

Background: Endoplasmic reticulum (ER) stress maintains cellular protein homeostasis.
Results: A novel ER stress-responsive element, ERSE-26, identified in 38 genes, is regulated by sXBP1 during ER stress.
Conclusion: ER stress increases levels of prion and other proteins not previously known to be involved in the ER stress response.
Significance: ERSE-26 implicates novel genes regulated by the ER stress response.
Keywords: Endoplasmic Reticulum Stress, ER Stress, Gene Regulation, Gene Transcription, Prions, ERSE, XBP-1
Abstract
Understanding the regulatory mechanisms mediating PRNP gene expression is highly relevant to elucidating normal cellular prion protein (PrP) function(s) and the transmissibility of prion protein neurodegenerative diseases. Here, luciferase reporter assays showed that an endoplasmic reticulum stress element (ERSE)-like element, CCAAT-N26-CCACG in the human PRNP promoter, is regulated by ER stress and X-box-binding protein 1 (XBP1) but not by activating transcription factor 6 α (ATF6α). Bioinformatics identified the ERSE-26 motif in 37 other human genes in the absence of canonical ERSE sites except for three genes. Several of these genes are associated with a synaptic function or are involved in oxidative stress. Brefeldin A, tunicamycin, and thapsigargin ER stressors induced gene expression of PRNP and four randomly chosen ERSE-26-containing genes, ERLEC1, GADD45B, SESN2, and SLC38A5, in primary human neuron cultures or in the breast carcinoma MCF-7 cell line, although the level of the response depends on the gene analyzed, the genetic background of the cells, the cell type, and the ER stressor. Overexpression of XBP1 increased, whereas siRNA knockdown of XBP1 considerably reduced, PRNP and ERLEC1 mRNA levels in MCF-7 cells. Taken together, these results identify a novel ER stress regulator, which implicates the ER stress response in previously unrecognized cellular functions.
Introduction
The regulation of gene expression from the human PRNP gene encoding the cellular prion protein (PrP)3 is not well characterized. Yet, the importance of the normal cellular PrP is undisputable. The levels of PrP influence prion disease transmission. The absence of PrP eliminates infection by transmissible prion diseases, and low levels of PRNP gene expression associated with promoter nucleotide polymorphisms decrease the risk of infection by bovine spongiform encephalopathy (1–3). In contrast, transgenic overexpression of PRNP accelerates transmissible prion disease progression (1, 4, 5). In addition, polymorphisms in the regulatory region of PRNP may present a risk for Creutzfeldt-Jakob disease (6, 7). Furthermore, accruing evidence implicates the normal cellular PrP in a number of essential cellular functions including neuroprotection against oxidative stress (8, 9), copper toxicity (10), Bax-mediated cell death (11), and ischemia (12–15). There is also evidence that PrP participates in normal synaptic function (16) and myelination (17, 18). Fully understanding the regulation of PRNP gene expression is essential to developing therapies against transmissible prion diseases and will also help elucidate the function of the normal cellular PrP.
PrP is developmentally and ubiquitously expressed in most organs and tissues, with a higher expression in brain, lung, heart, and muscle (19–25). The PRNP promoter contains GC-rich repeats, which are common to housekeeping genes. It does not contain the core promoter TATA box sequence but does contain Sp1, Ap-1, Ap-2, and CCAAT box transcriptional binding sites (26, 27). Regulatory elements include MyoD, heat shock elements (28), and metal regulatory element MTF-1 (29). Up-regulation of PRNP gene expression occurs via NGF (30) or hypoxic conditions in brain (15, 31), in HIV infection of astrocytes (32), during hematopoietic differentiation (24), and by cellular exposure to copper (29) and has been associated with a change in chromatin conformation (33).
We recently identified four endoplasmic reticulum stress response elements (ERSE) in the PRNP promoter and showed that ER stress transcriptionally up-regulates PRNP gene expression (34). The unfolded protein response is a well conserved process that cells use to restore ER homeostasis or enter apoptosis. In higher eukaryotes, three sensors of ER status exist (reviewed in Ref. 35). IRE1α (inositol-requiring enzyme 1) is a transmembrane ribonuclease that splices and activates X-box-binding protein (XBP1) mRNA when the foldase BiP (Grp78, encoded by HSPA5) dissociates from IRE1α and preferentially attaches to misfolded client proteins (35, 36). Concurrently, the transmembrane protein ATF6α (activating transcription factor 6), which is normally bound by BiP, is released and translocates to the Golgi complex, where it is cleaved by the transmembrane Site 1 and Site 2 proteases to form a highly active transcription factor (37, 38). The third branch of ER stress signal transduction is mediated by the transmembrane PKR-like ER localized kinase (PERK), which upon BiP dissociation phosphorylates eukaryotic translation initiation factor 2 (eIF2α). Phosphorylation of eIF2α arrests translation of most messages except for several with upstream activating open reading frames, such as activating transcription factor 4 (ATF4) mRNA (35, 39). Following ER signal transduction, spliced XBP1 (sXBP1), cleaved ATF6 (ΔATF6α), and ATF4 translocate to the nucleus where they are able to activate the expression of ER stress-responsive genes. ΔATF6α, sXBP1, and ATF4 interact with several promoter DNA motifs called ERSE, resulting in the transactivation of unfolded protein response target genes (35).
One of the PRNP ERSE, CCAAT, separated from CCACG by 26 nucleotides (CCAAT-N26-CCACG or ERSE-26), was of particular interest, as it resembled almost exactly the classical ERSE, CCAAT-N9-CCACG (ERSE-9), characterized by Yoshida et al. (37) in ER stress up-regulation of glucose-regulated proteins. ERSE-9 is transactivated by ΔATF6α (36, 37). Although XBP1 can bind to ERSE-9 and transactivate a luciferase reporter under the regulation of five tandem repeats of ERSE-9 (40), overexpression of XBP1 did not transactivate the glucose-regulated proteins (36). Separation of the CCACG from CCAAT with eight or 10–13 nucleotides instead of nine nucleotides abolished the ability of ΔATF6 to bind to the ERSE-9 motif (36, 37). Therefore, it would be expected that PRNP CCAAT-N26-CCACG might not be transactivated by ΔATF6α. Nevertheless, the homology of PRNP ERSE-26 with the classical ERSE-9 and the ability of ER stress to induce PRNP gene expression (34) prompted us to further investigate this motif. We found that ERSE-26 is transactivated by sXBP1 at the CCACG motif but is not transactivated by ΔATF6α. A bioinformatic analysis of the human genome revealed 37 other genes containing ERSE-26 within their promoter region. Several were confirmed to be up-regulated by ER stress in primary human neuron cultures or in the breast carcinoma MCF-7 cell line. These results identified a novel ER stress-regulated ERSE motif that is common to 38 human genes.
EXPERIMENTAL PROCEDURES
Genome-wide Search for Genes Bearing an ERSE-26
To find genes in the human genome with an ERSE-26 in their promoter, a program was written in Python. The sequences of the complete human genome, version GRC37.1 from the Genome Reference Commission (41), and the yeast genome (Saccharomyces cerevisiae, S288c-NCBI) were scanned using regular expressions representing the plus and minus strand versions of ERSE-26. The promoters of all human genes were searched. The region from −2000 bp upstream of the transcription start site to +500 bp downstream of the transcription start site was considered to be the promoter region of the gene. The regular expression pattern used was “ccaat. 26ccacg”, matching precisely (no deviations were permitted). Both ERSE-26 elements in the forward and reverse orientations were searched. Additionally, promoters in the positive and negative strands of the genome were examined. Source code is provided in a Git repository.
Cell Culture Conditions
Human primary neuron cultures, obtained from human fetal brains subject to McGill University ethical approval, were cultured as described previously (42). Briefly, brains were minced, trypsinized, plated on polylysine-coated flasks, and grown in the presence of 5-fluorodeoxyuridine, 1× penicillin-streptomycin, and minimum essential medium supplemented with 10% bovine calf serum in a 5% CO2 environment at 37 °C. MCF-7 cells (from ATCC) were grown in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) in a 5% CO2 environment at 37 °C. HEK293 cells were grown in DMEM supplemented with 1.7 g/liter NaHCO3 and 10% FBS in a 5% CO2 incubator at 37 °C.
Pharmacological Induction of ER Stress and Other Cell Treatments
Neurons and MCF-7 cells were subjected to ER stress under conditions that were optimized experimentally to minimize cell death and maximize ER stress induction. In neurons, ER stress was induced by 5 μg/ml brefeldin A (BFA), 0.5 μg/ml tunicamycin (TM), and 3.25 μg/ml thapsigargin (Thps) from Biomol. Drugs were dissolved in DMSO, and cells were treated for 6 h using the equivalent concentration of DMSO as control followed by immediate total RNA extraction. MCF-7 cells were treated for 18 h, with all drugs at a concentration of 5 μg/ml.
Total RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted from neurons using TRIzol (Invitrogen) as described by the manufacturer's protocol. For MCF-7 cells, the Chomczynski guanidine thiocyanate method was used (43). RNA was quantitated on an H4 plate reader (Biotek, Winooski, VT) for neurons or a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). cDNA was prepared using avian myeloblastosis reverse transcriptase (AMV-RT) following the manufacturer's protocol (Roche Applied Science). Briefly, 1 μg of total RNA was used for cDNA synthesis by AMV-RT with poly(dT) primers, and cDNA used as a template for subsequent PCRs.
Primer Design and PCR of ERSE-26 Genes
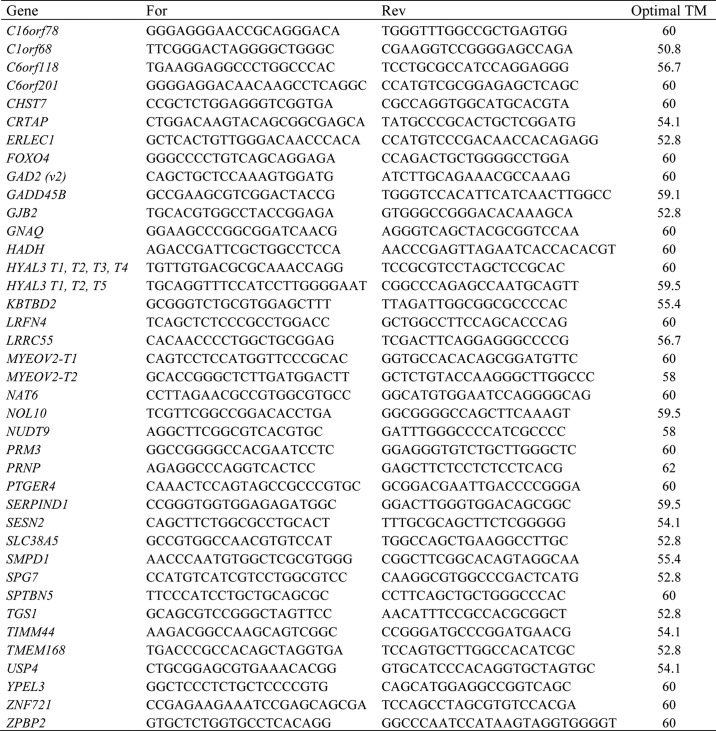
For PCRs, the primers were designed using the NCBI PrimerBlast software available online (44). Primers were designed to have an annealing temperature of 60 °C. All oligonucleotides were ordered from a commercial supplier (Invitrogen) and optimized with a gradient of annealing temperatures. Table 1 contains all primer sequences. During all subsequent PCRs, the optimal annealing temperature of a primer pair of a gene was always used. cDNA was diluted to 1:200, and PCR was performed with Taq DNA polymerase (New England Biolabs) following the manufacturer's protocol. Three biologically independent neuron preparations were used, and two technical replicates per gene per preparation were tested. For MCF-7 cells, three independent experiments were conducted in triplicate for all ER stress drug treatments and sXBP1 transfections.
TABLE 1.
Primers used for RT-PCR
Western Blotting of PrP, BiP, and β-Actin in ER Stress-treated Neurons
Cells were lysed for 20 min on ice in lysis buffer (150 mm NaCl, 2 mm EDTA, 0.5% Triton X-100 (v/v), 0.5% sodium deoxycholate (w/v), and 50 mm Tris-HCl, pH 7.5) containing fresh protease and phosphatase inhibitors (38 μg/ml AEBSF, 0.5 μg/ml leupeptin, 0.1 μg/ml pepstatin, 0.1 μg/ml N-α-p-tosyl-l-lysine chloromethyl ketone hydrochloride, 4 mm sodium orthovanadate, and 20 mm sodium fluoride). After centrifugation of the lysate at 11,500 × g for 10 min at 4 °C, the supernatant was collected as the detergent-soluble fraction. The protein concentration was determined with the BCA protein assay reagents (Fisher). One hundred μg of protein was precipitated with 4 volumes of ice-cold methanol overnight at −20 °C and centrifuged at 11,500 × g for 15 min at 4 °C; the dried pellet was solubilized in Laemmli sample buffer (2% SDS (w/v), 5% β-mercaptoethanol, (v/v), 10% glycerol (v/v), 0.01% bromphenol blue (w/v), and 62.5 mm Tris-HCl, pH 6.8). The proteins were boiled for 3 min, separated in a 10 or 15% SDS-PAGE, and transferred to PVDF membranes. PrP was detected with the polyclonal R155 antiserum (1/500, anti-PrP36–56) or 3F4 (1/2000, anti-PrP109–112). Other antibodies used were β-actin (1:5000, clone AC-15, Sigma) and BiP (1:250, clone H-129, Santa Cruz Biotechnology Inc.). Immunoreactivity was revealed with 1/5000 anti-rabbit IgG conjugated to horseradish peroxidase secondary antibodies (GE Healthcare) and chemiluminescence reagents from GE Healthcare or Millipore (Billerica, MA) and was detected with the Molecular Dynamics Storm 840 (GE Healthcare) or Kodak Biomax MR film (Carestream Health, Toronto, Ontario, Canada).
Cloning of ERSE-26 Fragment into pMetLuc2 Vector and Site-directed Mutagenesis
The pMetLuc2-Reporter construct (pML2, obtained from Clontech) is a plasmid that encodes a luciferase gene from the copepod Metrida longa. This gene contains a powerful endogenous signal peptide, causing the protein to be secreted into the growth medium. This allows for a no-lysis protocol for assaying luciferase activity in transfected cells by simply reading the luciferase activity (when a substrate is added) of the medium. To clone the PRNP promoter fragment into pML2, primers were designed to amplify the region from −391bp to −274bp, around the ERSE-26, which was obtained from the pGL3 PRNP promoter construct, kindly given to us by the laboratory of Dr. Collinge (26). The following primers were used (restriction sites in bold): forward, 5′-GAG CTC TCT CCA TTA TGT AAC GGG GA-3′; and reverse, 3′-GCG AAT TCT CAG TTG ATA CCG CCT GCG G-5′. Primers contained the restriction endonuclease sites for EcoRI and SacI. The PCR product and pMetLuc2-reporter vector were digested using EcoRI and SacI followed by ligation with T4 DNA ligase (Fermentas). DH5α Escherichia coli were transformed using standard protocols, and DNA was prepared for transfection using the alkaline lysis method (45). The resulting pML2-EL26 construct was sequenced. Following cloning of the PRNP promoter fragment into the pMetLuc2 vector, the ERSE-26 was mutated by PCR with mutagenic oligos. Primers were designed to target the second part of the ERSE-26, the putative XBP1/ATF6 binding site; CCACG was changed to ATCTA. Two additional base pairs after the XBP1/ATF6 site were also changed from TC to GA, yielding a final mutation of ATCTAGA. The following primers were used for mutagenesis, (with the mutation in bold and the former location of the CCACG site underlined): 5′-GAT TTT TAC AGT CAA TGA GAT CTA GAA GGG AGC GAT GGC ACC C-3′ and 5′-GGG TGC CAT CGC TCC CTT CTA GAT CTC ATT GAC TGT AAA AAT C-3′. In addition, two other mutants were generated by inserting 4 or 8 base pairs at the center of the ERSE-26 element. For the elongated ERSE-30 and ERSE-34, the following primers were used (with the inserted base pairs underlined): ERSE-30-For 5′-GGG CCG AAT TTC CAA TTA AAG ATG ATT TTA AAA TAC AGT CAA TGA GCC ACG TCA GGG AGC G-3′ and ERSE-30-Rev 5′-CGC TCC CTG ACG TGG CTC ATT GAC TGT ATT TTA AAA TCA TCT TTA ATT GGA AAT TCG GCC C-3′; and ERSE-34-For 5′-GGA GCT TTG GGC CGA ATT TCC AAT TAA AGA TGA TTT TAA AAA AAA TAC AGT CAA TGA GCC ACG TCA GGG AGC GAT GGC AC-3′ and ERSE-34-Rev 5′-GTG CCA TCG CTC CCT GAC GTG GCT CAT TGA CTG TAT TTT TTT TAA AAT CAT CTT TAA TTG GAA ATT CGG CCC AAA GCT CC-3′. DNA was synthesized using Pfu Turbo (Agilent Technologies) and subjected to DpnI digestion to remove non-mutated plasmid DNA. DNA was then transformed into DH5α E. coli using standard techniques and prepared using the alkaline lysis method. Transfection in HEK293 cells was done as described below.
Transfection of HEK293 Cells and Luciferase Assay
HEK293 cells were plated in 6-well plates at ∼500,000 cells/well. Twenty-four hours after plating, using the polyethyleneimine method (46), cells were transfected with pMetLuc2-Reporter-ERSE-26, pMetLuc2-Reporter-ERSE-30, or pMetLuc2-Reporter-ERSE-34 and plasmids pCGN-EGFP, pCGN-ATF6 encoding ΔATF6α (amino acids 1–373), or pCGN-sXBP1, kindly provided by Dr. Kaufman (40). Following 6 h of transfection, the medium was changed, and cells were allowed to grow for 20 h. After 20 h, the medium was collected. Luciferase assays were conducted on 50 μl of HEK293 growth medium. Substrate (Ready-to-Glow secreted luciferase substrate, Clontech) for the luciferase protein was prepared according to the manufacturer's protocols. Assays were conducted in a 96-well white, opaque plate (Costar), and total luminescence was determined using an H4 plate reader (Biotek, Winooski, VT). At least two reads were conducted per well in three biologically independent experiments. As a control, total RNA was extracted as described above to verify sXBP1 and ATF6α transcripts in transfected cells.
Western Blotting of XBP1 and ATF6α in HEK293 Cells
HEK293 cells were extracted with Nonidet P-40 lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, and 5 mm EDTA, pH 8.0). Extracts were separated by 10% SDS-PAGE, transferred to PVDF membrane using a Bio-Rad Transblot Turbo apparatus, blocked for 1 h in 5% milk, and then incubated with 1:100 anti-XBP1 (sc7160, Santa Cruz Biotechnology) or 1:100 anti-ATF6 (IMG-273, Imgenex) to confirm expression of transgenes. Membranes were detected using anti-mouse/rabbit IgG horseradish peroxidase at 1:5000 in 5% milk (Jackson Immunoresearch) and visualized with ECL Prime (GE Healthcare) on Kodak BioMax MR film.
Transfection of MCF-7 Cells
MCF-7 cells from ATCC (Manassas, VA) were cultured as described above and transfected with a Nucleofector Kit V following the manufacturer's protocol (AmaxaTM). Cells were transfected with either pCGN-EGFP-sXBP1 (pCGN-sXBP1) or vehicle (pCGN-EGFP), kindly given by the laboratory of Dr. Kaufman (40, 47). The siRNAs against XBP1 as well as scrambled siRNA controls were obtained from Santa Cruz Biotechnology (sc38627 and sc37007, respectively). Total RNA was extracted using Chomczynski's method, and cDNA was synthesized using AMV-RT as described above.
Quantitative PCR of ERSE-26 Genes
qPCR was conducted on cDNA from neurons and MCF-7 cells using SYBR Green Taq Master Mix (Quanta Biosciences). For several ERSE-26 genes, new primers had to be designed, and the additional qPCR-validated primers were obtained from the PrimerBank database (48), a repository of primer sequences synthesized by a commercial supplier (Invitrogen). These additional primers are as follows: GADD45B, For 5′-TACGAGTCGGCCAAGTTGATG-3′ and Rev 5′-GGATGAGCGTGAAGTGGATTT-3′; XBP1, For 5′-TGCTGAGTCCGCAGCAGGTG-3′ and Rev 5′-GCTGGCAGGCTCTGGGGAAG-3′; GAD2, For 5′-ATTGGGAATTGGCAGACCAAC-3′ and Rev 5′-TTGAAGTATCTAGGATGCCCTGT-3′; SESN2, For 5′-AAGGACTACCTGCGGTTCG-3′ and Rev 5′-CGCCCAGAGGACATCAGTG-3′; GAPDH, For 5′-TGCACCACCAACTGCTTAGC-3′ and Rev 5′-GGCATGGACTGTGGTCATGAG-3′; and HPRT1, For 5′-CCTGGCGTGGTGATTAGTGAT-3′ and Rev 5′-AGACGTTCAGTCCTGTCCATAA-3′. For neurons and MCF-7, three technical replicates for each of the three biologically distinct preparations were conducted. For MCF-7 cells, two technical replicates per biological replicate were done. Using ERSE-26-containing gene primers that produced a single unique amplicon, the standard curves and amplification constants were determined. An Applied Biosystems 7500Fast qPCR apparatus (Invitrogen) was used, and the manufacturer's default thermal cycle was used for all experiments. Primers were verified for specificity and performance for qPCR. All output was converted to -fold induction values compared with control treatments normalized with loading controls (HPRT1) using Pfaffl's method (49). In the siRNA experiments, we observed too much variability in the HPRT1 mRNA levels with siRNA and drug treatments, and therefore the data were normalized to GAPDH.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as described previously with some modifications (34). MCF-7 cells transfected with either pCGN-XBP1 or pCGN-ATF6 were incubated with 1% formaldehyde for 10 min at 37 °C. The cross-linking was stopped with glycine at a final concentration of 0.125 m. After 5 min at 37 °C, cells were lysed in swelling buffer (10 mm Tris-HCl, pH 8, 0.25 m sucrose, 0,5% Nonidet P-40, 2 mm DTT) containing fresh protease inhibitors (38 μg/ml AEBSF, 0.5 μg/ml leupeptin, 0.1 μg/ml pepstatin, and 0.1 μg/ml N-α-p-tosyl-l-lysine chloromethyl ketone hydrochloride) and incubated for 20 min on ice in sonication buffer (50 mm Hepes, pH 7.4, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1% SDS, and protease inhibitors). Nuclear lysates were sonicated to shear the genomic DNA into fragments of 200–1000 bp. The chromatin was precleared with protein G-Sepharose (Sigma) precoated with 1 μg/ml sonicated salmon sperm nuclei (S3126, Sigma) and 1 mg/ml BSA. Chromatin was then incubated overnight with precoated protein G-Sepharose and anti-ATF6 (IMG-273, Imgenex), anti-XBP1 (sc7160, Santa Cruz Biotechnology), control IgG antibodies (3 μg), or no antibody. After washing, the immunoprecipitates were eluted two times with 200 μl of elution buffer (50 mm NaHCO3, 1% SDS, 1 mm EDTA, and 50 mm Tris-HCl, pH 8.0). Cross-linking was then reversed overnight at 65 °C. The immunoprecipitated and whole cell extract DNA were purified and used for PCR analyses using primers designed to amplify only the ERSE-26 of PRNP: PRNP sense, 5′-CTGAGCCTTTCATTTTCTCG-3′, and antisense, 5′-GAGATTCGCTTGAACACTTG-3′; HSPA5 sense, 5′-GTGAACGTTAGAAACGAA TAGCAGCCA-3′, and antisense, 5′-GTCGAC CTCACCGTCGCCTA-3′; and ACTB sense, 5′-CTGGAACGGTGAAGGTGACA-3′, and antisense, 5′-AAGGGACTTCCTGTAACAATG CA-3′. The PCR products were visualized after electrophoresis on a 3% agarose gel containing ethidium bromide.
Data Mining of Existing Databases
Microarray databases in GeoDataset (NCBI) were explored for evidence of increased HSPA5 mRNA levels as evidence of ER stress. The GeoDataSet named “In vitro generation of long-lived human plasma cells” (50) compared Illumina microarray profiles (Illumina human HT-12, V4.0, expression beadchip) of human adult peripheral B cells differentiated into plasma cells. A significant -fold change was taken when genes identified as containing ERSE-26 had an adjusted p value of p < 0.05.
Statistical Analysis
For the luciferase assays, a one-way analysis of variance (ANOVA) followed by Tukey's Honest Significant Difference post hoc test was used to determine whether luminescence values were statistically significant. For qPCR data, a treatment by gene ANOVA followed by Tukey's Honest Significant Difference post hoc analyses, pairwise one-way t test or as indicated in the figure legends on individual means (using SPSS version 17) was conducted on -fold increases in cDNA amplicons.
RESULTS
sXBP1, Not ΔATF6α, Transactivates ERSE-26
To determine whether ERSE-26 responds to ER stress-regulated transcription factors ATF6α and sXBP1, human embryonic kidney HEK293 cells were co-transfected with the pML2-ERSE-26 luciferase reporter construct and pCGN-sXBP1 or pCGN-ATF6-(1–373) encoding ΔATF6α. Co-transfection of pML2-ERSE-26 and sXBP1 resulted in high luciferase activity (Fig. 1A), whereas non-transfected cells, mock transfected cells, or cells transfected with only pML2-ERSE-26, pCGN-sXBP1, or pCGN-ATF6-(1–373) showed little luciferase activity. Mutagenesis of the putative ATF6α/sXBP1 binding site, CCACG, to ATCTA in the ERSE-26 (pML2-ERSE26m) abolished transactivation by sXBP1 (Fig. 1B). RT-PCR (Fig. 1C) and Western blots (Fig. 1D) confirmed the expression of sXBP1 only in cells transfected with the pCGN-sXBP1 construct. In contrast to sXBP1, ΔATF6α did not activate ERSE-26-mediated transcription (Fig. 1, A and B). Again, RT-PCR (Fig. 1C) and Western blots (Fig. 1D) confirmed increased expression of ΔATF6α in the transfected cells. The length of the 26 interspersing nucleotides of ERSE-26 influence transcriptional regulation by sXBP1, as luciferase levels were decreased by almost 50% in ERSE-30 and ERSE-34 compared with ERSE-26 (Fig. 1E). Lastly, ChIP analyses confirmed the interaction of sXBP1 with ERSE-26 (Fig. 1F). In contrast, ATF6α did not co-immunoprecipitate the ERSE-26 (Fig. 1G). Together, these results indicate that sXBP1, not ΔATF6α, is the transactivating factor for the ERSE-26 motif.
FIGURE 1.

ERSE-26 is transactivated by sXBP1. A, relative luminescence units (RLU) from pML2 luciferase secreted from HEK293 cells transfected or co-transfected as indicated with the pML-2-ERSE-26 (ERSE-26), pCGN-ATF6α-(1–373), pCGN-sXBP1 (sXBP1), or the control pCGN-EGFP construct. The pML2 construct was used as a control (−, in all panels), and luminescence from non-transfected (NT) or mock transfected HEK293 cells is indicated in the last two bars. The results represent the mean ± S.E. of three independent experiments. *, p < 0.001 when compared with cells transfected with pML2-Empty and pCGN-sXBP1. B, relative luminescence units from pML2 luciferase secreted from transfected or co-transfected HEK293 cells, as indicated, to compare the transactivation of pML2-ERSE-26 versus the mutant pML2-ERSE-26m. The results represent the mean ± S.E. of three independent experiments. *, p < 0.01 when compared with cells transfected with pML2-EL26 and pCGN-sXBP1. C, ethidium bromide-agarose gel of sXBP1 and ATF6 amplicons obtained by RT-PCR of transfected cell mRNA. D, Western blot of sXBP1- and ΔATF6α-transfected HEK293 cells. E, relative luminescence units from HEK293 co-transfected with pML2-ERSE-26, pML2-ERSE-30, or pML2-ERSE-34 and pCGN-EGFP, pCGN-sXBP1 (sXBP1), or pCGN-ATF6α-(1–373). The results represent the mean ± S.E. of three independent experiments. *, p < 0.001 when comparing cells transfected with pCGN-sXBP1 to control (Ctl). #, p < 0.01 when comparing cells transfected with pML2-ERSE-30 or pML2-ERSE-34 with pML2-ERSE-26. Lower panel, ethidium bromide-agarose gel of XBP1, ATF6α, and HPRT1 amplicons obtained by RT-PCR of transfected cellular mRNA. F and G, agarose gel showing RT-PCR of genes from the ChIP assay in MCF-7 cells transfected with either pCGN-XBP1 (F) or pCGN-ATF6 (G). Cross-linked chromatin immunoprecipitated with anti-XBP1 (F) or anti-ATF6α antibody, rabbit IgG, or no antibody (G) was used for PCR amplification of the PRNP ERSE-26 motif. The HSPA5 promoter was used as a positive control, whereas PCR on the promoter of ACTB was used as an internal negative control.
ERSE-26 Motif Identified in the Promoters of 37 Additional Human Genes
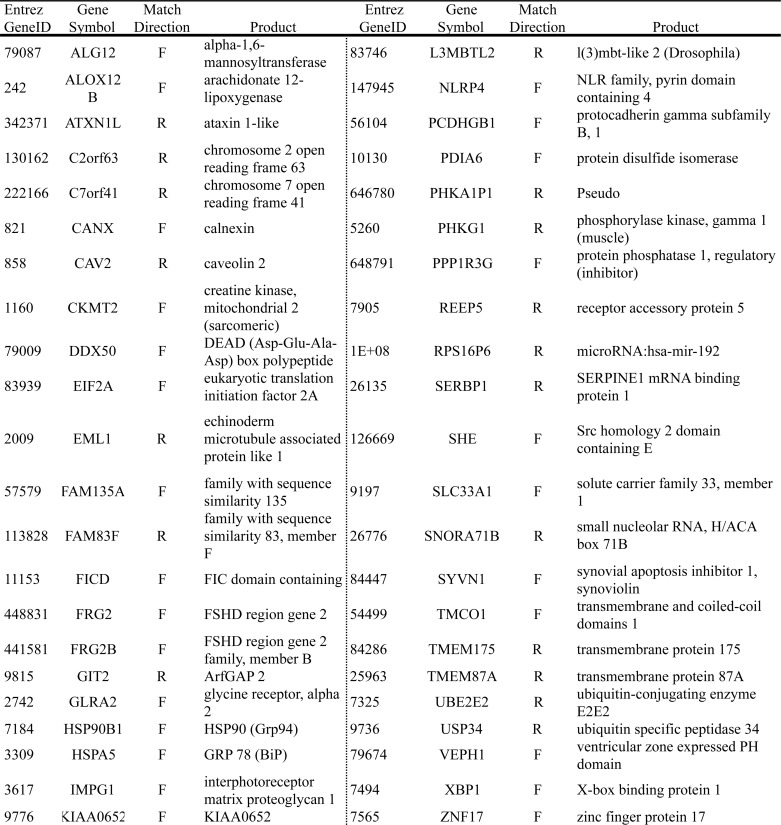
To assess whether ERSE-26 is present in the promoter of genes other than PRNP, a Python program was written to search the Genome Reference Consortium human genome (Build GRC37.1). In total, 38 genes contained an ERSE-26 within −2000 bp and +500 bp of the transcription start site of the gene (Table 2). None of these genes contained canonical ERSEs except C6orf20, which contains an ERSE-II motif, and GADD45B and LRRC55, which contain XBP1 binding sites in their promoters. Twenty-six ERSE-26 matches were found in the reverse (3′ to 5′) direction, and 12 were found in the forward direction. As a control for the search program, the search was repeated with the canonical ERSE-9 as a target pattern. Forty-four genes, including the previously known ERSE-containing genes HSPA5 (BiP, Grp78), XBP1, and PDIA6, contained an ERSE-9 within −2000 bp to +500 bp of the transcription start site of the gene (Table 3). These results show that the ERSE-26 motif is not unique to the PRNP promoter and suggest that ER stress may regulate a wide variety of genes previously unsuspected to be involved in the ER stress response.
TABLE 2.
Genes containing an ERSE-26 in their promoters
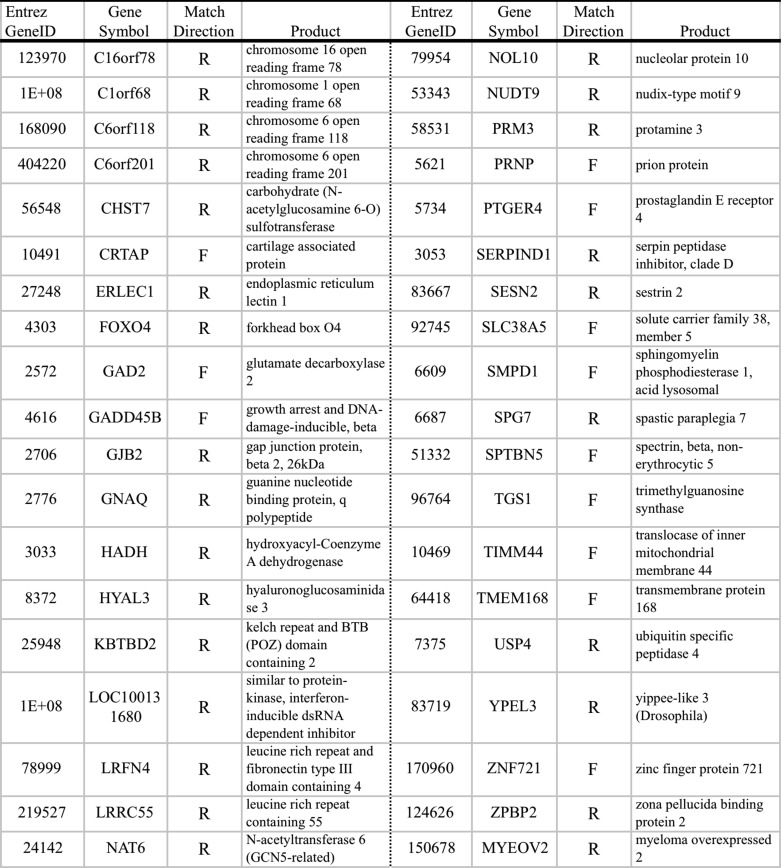
TABLE 3.
Genes containing an ERSE-9 in their promoters
To assess whether ERSE-26 is evolutionarily conserved, we searched the yeast genome. The search identified eight yeast genes containing ERSE-26 (Table 4), but none of these were the same as the mammalian ERSE-26 genes. However, three of the eight genes may be relevant to ER function. Fun12p functions as a translation initiation factor (51), Chs6p is involved in Golgi to plasma membrane protein trafficking (Saccharomyces Genome Database), and Hap1p is a zinc finger transcriptional factor responding to oxygen and heme levels (52). These data indicate that ERSE-26 exists in lower organisms but not necessarily in genes that are orthologous to the mammalian ERSE-26 genes.
TABLE 4.
Yeast genes containing an ERSE-26 in their promoters
| Match pattern | Distance from start | Match directiona | Gene name | Gene product | Function | |
|---|---|---|---|---|---|---|
| bp | ||||||
| gcacc | taacc | −609, −573 | F | FUN12 | Fun12p | Translation initiation Factor IF2 |
| ccaat | ccacg | −858, −822 | F | BUD9 | Bud9p | Controls yeast polarity and development. |
| ggtta | ggtgc | −1291, −1255 | R | MTC1 | Mtc1p | Maintenance of telomere-capping protein 1 |
| gcacc | taacc | −479, −443 | F | CHS6 | Chs6p | Export of cargo from Golgi to plasma membrane |
| gcacc | taacc | −1293, −1257 | F | SMX2 | Smx2p | Part of spiceosome |
| gcacc | taacc | −550, −514 | F | HAP1 | Hap1p | Zinc finger transcriptional factor responding to O2 and heme |
| gcacc | taacc | 378, 414 | F | RKM5 | Rkm5p | Protein-lysine methyltransferase |
| gcacc | taacc | −1053, −1017 | F | NHA1 | Nha1p | Na+/H+ antiporter |
a R, reverse; F, forward.
ERSE-26 Genes Are Up-regulated in Primary Human Neuron Cultures Treated with BFA, TM, or Thps
To confirm that the ERSE-26-containing promoters are transactivated during ER stress, primary human neurons were treated with the ER stressors BFA, TM, and Thps (Fig. 2). RT-PCR identified transcripts from 20 of the 38 ERSE-26 genes in primary human neuron cultures (Fig. 2A). ER stress induction under these conditions was confirmed by the presence of the sXBP1 amplicon (Fig. 2, A and B) or increased HSPA5 (BiP) mRNA (Fig. 2B) or protein (Fig. 2C) levels. The levels of ER stress-induced sXBP1 varied with the type of ER stress and also showed variable levels of induction in independent neuron cultures. Nevertheless, relative to the DMSO control, each of the pharmacological inducers of ER stress (BFA, TM, and Thps) induced expression of many of the ERSE-26 genes in at least one human neuron culture (Fig. 2A). To assess the levels of mRNA in a more quantitative manner, qPCR was conducted on a few ERSE-26-containing genes (Fig. 2B). Compared with the DMSO control, the three ER stressors significantly increased the levels of HSPA5 and XBP1. Although the levels of HSPA5 mRNA were similarly increased by each of three ER stressors, the -fold increase in XBP1 levels varied considerably (20–60-fold). PRNP mRNA levels increased by 3–8-fold with the three different ER stressors. ERLEC1 mRNA levels increased 4–6-fold with BFA and TM but not Thps. GADD45B mRNA increased 2-fold with Thps only. SESN2 levels increased significantly (8–12-fold) with TM and Thps, whereas SCL38A5 levels were not significantly increased with any of the three ER stressors. These variable results probably reflect inherent differences in mRNA stability for different genes or the presence of other transactivating or silencing factors in the promoter of these genes. Furthermore, the variability in response in different primary human neuron preparations indicates a potential influence of the genetic background on the ER stress response. As a negative control, neurons were submitted to serum deprivation as another stress. Compared with either the DMSO control or the non-treated cells, the mRNA levels of the ERSE-26-containing genes did not increase and neither did those of the ER stress-regulated genes HSPA5 and XBP1. Together, these results indicate that ER stress can modulate the expression of many ERSE-26-containing genes.
FIGURE 2.

Induction of ER stress increases ERSE-26 gene expression in cultured primary human neurons. A, ethidium bromide-stained agarose gel of ERSE-26-containing gene amplicons. Each panel is a representative example from each gene, respectively. B, -fold increase of the indicated gene expression by qRT-PCR over the level obtained in the DMSO-treated control cells and arbitrarily set at 1. In addition to treatment with BFA, TM, and Thps, serum deprivation (−S) and non-treated cells (NT) were analyzed. The data represent duplicates of three independent neuron cultures; mean ± S.E. C, Western blot analyses of MCF-7 cell protein extracts for PrP, BiP, and β-actin levels.
ERSE-26-containing Genes Are Up-regulated in ER-stressed Breast Carcinoma MCF-7 Cells
To determine whether the variability observed in different human neuron preparations is dependent on the genetic background of the cells, we conducted the same experiments on the breast carcinoma MCF-7 cell line. qPCR results showed a more consistent induction of the various genes studied in independent experiments (Fig. 3A). However, some of the genes responded differently to the three ER stressors. HSPA5 levels increased 35-fold with BFA but only 10–12-fold with TM and Thps treatments. Similar results were observed with PRNP and ERLEC1. In contrast, XBP1, GADD45B, SESN2, and SLC38A5 levels increased similarly with the three different ER stressors. Except for TM on SESN2, all ERSE-26 genes had a significant increase in gene expression with the three different ER stressors when compared with control DMSO-treated cells. Although the MCF-7 cells responded more consistently to ER stress than primary cultures of human neurons, there was also a differential response of these genes to different ER stressors. To determine whether this differential response was dependent on XBP1 levels, MCF-7 cells were transfected with pCGN-EGFP-XBP1 or control vector pCGN-EGFP. As expected, sXBP1 mRNA levels were increased 25-fold over pCGN-EGFP-transfected cells (Fig. 3B). HSPA5, PRNP, and ERLEC1 mRNA levels increased 2–3-fold with XBP1 expression, but neither the GADD45B nor the SESN2 mRNA level was modulated by XBP1 overexpression. Overexpression of XBP1 protein in transfected MCF-7 cells was confirmed by Western blotting (Fig. 3C).We then tested the regulation of these genes by XBP1 in MCF-7 cells treated with BFA in the absence or presence of scrambled siRNA (siCtl) or siRNA against XBP1 (siXBP1). BFA increased the levels of sXBP1 mRNA in MCF-7 cells and thus provided a model to determine whether sXBP1 can up-regulate ERSE-26-containing genes in live cells (Fig. 3D). Unexpectedly, the HPRT1 mRNA levels varied with the different treatments, and thus we had to use GAPDH mRNA levels to normalize instead of HPRT1. XBP1 mRNA level increased 2-fold in BFA-treated MCF-7 cells compared with the DMSO control-treated cells (Fig. 3E). The siXBP1 decreased these levels by 80%. PRNP, HSPA5, ERLEC1, and SESN2 mRNA levels increased with BFA treatment. In the presence of siXBP1, PRNP and ERLEC1 mRNA levels decreased by 50%, but HSPA5 was only slightly decreased. This was not unexpected, because HSPA5 is regulated also by ATF6α and ATF4 (36, 53). Furthermore, siXBP1 did not prevent BFA-induced SESN2 mRNA levels, consistent with the inability of the sXBP1 to up-regulate this gene in MCF-7 cells (Fig. 3B). GADD45B was not up-regulated by BFA when normalized to GAPDH, and basal levels were unaffected by the siXBP1. Lastly, we confirmed the down-regulation of XBP1 protein in siXBP1-treated cells (Fig. 3E). Together, these results show that ER stress-induced or overexpressed sXBP1 can transactivate ERSE-26-containing genes but that the ERSE-26 motif is not always sufficient for sXBP1 transactivation.
FIGURE 3.

MCF-7 cells show induced ERSE-26 gene expression following ER stress treatment or sXBP1 transfection. A, -fold induction of ERSE-26 genes in MCF-7 cells treated with BFA, TM, or Thps for 18 h as determined by qRT-PCR. ANOVA, p < 0.001 followed by planned paired t test using a Bonferroni correction between DMSO and BFA, DMSO, and TM and between DMSO and Thps. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, ERSE-26 gene expression levels in pCGN-EGFP-sXBP1-transfected MCF-7 cells compared with pCGN-EGFP-transfected cells (control (Ctl)). The results represent an average ± S.E. of three experiments done in triplicate. A paired t test was used to assess XBP1 levels (left panel), and ANOVA (p < 0.0001) and Tukey-Kramer multiple comparisons were used to assess the levels of gene expression in the right panel. ***, p < 0.001; *, p < 0.05. C, Western blot with anti-XBP1 or anti-β-actin antibodies in Nonidet P-40-soluble or -insoluble protein extracts from MCF-7 cells transfected with pCGN-EGFP (EGFP) or pCGN-sXBP1-IRES-EGFP (sXBP1/EGFP). D, ethidium bromide-stained agarose gel of XBP1 RT-PCR from MCF-7 cells treated with BFA, Thps, and TM. E, ERSE-26 gene expression levels in siRNA scrambled control (siCtl) or siRNA against XBP1 (siXBP1)-transfected MCF-7 cells submitted to BFA. The results represent the mean ± S.E. of four independent experiments. *, p < 0.05 comparing ±BFA. F, Western blot with anti-XBP1 or β-actin antibodies in Nonidet P-40-soluble and -insoluble protein extracts from MCF-7 cells treated with DMSO or BFA (5 μg/ml, 18 h) 24 h after transfection with control scrambled or XBP1-targeting siRNAs.
Evidence for ERSE-26 Physiological Function
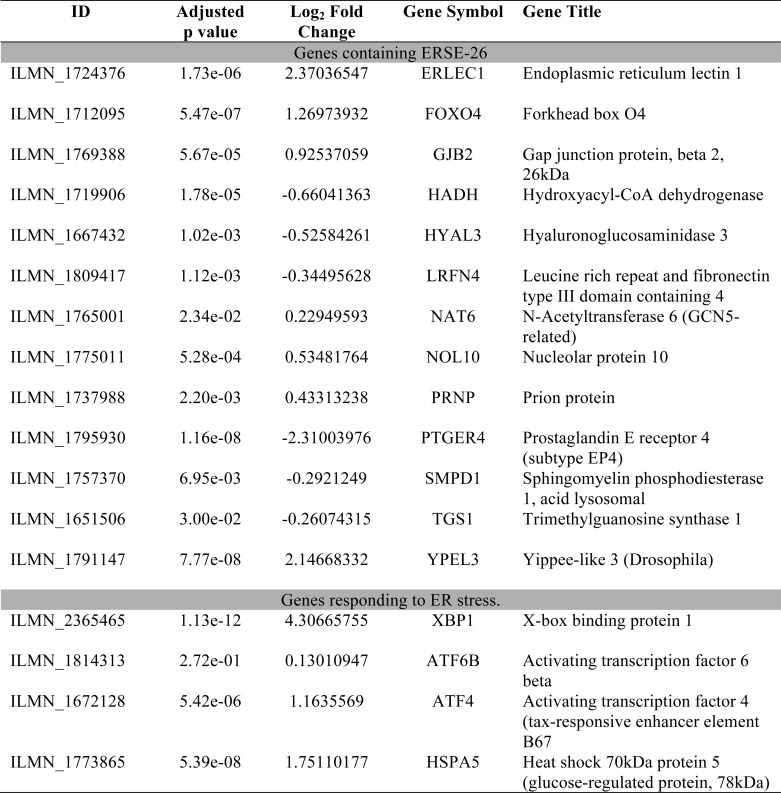
To determine whether the ERSE-26 motif is potentially transcribed by sXBP1 in a physiological condition, we examined available microarray data sets comparing the mRNA profiles of human XBP1-dependent B cell differentiation into plasma cells (Table 5) (54). In this array, XBP1 was up-regulated 4.3-fold (Log2). All ERSE-26 genes identified by our bioinformatics analysis were present in the data set, and 13 of 38 showed a significant (adjusted p < 0.05) Log2 -fold change in mRNA levels when activated B cells were differentiated into plasma cells. Of the 13 genes, ERLEC1 (2.4-fold), FOXO4 (1.29-fold), and YPEL3 (2.1-fold) were up-regulated, and PTGER4 was down-regulated (−2.3-fold). Foxo4 negatively regulates cell cycle and thus could be up-regulated to stop proliferation and allow differentiation of the B cells into plasma cells (55). ERLEC1 is an ER-resident protein that has been implicated in metastasis and the survival of lung cancer cells, in the regulation of the unfolded protein response, and in trafficking of glycoprotein in the ER quality control system (56). Ypel3 is associated with cellular senescence and is repressed in colon and mammary tumors, suggesting a novel role as a tumor suppressor (57). Ptger4 promotes proliferation in endometrial cancers (58). Therefore, these ERSE-26-containing genes could be implicated in differentiated plasma cells via XBP1.
TABLE 5.
Differential mRNA gene expression of ERSE-26 genes between activated B cells and plasma cells (50)
DISCUSSION
The role of ER stress in disease is broad, and many conditions that disturb ER homeostasis by affecting protein folding can have a large impact on the physiological state of cells and organisms (2). Here, we added ERSE-26, an ER stress-dependent regulatory motif, to the already complex ER stress response. We show that ERSE-26 (CCAAT-N26-CCACG), first identified in the human PRNP promoter (34), is transactivated by sXBP1 in a luciferase reporter system and that mutagenesis of CCACG to ATCTA abolishes the ability of sXBP1 to transactivate ERSE-26. Furthermore, we show that ER stressors in mammalian cells increased the levels of mRNA from genes containing ERSE-26 and other ER stress-regulatory elements (PRNP and GADD45B) and in genes with no other known ER stress regulatory elements (ERLEC1 and SESN2). We also show that expression of sXBP1 in MCF-7 cells is sufficient to significantly increase PRNP gene expression and that siRNA against XBP1 stunts the BFA-mediated increase of PRNP mRNA levels. This response is surprising because the canonical ERSE CCAAT-N9-CCACG is transactivated by ΔATF6α, not sXBP1, and disruption of N9 abolishes ΔATF6α transactivation of ERSE (36, 37). In contrast, we found that ATF6α does not interact with or transactivate ERSE-26. Our results suggest that CCAAT-NX-CCACG motifs present in gene promoters may have greater functionality in the response to ER stress than believed previously.
The bioinformatic search for gene promoters containing ERSE-26 identified 38 genes, including PRNP. Except for C6orf20, GADD45B, and LRRC55, none of these genes had been identified previously as ERSE-containing or ER stress-responsive genes. The function of the ERSE-26 genes was studied to determine whether these could link the ER stress response via the ERSE-26 motifs with specific functions. One notable functional group was genes related to cell adhesion and synapse function. GAD2, LRFN4, LRRC55, and SLC38A5 all play a role in this process. GAD2 is one of two terminal rate-limiting enzymes that cleave glutamic acid into γ-aminobutyric acid (59). LRFN4, also known as SALM3, binds post-synaptic density 95 protein and induces neurite outgrowth (60). LRRC55 enhances the activity of calcium-activated potassium channels (61). Finally, SLC38A5, also known as SN2 or SNAT5, is an astrocyte-expressed amino acid transporter that permits astrocytes to release glutamic acid for neurons to cleave to γ-aminobutyric acid (62). Together with the suspected role of PrP at the synapse (reviewed in Ref. 63), these results raise the possibility that ER stress regulates synaptic function in neurons through the ERSE-26 motif.
Another group has identified oxidative stress function. Because protein folding is a redox state-dependent, energy-requiring process, an overlap between the ER stress response and the oxidative stress response is logical. The genes NUDT9, TIMM44, SESN2, and GADD45B are localized in the mitochondria and respond to oxidative stress (64–67). GADD45B is an activator of pro-survival p38 MAPK signaling (66, 68, 69). Both TIMM44 and NUDT9 localize to the mitochondria, and TIMM44 acts as a translocase to allow import of peptides/proteins to the inner mitochondrial membrane (70). SESN2 and GADD45B are both regulated by p53 in response to environmental stresses (66, 71). Interestingly, our results included a transcription factor, FOXO4. The FoxO family of transcription factors regulates sestrin family members, which suggests a possible feedback loop in ERSE-26 genes (64). Finally, PRNP contains a p53 binding site, is induced in oxidative stress, and protects cells against oxidative stress (8–10, 72–74). Therefore, ER stress regulation via ERSE-26 in these genes may allow functions related to oxidative stress.
There is also evidence that some of the ERSE-26 genes are up-regulated in an XBP1-dependent manner during B cell differentiation into plasma cells. These results support a role for ERSE-26-dependent gene expression in a human physiological system.
Our results show that the -fold increase in ER stress-mediated gene expression is highly variable and can be regulated by genetic background, cell type, and ER stressor. Furthermore, despite clear evidence that XBP1 regulates ERSE-26 in the PRNP and ERLEC1 promoters, not all ERSE-26-containing genes are up-regulated by XBP1 only. Elucidating how and why the ER stress response is so variable remains a future goal, but these results illustrate once again the complexity of the ER stress response. Nevertheless, our data identify a novel XBP1-regulated ERSE and reveal previously unknown ER stress-regulated genes.
Acknowledgments
We gratefully acknowledge Dr. John Collinge for providing the PRNP promoter construct and Dr. Kaufman for the XBP1 and ATF6 constructs. We are also very grateful to Dr. Veronica Afonso for statistical analyses. We thank the Birth Defects Research Laboratory (University of Washington, Seattle) for providing conceptal tissue for research and Dr. Vikas Kaushal for help in the culture of the neurons.
This work was supported by Canadian Institutes of Health Research Operating Grants MOP89376 and MOP102738 (to A. C. L).
- PrP
- prion protein
- ER
- endoplasmic reticulum
- ERSE
- endoplasmic reticulum stress response element
- sXBP1
- spliced X-box-binding protein 1
- BFA
- brefeldin A
- TM
- tunicamycin
- Thps
- thapsigargin
- AMV-RT
- avian myeloblastosis reverse transcriptase
- AEBSF
- 4-(2-aminoethyl)benezenesulfonyl fluoride hydrochloride
- ANOVA
- analysis of variance.
REFERENCES
- 1. Büeler H., Aguzzi A., Sailer A., Greiner R. A., Autenried P., Aguet M., Weissmann C. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347 [DOI] [PubMed] [Google Scholar]
- 2. Sander P., Hamann H., Drögemüller C., Kashkevich K., Schiebel K., Leeb T. (2005) Bovine prion protein gene (PRNP) promoter polymorphisms modulate PRNP expression and may be responsible for differences in bovine spongiform encephalopathy susceptibility. J. Biol. Chem. 280, 37408–37414 [DOI] [PubMed] [Google Scholar]
- 3. Sander P., Hamann H., Pfeiffer I., Wemheuer W., Brenig B., Groschup M. H., Ziegler U., Distl O., Leeb T. (2004) Analysis of sequence variability of the bovine prion protein gene (PRNP) in German cattle breeds. Neurogenetics 5, 19–25 [DOI] [PubMed] [Google Scholar]
- 4. Prusiner S. B., Scott M., Foster D., Pan K. M., Groth D., Mirenda C., Torchia M., Yang S. L., Serban D., Carlson G. A., et al. (1990) Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63, 673–686 [DOI] [PubMed] [Google Scholar]
- 5. Kercher L., Favara C., Chan C. C., Race R., Chesebro B. (2004) Differences in scrapie-induced pathology of the retina and brain in transgenic mice that express hamster prion protein in neurons, astrocytes, or multiple cell types. Am. J. Pathol. 165, 2055–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCormack J. E., Baybutt H. N., Everington D., Will R. G., Ironside J. W., Manson J. C. (2002) PRNP contains both intronic and upstream regulatory regions that may influence susceptibility to Creutzfeldt-Jakob disease. Gene 288, 139–146 [DOI] [PubMed] [Google Scholar]
- 7. Sanchez-Juan P., Bishop M. T., Croes E. A., Knight R. S., Will R. G., van Duijn C. M., Manson J. C. (2011) A polymorphism in the regulatory region of PRNP is associated with increased risk of sporadic Creutzfeldt-Jakob disease. BMC Med. Genet. 12, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown D. R., Schulz-Schaeffer W. J., Schmidt B., Kretzschmar H. A. (1997) Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 146, 104–112 [DOI] [PubMed] [Google Scholar]
- 9. Brown D. R., Wong B. S., Hafiz F., Clive C., Haswell S. J., Jones I. M. (1999) Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 344, 1–5 [PMC free article] [PubMed] [Google Scholar]
- 10. Qin K., Zhao L., Ash R. D., McDonough W. F., Zhao R. Y. (2009) ATM-mediated transcriptional elevation of prion in response to copper-induced oxidative stress. J. Biol. Chem. 284, 4582–4593 [DOI] [PubMed] [Google Scholar]
- 11. Bounhar Y., Zhang Y., Goodyer C. G., LeBlanc A. (2001) Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 276, 39145–39149 [DOI] [PubMed] [Google Scholar]
- 12. Weise J., Crome O., Sandau R., Schulz-Schaeffer W., M., Zerr I. (2004) Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 372, 146–150 [DOI] [PubMed] [Google Scholar]
- 13. Mitteregger G., Vosko M., Krebs B., Xiang W., Kohlmannsperger V., Nölting S., Hamann G. F., Kretzschmar H. A. (2007) The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 17, 174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeong J. K., Seo J. S., Moon M. H., Lee Y. J., Seol J. W., Park S. Y. (2012) Hypoxia-inducible factor-1α regulates prion protein expression to protect against neuron cell damage. Neurobiol. Aging 33, 1006.e1–10 [DOI] [PubMed] [Google Scholar]
- 15. McLennan N. F., Brennan P. M., McNeill A., Davies I., Fotheringham A., Rennison K. A., Ritchie D., Brannan F., Head M. W., Ironside J. W., Williams A., Bell J. E. (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 165, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herms J., Tings T., Gall S., Madlung A., Giese A., Siebert H., Schürmann P., Windl O., Brose N., Kretzschmar H. (1999) Evidence of presynaptic location and function of the prion protein. J. Neurosci. 19, 8866–8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A. D., Toyka K. V., Nave K. A., Weis J., Aguzzi A. (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318 [DOI] [PubMed] [Google Scholar]
- 18. Nishida N., Tremblay P., Sugimoto T., Shigematsu K., Shirabe S., Petromilli C., Erpel S. P., Nakaoke R., Atarashi R., Houtani T., Torchia M., Sakaguchi S., DeArmond S. J., Prusiner S. B., Katamine S. (1999) A mouse prion protein transgene rescues mice deficient for the prion protein gene from Purkinje cell degeneration and demyelination. Lab. Invest. 79, 689–697 [PubMed] [Google Scholar]
- 19. Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith J. M., et al. (1985) Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 315, 331–333 [DOI] [PubMed] [Google Scholar]
- 20. Kretzschmar H. A., Stowring L. E., Westaway D., Stubblebine W. H., Prusiner S. B., Dearmond S. J. (1986) Molecular cloning of a human prion protein cDNA. DNA 5, 315–324 [DOI] [PubMed] [Google Scholar]
- 21. Oesch B., Westaway D., Wälchli M., McKinley M. P., Kent S. B., Aebersold R., Barry R. A., Tempst P., Teplow D. B., Hood L. E., et al. (1985) A cellular gene encodes scrapie PrP 27–30 protein. Cell 40, 735–746 [DOI] [PubMed] [Google Scholar]
- 22. Robakis N. K., Sawh P. R., Wolfe G. C., Rubenstein R., Carp R. I., Innis M. A. (1986) Isolation of a cDNA clone encoding the leader peptide of prion protein and expression of the homologous gene in various tissues. Proc. Natl. Acad. Sci. U.S.A. 83, 6377–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manson J., West J. D., Thomson V., McBride P., Kaufman M. H., Hope J. (1992) The prion protein gene: a role in mouse embryogenesis? Development 115, 117–122 [DOI] [PubMed] [Google Scholar]
- 24. Liu T., Li R., Wong B. S., Liu D., Pan T., Petersen R. B., Gambetti P., Sy M. S. (2001) Normal cellular prion protein is preferentially expressed on subpopulations of murine hemopoietic cells. J. Immunol. 166, 3733–3742 [DOI] [PubMed] [Google Scholar]
- 25. Makrinou E., Collinge J., Antoniou M. (2002) Genomic characterization of the human prion protein (PrP) gene locus. Mamm. Genome 13, 696–703 [DOI] [PubMed] [Google Scholar]
- 26. Mahal S. P., Asante E. A., Antoniou M., Collinge J. (2001) Isolation and functional characterisation of the promoter region of the human prion protein gene. Gene 268, 105–114 [DOI] [PubMed] [Google Scholar]
- 27. Puckett C., Concannon P., Casey C., Hood L. (1991) Genomic structure of the human prion protein gene. Am. J. Hum. Genet. 49, 320–329 [PMC free article] [PubMed] [Google Scholar]
- 28. Shyu W. C., Harn H. J., Saeki K., Kubosaki A., Matsumoto Y., Onodera T., Chen C. J., Hsu Y. D., Chiang Y. H. (2002) Molecular modulation of expression of prion protein by heat shock. Mol. Neurobiol. 26, 1–12 [DOI] [PubMed] [Google Scholar]
- 29. Bellingham S. A., Coleman L. A., Masters C. L., Camakaris J., Hill A. F. (2009) Regulation of prion gene expression by transcription factors SP1 and metal transcription factor-1. J. Biol. Chem. 284, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 30. Mobley W. C., Neve R. L., Prusiner S. B., McKinley M. P. (1988) Nerve growth factor increases mRNA levels for the prion protein and the β-amyloid protein precursor in developing hamster brain. Proc. Natl. Acad. Sci. U.S.A. 85, 9811–9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kikuchi Y., Kakeya T., Nakajima O., Sakai A., Ikeda K., Yamaguchi N., Yamazaki T., Tanamoto K., Matsuda H., Sawada J., Takatori K. (2008) Hypoxia induces expression of a GPI-anchorless splice variant of the prion protein. FEBS J. 275, 2965–2976 [DOI] [PubMed] [Google Scholar]
- 32. Müller W. E., Pfeifer K., Forrest J., Rytik P. G., Eremin V. F., Popov S. A., Schröder H. C. (1992) Accumulation of transcripts coding for prion protein in human astrocytes during infection with human immunodeficiency virus. Biochim. Biophys. Acta 1139, 32–40 [DOI] [PubMed] [Google Scholar]
- 33. Cabral A. L., Lee K. S., Martins V. R. (2002) Regulation of the cellular prion protein gene expression depends on chromatin conformation. J. Biol. Chem. 277, 5675–5682 [DOI] [PubMed] [Google Scholar]
- 34. Déry M. A., Jodoin J., Ursini-Siegel J., Aleynikova O., Ferrario C., Hassan S., Basik M., Leblanc A. C. (2013) Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 15, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 36. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., Mori K. (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida H., Haze K., Yanagi H., Yura T., Mori K. (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749 [DOI] [PubMed] [Google Scholar]
- 38. Haze K., Yoshida H., Yanagi H., Yura T., Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 [DOI] [PubMed] [Google Scholar]
- 40. Lee K., Tirasophon W., Shen X., Michalak M., Prywes R., Okada T., Yoshida H., Mori K., Kaufman R. J. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Church D. M., Schneider V. A., Graves T., Auger K., Cunningham F., Bouk N., Chen H. C., Agarwala R., McLaren W. M., Ritchie G. R., Albracht D., Kremitzki M., Rock S., Kotkiewicz H., Kremitzki C., Wollam A., Trani L., Fulton L., Fulton R., Matthews L., Whitehead S., Chow W., Torrance J., Dunn M., Harden G., Threadgold G., Wood J., Collins J., Heath P., Griffiths G., Pelan S., Grafham D., Eichler E. E., Weinstock G., Mardis E. R., Wilson R. K., Howe K., Flicek P., Hubbard T. (2011) Modernizing reference genome assemblies. PLoS Biol. 9, e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. LeBlanc A. (1995) Increased production of 4 kDa amyloid β peptide in serum-deprived human primary neuron cultures: possible involvement of apoptosis. J. Neurosci. 15, 7837–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chomczynski P., Sacchi N. (2006) The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat. Protoc. 1, 581–585 [DOI] [PubMed] [Google Scholar]
- 44. Sayers E. W., Barrett T., Benson D. A., Bolton E., Bryant S. H., Canese K., Chetvernin V., Church D. M., Dicuccio M., Federhen S., Feolo M., Fingerman I. M., Geer L. Y., Helmberg W., Kapustin Y., Krasnov S., Landsman D., Lipman D. J., Lu Z., Madden T. L., Madej T., Maglott D. R., Marchler-Bauer A., Miller V., Karsch-Mizrachi I., Ostell J., Panchenko A., Phan L., Pruitt K. D., Schuler G. D., Sequeira E., Sherry S. T., Shumway M., Sirotkin K., Slotta D., Souvorov A., Starchenko G., Tatusova T. A., Wagner L., Wang Y., Wilbur W. J., Yaschenko E., Ye J. (2012) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Birnboim H. C., Doly J. (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boussif O., Lezoualc'h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 92, 7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bommiasamy H., Back S. H., Fagone P., Lee K., Meshinchi S., Vink E., Sriburi R., Frank M., Jackowski S., Kaufman R. J., Brewer J. W. (2009) ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 122, 1626–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Spandidos A., Wang H., Seed B. (2012) PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 40, D1144–D1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cocco M., Stephenson S., Care M. A., Newton D., Barnes N. A., Davison A., Rawstron A., Westhead D. R., Doody G. M., Tooze R. M. (2012) In vitro generation of long-lived human plasma cells. J. Immunol. 189, 5773–5785 [DOI] [PubMed] [Google Scholar]
- 51. Sutrave P., Shafer B. K., Strathern J. N., Hughes S. H. (1994) Isolation, identification and characterization of the FUN12 gene of Saccharomyces cerevisiae. Gene 146, 209–213 [DOI] [PubMed] [Google Scholar]
- 52. Schjerling P., Holmberg S. (1996) Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res. 24, 4599–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luo S., Baumeister P., Yang S., Abcouwer S. F., Lee A. S. (2003) Induction of Grp78/BiP by translational block. Activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 278, 37375–37385 [DOI] [PubMed] [Google Scholar]
- 54. Reimold A. M., Iwakoshi N. N., Manis J., Vallabhajosyula P., Szomolanyi-Tsuda E., Gravallese E. M., Friend D., Grusby M. J., Alt F., Glimcher L. H. (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307 [DOI] [PubMed] [Google Scholar]
- 55. Huang H., Tindall D. J. (2007) Dynamic FoxO transcription factors. J. Cell Sci. 120, 2479–2487 [DOI] [PubMed] [Google Scholar]
- 56. Yanagisawa K., Konishi H., Arima C., Tomida S., Takeuchi T., Shimada Y., Yatabe Y., Mitsudomi T., Osada H., Takahashi T. (2010) Novel metastasis-related gene CIM functions in the regulation of multiple cellular stress-response pathways. Cancer Res. 70, 9949–9958 [DOI] [PubMed] [Google Scholar]
- 57. Kelley K. D., Miller K. R., Todd A., Kelley A. R., Tuttle R., Berberich S. J. (2010) YPEL3, a p53-regulated gene that induces cellular senescence. Cancer Res. 70, 3566–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Catalano R. D., Wilson M. R., Boddy S. C., McKinlay A. T., Sales K. J., Jabbour H. N. (2011) Hypoxia and prostaglandin E receptor 4 signalling pathways synergise to promote endometrial adenocarcinoma cell proliferation and tumour growth. PloS One 6, e19209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Erlander M. G., Tillakaratne N. J., Feldblum S., Patel N., Tobin A. J. (1991) Two genes encode distinct glutamate decarboxylases. Neuron 7, 91–100 [DOI] [PubMed] [Google Scholar]
- 60. Morimura N., Inoue T., Katayama K., Aruga J. (2006) Comparative analysis of structure, expression and PSD95-binding capacity of Lrfn, a novel family of neuronal transmembrane proteins. Gene 380, 72–83 [DOI] [PubMed] [Google Scholar]
- 61. Yan J., Aldrich R. W. (2012) BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 7917–7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nakanishi T., Kekuda R., Fei Y. J., Hatanaka T., Sugawara M., Martindale R. G., Leibach F. H., Prasad P. D., Ganapathy V. (2001) Cloning and functional characterization of a new subtype of the amino acid transport system N. Am. J. Physiol. Cell Physiol. 281, C1757–C1768 [DOI] [PubMed] [Google Scholar]
- 63. Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008) Physiology of the prion protein. Physiol. Rev. 88, 673–728 [DOI] [PubMed] [Google Scholar]
- 64. Budanov A. V., Lee J. H., Karin M. (2010) Stressin' sestrins take an aging fight. EMBO Mol. Med. 2, 388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Perraud A. L., Shen B., Dunn C. A., Rippe K., Smith M. K., Bessman M. J., Stoddard B. L., Scharenberg A. M. (2003) NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J. Biol. Chem. 278, 1794–1801 [DOI] [PubMed] [Google Scholar]
- 66. Takekawa M., Saito H. (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95, 521–530 [DOI] [PubMed] [Google Scholar]
- 67. Bauer M. F., Gempel K., Reichert A. S., Rappold G. A., Lichtner P., Gerbitz K. D., Neupert W., Brunner M., Hofmann S. (1999) Genetic and structural characterization of the human mitochondrial inner membrane translocase. J. Mol. Biol. 289, 69–82 [DOI] [PubMed] [Google Scholar]
- 68. Engelmann A., Speidel D., Bornkamm G. W., Deppert W., Stocking C. (2008) Gadd45β is a pro-survival factor associated with stress-resistant tumors. Oncogene 27, 1429–1438 [DOI] [PubMed] [Google Scholar]
- 69. Takekawa M., Tatebayashi K., Itoh F., Adachi M., Imai K., Saito H. (2002) Smad-dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF-β. EMBO J. 21, 6473–6482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Moro F., Sirrenberg C., Schneider H. C., Neupert W., Brunner M. (1999) The TIM17.23 preprotein translocase of mitochondria: composition and function in protein transport into the matrix. EMBO J. 18, 3667–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Budanov A. V. (2011) Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 15, 1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vincent B., Sunyach C., Orzechowski H. D., St George-Hyslop P., Checler F. (2009) p53-Dependent transcriptional control of cellular prion by presenilins. J. Neurosci. 29, 6752–6760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wong B. S., Liu T., Li R., Pan T., Petersen R. B., Smith M. A., Gambetti P., Perry G., Manson J. C., Brown D. R., Sy M. S. (2001) Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J. Neurochem. 76, 565–572 [DOI] [PubMed] [Google Scholar]
- 74. Shyu W. C., Lin S. Z., Saeki K., Kubosaki A., Matsumoto Y., Onodera T., Chiang M. F., Thajeb P., Li H. (2004) Hyperbaric oxygen enhances the expression of prion protein and heat shock protein 70 in a mouse neuroblastoma cell line. Cell. Mol. Neurobiol. 24, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]