusively to the cytoplasm (Fig. 6, A and B, top rows of images). In addition, strong signals were detected at the luminal centers of seminiferous tubules (Fig. 6, A and B, top rows of images). The central regions, which were devoid of DAPI staining and lack of nuclei (Fig. 6, A and B, top rows of images), correspond to structures formed by sperm flagella. Under the regular light microscope, there were no obvious differences on the sperm flagella of the wild-type and mutant seminiferous tubules. Consistent with this, H&E staining revealed the presence of sperm flagella at the luminal centers of both wild-type and Atat1−/− seminiferous tubules (Fig. 6C).
FIGURE 5.

β-Galactosidase expression in testis and brain sections. A, β-galactosidase activity detection in frozen sections of wild-type and Atat1−/− testes. β-Galactosidase staining showed that Atat1 was mainly expressed in the internal cell layers of seminiferous tubules, where spermatocytes and spermatids are located. B–D, β-galactosidase staining of frozen wild-type and Atat1−/− brain sections. High level β-galactosidase expression was detected in the cerebral cortex (B), cerebellum (C), and hippocampus (D). This is consistent with results from whole-mount staining of the corresponding tissues (Fig. 4, L–N). At the mutant sagittal plane in D, the dentate gyrus did not display a bulge (refer to Figs. 7 and 8). See Fig. 4 for abbreviations of different structures in the hippocampus; Sb, subiculum; PSb, parasubiculum.
FIGURE 6.

Comparison of testis sections from wild-type and Atat1−/− mice. A and B, immunofluorescence microscopic analysis with antibodies recognizing regular (A) and acetylated (B) α-tubulin. Probing with the anti-α-tubulin antibody detected α-tubulin in almost every cell within seminiferous tubules of wild-type and Atat1−/− testis sections. In addition, strong signals were present at the luminal centers of the tubules (denoted with green arrows; A and B, top rows of images). The centers correspond to the areas where sperm flagella reside. Staining with the antibody against acetylated α-tubulin revealed the presence of α-tubulin acetylation at the central regions of seminiferous tubules in the wild-type testis section (A, bottom three images). Importantly, this signal was no longer observed in the testis section of the Atat1−/− mouse (B, bottom three images). Scale bar, 40 μm. C, H&E staining of testis sections. The sections were fixed in Bouin's solution and subjected to H&E staining. Green arrows denote sperm flagella. Scale bar, 100 μm.
Interestingly, acetylated α-tubulin was highly abundant at the luminal centers of the wild-type seminiferous tubules (Fig. 6B, bottom three images). Importantly, the α-tubulin acetylation signals were absent at the central regions of the mutant seminiferous tubules (Fig. 6B, bottom three images). These results indicate that Atat1 is required for α-tubulin acetylation of sperm flagella at the luminal centers of seminiferous tubules.
Histological Analysis of Atat1 Expression in the Brain
As the β-galactosidase activity resulting from the knock-in lacZ gene (Fig. 1A) was very high in the whole-mount brain (Fig. 4, L–N), we performed β-galactosidase staining in thin brain sections to gain information on Atat1 expression at the histological level. The staining revealed that the β-galactosidase activity was wide in the mutant brain, including the cerebral cortex and cerebellum (Fig. 5, B and C). Moreover, consistent with the results from whole-mount staining (Fig. 4, M and N), specific and strong β-galactosidase staining was also detected in different regions of the mutant hippocampus, including the pyramidal layers in CA1 and CA3, as well as the granular cell layers in the lateral blade (suprapyramidal portion) and the medial blade (infrapyramidal portion) of the dentate gyrus (Fig. 5D). The wide β-galactosidase staining in the brain is consistent with the result that α-tubulin acetylation is the highest in this organ (Fig. 2A). Wide expression of Atat1 in different regions of the brain suggests a potential physiological role of Atat1 in this important organ.
Dentate Gyrus Distortion in Mutant Mice
During analysis of Atat1 expression in the brain, we noticed a bulge in the suprapyramidal blade (Fig. 4N, marked by red arrowhead). H&E staining confirmed that this bulge was more prominent in the mutant dentate gyrus than the wild-type counterpart (data not shown). Cresyl violet is frequently used to stain Nissl bodies (corresponding to rough endoplasmic reticulum) in neurons for histological analysis of various structures in the brain, so we performed Nissl staining. As shown in Fig. 7, compared with the wild-type counterpart, the mutant dentate gyrus was slightly deformed, especially for the possession of a prominent bulge in the lateral blade of the granular cell layers. In addition to this difference, the lateral ventricle appeared to be dilated, which is currently under investigation. Although more thorough and careful analyses are needed, other regions of the brain appeared to be normal.
FIGURE 7.

Histological analysis of wild-type and Atat1−/− brain sections. A, Nissl staining of a sagittal section from the wild-type mouse brain. B, black/white copy of the color image shown in A, with different brain regions annotated. Structures of the brain were labeled according to published atlases (71, 72). Abbreviations used are as follows: 3V, third ventricle; 4V, 4th ventricle; II–X, nine lobes of the cerebellum, with lobe I missing in the section; CC, corpus callosum; Gr, granular cell layers in the olfactory bulb; Gl, glomerulus of the olfactory bulb; LV, lateral ventricle; MB, midbrain; Mc, mitral cell layer of the olfactory bulb; MO, medulla oblongata; HP, hippocampus; HT, hypothalamus; Ic, inferior colliculus; OB, olfactory bulb; OT, olfactory tubercle; Sc, superior colliculus; TH, thalamus. C, Nissl staining of a sagittal section from the Atat1−/− mouse brain. The position of this section corresponds roughly to that shown in A. The red arrowhead in C marks a bulge in the lateral or suprapyramidal blade of the mutant dentate gyrus. The green asterisk denotes the dilated lateral ventricle, and dark asterisks indicate two broken areas due to section processing. Scale bar, 500 μm.
Anatomically, the size and morphology of the mouse hippocampus vary at different sagittal planes (71, 72). To determine alteration of the dentate gyrus at different section planes, we systematically performed Nissl staining with sagittal brain sections from the wild-type and mutant mice. Shown in Fig. 8, A–C and E, are hippocampal images taken from four pairs of representative sagittal brain sections at the medial-to-lateral direction. Comparison of the wild-type and mutant dentate gyri confirmed the difference in the lateral blade of granular cell layers in the dorsal hippocampus (Fig. 8, A–C and E), whereas the dentate gyrus in the ventral hippocampus was not affected (Fig. 8D). The results presented in Fig. 8 were taken from one pair of the wild-type and mutant mice, and two other pairs of mice displayed similar differences in the dentate gyrus (data not shown).
FIGURE 8.
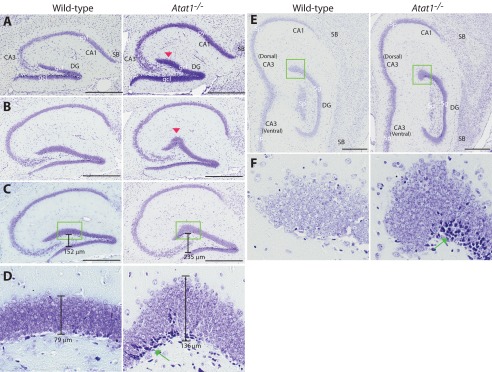
Histological analysis of wild-type and Atat1−/− hippocampi. A–C and E, representative hippocampal images taken from serial sections on four different sagittal planes in the medial-to-lateral direction. Nissl staining was performed as in Fig. 7, the two sections of which correspond to a sagittal position between those in A and B shown here. Red arrowheads in A and B mark the bulge in the lateral or suprapyramidal blade of the mutant dentate gyrus. Note that at the sagittal plane corresponding to that shown in Fig. 5D (middle and right), no bulge was found. D and F, high resolution images of the boxed areas in C and E, respectively. Green arrows denote darkly stained cell populations within the subgranular zone of the mutant dentate gyrus. D, the peak width of the granular cell layer is shown; the width was measured with the ruler tool of the Spectrum software used to analyze images digitized by the ScanScope. Abbreviations used are as follows: CA1 and CA3, cornu ammonis areas 1 and 3 of the hippocampus, respectively; DG, dentate gyrus; gcl, granular cell layer of the dentate gyrus; pyl, pyramidal layer of CA1 or CA3; SB, subiculum. Scale bar, 500 μm.
Normally, the wild-type mouse dorsal dentate gyrus is V-shaped in the granular cell layer and possesses one crest (Fig. 8), whereas the human counterpart is wavy and has multiple bulges or crests. In light of the peculiar bulge in the Atat1−/− dentate gyrus, we exhaustively searched the literature for mutant mouse strains with similar distortion in the dentate gyrus. This search uncovered two early studies showing that mice lacking the Src-related tyrosine kinase Fyn display two similar bulges in the lateral blade of the dentate gyrus (73, 74). Strikingly, one of the bulges is located at a position close to that in the Atat1−/− dentate gyrus. Fyn is required for learning and memory (73, 74), but how the two bulges are developed remains to be elucidated.
To gain mechanistic insights into how the bulge in the Atat1−/− dentate gyrus is formed, we analyzed the structure at the cellular level. For this, we analyzed the digitized images of Nissl-stained brain sections using the ruler tool of the Spectrum software to measure the size and dimensions of different structures in the hippocampus. This analysis revealed two major differences. First, the space between the two blades was wider in the mutant dentate gyrus, especially in the bulged region (Fig. 8C). Of relevance, the peak width of the bulge was 136 μm in the mutant dentate gyrus, as compared with 79 μm in the corresponding region of the wild-type dentate gyrus (Fig. 8D). Second, in the bulge, there were more granular cells. According to the ruler tool of the Spectrum software, the diameter of the round granular cell soma remained the same in the mutant (10–12 μm). There were 6–7 layers of tightly packed granular cells in the wild type, whereas there were 11–13 cell layers in the bulge of the mutant dentate gyrus (Fig. 8D). Although to a different extent at various sagittal planes (e.g. Fig. 8F), cell hyperplasia was apparent in the bulge of the mutant dentate gyrus. Therefore, Atat1 deletion caused hyperplasia in a specific region of the dentate gyrus, subsequently leading to bulge formation there.
Colocalization of Atat1 with Doublecortin in Cultured Cells
To gain further mechanistic insights into the overgrowth in the lateral blade of the Atat1−/− dentate gyrus, we first asked whether Atat1 plays a role in regulating cell cycle progression. For this, we performed FACS analysis of wild-type and mutant MEFs. The analysis revealed that Atat1 deletion did not affect cell proliferation (G0-G1/S/G2-M, 51.6:10.1:38.3% and 48.5:11.0:39.9% for Atat1+/− and Atat1−/− MEFs, respectively). However, when coexpressed in HEK293 cells, human ATAT1 colocalized nicely with nuclear mitotic apparatus antigen in mitotic spindles (data not shown), which is consistent with a recent report that ATAT1 associates with the mitotic spindle in CHO cells (75). Thus, it is still possible that ATAT1 fine-tunes mitotic programs under certain conditions.
From Nissl staining, we noticed a darkly stained cell population in the subgranular zone of the dentate gyrus (Fig. 8, D and F). This population appeared to be expanded in the bulge of the mutant dentate gyrus (Fig. 8, D and F). The subgranular zones of mouse and human dentate gyri contain adult neural stem cells (76). These stem cells are positive for doublecortin (DCX), a microtubule-associated protein (77–79). DCX is important for neurogenesis, and its gene is mutated in two genetic disorders, X-linked lissencephaly (smooth brain) and double cortex, both of which are characterized with structural abnormalities in the brain (80, 81). Mechanistically, DCX binds selectively to and stabilizes 13-protofilament microtubules (82). Intriguingly, Mec-17 in C. elegans is required for formation of predominantly 15-protofilament microtubules in touch receptor neurons (83, 84). In light of this exciting analogy between human DCX and C. elegans Mec-17, we wondered whether human ATAT1 interacts with DCX.
Immunoprecipitation revealed no detectable interaction in soluble protein extracts (data not shown). Because DCX induces microtubule bundling (77, 78), we investigated whether Atat1 associates with DCX in cells. To avoid complication from potential fixation artifacts, live fluorescence microscopy was performed. When expressed alone, GFP-α-tubulin distributed evenly in the cytoplasm, whereas RFP-DCX associated with bundled microtubules (Fig. 9A, left and middle panels) (77, 78). As reported (77, 78), when coexpressed, GFP-α-tubulin colocalized with RFP-DCX in bundled microtubules (Fig. 9B). When expressed alone, GFP-tagged and mCherry-tagged human ATAT1 proteins were pan-cellular or slightly enriched in the cytoplasm (Fig. 9A, right panel, and data not shown). Strikingly, when coexpressed, GFP-ATAT1 colocalized with RFP-DCX on bundled microtubules (Fig. 9C). Similar colocalization was also found between mCherry-ATAT1 and GFP-DCX (data not shown). These results suggest a potential mechanism through which ATAT1 associates with DCX and regulates neuronal properties in vivo.
FIGURE 9.

Human ATAT1 colocalizes with doublecortin-bundled microtubules. A, HEK293 cells were transfected with an expression plasmid for GFP-α-tubulin, RFP-DCX, or GFP-ATAT1 for live green or red fluorescence microscopy. B, HEK293 cells were cotransfected with the expression plasmids for GFP-α-tubulin and RFP-DCX for live green and red fluorescence microscopy. Cross-talk between different channels was none or minimal (data not shown). C, same as B except that GFP-ATAT1 was expressed instead of GFP-α-tubulin. Similar results were obtained with mCherry-ATAT1 and GFP-DCX, although mCherry-ATAT1 was more enriched in the cytoplasm than GFP-ATAT1 (data not shown). Scale bar, 40 μm.
DISCUSSION
Mouse Atat1 Is the Predominant α-Tubulin Acetyltransferase in Vivo
In this study, we have taken a genetic approach and demonstrated that Atat1−/− mice exhibited dramatic loss of α-tubulin acetylation in a majority of adult tissues, including the testis, kidney, liver, and brain, as well as in embryonic fibroblasts (Fig. 2 and data not shown). In the mutant lung, there was only a minimal level of α-tubulin acetylation (<5%; Fig. 2A, lanes 7 and 8). Thus, although they acetylate α-tubulin acetylation in vitro, the acetyltransferases ARD1-NAT1 (53), ELP3 (43, 54), and GCN5 (55) may not contribute significantly to α-tubulin acetylation in these tissues and cells in vivo. However, the acetylation was not so much affected in the spleen and fat of Atat1−/− mice (Fig. 2A). There was a residual level of α-tubulin acetylation in the lung (Fig. 2). ARD1-NAT1 (53), ELP3 (43, 54), and GCN5 (55) may be responsible for the retained acetylation in these organs of the mutant mice.
This is the first knock-out analysis of mouse Atat1 and provides direct evidence that Atat1 is the predominant α-tubulin acetyltransferase in mouse embryos and different adult organs. By RNAi, it has been shown that Atat1 contributes significantly to α-tubulin acetylation in HeLa and immortalized retinal pigment epithelial cells (32, 57). Our knock-out analysis provides unequivocal genetic support for the knockdown studies based on cultured cells. This knock-out analysis also yields the first unexpected picture about the functional impact of mouse Atat1 deletion at the organismal level.
Roles of Atat1 in Regulating Advanced Functions of the Brain, Testis, and Other Organs
Surprisingly, Atat1−/− mice exhibited no overt phenotypes: viable, fertile, and healthy as their wild-type littermates (Fig. 1A and data not shown). The viability and fertility of mutant mice are really unexpected in light of the reported importance of Atat1 in zebrafish development and mammalian cilium assembly (32, 57). The primary cilium is a unique organelle important for cellular signaling and animal development (61). However, our results are consistent with the findings that Atat1 homologs play nonessential roles in Tetrahymena and C. elegans (32, 57). Related to this, Lys-40 is not required for survival of Chlamydomonas, Tetrahymena, and C. elegans (39–41), and arginine substitution exerts little effect on the function of a plant α-tubulin (42). Our results are also in agreement with the report that Hdac6-null mice are viable and develop rather normally (48). Our findings indicate that mouse Atat1 is not required for animal survival, thereby shedding light on functions of α-tubulin acetylation in vivo.
Expression analysis using the knock-in lacZ reporter revealed high expression of Atat1 in the brain, testis, and gastrointestinal tract (Fig. 4). The high expression in the brain is consistent with the highest level of α-tubulin acetylation in this organ (Fig. 2A). In line with our results, recent in situ hybridization studies revealed high and dynamic expression of Atat1 in the mouse and rat brains (44, 75). Based on the high Atat1 expression in the mouse brain, testis, and gastrointestinal tract, it is tempting to speculate that Atat1 and perhaps α-tubulin acetylation may fine-tune microtubule-dependent processes in the brain, testis, and gastrointestinal tract. Because of dramatic loss of α-tubulin acetylation (Fig. 2 and data not shown) and the presence of the knock-in lacZ reporter (Fig. 1A), Atat1−/− mice are invaluable for investigating whether this is indeed the case.
To detect subtle changes in the organs where Atat1 is highly expressed, we performed histological analyses, which revealed that Atat1 was highly and specifically expressed in the cerebral cortex and the hippocampus (Fig. 5, B and D). Related to the hippocampus, the mutant dentate gyrus was deformed, displaying a bulge in the lateral blade of granular cell layers (Figs. 7 and 8). Of relevance, the dorsal dentate gyrus of the wild-type mouse is normally V-shaped in the granular cell layer and possesses one crest (Fig. 8) (71, 72), whereas the human counterpart is wavy and has multiple bulges or crests. In terms of morphology, the bulged dentate gyrus in the mutant mouse is more similar to the rat dentate gyrus. Intriguingly, mice lacking the Src-related tyrosine kinase Fyn display two similar bulges in the lateral blade of the dentate gyrus, with one of them located at a position close to that in the Atat1−/− dentate gyrus (73, 74). How the bulges are developed in the Fyn−/− dentate gyrus remains to be determined. Analysis of the Atat1−/− dentate gyrus revealed that Atat1 deletion caused cell hyperplasia in the specific region of the dentate gyrus, subsequently leading to bulge formation (Fig. 8, D and F). Although no morphologic differences have been found in other regions of the hippocampus and within the cerebral cortex, it will be important to carry out more thorough and systematic analyses by using different molecular markers. This is particularly important in light of recent studies showing that α-tubulin acetylation and Atat1 regulate neuronal migration (43, 44). The hippocampus is crucial for learning and memory, so it will be exciting to analyze Atat1−/− mice by various behavioral tests, such as navigation in the Morris water maze, to gain insights into how advanced functions like learning and memory are affected by Atat1 deletion.
Lateral ventricles of the mutant brain seemed to be dilated (Fig. 7), so it will be important to systematically analyze this dilation, which may be related to potential ciliary defects (32, 57). Of relevance, a survey of 50 genomes of evolutionarily diverse organisms revealed that the presence of Mec-17 orthologs correlates perfectly with the possession of cilia or flagella (32), suggesting the importance of such proteins in cilia or flagella. This is also related to defective α-tubulin acetylation on Atat1−/− sperm flagella (see Fig. 6 and below).
In the mouse testis, Atat1 expression was detected in the internal cell layers of seminiferous tubules (Fig. 5). Indirect immunofluorescence microscopy revealed high α-tubulin acetylation at the luminal centers of the wild-type but not mutant seminiferous tubules (Fig. 6, A and B). Related to this, Western blotting (Fig. 2) identified Atat1 as the sole α-tubulin acetyltransferase in the testis. The luminal centers of seminiferous tubules are devoid of nuclei but contain sperm flagella. Thus, Atat1 is required for α-tubulin acetylation of sperm flagella. As for the functional impact, it will be necessary to systematically analyze sperm development and maturation. Even though the mutant male mice are fertile, it is still possible that they have subtle defects. It is noteworthy that the lumens of seminiferous tubules appeared to be dilated when compared with the wild-type counterparts (Fig. 6C), so it will be important to carry out systematic analysis to verify this.
In addition to the brain and testis, it will be important to systematically analyze other organs where cilia and related structures are important. The oldest Atat1−/− mutant mice that we have examined are about 18 months old, so it still remains possible that Atat1 has important roles at later stages of life. Moreover, Atat1 may play a role under special conditions. Treatment for spinal cord injury depends on axon regeneration and appropriate synapse formation. Several reports implicate microtubule stabilization in facilitating axon regeneration (85, 86). Our results showed that Atat1 is the sole α-tubulin acetyltransferase in the brain (Fig. 2) and is highly expressed in the spinal cord (Fig. 4K). It has been recently reported that Atat1 and α-tubulin acetylation regulate microtubule structure in C. elegans (83, 84). Thus, it will be important to investigate how Atat1 may play a role in axon regeneration. Furthermore, α-tubulin acetylation decreases in response to intestinal inflammation induced by the bacterium Clostridium difficile, suggesting a need to investigate how Atat1 and α-tubulin acetylation may be involved in this and related pathogenic conditions (87).
Regulation of Microtubule Architecture and Function by Atat1
α-Tubulin acetylation at Lys-40 has been used as a routine marker for stable microtubules in cilia, flagella, axons, midbodies, and other subcellular structures (23, 24, 29). It still remains debatable whether the acetylation is causal in microtubule stabilization (30–33). The acetylation has also been linked to microtubule-mediated transport of proteins and organelles (31, 34–38), but it is mysterious how a luminal modification affects transport processes that occur on the external surface of microtubules (29). Further analysis of cell and tissues from Atat1−/− mice will shed light on the precise role of α-tubulin acetylation in these processes.
In C. elegans, Mec-17 was recently discovered to be important for maintaining unique microtubule architecture (83, 84). In this organism, the touch receptor neurons have mainly 15-protofilament microtubules, whereas the other cells possess 11-protofilament microtubules (84). In the touch receptor neurons lacking Mec-17, there are fewer and shorter microtubules, containing 11–15 protofilaments as compared with the predominantly 15 protofilaments in microtubules from the wild type (83, 84). Mec-17 increases cohesion between tubulin subunits in the microtubule lattice and constrains the protofilament number by acetylating Lys-40 of α-tubulin. This modification promotes formation of inter-protofilament salt bridges, which then stabilize lateral interactions between protofilaments and constrain quinary structure to produce stable and structurally uniform 15-protofilament microtubules in the wild-type touch receptor neurons (83).
An important question is how to translate these intriguing findings to microtubules in mammals. It will be interesting to investigate how the microtubule architecture is affected in tissues and cells from Atat1−/− mice. Notably, different from C. elegans, most microtubules in other organisms (including mammals) have 13 protofilaments (84); to compensate for this difference, Atat1 in other organisms may need to have roles distinct from that of Mec-17 in C. elegans. Of relevance, the mammalian neural stem cell marker DCX binds selectively to and stabilizes 13-protofilament microtubules (82). Importantly, when coexpressed, DCX colocalized with ATAT1 in bundled microtubules (Fig. 9), suggesting that these two proteins may cooperate with each other in regulating microtubule structure and function. One exception to the general 13-protofilament rule is that like touch receptor neurons in C. elegans, mammalian pillar cells also have 15-protofilament microtubules (84). It will be particularly interesting to analyze how the microtubule architecture is affected in such cells from Atat1−/− mice. Thus, these mice should be useful for analyzing microtubule structure, function, and regulation in various cells and tissues in vivo.
Conclusion
The genetic and molecular analyses presented herein provide direct evidence that Atat1 is the predominant α-tubulin acetyltransferase in mouse embryos and tissues. This is the first demonstration that mammalian Atat1 acts as a bona fide α-tubulin acetyltransferase in vivo. The viability and grossly normal development of Atat1−/− embryos and mice indicate that Atat1 is not essential for basic functions such as animal survival and development, but it may be important for more advanced functions, including learning and memory. It is highly expressed in the brain, testis, renal pelvis, and gastrointestinal tract, so further studies should focus on how mammalian Atat1 fine-tunes advanced functions in the organs under various physiological and pathological conditions. The mutant mice should also be valuable for studying how mammalian α-tubulin acetylation regulates microtubule structure and function in vivo.
This work was supported by research grants from the Canadian Institutes of Health Research, the Canadian Cancer Society, the Canada Foundation for Innovation, and the Ministère du Développement Économique, Innovation, et Exportation du Québec (to X. J. Y.).
- DEPC
- diethyl pyrocarbonate
- MEF
- mouse embryonic fibroblast
- PFA
- paraformaldehyde
- DCX
- doublecortin
- RFP
- red fluorescent protein.
REFERENCES
- 1. Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang X. J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allfrey V., Faulkner R. M., Mirsky A. E. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 6. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 7. Berger S. L. (2007) The complex language of chromatin regulation during transcription. Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 8. Latham J. A., Dent S. Y. (2007) Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 14, 1017–1024 [DOI] [PubMed] [Google Scholar]
- 9. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 10. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 11. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaluarachchi Duffy S., Friesen H., Baryshnikova A., Lambert J. P., Chong Y. T., Figeys D., Andrews B. (2012) Exploring the yeast acetylome using functional genomics. Cell 149, 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C. F., Grishin N. V., Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X. J., Seto E. (2007) HATs and HDACs: from structure, function, and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 [DOI] [PubMed] [Google Scholar]
- 16. Allis C. D., Berger S. L., Cote J., Dent S., Jenuwien T., Kouzarides T., Pillus L., Reinberg D., Shi Y., Shiekhattar R., Shilatifard A., Workman J., Zhang Y. (2007) New nomenclature for chromatin-modifying enzymes. Cell 131, 633–636 [DOI] [PubMed] [Google Scholar]
- 17. Lee K. K., Workman J. L. (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295 [DOI] [PubMed] [Google Scholar]
- 18. Khochbin S., Verdel A., Lemercier C., Seigneurin-Berny D. (2001) Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 11, 162–166 [DOI] [PubMed] [Google Scholar]
- 19. Grozinger C. M., Schreiber S. L. (2002) Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9, 3–16 [DOI] [PubMed] [Google Scholar]
- 20. Verdin E., Dequiedt F., Kasler H. G. (2003) Class II histone deacetylases: versatile regulators. Trends Genet. 19, 286–293 [DOI] [PubMed] [Google Scholar]
- 21. Yang X. J., Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Westermann S., Weber K. (2003) Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 4, 938–947 [DOI] [PubMed] [Google Scholar]
- 24. Janke C., Bulinski J. C. (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773–786 [DOI] [PubMed] [Google Scholar]
- 25. L'Hernault S. W., Rosenbaum J. L. (1985) Chlamydomonas α-tubulin is posttranslationally modified by acetylation on the ϵ-amino group of a lysine. Biochemistry 24, 473–478 [DOI] [PubMed] [Google Scholar]
- 26. Piperno G., Fuller M. T. (1985) Monoclonal antibodies specific for an acetylated form of α-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J. Cell Biol. 101, 2085–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Piperno G., LeDizet M., Chang X. J. (1987) Microtubules containing acetylated α-tubulin in mammalian cells in culture. J. Cell Biol. 104, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. LeDizet M., Piperno G. (1987) Identification of an acetylation site of Chlamydomonas α-tubulin. Proc. Natl. Acad. Sci. U.S.A. 84, 5720–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soppina V., Herbstman J. F., Skiniotis G., Verhey K. J. (2012) Luminal localization of α-tubulin K40 acetylation by cryo-EM analysis of Fab-labeled microtubules. PLoS One 7, e48204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palazzo A., Ackerman B., Gundersen G. G. (2003) Cell biology: tubulin acetylation and cell motility. Nature 421, 230. [DOI] [PubMed] [Google Scholar]
- 31. Zilberman Y., Ballestrem C., Carramusa L., Mazitschek R., Khochbin S., Bershadsky A. (2009) Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J. Cell Sci. 122, 3531–3541 [DOI] [PubMed] [Google Scholar]
- 32. Shida T., Cueva J. G., Xu Z., Goodman M. B., Nachury M. V. (2010) The major α-tubulin K40 acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. U.S.A. 107, 21517–21522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sudo H., Baas P. W. (2010) Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J. Neurosci. 30, 7215–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 [DOI] [PubMed] [Google Scholar]
- 35. Dompierre J. P., Godin J. D., Charrin B. C., Cordelières F. P., King S. J., Humbert S., Saudou F. (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 27, 3571–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Witte H., Neukirchen D., Bradke F. (2008) Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 180, 619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Friedman J. R., Webster B. M., Mastronarde D. N., Verhey K. J., Voeltz G. K. (2010) ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hammond J. W., Huang C. F., Kaech S., Jacobson C., Banker G., Verhey K. J. (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell 21, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gaertig J., Cruz M. A., Bowen J., Gu L., Pennock D. G., Gorovsky M. A. (1995) Acetylation of lysine 40 in α-tubulin is not essential in Tetrahymena thermophila. J. Cell Biol. 129, 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kozminski K. G., Diener D. R., Rosenbaum J. L. (1993) High level expression of nonacetylatable α-tubulin in Chlamydomonas reinhardtii. Cell Motil. Cytoskeleton 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 41. Fukushige T., Siddiqui Z. K., Chou M., Culotti J. G., Gogonea C. B., Siddiqui S. S., Hamelin M. (1999) MEC-12, an α-tubulin required for touch sensitivity in C. elegans. J. Cell Sci. 112, 395–403 [DOI] [PubMed] [Google Scholar]
- 42. Xiong X., Xu D., Yang Z., Huang H., Cui X. (2013) A single amino-acid substitution at lysine 40 of an Arabidopsis thaliana α-tubulin causes extensive cell proliferation and expansion defects. J. Integr. Plant Biol. 55, 209–220 [DOI] [PubMed] [Google Scholar]
- 43. Creppe C., Malinouskaya L., Volvert M. L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S., Belachew S., Malgrange B., Chapelle J. P., Siebenlist U., Moonen G., Chariot A., Nguyen L. (2009) Elongator controls the migration and differentiation of cortical neurons through acetylation of α-tubulin. Cell 136, 551–564 [DOI] [PubMed] [Google Scholar]
- 44. Li L., Wei D., Wang Q., Pan J., Liu R., Zhang X., Bao L. (2012) MEC-17 deficiency leads to reduced α-tubulin acetylation and impaired migration of cortical neurons. J. Neurosci. 32, 12673–12683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 [DOI] [PubMed] [Google Scholar]
- 46. Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., Horinouchi S., Yoshida M. (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 21, 6820–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., Cao C., Li N., Cheng H. L., Chua K., Lombard D., Mizeracki A., Matthias G., Alt F. W., Khochbin S., Matthias P. (2008) Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 28, 1688–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 50. Bobrowska A., Donmez G., Weiss A., Guarente L., Bates G. (2012) SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis, or the progression of Huntington's disease phenotypes in vivo. PLoS One 7, e34805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fukada M., Hanai A., Nakayama A., Suzuki T., Miyata N., Rodriguiz R. M., Wetsel W. C., Yao T. P., Kawaguchi Y. (2012) Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS One 7, e30924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sadoul K., Wang J., Diagouraga B., Vitte A. L., Buchou T., Rossini T., Polack B., Xi X., Matthias P., Khochbin S. (2012) HDAC6 controls the kinetics of platelet activation. Blood 120, 4215–4218 [DOI] [PubMed] [Google Scholar]
- 53. Ohkawa N., Sugisaki S., Tokunaga E., Fujitani K., Hayasaka T., Setou M., Inokuchi K. (2008) N-Acetyltransferase ARD1-NAT1 regulates neuronal dendritic development. Genes Cells 13, 1171–1183 [DOI] [PubMed] [Google Scholar]
- 54. Solinger J. A., Paolinelli R., Klöss H., Scorza F. B., Marchesi S., Sauder U., Mitsushima D., Capuani F., Stürzenbaum S. R., Cassata G. (2010) The Caenorhabditis elegans elongator complex regulates neuronal α-tubulin acetylation. PLoS Genet 6, e1000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Conacci-Sorrell M., Ngouenet C., Eisenman R. N. (2010) Myc-nick: a cytoplasmic cleavage product of Myc that promotes α-tubulin acetylation and cell differentiation. Cell 142, 480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steczkiewicz K., Kinch L., Grishin N. V., Rychlewski L., Ginalski K. (2006) Eukaryotic domain of unknown function DUF738 belongs to Gcn5-related N-acetyltransferase superfamily. Cell Cycle 5, 2927–2930 [DOI] [PubMed] [Google Scholar]
- 57. Akella J. S., Wloga D., Kim J., Starostina N. G., Lyons-Abbott S., Morrissette N. S., Dougan S. T., Kipreos E. T., Gaertig J. (2010) MEC-17 is an α-tubulin acetyltransferase. Nature 467, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kormendi V., Szyk A., Piszczek G., Roll-Mecak A. (2012) Crystal structures of tubulin acetyltransferase reveal a conserved catalytic core and the plasticity of the essential N terminus. J. Biol. Chem. 287, 41569–41575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Taschner M., Vetter M., Lorentzen E. (2012) Atomic resolution structure of human α-tubulin acetyltransferase bound to acetyl-CoA. Proc. Natl. Acad. Sci. U.S.A. 109, 19649–19654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Friedmann D. R., Aguilar A., Fan J., Nachury M. V., Marmorstein R. (2012) Structure of the α-tubulin acetyltransferase, αTAT1, and implications for tubulin-specific acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 19655–19660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goetz S. C., Anderson K. V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nagy A., Gertsenstein M., Vintersten K., Behringer R. (2003) Manipulating the Mouse Embryo, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 63. Walkinshaw D. R., Weist R., Xiao L., Yan K., Kim G. W., Yang X. J. (2013) Dephosphorylation at a conserved SP motif governs cAMP sensitivity and nuclear localization of class IIa histone deacetylases. J. Biol. Chem. 288, 5591–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Takahashi K., Okita K., Nakagawa M., Yamanaka S. (2007) Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2, 3081–3089 [DOI] [PubMed] [Google Scholar]
- 65. Tahmasebi S., Ghorbani M., Savage P., Yan K., Gocevski G., Xiao L., You L., Yang X. J. (2013) Sumoylation of Kruppel-like factor 4 inhibits pluripotency induction but promotes adipocyte differentiation. J. Biol. Chem. 288, 12791–12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Walkinshaw D. R., Weist R., Kim G. W., You L., Xiao L., Nie J., Li C. S., Zhao S., Xu M., Yang X. J. (2013) The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 288, 9345–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. LeDizet M., Piperno G. (1986) Cytoplasmic microtubules containing acetylated α-tubulin in Chlamydomonas reinhardtii: spatial arrangement and properties. J. Cell Biol. 103, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baas P. W., Black M. M. (1990) Individual microtubules in the axon consist of domains that differ in both composition and stability. J. Cell Biol. 111, 495–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith C. L. (1994) The initiation of neurite outgrowth by sympathetic neurons grown in vitro does not depend on assembly of microtubules. J. Cell Biol. 127, 1407–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fukushima N., Furuta D., Hidaka Y., Moriyama R., Tsujiuchi T. (2009) Post-translational modifications of tubulin in the nervous system. J. Neurochem. 109, 683–693 [DOI] [PubMed] [Google Scholar]
- 71. Dong H. W. (2008) Allen Reference Atlas, A Digital Brain Atlas of the C57BL/6J Male Mouse, John Wiley & Sons, Inc, Hoboken, NJ [Google Scholar]
- 72. Paxinos G., Franklin K. B. (2008) The Mouse Brain in Stereotaxic Coordinates, 3rd Ed., Academic Press, New York [Google Scholar]
- 73. Grant S. G., O'Dell T. J., Karl K. A., Stein P. L., Soriano P., Kandel E. R. (1992) Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258, 1903–1910 [DOI] [PubMed] [Google Scholar]
- 74. Kojima N., Wang J., Mansuy I. M., Grant S. G., Mayford M., Kandel E. R. (1997) Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc. Natl. Acad. Sci. U.S.A. 94, 4761–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kalebic N., Martinez C., Perlas E., Hublitz P., Bilbao-Cortes D., Fiedorczuk K., Andolfo A., Heppenstall P. A. (2013) The tubulin acetyltransferase αTAT1 destabilizes microtubules independently of its acetylation activity. Mol. Cell. Biol. 33, 1114–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhao C., Deng W., Gage F. H. (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660 [DOI] [PubMed] [Google Scholar]
- 77. Francis F., Koulakoff A., Boucher D., Chafey P., Schaar B., Vinet M. C., Friocourt G., McDonnell N., Reiner O., Kahn A., McConnell S. K., Berwald-Netter Y., Denoulet P., Chelly J. (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23, 247–256 [DOI] [PubMed] [Google Scholar]
- 78. Gleeson J. G., Lin P. T., Flanagan L. A., Walsh C. A. (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23, 257–271 [DOI] [PubMed] [Google Scholar]
- 79. Magavi S. S., Leavitt B. R., Macklis J. D. (2000) Induction of neurogenesis in the neocortex of adult mice. Nature 405, 951–955 [DOI] [PubMed] [Google Scholar]
- 80. des Portes V., Pinard J. M., Billuart P., Vinet M. C., Koulakoff A., Carrié A., Gelot A., Dupuis E., Motte J., Berwald-Netter Y., Catala M., Kahn A., Beldjord C., Chelly J. (1998) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92, 51–61 [DOI] [PubMed] [Google Scholar]
- 81. Gleeson J. G., Allen K. M., Fox J. W., Lamperti E. D., Berkovic S., Scheffer I., Cooper E. C., Dobyns W. B., Minnerath S. R., Ross M. E., Walsh C. A. (1998) Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72 [DOI] [PubMed] [Google Scholar]
- 82. Moores C. A., Perderiset M., Francis F., Chelly J., Houdusse A., Milligan R. A. (2004) Mechanism of microtubule stabilization by doublecortin. Mol. Cell 14, 833–839 [DOI] [PubMed] [Google Scholar]
- 83. Cueva J. G., Hsin J., Huang K. C., Goodman M. B. (2012) Posttranslational acetylation of α-tubulin constrains protofilament number in native microtubules. Curr. Biol. 22, 1066–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Topalidou I., Keller C., Kalebic N., Nguyen K. C., Somhegyi H., Politi K. A., Heppenstall P., Hall D. H., Chalfie M. (2012) Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr. Biol. 22, 1057–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hellal F., Hurtado A., Ruschel J., Flynn K. C., Laskowski C. J., Umlauf M., Kapitein L. C., Strikis D., Lemmon V., Bixby J., Hoogenraad C. C., Bradke F. (2011) Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 331, 928–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sengottuvel V., Fischer D. (2011) Facilitating axon regeneration in the injured CNS by microtubules stabilization. Commun. Integr. Biol. 4, 391–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nam H. J., Kang J. K., Kim S. K., Ahn K. J., Seok H., Park S. J., Chang J. S., Pothoulakis C., Lamont J. T., Kim H. (2010) Clostridium difficile toxin A decreases acetylation of tubulin, leading to microtubule depolymerization through activation of histone deacetylase 6, and this mediates acute inflammation. J. Biol. Chem. 285, 32888–32896 [DOI] [PMC free article] [PubMed] [Google Scholar]