

Background: The mechanism by which CX50fs, a mutant connexin50 containing a frameshift after amino acid 255, causes cataracts is unknown.
Results: CX50fs was unstable in HeLa cells, but epoxomicin treatment restored gap junction abundance and intercellular communication.
Conclusion: CX50fs undergoes enhanced endoplasmic reticulum-associated proteasomal degradation leading to decreased function.
Significance: The results suggest protease inhibition as a novel therapeutic approach to treat connexin mutant-associated diseases.
Keywords: Cataract, Cell Junctions, Connexin, Gap Junctions, Proteasome, Intercellular Communication
Abstract
The mechanisms by which mutant connexins lead to disease are diverse, including those of connexin50 (CX50) encoded by the GJA8 gene. We investigated the cellular and functional behavior of CX50fs, a mutant CX50 that has a frameshift after amino acid 255 and causes recessive congenital cataracts. Cellular levels of CX50fs were much lower than those of wild type CX50 in stably transfected HeLa cells. Whereas CX50 localized at distinct gap junction plaques and supported extensive intercellular transfer of Neurobiotin, CX50fs gap junctions were rare, and their support of Neurobiotin transfer was reduced by >90%. After inhibition of new protein synthesis with cycloheximide, CX50fs disappeared much more rapidly than CX50, suggesting increased degradation of the mutant. Treatment of cells with epoxomicin (a proteasomal inhibitor) led to a dramatic increase in CX50fs levels and in the abundance of gap junctions. Epoxomicin treatment also rescued intercellular transfer of Neurobiotin to levels similar to those in cells expressing the wild type protein. Treatment with eeyarestatin I (an inhibitor of p97-dependent protein degradation) resulted in many abundant slowly migrating CX50 and CX50fs bands consistent with polyubiquitination of the proteins. These results demonstrate that the CX50fs mutant is rapidly degraded by endoplasmic reticulum-associated degradation in mammalian cells. This accelerated degradation reduces the abundance of gap junctions and the extent of intercellular communication, potentially explaining the pathogenesis of cataracts linked to this mutant. The efficacy of epoxomicin in restoring function suggests that protease inhibition might have therapeutic value for this and other diseases caused by mutants with similar defects.
Introduction
Connexins (CXs)2 are members of a family of transmembrane proteins that oligomerize to form gap junction channels and hemichannels. Gap junction channels allow direct transfer of cytoplasmic ions and molecules between adjacent cells, and hemichannels allow transfer of ions and molecules between the cytoplasm and the extracellular milieu (for review, see Ref. 1). Identification of connexin mutants has linked alterations of connexins and their functions to human diseases including demyelinating neuropathies, oculodentodigital dysplasia, skin disorders, deafness, arrhythmias, and cataracts (2–9).
A number of the connexin mutants have been studied by in vitro expression to characterize abnormalities that might explain their contributions to pathogenesis. Many mutants have reduced or absent channel function or form channels/hemichannels with altered gating or permeability (2, 3, 5–9). Many mutants show alterations in their cellular behaviors including impaired trafficking or degradation that can lead to accumulation of the abnormal protein (2, 3, 5–9). Several of the CX46 and CX50 mutants linked to cataracts have been thoroughly studied (10–17). Most of these mutants are associated with congenital cataracts that are inherited as autosomal dominant traits.
In this study, we characterized the behavior of a mutant CX50 associated with a triangular cataract that is inherited recessively (18). In this mutant, the insertion of a G in the GJA8 gene that encodes CX50 leads to a translational frameshift that changes the reading frame after amino acid 255. The altered CX50 polypeptide contains 123 C-terminal amino acids (amino acids 256–378) that differ from the wild type CX50 sequence. Although Schmidt et al. (18) termed the mutant c776insG, we refer to the mutant protein as CX50fs for simplicity. Studies of this mutant could not be performed in lenses from affected individuals because they are very rare and the integrity of the lens is destroyed during surgical extraction. Moreover, epithelial cells lose their organelles during differentiation into fiber cells (which form the bulk of the lens) making studies on cultured lens fiber cells impossible. Therefore, we have used an exogenous system for cellular, biochemical, and physiological characterization of this mutant. The data presented suggest that CX50fs causes cataracts because of its instability.
EXPERIMENTAL PROCEDURES
Chemicals
All chemicals were obtained from Sigma unless otherwise specified.
Generation of CX50 Constructs
DNAs encoding CX50fs and CX50Tr (a mutant CX50 truncated after amino acid 255) were obtained by polymerase chain reaction using Phusion DNA polymerase (New England BioLabs), plasmid template containing wild type CX50 in pcDNA 3.1/Hygro(+) (Invitrogen) and oligonucleotides encoding the nucleotide insertion (CX50fs) or an early stop codon (CX50Tr) using the strategy published by Minogue et al. (13). The coding regions of all constructs were fully sequenced at the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility to ensure that PCR amplification did not introduce random mutations.
Cell Culture
HeLa cells were grown in minimum essential medium supplemented with nonessential amino acids, 10% fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin G, and 10 μg/ml streptomycin sulfate. Stably transfected cell clones expressing CX50fs or CX50Tr were obtained by selection in 400 μg/ml hygromycin. For HeLa-CX50 cells, the growth medium was supplemented with 1 mg/ml Geneticin (Invitrogen) similar to Lichtenstein et al. (15).
Cell Treatments
Cells were grown to approximately 80% confluence. Then, the culture growth medium was replaced with normal growth medium alone or normal growth medium containing 2 μm epoxomicin or 100 μm chloroquine and incubated for 4 h. To study the temporal course of disappearance of CX50 and CX50fs, cells were treated with 40 μg/ml cycloheximide for 4, 6, 8, and 18 h. To study the participation of endoplasmic reticulum-associated degradation (ERAD) in the degradation of CX50fs, the culture growth medium was replaced with normal growth medium containing DMSO (used as a solvent for eeyarestatin I) or 10 μm eeyarestatin I (EMD Millipore) and incubated for 8 h.
Generation of Affinity-purified Anti-CX50IL Antibodies
Anti-CX50IL antibodies were generated in rabbits using a unique amino acid sequence from the intracellular loop of human CX50 (amino acids 113–132) coupled to keyhole limpet hemocyanin as immunogen. Their ability to recognize the immunogenic peptide and their titer were determined by ELISA. Anti-CX50IL antibodies were subsequently affinity purified by YenZym Antibodies (South San Francisco, CA) using an immunogenic peptide-conjugated affinity matrix.
Immunofluorescence
Cells cultured on glass coverslips or on 4-well LAB-TEK Nalge Nunc International chamber slides (Thermo Fisher Scientific) were fixed in 4% paraformaldehyde in PBS for 15 min and subjected to immunofluorescence as described previously (12) using rabbit polyclonal anti-CX50IL antibodies and Cy3-conjugated goat anti-rabbit IgG antibodies (Jackson ImmunoResearch). Specimens were studied with a Zeiss Plan Apochromat 40× objective (n.a., 1.0) in an Axioplan 2 microscope (Carl Zeiss) equipped with a mercury lamp. Images were acquired with a Zeiss AxioCam digital camera using Zeiss AxioVision software. Figures were assembled using Photoshop CS3 Extended (Adobe Systems).
Immunoblotting
Cells at 80–90% confluence cultured on 100-mm plastic dishes were harvested in PBS, 4 mm EDTA, 2 mm PMSF, and cOmplete EDTA-free protease inhibitor mixture (Roche Applied Science) and centrifuged at 13,000 × g for 5 min. Pellets were resuspended in harvesting buffer and sonicated. Aliquots from cell homogenates containing 20 μg of protein were subjected to immunoblotting as described previously (16) using rabbit polyclonal anti-CX50IL antibodies and peroxidase-conjugated goat anti-rabbit IgG antibodies. Binding of the secondary antibodies was detected using ECL (GE Healthcare).
Assessment of Intercellular Communication
Cells were plated in 100-mm dishes containing glass coverslips and allowed to grow to approximately 90% confluence. Then, the coverslips were transferred to a dish containing F12 medium buffered with 15 mm HEPES, pH 7.4. Cells were microinjected for 1 min with a solution containing 5% Lucifer yellow and 9% Neurobiotin (Vector Laboratories) or 1 mg/ml propidium iodide (Sigma) using a picospritzer (model PLI-188; Nikon Instruments Inc., Melville, NY). The gap junction tracers were allowed to transfer for at least 5 min before counting the number of cells to which propidium transferred.
To visualize Neurobiotin transfer, 5 min after microinjection of the gap junction tracers, cells were fixed in 4% formaldehyde for 15 min and rinsed three times in PBS. Fixed cells were incubated in 10% normal goat serum, 1% Triton X-100 in PBS, pH 7.4 (blocking solution) for 1 h. Then, the cells were incubated for 45 min in Cy3-streptavidin conjugate diluted in blocking solution followed by six 7-min rinses in PBS, pH 7.4. The extent of intercellular transfer was determined by counting the number of cells surrounding the microinjected cell that contained Neurobiotin. Because CX50 channels allow very limited transfer of Lucifer yellow, co-injection of this tracer was used to facilitate identification of the microinjected cell. Results are reported as means ± S.E. Statistical analysis was performed using Student's t test.
RESULTS
The Cataract-associated Mutant, CX50fs, Forms Small and Infrequent Gap Junction Plaques
To compare the behavior of CX50fs with that of wild type CX50, we stably transfected HeLa cells with DNA constructs encoding these proteins (HeLa-CX50 and HeLa-CX50fs). The wild type and mutant CX50 were detected using a newly developed antibody raised against the intracellular loop of CX50 because this region is identical in both proteins. A major immunoreactive band was detected by immunoblotting in homogenates from HeLa-CX50 cells whereas no bands were detected in homogenates from untransfected cells. Immunoblots of homogenates from HeLa-CX50fs cells showed a band of faster electrophoretic mobility than wild type CX50 and occasionally, a much lower intensity band of a slower electrophoretic mobility than wild type CX50. Levels of immunoreactive CX50 were much lower in HeLa-CX50fs cells than in HeLa-CX50 cells (Fig. 1A).
FIGURE 1.
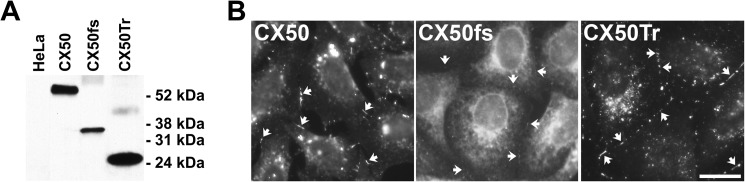
Levels and distribution of wild type and CX50 mutants in mammalian cells. A, homogenates of untransfected HeLa cells or HeLa cells stably transfected with CX50, CX50fs, or CX50Tr were subjected to immunoblotting using anti-CX50IL antibodies. The positions of the molecular mass standards are indicated. B, photomicrographs show the distribution of immunoreactive CX50, CX50fs, and CX50Tr in stably transfected HeLa cells. In this composite image, exposure times and brightness were optimized to allow visualization of gap junctions plaques (arrows). Scale bar, 21 μm.
To test whether the reduction in CX50fs levels resulted from the absence of the last 178 amino acids of CX50, we expressed a mutant CX50 truncated after amino acid 255 where the frameshift occurs in CX50fs (CX50Tr). In HeLa cells transfected with CX50Tr (HeLa-CX50Tr), levels of immunoreactive CX50 were not reduced; rather, they were higher than in HeLa-CX50 cells (Fig. 1A). These results suggest that the presence of the abnormal C-terminal sequence in the mutant was responsible for the decreased levels of CX50fs (and not the absence of amino acids 256–433).
We also assessed the distribution of CX50 in these cells. CX50fs rarely formed gap junction plaques, and they were barely detectable (Fig. 1B). In contrast, CX50Tr localized in gap junctional plaques that were more abundant and as large as (if not larger than) those found between cells expressing wild type CX50 (Fig. 1B). No immunoreactive CX50 was detected in untransfected cells (data not shown).
HeLa-CX50fs Cells Show Very Low Levels of Intercellular Communication
We assessed the abilities of CX50fs and CX50Tr to support intercellular transfer of Neurobiotin and compared them with that of wild type CX50. Neurobiotin transferred to very few or no untransfected HeLa cells (0.8 ± 0.2), whereas in HeLa-CX50 cells it transferred to 119 ± 5.7 neighboring cells (Fig. 2). In contrast, CX50fs supported Neurobiotin transfer to 10.9 ± 0.9 cells, a number that was significantly smaller than that between HeLa-CX50 cells but higher than that between untransfected cells (Fig. 2). Cells expressing CX50Tr transferred Neurobiotin to 295.8 ± 11.2 neighboring cells, a number that was significantly higher than that of HeLa-CX50 cells (Fig. 2).
FIGURE 2.

Cells transfected with CX50fs showed decreased intercellular transfer of Neurobiotin. A, photomicrographs show intercellular transfer of Neurobiotin from the microinjected cell (*) to the neighboring cells in untransfected HeLa cells or in HeLa cells stably transfected with CX50, CX50fs, or CX50Tr. Scale bar, 161 μm. B, graph shows the quantification of these experiments. The results are presented as mean + S.E. (error bars; p < 10−8 for CX50 versus CX50fs or CX50Tr; p < 10−8 for untransfected versus any of the transfected cells).
We also observed decreased intercellular communication between cells transfected with CX50fs when using propidium iodide as the gap junction tracer. Propidium transferred to 0.8 ± 0.4 untransfected cells, 5.7 ± 0.3 HeLa-CX50 cells, and 1.3 ± 0.4 HeLa-CX50fs cells. The extent of propidium transfer between HeLa-CX50fs cells was significantly lower than that between cells expressing wild type CX50 (p < 10−6), but not significantly different from that between untransfected cells.
Degradation of CX50fs Is Increased
The reduced levels of CX50fs suggested faster degradation of the mutant protein than of wild type CX50 in HeLa cells. Therefore, we treated HeLa-CX50 and HeLa-CX50fs with 40 μg/ml cycloheximide to inhibit de novo protein synthesis and assessed CX50 levels after different treatment durations. Levels of CX50fs were almost undetectable after 4 h of treatment whereas wild type CX50 was still detected even after 18 h of treatment (Fig. 3).
FIGURE 3.

After inhibition of de novo protein synthesis, CX50fs disappeared much more rapidly than wild type CX50. A, HeLa cells stably transfected with CX50 or CX50fs were treated with 40 μg/ml cycloheximide for 0–18 h as indicated. Then, cells were harvested and homogenized. Homogenate aliquots containing 20 μg of protein were subjected to immunoblotting using anti-CX50IL antibodies. B, graph shows the quantification of the results obtained in three independent experiments performed as described in A. The relative levels of CX50 (filled circles) and Cx50fs (open circles) were quantified by densitometry and calculated as a percentage of the densitometric values obtained at time = 0. The curves (CX50, solid line; CX50fs, dotted line) correspond to the best fit for a single exponential.
To examine degradation pathways that might be responsible for the rapid disappearance of CX50fs, we treated HeLa-CX50fs cells with an inhibitor of the lysosome (chloroquine) or the proteasome (epoxomicin) and compared immunoblot results with those obtained from HeLa-CX50 cells. Treatment of stably transfected cells with chloroquine led to a modest increase in levels of wild type CX50 (on average, 32%; Fig. 4), but did not increase levels of CX50fs (on average a 30% decrease; Fig. 4). In contrast, treatment with epoxomicin significantly increased levels of CX50fs (on average, 6.4 times the values in control DMSO-treated cells; Fig. 4); this is likely an underestimation, because the exposure time required to detect the CX50fs band in control DMSO-treated cells resulted in overexposure of the band in epoxomicin-treated HeLa-CX50fs cells. Epoxomicin treatment had little effect on levels of wild type CX50 (on average, a 9% decrease; Fig. 4).
FIGURE 4.

Epoxomicin dramatically increased levels of CX50fs. A, immunoblot shows the levels of immunoreactive CX50 in homogenates from HeLa-CX50 and HeLa-CX50fs cells left untreated or treated with DMSO (solvent for epoxomicin), 100 μm chloroquine, or 2 μm epoxomicin for 4 h. B, graph shows the quantification of the results obtained in three independent experiments performed as described in A. For each condition, the levels of CX50 and CX50fs were quantified by densitometry. For chloroquine, the -fold change in levels was calculated as the ratio between the densitometric values obtained after treatment with chloroquine and those obtained in Control. For epoxomicin, the -fold change in levels was calculated as the ratio between the densitometric values obtained after treatment with epoxomicin and those obtained in DMSO.
Proteasomal Inhibition Increases the Abundance of Gap Junctional Plaques and Rescues Intercellular Communication in HeLa-CX50fs Cells
Because of its effects on CX50fs levels (likely abolishing its accelerated degradation), we hypothesized that treatment with a proteasome inhibitor might also restore gap junction plaque formation and function in cells expressing this mutant. We treated HeLa-CX50 and HeLa-CX50fs cells with 2 μm epoxomicin for 4 h and evaluated the cellular distribution of immunoreactive CX50. This treatment did not dramatically affect the frequency or size of gap junctions between cells expressing wild type CX50, although some variability in the amount of intracellular staining was observed (Fig. 5A). We observed on average 0.69 interfaces with immunopositive gap junction plaques per cell in control (DMSO-treated) cultures versus 0.79 immunoreactive interfaces per cell in epoxomicin-treated cultures of HeLa-CX50 cells (Fig. 5B). In contrast, epoxomicin treatment greatly increased cytoplasmic immunoreactivity and the abundance of gap junction plaques in HeLa-CX50fs cells such that obvious plaques were observed between nearly all cells (Fig. 5A). This difference was clearly reflected in the number of interfaces containing immunoreactive gap junction plaques per cell; this value increased from an average of <0.02 in control HeLa-CX50fs cells to an average of 0.52 in epoxomicin-treated cells (Fig. 5B).
FIGURE 5.

Treatment of HeLa-CX50fs cells with epoxomicin increased gap junction plaques and rescued intercellular communication. A, photomicrographs show the distribution of CX50 and CX50fs in cells that were treated with DMSO (Control) or with 2 μm epoxomicin for 4 h. Arrows point to gap junction plaques between untreated HeLa-CX50fs cells. For either HeLa-CX50 or HeLa-CX50fs cells, the images of DMSO-treated and epoxomicin-treated cells were obtained with the same exposure time. Scale bar, 34 μm. B, the vertical point graph shows the quantification of the results presented in A. For each treatment, the total number of cell interfaces with immunopositive gap junction plaques and the total number of cells were counted in 10 different fields of view (containing confluent, but well spread cells). The results are presented as the number of immunoreactive interfaces per cell. The average for each treatment is indicated by the short horizontal line. C, the bar graph shows the quantification of intercellular transfer of Neurobiotin in HeLa-CX50 and HeLa-CX50fs cells after DMSO (−) or epoxomicin (+) treatment. The results are presented as mean + S.E. (error bars; p < 10−5 for untreated CX50fs versus CX50 or epoxomicin-treated CX50fs).
To assess the effects of proteasome inhibition on gap junction function, we also examined Neurobiotin transfer between HeLa-CX50 and HeLa-CX50fs cells under control conditions and after treatment with 2 μm epoxomicin for 4 h. Epoxomicin treatment did not significantly affect the extent of intercellular communication among HeLa-CX50 cells (Fig. 5C). In contrast, treatment of HeLa-CX50fs with epoxomicin resulted in a very large and significant increase (nearly 9-fold) in the number of Neurobiotin-coupled neighbors. Indeed, the extent of coupling of HeLa-CX50fs cells under these conditions was not significantly different from that determined in HeLa cells expressing the wild type connexin (Fig. 5B).
Inhibition of p97-dependent Protein Degradation Increases Levels, but Not the Abundance of CX50fs Gap Junction Plaques
The fast degradation of CX50fs and its dependence on the activity of the proteasome suggested that CX50fs was being degraded by ERAD. To test the role of ERAD in the degradation of CX50fs compared with that of wild type CX50, we treated HeLa-CX50 and HeLa-CX50fs cells with eeyarestatin I, a known ERAD inhibitor that targets p97, a central element in ubiquitin-dependent processes, leading to accumulation of polyubiquitinated proteins (19, 20). Treatment of these cells with eeyarestatin I changed the banding pattern and total intensity of immunoreactive CX50 (Fig. 6A). In HeLa-CX50 cells, a fraction of the protein was detected at the same electrophoretic mobility as in control (DMSO-treated) cells, but most CX50 was detected at much higher apparent molecular masses. In HeLa-CX50fs cells treated with eeyarestatin I, CX50 immunoreactivity was not detected at the position corresponding to the CX50fs band identified in control cells; rather, CX50fs was detected as a ladder of bands of higher apparent molecular masses, the smallest of which corresponded to the band occasionally detected in immunoblots of untreated HeLa-CX50fs cells. The eeyarestatin I-induced increase of total Cx50 immunoreactivity (compared with DMSO-treated controls) was much more pronounced for CX50fs than for wild type CX50 cells (on average, 11.2-fold in HeLa-CX50fs compared with 3.5-fold in HeLa-CX50; n = 3) (Fig. 6B).
FIGURE 6.

Treatment with a p97 inhibitor led to accumulation of slower migrating forms of CX50 and CX50fs. A, immunoblot shows the levels of immunoreactive CX50 in homogenates from HeLa-CX50 and HeLa-CX50fs cells treated with DMSO (solvent for eeyarestatin I) or treated with 10 μm eeyarestatin I for 8 h. Note that the CX50fs band appears almost as intense as that of wild type CX50 in the Control samples, because the CX50fs data come from a different blot with a longer exposure time of the film to chemiluminescence than the data for wild type CX50. B, graph shows the quantification of the results obtained in three independent experiments performed as described in A. For each condition, the levels of CX50 and CX50fs were quantified by densitometry. The -fold change in levels was calculated as the ratio between the densitometric values obtained after treatment with eeyarestatin I and those obtained in homogenates from DMSO-treated cultures (Control). C, photomicrographs show the distribution of CX50 and CX50fs in cells that were treated with DMSO (Control) or with 10 μm eeyarestatin I for 8 h. For either HeLa-CX50 or HeLa-CX50fs cells, the images of DMSO-treated and eeyarestatin I-treated cells were acquired with the same exposure time. The arrow points to a gap junction plaque between Control HeLa-CX50fs cells. Scale bar, 8.3 μm.
We also assessed the distribution of the proteins in cells treated with eeyarestatin I. With the exception of the perinuclear CX50 immunostaining which was not apparent in treated cells, wild type CX50 localized in gap junction plaques and in bright intracellular structures in eeyarestatin I-treated HeLa-CX50 cells similar to its distribution in DMSO-treated control cells (Fig. 6C). Treatment of HeLa-CX50fs cells with eeyarestatin I led to a major increase in the intensity of intracellular immunoreactivity with no obvious changes in CX50fs distribution compared with DMSO-treated control cells (Fig. 6C).
DISCUSSION
In this paper, we have elucidated defects of the cataract-linked GJA8 mutant, c776insG, which encodes an abnormal lens connexin that we have termed CX50fs. When stably expressed in HeLa cells, CX50fs has a severely reduced ability to support intercellular communication compared with wild type CX50. This defect is not due to the absence of amino acids 256–433 from the C terminus of CX50fs, because a truncated form of CX50 lacking these amino acids supports transfer of Neurobiotin even better than wild type CX50. Rather, the reduced function of CX50fs must result from the abnormal amino acids that follow the frameshift.
Our data regarding the truncated form of CX50 complement previous studies and emphasize the complexities of defining the roles of amino acid domains in the C terminus of CX50 for gap junction plaque formation and function. Previous studies have shown that mouse CX50 truncated at amino acid 290 (corresponding to amino acid 283 of human CX50) forms gap junctions plaques (21); however, when three FLAG epitopes are appended to its C terminus, it localizes in intracellular membranes (22). A shorter truncated form of mouse CX50 lacking amino acids 246–440 containing the tandem of FLAG epitopes localized in sparse gap junction plaques (22). The reported effects of partial deletion of the C terminus on gap junction function are diverse. Mouse, ovine, or human CX50 carrying deletions from the C terminus of different lengths and up to a site corresponding to amino acid 284 of human CX50 (291 in mouse and ovine CX50) supported little (or no) macroscopic gap junctional conductance and no intercellular transfer of Neurobiotin (21–25), but ovine CX50 truncated at amino acid 290 induced similar macroscopic junctional conductance to the full-length protein in Xenopus oocytes (26). The Δ246–440 mouse CX50 truncation (containing a longer deletion) supported a reduced transfer of Neurobiotin (22); although no data on gap junctional conductance for this truncated form are available, based on the size difference it is likely that gap junctional (ionic) conductance will be decreased to a lesser extent than the transfer of Neurobiotin. By sequence alignment, the C-terminal domain of CX50Tr (which formed abundant gap junction plaques and showed increased transfer of Neurobiotin in our study) is longer than that of Δ246–440 mouse CX50, but shorter than that of mouse CX50 truncated at amino acid 290. Thus, our data on CX50Tr suggest that amino acids 256–433 of human CX50 are dispensable for protein stability, formation of gap junctions, and gap junction-mediated intercellular communication. These results raise the possibility that truncation of CX50 after isoleucine 255 may have exposed a normally hidden domain in the C terminus of CX50 or generated a novel binding site for a scaffolding protein (e.g. ZO-1) that allowed formation of gap junctional plaques in the absence of the ZO-1 binding sequence identified by Chai et al. (22) in mouse CX50. Because studying scaffolding-interacting domains in a truncated form of CX50 generated by site-directed mutagenesis is tangential to the main purpose of this report, these studies were not pursued further.
Our results demonstrate that CX50fs is very unstable compared with wild type CX50 based on its rapid disappearance after blocking de novo protein synthesis with cycloheximide. This conclusion is further supported by the large increase in CX50fs levels observed after inhibiting the proteasome with epoxomicin. Proteasomal degradation at the level of the ER has been described for some CX32 mutants associated with X-linked Charcot-Marie-Tooth disease (27). Many proteasomal substrates are targeted for degradation by modification with ubiquitin chains through a series of enzymatic reactions (28); p97 is involved in their dislocation from the ER before they can be degraded (20). The p97 inhibitor eeyarestatin I-induced accumulation of CX50fs modified forms (that are likely polyubiquitinated) and the major increase in CX50fs immunoreactivity (in a somewhat reticular ER-like pattern) suggest that the protein is polyubiquitinated and targeted for proteasomal degradation before reaching the plasma membrane (and forming gap junction plaques). Taken together, these results imply that most (if not all) of the accelerated degradation of CX50fs occurs via proteasome-dependent ERAD.
In contrast to the CX50 mutant, levels of wild type CX50 were not affected by inhibition of the proteasome; they were increased by inhibition of the lysosome. The eeyarestatin I-induced accumulation of unmodified and modified (most likely by ubiquitin) forms of CX50 implicates the participation of p97 in the degradation of this connexin. Ubiquitination of chicken CX50 has been demonstrated (29). Because p97 is involved in autophagy (a degradation pathway that depends on the activity of lysosomal enzymes), and CX50 is a substrate of autophagy (30), these results imply that CX50 is subject to post-translational modification before its degradation by the lysosome.
Similar to the CX50fs levels, the abundance of gap junctional plaques was greatly increased when proteasomal degradation was blocked by epoxomicin treatment. An increase in the abundance of gap junctions formed of other connexins (e.g. CX43) has previously been observed in response to proteasomal inhibition (31–35). The increase in CX43 gap junctional plaques induced by MG132 treatment has been ascribed to proteasome inhibition-induced Akt activation (35). However, Akt is unlikely to have a substantial participation in the mechanism by which proteasome inhibitors exert their effects on CX50fs because we still observed a major increase in CX50fs plaques when Akt was inhibited prior to epoxomicin treatment (data not shown). The most reasonable conclusion is that the effects induced by proteasome inhibition on CX50fs result from blockage of ERAD.
Most dramatically, epoxomicin treatment restored intercellular transfer of Neurobiotin in cells expressing CX50fs to an extent similar to that in cells expressing wild type CX50. Although involvement of the proteasome in the degradation of wild type and mutant connexins has been reported previously (27, 31–34, 36–39), only the studies by Musil et al. of CHO cells (31) and Girǎo and Pereira using bovine lens epithelial cells (32) that express wild type Cx43 have reported increased gap junctional communication following proteasomal inhibition. Although Musil et al. (31) attributed the increase to an improvement in the inefficient capacity of CHO cells to assemble gap junctions, a similar hypothesis cannot explain our results, because HeLa cells efficiently assemble wild type connexins (and some mutants) into gap junction plaques. Thus, our results imply that the reduced intercellular communication between HeLa cells expressing CX50fs was due to the instability of the mutant protein and that the aberrant sequence had minimal (if any) effect on gap junction function.
It is a reasonable extrapolation that ERAD of CX50fs can explain the development of cataracts in individuals carrying this mutation. Components of the ubiquitin-proteasome system are present, and the function of this system has been documented in the lens (40, 41), where CX50 is normally expressed. Therefore, it is likely that most of this CX50 mutant would be rapidly degraded by ERAD in vivo. Considering the results of our study, it would be expected that the CX50fs (c776insG) allele behaves like a CX50-null allele, explaining the recessive inheritance pattern of cataracts associated with this CX50 mutant. Thus, individuals that are heterozygous for this mutation should have approximately 50% of normal CX50 function, and like mice that are heterozygous for the CX50 knock-out, they should have normal lenses (42, 43). This appears to be true, because visual acuity was unaffected in adult individuals carrying one mutant allele; they only had discrete, symmetric opacities in the fetal lens nucleus (18). In contrast, individuals that are homozygous for the mutant allele should have almost no CX50 gap junctions or function leading to cataracts as in the homozygous null mice.
The remarkable rescue of CX50fs gap junction function by epoxomicin suggests that a proteasomal inhibitor could be used to ameliorate or prevent the formation of cataracts. Proteasomal inhibitors are currently undergoing clinical trials or being incorporated into therapeutic protocols for people (44). In fact, one drug of this class, bortezomib, is being used for treatment of some hematologic malignancies. Proteasomal inhibitors have also been reported to rescue targeting to the plasma membrane and function of some proteasomally degraded mutants associated with other diseases (45–48). Other inhibitors of the proteasome that may have reduced secondary effects (49, 50) are being developed, and they may eventually allow chronic application to prevent or ameliorate disease (like cataracts). Indeed, inhibition of other proteases may also have benefits for lens disease due to connexin abnormalities, because the formation of nuclear cataracts in lenses from CX46 knock-out mice can be blocked by in vitro incubation of the lenses with the irreversible cysteine protease inhibitor, E-64 (51).
In summary, we report a frameshift mutant of CX50 that is unstable but can be rescued by treatment with a proteasomal inhibitor. These results suggest a novel therapeutic approach to treat some forms of cataract. They also suggest that the use of proteasomal inhibitors might be beneficial in the treatment of other diseases caused by connexin mutants with similar defects.
Acknowledgment
We thank Wenji Guo for participation in the initial characterization of this mutant.
This work was supported by National Institutes of Health Grant R01EY08368.
- CX
- connexin
- CX50fs
- frameshift mutant CX50
- CX50IL
- intracellular loop of CX50
- CX50Tr
- truncated form of CX50 containing the first 255 amino acids of wild type CX50
- DMSO
- dimethyl sulfoxide
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation.
REFERENCES
- 1. Sáez J. C., Berthoud V. M., Braňes M. C., Martínez A. D., Beyer E. C. (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400 [DOI] [PubMed] [Google Scholar]
- 2. Gollob M. H., Jones D. L., Krahn A. D., Danis L., Gong X. Q., Shao Q., Liu X., Veinot J. P., Tang A. S., Stewart A. F., Tesson F., Klein G. J., Yee R., Skanes A. C., Guiraudon G. M., Ebihara L., Bai D. (2006) Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688 [DOI] [PubMed] [Google Scholar]
- 3. Laird D. W. (2006) Life cycle of connexins in health and disease. Biochem. J. 394, 527–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hejtmancik J. F. (2008) Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 19, 134–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paznekas W. A., Karczeski B., Vermeer S., Lowry R. B., Delatycki M., Laurence F., Koivisto P. A., Van Maldergem L., Boyadjiev S. A., Bodurtha J. N., Jabs E. W. (2009) GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum. Mutat. 30, 724–733 [DOI] [PubMed] [Google Scholar]
- 6. Thibodeau I. L., Xu J., Li Q., Liu G., Lam K., Veinot J. P., Birnie D. H., Jones D. L., Krahn A. D., Lemery R., Nicholson B. J., Gollob M. H. (2010) Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 122, 236–244 [DOI] [PubMed] [Google Scholar]
- 7. Abrams C. K., Scherer S. S. (2012) Gap junctions in inherited human disorders of the central nervous system. Biochim. Biophys. Acta 1818, 2030–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pfenniger A., Wohlwend A., Kwak B. R. (2011) Mutations in connexin genes and disease. Eur. J. Clin. Invest. 41, 103–116 [DOI] [PubMed] [Google Scholar]
- 9. Scott C. A., Kelsell D. P. (2011) Key functions for gap junctions in skin and hearing. Biochem. J. 438, 245–254 [DOI] [PubMed] [Google Scholar]
- 10. Pal J. D., Berthoud V. M., Beyer E. C., Mackay D., Shiels A., Ebihara L. (1999) Molecular mechanism underlying a Cx50-linked congenital cataract. Am. J. Physiol. 276, C1443–C1446 [DOI] [PubMed] [Google Scholar]
- 11. Pal J. D., Liu X., Mackay D., Shiels A., Berthoud V. M., Beyer E. C., Ebihara L. (2000) Connexin46 mutations linked to congenital cataract show loss of gap junction channel function. Am. J. Physiol. Cell Physiol. 279, C596–C602 [DOI] [PubMed] [Google Scholar]
- 12. Berthoud V. M., Minogue P. J., Guo J., Williamson E. K., Xu X., Ebihara L., Beyer E. C. (2003) Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur. J. Cell Biol. 82, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Minogue P. J., Liu X., Ebihara L., Beyer E. C., Berthoud V. M. (2005) An aberrant sequence in a connexin46 mutant underlies congenital cataracts. J. Biol. Chem. 280, 40788–40795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeRosa A. M., Xia C. H., Gong X., White T. W. (2007) The cataract-inducing S50P mutation in Cx50 dominantly alters the channel gating of wild-type lens connexins. J. Cell Sci. 120, 4107–4116 [DOI] [PubMed] [Google Scholar]
- 15. Lichtenstein A., Gaietta G. M., Deerinck T. J., Crum J., Sosinsky G. E., Beyer E. C., Berthoud V. M. (2009) The cytoplasmic accumulations of the cataract-associated mutant, Connexin50P88S, are long-lived and form in the endoplasmic reticulum. Exp. Eye Res. 88, 600–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minogue P. J., Tong J. J., Arora A., Russell-Eggitt I., Hunt D. M., Moore A. T., Ebihara L., Beyer E. C., Berthoud V. M. (2009) A mutant connexin50 with enhanced hemichannel function leads to cell death. Invest. Ophthalmol. Vis. Sci. 50, 5837–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tong J. J., Minogue P. J., Guo W., Chen T. L., Beyer E. C., Berthoud V. M., Ebihara L. (2011) Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am. J. Physiol. Cell Physiol. 300, C1055–C1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmidt W., Klopp N., Illig T., Graw J. (2008) A novel GJA8 mutation causing recessive triangular cataract. Mol. Vis. 14, 851–856 [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Q., Li L., Ye Y. (2008) Inhibition of p97-dependent protein degradation by eeyarestatin I. J. Biol. Chem. 283, 7445–7454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meyer H., Bug M., Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 21. DeRosa A. M., Mui R., Srinivas M., White T. W. (2006) Functional characterization of a naturally occurring Cx50 truncation. Invest. Ophthalmol. Vis. Sci. 47, 4474–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chai Z., Goodenough D. A., Paul D. L. (2011) Cx50 requires an intact PDZ-binding motif and ZO-1 for the formation of functional intercellular channels. Mol. Biol. Cell 22, 4503–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stergiopoulos K., Alvarado J. L., Mastroianni M., Ek-Vitorin J. F., Taffet S. M., Delmar M. (1999) Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ. Res. 84, 1144–1155 [DOI] [PubMed] [Google Scholar]
- 24. Eckert R. (2002) pH gating of lens fibre connexins. Pflügers Archiv. 443, 843–851 [DOI] [PubMed] [Google Scholar]
- 25. Xu X., Berthoud V. M., Beyer E. C., Ebihara L. (2002) Functional role of the carboxyl terminal domain of human connexin 50 in gap junctional channels. J. Membr. Biol. 186, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin J. S., Eckert R., Kistler J., Donaldson P. (1998) Spatial differences in gap junction gating in the lens are a consequence of connexin cleavage. Eur. J. Cell Biol. 76, 246–250 [DOI] [PubMed] [Google Scholar]
- 27. VanSlyke J. K., Deschenes S. M., Musil L. S. (2000) Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol. Biol. Cell 11, 1933–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kravtsova-Ivantsiv Y., Ciechanover A. (2012) Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 125, 539–548 [DOI] [PubMed] [Google Scholar]
- 29. Yin X., Liu J., Jiang J. X. (2008) Lens fiber connexin turnover and caspase-3-mediated cleavage are regulated alternately by phosphorylation. Cell Commun. Adhes. 15, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lichtenstein A., Minogue P. J., Beyer E. C., Berthoud V. M. (2011) Autophagy: a pathway that contributes to connexin degradation. J. Cell Sci. 124, 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Musil L. S., Le A. C., VanSlyke J. K., Roberts L. M. (2000) Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J. Biol. Chem. 275, 25207–25215 [DOI] [PubMed] [Google Scholar]
- 32. Girǎo H., Pereira P. (2003) Phosphorylation of connexin 43 acts as a stimulus for proteasome-dependent degradation of the protein in lens epithelial cells. Mol. Vis. 9, 24–30 [PubMed] [Google Scholar]
- 33. Thomas M. A., Zosso N., Scerri I., Demaurex N., Chanson M., Staub O. (2003) A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J. Cell Sci. 116, 2213–2222 [DOI] [PubMed] [Google Scholar]
- 34. Qin H., Shao Q., Igdoura S. A., Alaoui-Jamali M. A., Laird D. W. (2003) Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J. Biol. Chem. 278, 30005–30014 [DOI] [PubMed] [Google Scholar]
- 35. Dunn C. A., Su V., Lau A. F., Lampe P. D. (2012) Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 287, 2600–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laing J. G., Beyer E. C. (1995) The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 270, 26399–26403 [DOI] [PubMed] [Google Scholar]
- 37. Laing J. G., Tadros P. N., Westphale E. M., Beyer E. C. (1997) Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 236, 482–492 [DOI] [PubMed] [Google Scholar]
- 38. Beardslee M. A., Laing J. G., Beyer E. C., Saffitz J. E. (1998) Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629–635 [DOI] [PubMed] [Google Scholar]
- 39. Laing J. G., Tadros P. N., Green K., Saffitz J. E., Beyer E. C. (1998) Proteolysis of connexin43-containing gap junctions in normal and heat-stressed cardiac myocytes. Cardiovasc. Res. 38, 711–718 [DOI] [PubMed] [Google Scholar]
- 40. Shang F., Gong X., Taylor A. (1997) Activity of ubiquitin-dependent pathway in response to oxidative stress: ubiquitin-activating enzyme is transiently up-regulated. J. Biol. Chem. 272, 23086–23093 [DOI] [PubMed] [Google Scholar]
- 41. Pereira P., Shang F., Hobbs M., Girão H., Taylor A. (2003) Lens fibers have a fully functional ubiquitin-proteasome pathway. Exp. Eye Res. 76, 623–631 [DOI] [PubMed] [Google Scholar]
- 42. White T. W., Goodenough D. A., Paul D. L. (1998) Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J. Cell Biol. 143, 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rong P., Wang X., Niesman I., Wu Y., Benedetti L. E., Dunia I., Levy E., Gong X. (2002) Disruption of Gja8 (α8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development 129, 167–174 [DOI] [PubMed] [Google Scholar]
- 44. Voorhees P. M., Dees E. C., O'Neil B., Orlowski R. Z. (2003) The proteasome as a target for cancer therapy. Clin. Cancer Res. 9, 6316–6325 [PubMed] [Google Scholar]
- 45. Danilov S. M., Kalinin S., Chen Z., Vinokour E. I., Nesterovitch A. B., Schwartz D. E., Gribouval O., Gubler M. C., Minshall R. D. (2010) Angiotensin I-converting enzyme Gln1069Arg mutation impairs trafficking to the cell surface resulting in selective denaturation of the C-domain. PLoS One 5, e10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deuquet J., Lausch E., Guex N., Abrami L., Salvi S., Lakkaraju A., Ramirez M. C., Martignetti J. A., Rokicki D., Bonafe L., Superti-Furga A., van der Goot F. G. (2011) Hyaline fibromatosis syndrome inducing mutations in the ectodomain of anthrax toxin receptor 2 can be rescued by proteasome inhibitors. EMBO Mol. Med. 3, 208–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lenk G. M., Ferguson C. J., Chow C. Y., Jin N., Jones J. M., Grant A. E., Zolov S. N., Winters J. J., Giger R. J., Dowling J. J., Weisman L. S., Meisler M. H. (2011) Pathogenic mechanism of FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet. 7, e1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilke M., Bot A., Jorna H., Scholte B. J., de Jonge H. R. (2012) Rescue of murine F508del CFTR activity in native intestine by low temperature and proteasome inhibitors. PLoS One 7, e52070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruschak A. M., Slassi M., Kay L. E., Schimmer A. D. (2011) Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 103, 1007–1017 [DOI] [PubMed] [Google Scholar]
- 50. Kaffy J., Bernadat G., Ongeri S. (2013) Non-covalent proteasome inhibitors. Curr. Pharm. Des. 19, 4115–4130 [DOI] [PubMed] [Google Scholar]
- 51. Baruch A., Greenbaum D., Levy E. T., Nielsen P. A., Gilula N. B., Kumar N. M., Bogyo M. (2001) Defining a link between gap junction communication, proteolysis, and cataract formation. J. Biol. Chem. 276, 28999–29006 [DOI] [PubMed] [Google Scholar]