

Background: Among the eight subunits of the COP9 signalosome (CSN), Csn8 is only present in higher eukaryotes.
Results: Decreases in Csn8 accelerate cell growth and shorten G1 duration, whereas decreases in Csn5 impair cell proliferation.
Conclusion: Although Csn5 promotes cell proliferation, Csn8 negatively regulates G1 progression.
Significance: The dynamics of CSN are an important factor influencing cell division and differentiation.
Keywords: Cell Biology, Cell Cycle, Cell Division, Cell Proliferation, Protein Complexes, COP9 Signalosome, CSN5 or Jab1, Csn8, G1 Phase
Abstract
The COP9 signalosome (CSN) is a conserved protein complex known to be involved in developmental processes of eukaryotic organisms. Genetic disruption of a CSN gene causes arrest during early embryonic development in mice. The Csn8 subunit is the smallest and the least conserved subunit, being absent from the CSN complex of several fungal species. Nevertheless, Csn8 is an integral component of the CSN complex in higher eukaryotes, where it is essential for life. By characterizing the mouse embryonic fibroblasts (MEFs) that express Csn8 at a low level, we found that Csn8 plays an important role in maintaining the proper duration of the G1 phase of the cell cycle. A decreased level of Csn8, either in Csn8 hypomorphic MEFs or following siRNA-mediated knockdown in HeLa cells, accelerated cell growth rate. Csn8 hypomorphic MEFs exhibited a shortened G1 duration and affected expression of G1 regulators. In contrast to Csn8, down-regulation of Csn5 impaired cell proliferation. Csn5 proteins were found both as a component of the CSN complex and outside of CSN (Csn5-f), and the amount of Csn5-f relative to CSN was increased in the Csn8 hypomorphic cells. We conclude that CSN harbors both positive and negative regulators of the cell cycle and therefore is poised to influence the fate of a cell at the crossroad of cell division, differentiation, and senescence.
Introduction
The COP9 signalosome (CSN)7 is a heteromeric protein complex consisting of eight subunits (Csn1 to Csn8) in higher eukaryotes (1, 2). Gene targeting studies in mice showed that CSN is involved in diverse developmental and physiological processes including but not limited to embryonic development, immune responses, cell cycle control, and cardiovascular functions (3–9). At the molecular level, CSN has been best characterized as a regulator of cullin-RING family of ubiquitin E3 ligases (CRLs). The CSN regulates CRL activities by at least three different means: by its isopeptidase activity that removes covalent modification of a ubiquitin-like protein, Nedd8, from the cullin proteins of the CRLs (also known as deneddylation) (10, 11); by recruiting a deubiquitinating enzyme (12, 13); and by nonenzymatic mechanisms (14–17). In addition to CRL E3s, CSN may have other targets as suggested from its association with kinase activities and its regulation of COP1, a RING-finger E3 (18–21).
Among the eight canonical components of CSN, Csn8 is the smallest and the least conserved subunit (Table 1). Csn8 is absent from the CSN assembly of Caenorhabditis elegans and some of the fungal species (22–26). Meanwhile, it has become increasingly clear that the functions of the eight CSN subunits are different because ablation or knockdown of different CSN subunit can lead to different functional consequences. Different phenotypes among loss-of-function csn mutants have been noted in Drosophila melanogaster (27) and fission yeast Schizosaccharomyces pombe (28). In mammalian cells, Csn5 (also known as Jab1) and Csn6 have been shown to promote cell growth; overexpression of Csn5 or Csn6 has been linked to cancers, whereas their knockdown reduces cell proliferation (7, 29–31). In contrast, Csn3 appears to negatively regulate cell proliferation as its knockdown accelerates cell growth in cultured cells (20). In addition, Csn3 and Csn6 affect human COP1 levels in different ways (20, 21). In Neurospora crassa, which lacks Csn8, knock-out of Csn3 results in a faster growth rate, whereas knock-out of other CSN genes reduces growth (23).
TABLE 1.
Subunit composition of the CSN in various eukaryotic organisms and corresponding paralog of the CSN subunit in the Lid subcomplex of the proteasome
Check mark indicates the presence of the indicated protein. Bold lettering and italicized lettering are used to emphasize the level of differences.
| Csn1 | Csn2 | Csn3 | Csn4 | Csn5 (Jab1) | Csn6 | Csn7 | Csn8 | References | |
|---|---|---|---|---|---|---|---|---|---|
| Homo sapiens and Mus musculus | 57 | ||||||||
| Arabidopsis thaliana | 58 | ||||||||
| Drosophila melanogaster | 59 | ||||||||
| Caenorhabditis elegans | CIF-1 | Absent | 22 | ||||||
| Dictyostelium discoideum | 60 | ||||||||
| Aspergillus nidulans | 61 | ||||||||
| Neurospora crassa | Absent | 23 | |||||||
| Schizosaccharomyces pombe | Absent | Absent | 26 | ||||||
| Saccharomyces cerevisiae | Csn11 (Pci8) | Csn10 | Absent | Rpn5 | Csn5 (Rri1) | Csi1 | Csn9 | Absent | 24 |
| Paralog in the proteasome Lid | Rpn7 | Rpn6 | Rpn3 | Rpn5 | Rpn11 | Rpn8 | Rpn9 | Rpn12 | 24 |
CSN normally exists as a stable eight-subunit protein complex, whereas its Csn5 subunit exists both as part of the complex and outside of the CSN, a feature that is highly conserved from yeast to human (1, 2). Here we use the term “Csn5-f” to refer to the “free form” of Csn5 protein that is not associated with the CSN holocomplex, regardless of whether Csn5 is monomeric or in association with other effector proteins. Csn5-f has recently been shown to interact with cyclin-dependent kinase CDK2 and act to prevent cell senescence (32). Although small subcomplexes composed of various subsets of CSN subunits have also been reported, their functional relevance remains to be established.
We previously reported germline and conditional disruption of mouse Csn8 (4). Complete ablation of Csn8 in mice led to the destruction of the CSN complex and instability of other subunits (4), similar to the results in higher plants (33, 34) and Drosophila (35). These observations indicate that Csn8 has an important role in maintaining the structural integrity of the complex despite its peripheral position in the complex (16). Germline deletion of Csn8, like germline deletion of other CSN genes in mouse, caused early embryonic lethality (4). Restricted deletion of Csn8 in peripheral T cells abolished the ability of the quiescent cells to enter the cell cycle in response to stimulation, but in cycling T-cells, Csn8 was not required for continued cell division (4). In addition, restricted disruption of Csn8 in liver caused massive cell death and aberrant cell proliferation (8), whereas its disruption in cardiomyocyte of postnatal heart caused heart failure and severe defects in autophagosomal maturation (5, 9).
During the process of generating the Csn8 conditional allele, we have maintained an intermediate allele, Csn8Neoflox or Csn8h. Here we report that this genomic allele expresses Csn8 at a lower level, therefore representing a hypomorphic allele. Through biochemical and cell cycle analyses of this weak Csn8 mutant, we show that Csn8 is involved in maintaining the duration of the G1 phase of the cell cycle. With Csn5 acting to promote cell proliferation, the CSN thus contains both positive and negative regulators of cell cycle and therefore is poised to influence the fate of the cell at the junction of cell division and differentiation during development.
MATERIALS AND METHODS
Primary MEF Cell Culture and Genotyping
To generate h/− mouse embryonic fibroblast (MEF) cells, timed mating between Csn8+/− and Csn8h/h mice was carried out. The resulting embryos, with expected genotype of h/− and h/+ in 1:1 ratio, were used for MEF lines as described previously (4). Genotyping and the generation of Csn8flox/flox conditional and Csn8−/− MEF cells have been described (4). MEF cells from passages 2–4 were used in all of the experiments.
Antibodies
The following antibodies were used in this study: anti-Csn1, anti-Csn2, anti-Csn3, anti-Csn8, and anti-Skp2 have been described previously (36); anti-Csn5, anti-Cul1, anti-Cul2, anti-BrdU, anti-CycD1, CycE, CDK4, and p16 were from Santa Cruz Biotechnology; anti-Cul4A has been described previously (37); anti-α-tubulin (DM1A) and anti-Prmt5 (Prmt5–21) were from Sigma-Aldrich; and anti-p27 was from BD Transduction Laboratories.
Tet-inducible Knockdown and Growth Curve Determination
We used the pSuper series of vector (OligoEngine, Seattle, WA) for Tet-dependent shRNA-mediated knockdown of Csn5 and Csn8 in HeLa cells (38). The shCsn5 vector was kindly provided by the Dr. Deshaies laboratory (38). The same vector was used to express the Csn8-tageting small RNA (GGC UGU GAA AGG CAU AUU A) as described previously (39).
HeLa cells were transfected using Lipofectamine-2000 reagent (Life Technologies) with pshCsn8, pshCsn5, or empty vector together with pcDNA6/TR (Invitrogen/Life Technologies) in a 3:1 ratio. On day 2 after transfection, cells were split and amplified. On day 3, blasticidin (10 μg/ml) was added to the medium to select for transfected cells. After 4 days in the selection medium, half of the dishes were treated with doxycycline (1 μg/ml) to induce shRNA expression, whereas the other dishes were kept as −Dox control. On the following day, a growth curve experiment (see Fig. 4) was started. Both induced (+Dox) and uninduced (−Dox) cells were counted and seeded on multiple 6-cm dishes at 2 × 105 cells/dish in full medium containing blasticidin (5 μg/ml) with or without Dox. Csn5 and Csn8 proteins started to decline from around day 5, just before the log phase of cell proliferation under our experimental conditions. Cells were counted daily for up to 9 days with a hemocytometer.
FIGURE 4.

Silencing of Csn8 caused an increased growth rate, whereas silencing of Csn5 impaired proliferation in HeLa cells. Dox-dependent vectors expressing small hairpin RNA targeting Csn8 (shCsn8) or Csn5 (shCsn5) were transfected to HeLa cells. A–D, growth curves of the cells containing pshCsn8 (A and B) or pshCsn5 (C and D) with or without induction by Dox were determined (A and C). Equal numbers of the cells taken from the growth curve analyses were lysed for Western blots for confirmation of Dox-dependent silencing of Csn8 (B) or Csn5 (D). Tubulin (anti-Tub) was used as loading control. The experiment was repeated three times.
Cell Proliferation Assays
Cells of defined number were seeded in a 6-cm dish for growth curve or in 96-well/48-well dishes for ATPLite assays. Controls and Csn8 hypomorphic lines were manipulated identically. At the indicated day after seeding, cells were trypsinized, and the cell numbers were determined by counting with a hemocytometer. The ATPLite (PerkinElmer Life Sciences) assay was performed according to the manufacturer's instructions. For the result shown in Fig. 3B, cells were seeded at 50,000/well.
FIGURE 3.
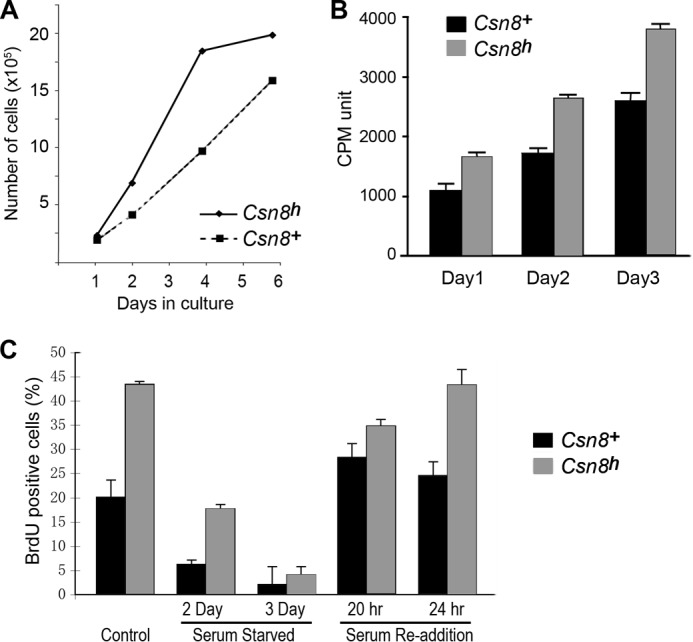
Characterization of cell proliferation properties of Csn8 hypomorphic MEFs. A, the Csn8h MEFs exhibit a faster growth rate in culture. Growth curves of Csn8h and Csn8+ control MEFs at passage 2 were measured by counting of cell numbers at the indicated days following seeding. Similar results were repeated more than four times with different MEF lines. B, the ATPLite assay was uses to compare the cell proliferation rates of the Csn8h and Csn8+ MEF cells. Experiments were repeated at least twice at different seeding densities. C, the percentage of BrdU incorporation was used to monitor proliferation of Csn8h and the Csn8+ MEFs grown under: normal growth condition (Control), serum starvation for the indicated days (Serum-Starved), and at the indicated hours after the readdition of serum following a 3-day serum starvation (Serum Re-addition). Cells were pulse-labeled with BrdU for 3 h. The Csn8+ is indicated by solid black bars, and the hypomorphic Csn8h is indicated by gray bars in both B and C. Error bars in B and C indicate S.D.
Flow Cytometry, BrdU Labeling, and G1 Length Determination
To determine G1 duration (see Fig. 5C), MEF cells were treated with 100 ng/ml nocodazole (Sigma) for 12 h. The mitotic cells were shaken off, washed to remove the drug, and seeded on coverslips in fresh medium containing 10 μm BrdU (Aldrich). At the indicated hours after replating, cells were fixed with 3.5% paraformaldehyde for 15 min followed by 20 min of treatment with 1.5 n HCl. Cells were immunostained with anti-BrdU monoclonal antibody (BD Pharmingen) followed by FITC-conjugated secondary antibody to identify the BrdU-positive cells. Cells were double-stained with DAPI, which served to identify all viable cells. The percentages of BrdU-positive cells among the total number of cells within a field were determined. We found that many MEF cells died or stopped dividing after M/G1 synchronization and that only 28.7% of Csn8+ cells were BrdU-positive by 24 h, in contrast to HeLa cells, of which ∼80% were active after the synchronization (36). We considered that those cells represented the pool of active continuously cycling cells, and accordingly set the value of BrdU-positives of Csn8+ at 24 h as 100%. The percentages of BrdU-positives in earlier time points were calculated relative to the 24-h value.
FIGURE 5.

The Csn8 hypomorphic MEF cells exhibit an accelerated G1 progression. A and B, graphs showing cell cycle analysis by FACS of Csn8h (h) and the Csn8+ control (+) MEFs (A) and Csn8flox/flox and csn8−/− MEFs (B). C, the upper panel shows a diagram illustrating the timeline procedure used to determine the length of the G1 phase in Csn8h MEF line. The asterisks mark the S phase entry points in Csn8h and Csn8+ lines based on the data shown in the lower panel. The graph in the lower panel shows the percentage of BrdU-positive cells in the time course as illustrated in the upper panel. Based on the rate of BrdU incorporation into the G1 synchronized cells, the timing of S phase entry, which was marked by peaked DNA synthesis, was determined (indicated by arrowheads). All experiments were repeated twice. Error bars in indicate S.D.
For flow cytometry analysis of cell cycle status (see Fig. 5, A and B), MEF cells were stained with propidium iodide. The fluorescence from the propidium iodide-DNA complex was measured with a FACSCalibur flow cytometer (BD Biosciences).
RESULTS
The Csn8 Hypomorphic Allele and MEF Line
We previously described generation and characterization of a Csn8 null mutant (Csn8−/−) (4) as well as the Cre/Lox-based Csn8 conditional mouse strain (Csn8flox/−, Csn8flox/flox) (4, 5, 8, 9). During the generation of the flox conditional allele, we have also obtained the intermediate Neoflox allele, which contains a PGK-NEO selection cassette flanked by a pair of Flp recombination (FRT) sites in intron 3, in addition to the LoxP-flanked exons 4–6 (Fig. 1A). The Neoflox allele was subsequently used to generate the flox conditional allele upon removal of the Neo cassette by Flp-mediated recombination. Correct recombination and deletion were monitored by Southern blot and PCR analysis (4). The Csn8Neoflox/+ mice appeared healthy and fertile. Like the Csn8+/− mice, Csn8Neoflox/+ mice contained normal levels of Csn8 protein due to the presence of a wild type (+) allele (not shown). Because the Neo cassette can sometimes interfere with the expression of the gene in situ, it could cause low expression of Csn8 in the Csn8Neoflox/− mice. Here we examined Csn8 gene expression in Csn8Neoflox/− mice in comparison with its Csn8Neoflox/+ siblings.
FIGURE 1.

Genotype characterization of the Csn8 hypomorphic line. A, graphic illustration of the Csn8 genomic locus in wild type (Csn8+), Csn8Neoflox (Csn8h), and Csn8− alleles. The Csn8Neoflox (Csn8h) allele contains the PGK-NEO cassette flanked by a pair of FRT sites in intron 3. The null allele, Csn8−, was generated after deletions of the FRT- and LoxP-flanked DNA segments by the Flp and Cre recombinases, respectively. Arrows indicate the PCR primers designed to distinguish the three different alleles. B, A DNA gel showing different PCR bands using primer pairs A and B on the genomic DNA of indicated genotypes.
The Csn8Neoflox/− mice were also viable and fertile. We first examined the Csn8 protein levels in representative organs: brain, heart, and spleen of an adult Csn8Neoflox/− mouse by Western blotting (Fig. 2A), and found that Csn8 protein amounts were decreased in Csn8Neoflox/− when compared with Csn8Neoflox/+ mice. Consistent with previous studies reporting that Csn8 deletion or knockdown can cause instability of other CSN subunits (4, 5, 8, 39), we found that Csn1, Csn3, and Csn5 protein amounts were also decreased to various degrees (Fig. 2A). Cullin proteins are substrates of the CSN deneddylase, and its modification has been widely used as an indicator of CSN activity. Western blots of Cul1 showed that the neddylation or protein accumulation of Cul1 was notably affected in Csn8Neoflox/− mice (Fig. 2A). This result shows that Csn8Neoflox/− mice express a lower level of Csn8 and can be considered a hypomorphic strain. Hereafter, we will use “Csn8h” or “h” to refer to the Csn8Neoflox/− hypomorphic (h) genotype for simplicity.
FIGURE 2.
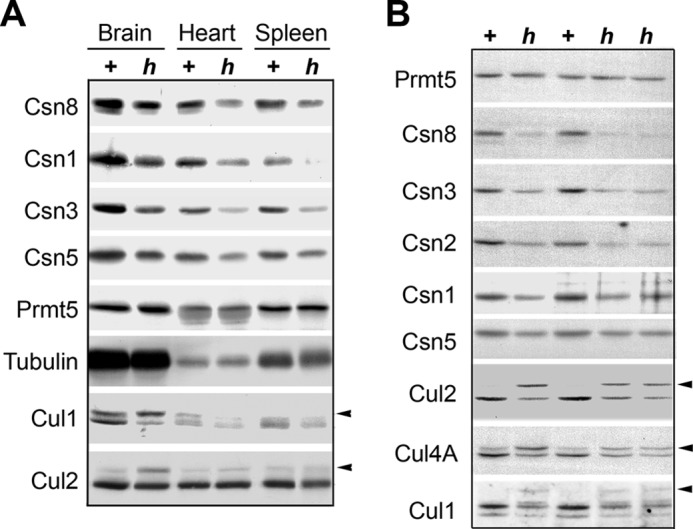
Csn8h lines expressed Csn8 at lower levels and exhibited altered cullin deneddylation pattern. A, representative organs such as brain, heart, and spleen from Csn8h (h) and its sibling control Csn8+ (+) mice were examined by Western blot using the indicated antibodies. Tubulin and Prmt5 are the loading controls. B, Western blot of Csn8h (h) and its sibling control Csn8+ (+) MEF lines using the indicated antibodies. Prmt5 is the loading control. In both panels, the arrows indicate the neddylated form of the respective cullin proteins.
To generate Csn8h MEFs, we first allowed mating between Csn8+/− and Csn8h/h mice, which would give rise to progenies with the genotype of Csn8h/− (Csn8h) and Csn8h/+ (Csn8+) in a 1:1 ratio, and the latter (Csn8+) was conveniently used as a sibling control for the former (Csn8h) in all experiments. The embryos were isolated at 13.5 embryonic days, and MEF lines were generated from each embryo, which were subsequently genotyped (Fig. 1B). Western blot analyses confirmed that Csn8 protein was expressed at a lower level in Csn8h (h) cells when compared with Csn8+ (+) cells (Fig. 2B). Similar to the tissues, most other CSN subunits were found at reduced levels in Csn8h MEFs, further supporting the notion that Csn8 is necessary for the stability of the CSN complex and hence stability of most CSN subunits in mice. Likewise, Western blot of cullin proteins showed that neddylation of Cul1, Cul2, and Cul4A and/or protein accumulation were strongly affected in Csn8h MEFs, with hyperneddylation of Cul2 being most noticeable. This result confirmed that Csn8h MEFs had weaker cullin deneddylation activity (Fig. 2B). We conclude from these results that the Csn8h MEFs are clearly hypomorphic for Csn8 and therefore can be used to study the biochemical and functional characteristics of Csn8 hypomorphism.
Csn8 Hypomorphic MEF Cells Exhibit a Faster Growth Rate in Culture
We noticed that the Csn8h MEF cells tended to grow faster than their paired Csn8+ cells because Csn8h cells regularly required greater dilutions on each replating. The growth curve determined by counting the number of cells following reseeding showed that indeed Csn8h cells produced greater numbers of cells in shorter times in culture (Fig. 3A). Similarly, the ATPLite cell proliferation assay showed higher proliferation activity of the Csn8h cells than Csn8+ cells (Fig. 3B). To determine whether Csn8h MEFs exhibit a normal response to growth factor starvation and stimulation, we carried out a serum starvation and readdition experiment and monitored proliferation (S-phase and DNA synthesis) using BrdU incorporation as a read-out (Fig. 3C). The Csn8 hypomorphic MEFs were able to undergo quiescence (<10% BrdU incorporation) after 3 days of serum starvation. This was slower when compared with the Csn8+ control cells, which largely went quiescent after just 2 days of serum removal. Upon the readdition of serum, the Csn8h cells, like the Csn8+ control, were able to re-enter the cell cycle as indicated by increased DNA synthesis activity (Fig. 3C). Consistent with the accelerated growth profile, the Csn8h cells (gray bars) displayed significantly higher BrdU incorporation rates than the Csn8+ control cells (black bars). Taken together, we found that Csn8 hypomorphic MEF cells proliferate faster, require a longer period of serum starvation to exit the cell cycle, and are capable of re-entering the cell cycle upon the readdition of serum. Because the Csn8−/− (null) cells are completely defective in cell cycle re-entry (4), these results indicate that the amount of Csn8 in the Csn8h cell, although lower than normal cells, is nonetheless sufficient to allow cell cycle reinitiation.
Decreased Expression of Csn8 or Csn5 Causes Opposite Cell Growth Profiles
Contrary to Csn8, hypomorphism of Csn5, either by RNA silencing (31, 40) or due to genetic heterozygosity (7, 41), has been reported to impair cell proliferation. To confirm that these two subunits indeed behave differently in cell proliferation, we conducted a parallel knockdown study of Csn5 or Csn8 in HeLa cells (Fig. 4). This experiment was to ensure that the results from silencing each of the two different CSN subunits could be directly compared. A tetracycline/doxycycline (Tet/Dox)-inducible shRNA-mediated knockdown system was utilized (38). In our procedure, Western blot of the respective targeted gene products showed that the silencing started around day 6 of Dox induction or days 4–5 after plating the cells for the growth curve assay (Fig. 4, B and D). As clearly shown in Fig. 4, the growth curves of Csn8 knockdown and Csn5 knockdown were drastically different. Upon Dox induction of shCsn8-mediated silencing, the cells exhibited an accelerated growth pattern when compared with the uninduced (−Dox) cells (Fig. 4A), similar to the profile of Csn8h MEFs (Fig. 3A). In contrast, the Csn5 knockdown cells could hardly grow (Fig. 4C), which is in agreement with published studies reporting that a decrease in Csn5 leads to severe cell proliferation defects (7, 31, 41). This result clearly showed that, under identical experimental conditions and in the same cell line, decreases in two different CSN subunits caused contrasting effects on cell proliferation; a decrease in Csn5 drastically diminished cell proliferation, whereas a decrease in Csn8 accelerated the cell cycle progression.
Csn8 Hypomorphic Cells Have a Shorter G1 Phase
To further understand the Csn8 function, it is necessary to delineate the specific cell cycle phase that is affected by decreased expression of Csn8. FACS analysis showed that when compared with the Csn8+ control, the Csn8h MEF cells exhibited a smaller G1 population and a correspondingly greater S phase population (Fig. 5A). The reduction of G1 population, although not drastic, was highly consistent between experiments (not shown). As shown in Fig. 5B, the same reduction of the G1 population was also observed in the Csn8 knock-out MEFs (Csn8−/−), in which Csn8 was deleted in culture from a Csn8 conditional line, Csn8flox/flox (4). In addition, Csn8−/− cells also displayed a larger G2 population, which could suggest a possible G2 delay in those cells (Fig. 5B). However, the potential G2 abnormality observed in Csn8−/− cells was not a significant issue in Csn8h MEFs (Fig. 5A). The diminished G1 population could indicate a shortened G1 phase in Csn8h cells, which would also explain its greater S phase population and the faster growth rate.
To measure the length of G1 phase in Csn8h and Csn8+ MEFs, cells were first arrested at M phase by nocodazole treatment in a procedure previously described (36) (Fig. 5C). M phase cells were collected and replated in fresh BrdU-containing medium, allowing the cells to enter the G1 phase in a synchronized manner and progress into the S phase. The period from replating of the cells to the time when most of the cells undergo DNA synthesis, or become BrdU-positive, would be considered the length of G1. To determine how long it took the Csn8+ and Csn8h MEFs to start synthesizing DNA after replating, we collected the cells in a time course from 6 to 24 h after replating and assayed the percentage of cells with BrdU incorporation. After disregarding dead cells that failed to take up BrdU in 24 h, we found that 82% of Csn8h MEFs entered the S phase at 10 h after replating, whereas the normal Csn8+ MEFs entered the S phase at about 12 h (82%, Fig. 5B, dotted line). This result demonstrates that Csn8 hypomorphic cells have a shorter G1 phase during their cell cycle progression, which explains why the Csn8h cells display an accelerated growth rate.
We next examined the expression level of a number of G1 regulators in Csn8 hypomorphic cells by Western blots (Fig. 6). When comparing different Csn8h lines with the Csn8+ sibling control lines, we found that the level of Skp2 F-box protein, a G1-promoting factor, appeared slightly but consistently up-regulated in Csn8h lines (Fig. 6A, h lanes). This was clearer upon quantification of Skp2 band intensity shown at the bottom panel of Fig. 6A. This result is opposite to Csn5 knockdown cells, which down-regulate Skp2 (31, 38). On the other hand, we could not definitively detect consistent differences in the level of cyclin E or CDK inhibitor p27kip1, both of which are sensitive to Csn5/Jab1 levels (7, 31, 38). Notably, both cyclin E and p27kip1 can be regulated by multiple mechanisms. We then compared the Csn8 MEFs side-by-side with Csn5+/− MEF cells, which express a lower level of Csn5 (Fig. 6B, lane 5). In Csn5+/− MEFs, cyclin-dependent kinase CDK4, cyclin D1, and cyclin E levels were all drastically decreased. In Csn8 hypomorphic MEFs, we found that in later passages (passage of 3 or greater), CDK4 and cyclin D1 were slightly up-regulated (Fig. 6B). G1 inhibitor p16Ink4a plays an important role in cell senescence and has been shown to be up-regulated in Csn5+/− cells (42). Western blot showed that p16, which increased with each passage of MEFs as anticipated, appeared suppressed in Csn8 hypomorphic MEFs (Fig. 6C), although there were variations on the extents of these changes. Overall, the changes in the abundance of the G1 activators and inhibitors are in agreement with the accelerated G1 profile of Csn8 hypomorphism and impaired proliferation of Csn5 hypomorphism.
FIGURE 6.

Expression of G1 regulators in Csn8 hypomorphic MEFs. A, Western blot analysis of Skp2, cyclin E, and p27kip1 in multiple Csn8h (h) and the Csn8+ control (+) MEF lines. Prmt5 was used as a loading control. Quantification of the Skp2 protein relative intensities by ImageJ was shown in the bottom panel. B, Western blot analysis of G1 activators CDK4 and cyclin D and E in Csn5+/− (Csn5 h) MEFs with its control (+) or Csn8h (h) MEFs and its control (+). The passage numbers of the MEF cells are indicated on the top. Tubulin (Tub) was used as a loading control. C, Western analysis of p16Ink4a in MEFs of different passages for Csn8h (h) and its control, Csn8+ (+). A nonspecific band was shown as an internal loading control.
Csn8 Hypomorphic Cells Have a Higher Ratio of Csn5-f Relative to CSN
Csn5 is unique in partitioning both as part of the 500-kDa CSN holocomplex and as a free form outside of CSN (Csn5-f). Csn5-f was recently shown to interact with cyclin-dependent kinase CDK2 (32). It was reported that in Csn5 hypomorphic cells such as siRNA-mediated Csn5 knockdown cells, Csn5-f- or Csn5-containing small complexes were decreased more drastically than the CSN-associated form of Csn5 (31, 43, 44). To investigate how Csn8 hypomorphism affects the distribution of Csn5 proteins between CSN-associated and Csn5-f populations, the Csn8h or Csn8+ MEF cell extracts were fractionated through a Superose-6 gel filtration column (Fig. 7A). Western blot analyses showed that Csn8 was detected only in 500-kDa fractions containing the CSN complex (Fig. 7, lanes 11–14, Peak-1) in both Csn8h and Csn8+ cells, although a lesser amount of Csn8 was present in Csn8h cells. Csn5, on the other hand, was found both in CSN-containing fractions (Peak-1) and in smaller molecular weight fractions (lanes 18–21, Peak-2), which denote the pool of Csn5-f. Quantification of the Csn5 protein band, as shown in the bottom panel of Fig. 7A, indicates that Csn8h cells exhibited a clear reduction in CSN-associated Csn5 relative to Csn5-f. Accordingly, a biochemical consequence of Csn8 hypomorphism is the increase in the ratio of Csn5-f (Peak-2) over the CSN complex (Peak-1) (Fig. 7B). In addition, Csn5 ectopic expression has been reported to predominantly increase Csn5-f relative to CSN (45) or the ratio of Csn5-f to CSN, whereas the Csn5 hypomorphism, which has a more drastic reduction in Csn5-f than CSN, decreases the Csn5-f/CSN ratio (31, 44) (Fig. 7B). In all of those cases, the ratio of Csn5-f/CSN positively correlates with cell proliferation activity.
FIGURE 7.

Gel filtration analysis of relative amounts of CSN and Csn5-f form in Csn8 hypomorphic cells. A, total protein extracts from Csn8+ or Csn8h MEFs were size-fractionated in a Superose-6 gel filtration column. The fractions were analyzed by anti-Csn5 and anti-Csn8 Western blotting. Csn8 was found in large molecular weight fractions around Peak-1 (fractions 11–14), whereas Csn5 was distributed in two peaks. The low molecular weight Peak-2 (fractions 18–21) represents the Csn5-f form. The bottom panel shows the quantification of the intensity of the Csn5 bands in the Western blots above. Solid black bars indicate Csn8+, whereas the gray bars indicate Csn8h hypomorphic MEFs. B, the table qualitatively summarizes the changes in the ratio of Csn5-f/CSN in Csn8h cells (data from panel A), in comparison with the reported studies of Csn5 ectopic overexpression (Csn5 OX) and Csn5 hypomorphic cells from the literature.
DISCUSSION
Germline ablation of mouse Csn2, Csn3, Csn5, Csn6, and Csn8 all leads to early embryonic arrest and death (3, 4, 7, 30, 46). Similarly, in Arabidopsis, complete loss-of-function mutation of each of the CSN subunits results in early seedling lethality (47, 48). This well documented phenotype can give an illusion that all CSN subunits affect cell proliferation identically and positively. However, because complete ablation of each of the CSN subunits results in destruction of the entire CSN complex, the prevalent pleiotropic developmental phenotypes from losing the complex can obscure the specific functional roles of individual subunits. Through a reduction-of-function approach, we think we have identified a specific function of Csn8: that it negatively regulates cell cycle in MEFs and HeLa cells. Csn8 is probably involved in a brake function in the G1 phase because decreased Csn8 expression resulted in premature transition to the S phase. Nevertheless, Csn8 may not have the same effect on all cell types due to the variations in the regulation and kinetics of the cell cycle.
Csn8 and Csn5 Act Antagonistically in Cell Cycle Regulation
We have previously shown that the Csn8−/− MEFs and T-cells can undergo cell divisions in cycling cells but lack the ability to re-enter the cell cycle from the G0 state (4). In the Csn8h hypomorphic cells, cell cycle re-entry from the G0 state was normal, but the cells displayed a faster growth rate, smaller G1 population, and a shortened G1 phase (Fig. 3). Small RNA-mediated silencing of Csn8 in HeLa cells caused a faster growth rate, whereas silencing of Csn5 by the same procedure caused a severe growth inhibition (Fig. 4). Thus the two different subunits of the CSN display opposing functionalities in how they affect cell proliferation. In addition to Csn8, Csn3 has also been implicated in negative regulation of cell growth in mammalian cells (20) and in N. crassa (23).
Csn5 interacts with many cell cycle regulators and transcription factors involved in cancer (1, 29). Notably, the Csn5-f form, rather than the CSN-associated form of Csn5, was shown to directly interact with cyclin-dependent kinase CDK2 (32). Csn5-f may also be the form that mediates nuclear export and the subsequent degradation of CDK inhibitor p27kip1 (44). In addition, abnormal overexpression of Csn5, rather than elevation of the deneddylation activity of the CSN, is linked to various cancers (1, 29). These findings are consistent with the notion that pro-proliferation activity is attributable to Csn5-f.
Csn8, CSN Deneddylation Activity, and Cell Proliferation
CSN is a multisubunit deneddylase, an activity that is highly conserved from yeast to human. Recently, Pick et al. (24) showed that purified budding yeast CSN complex, which lacks Csn8, can effectively deneddylate human cullins, indicating that Csn8 is probably not required for the deneddylase activity. We believe that the deneddylation defect in Csn8h cells is most likely due to decreased CSN level (Fig. 2). Similar deneddylation defects have been observed upon reduction of other CSN subunits including Csn5 (31, 38). So far, both accelerated and stalled cell proliferation have been observed in cells with decreased CSN deneddylation activity, which seems to suggest that the changes in cullin neddylation state may not account for the cell cycle phenotype in those cells. Nonetheless, quantitative analysis on the loss of deneddylation activity and the associated phenotypes in different cell cycle phases has not been conducted. We have previously shown that microinjection of a purified CSN complex into early G1 HeLa cells impedes the G1-to-S progression, and this activity, along with the deneddylation activity, can be blocked by anti-Csn2 polyclonal antibodies (36). On the other hand, there is evidence suggesting that an intact JAMM motif of Csn5, which is the catalytic center of the CSN deneddylase, is important for proliferation (40, 49). Complete loss of CSN has been shown to cause a G2 arrest in Arabidopsis (50). We found that a G2 phenotype was associated with Csn8−/−, but not Csn8h MEFs (Fig. 5, A and B), suggesting that the G2 function is unlikely a primary function of Csn8, but is more likely to be related to the CSN complex, similar to the study on Arabidopsis CSN (50).
Possible Mechanisms Underlying Accelerated Cell Cycle Progression of Csnh Cells
Given the function of Csn5-f in promoting cell proliferation, one hypothesis is that the accelerated cell cycle progression of Csn8 hypomorphic cells could be caused by increased ratio of Csn5-f to CSN. Csn8h cells show reduced CSN levels without a detectable effect on Csn5-f, thereby resulting in an increased Csn5-f to CSN ratio (Fig. 7). The increased level of Csn5-f relative to CSN might propel the accelerated cell proliferation observed in Csn8 hypomorphic cells. In this scenario, the way Csn8 regulates cell cycle is to influence the equilibrium of Csn5-f relative to the CSN complex, and the mitogenic activity of Csn5-f would ultimately determine the proliferation status of the cell.
Another hypothesis, which may or may not work together with the Csn5-f/CSN equilibrium theory, is that Csn8 suppresses cell proliferation independently of Csn5-f. Csn8 and Csn5 appear to be peripheral subunits of the CSN complex (16). Although Csn8 has not been detected to stably exist outside of the CSN under our mild experimental condition, a low abundant small Csn8 complex has been found by native gel electrophoresis (39) and by gel filtration chromatography (31). Either directly or indirectly, Csn8 seems to affect the expression of cell cycle regulators such as p16Ink4a, cyclin D1, Cdk4, and Skp2 (Fig. 6), which control the progression of G1. Unlike Csn5+/− or shCsn5 cells, the growth phenotype of the Csn8h was not robust. As a result, the amplitude of the observed molecular changes was not dramatic, and it varied depending on the extent of Csn8 reduction and the passage numbers of MEFs. At this point, the precise molecular mechanism of how Csn8 modulates these genes remains to be determined.
Modulation of G1 Length in Developmental Regulation
In mammalian cells, G1 phase is when the fate of the cell is determined; the cell may undergo quiescence or senescence (G0), differentiation, or cell size expansion or commit to division (51). In neural, hematopoietic, and embryonic stem cells, shorter G1 correlates with division, whereas long G1 correlates with differentiation (52–54). In fact, lengthening of G1 is not only required, but can also trigger cell differentiation. Overexpression of Cdk4/cyclin D1 is shown to shorten G1 and delay neurogenesis (55), whereas loss of Cdk2 and Cdk4 induces a switch from proliferation to differentiation in neural stem cells (56). In this context, Csn8, because of its function in prolonging G1 progression, may be relevant in facilitating cell differentiation.
Cell differentiation can be considered as a hallmark of organismal complexity because lower eukaryotes such as unicellular yeast have little cell differentiation. Csn8 is apparently associated with developmental complexity, considering its conservation profile in evolution; Csn8 is not in the CSN complex of budding yeast, fission yeast, N. crassa, and C. elegans, but is an integral subunit in mammals, higher plants, insects, social amoeba Dictyostelium discoideum, and interestingly, filamentous fungus Aspergillus nidulans (Table 1). It seems plausible that in lower organisms, cell differentiation is so primitive that Csn8 function either is not required or is not linked to CSN. We speculate that, equipped with both pro-proliferative (Csn5 and Csn6) and anti-proliferative factors (Csn3 and Csn8), the CSN plays a dynamic role in the homeostasis of cell differentiation and division during development of higher eukaryotes.
Acknowledgments
We thank the Francois X. Claret laboratory for the generous gift of Csn5+/− MEF cells and the Raymond Deshaies laboratory for the generous gift of shCsn5 and the control vectors.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL085629 (to X. J. W. and N. W,) and GM47850 (to X. W. D.). This work was also supported by the National Science Foundation (NSF) 2010 Program MCB-0929100 (to X. W. D.).
- CSN
- COP9 signalosome
- CRL
- cullin-RING family of ubiquitin E3 ligase
- MEF
- mouse embryonic fibroblast
- Dox
- doxycycline
- Tet
- tetracycline
- FRT
- Flp recombination
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Kato J. Y., Yoneda-Kato N. (2009) Mammalian COP9 signalosome. Genes Cells 14, 1209–1225 [DOI] [PubMed] [Google Scholar]
- 2. Wei N., Serino G., Deng X. W. (2008) The COP9 signalosome: more than a protease. Trends Biochem. Sci. 33, 592–600 [DOI] [PubMed] [Google Scholar]
- 3. Lykke-Andersen K., Schaefer L., Menon S., Deng X. W., Miller J. B., Wei N. (2003) Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol. Cell Biol. 23, 6790–6797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Menon S., Chi H., Zhang H., Deng X. W., Flavell R. A., Wei N. (2007) COP9 signalosome subunit 8 is essential for peripheral T cell homeostasis and antigen receptor-induced entry into the cell cycle from quiescence. Nat. Immunol. 8, 1236–1245 [DOI] [PubMed] [Google Scholar]
- 5. Su H., Li J., Menon S., Liu J., Kumarapeli A. R., Wei N., Wang X. (2011) Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ. Res. 108, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Panattoni M., Sanvito F., Basso V., Doglioni C., Casorati G., Montini E., Bender J. R., Mondino A., Pardi R. (2008) Targeted inactivation of the COP9 signalosome impairs multiple stages of T cell development. J. Exp. Med. 205, 465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tomoda K., Yoneda-Kato N., Fukumoto A., Yamanaka S., Kato J. Y. (2004) Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J. Biol. Chem. 279, 43013–43018 [DOI] [PubMed] [Google Scholar]
- 8. Lei D., Li F., Su H., Tian Z., Ye B., Wei N., Wang X. (2011) COP9 signalosome subunit 8 is required for postnatal hepatocyte survival and effective proliferation. Cell Death Differ. 18, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Su H., Li F., Ranek M. J., Wei N., Wang X. (2011) COP9 signalosome regulates autophagosome maturation. Circulation 124, 2117–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lyapina S., Cope G., Shevchenko A., Serino G., Tsuge T., Zhou C., Wolf D. A., Wei N., Shevchenko A., Deshaies R. J. (2001) Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 292, 1382–1385 [DOI] [PubMed] [Google Scholar]
- 11. Schwechheimer C., Serino G., Callis J., Crosby W. L., Lyapina S., Deshaies R. J., Gray W. M., Estelle M., Deng X. W. (2001) Interactions of the COP9 signalosome with the E3 ubiquitin ligase SCFTIRI in mediating auxin response. Science 292, 1379–1382 [DOI] [PubMed] [Google Scholar]
- 12. Zhou C., Wee S., Rhee E., Naumann M., Dubiel W., Wolf D. A. (2003) Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol. Cell 11, 927–938 [DOI] [PubMed] [Google Scholar]
- 13. Hetfeld B. K. J., Helfrich A., Kapelari B., Scheel H., Hofmann K., Guterman A., Glickman M., Schade R., Kloetzel P. M., Dubiel W. (2005) The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr. Biol. 15, 1217–1221 [DOI] [PubMed] [Google Scholar]
- 14. Zhou Z., Wang Y., Cai G., He Q. (2012) Neurospora COP9 signalosome integrity plays major roles for hyphal growth, conidial development, and circadian function. Plos Genet. 8, e1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer E. S., Scrima A., Böhm K., Matsumoto S., Lingaraju G. M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F., Iwai S., Gut H., Sugasawa K., Thomä N. H. (2011) The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039 [DOI] [PubMed] [Google Scholar]
- 16. Enchev R. I., Scott D. C., da Fonseca P. C., Schreiber A., Monda J. K., Schulman B. A., Peter M., Morris E. P. (2012) Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emberley E. D., Mosadeghi R., Deshaies R. J. (2012) Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J. Biol. Chem. 287, 29679–29689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uhle S., Medalia O., Waldron R., Dumdey R., Henklein P., Bech-Otschir D., Huang X., Berse M., Sperling J., Schade R., Dubiel W. (2003) Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. 22, 1302–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Li W., Piqueras R., Cao K., Deng X. W., Wei N. (2009) Regulation of COP1 nuclear localization by the COP9 signalosome via direct interaction with CSN1. Plant J 58, 655–667 [DOI] [PubMed] [Google Scholar]
- 20. Yoneda-Kato N., Tomoda K., Umehara M., Arata Y., Kato J. (2005) Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. EMBO J. 24, 1739–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choi H. H., Gully C., Su C. H., Velazquez-Torres G., Chou P. C., Tseng C., Zhao R., Phan L., Shaiken T., Chen J., Yeung S. C., Lee M. H. (2011) COP9 signalosome subunit 6 stabilizes COP1, which functions as an E3 ubiquitin ligase for 14–3-3σ. Oncogene 30, 4791–4801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luke-Glaser S., Roy M., Larsen B., Le Bihan T., Metalnikov P., Tyers M., Peter M., Pintard L. (2007) CIF-1, a shared subunit of the COP9/signalosome and eukaryotic initiation factor 3 complexes, regulates MEL-26 levels in the Caenorhabditis elegans embryo. Mol. Cell Biol. 27, 4526–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Hu Q., Chen H., Zhou Z., Li W., Wang Y., Li S., He Q. (2010) Role of individual subunits of the Neurospora crassa CSN complex in regulation of deneddylation and stability of cullin proteins. Plos Genet. 6, e1001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pick E., Golan A., Zimbler J. Z., Guo L., Sharaby Y., Tsuge T., Hofmann K., Wei N. (2012) The minimal deneddylase core of the COP9 signalosome excludes the Csn6 MPN- domain. Plos One 7, e43980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wee S., Hetfeld B., Dubiel W., Wolf D. A. (2002) Conservation of the COP9/signalosome in budding yeast. BMC Genet. 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu C., Powell K. A., Mundt K., Wu L., Carr A. M., Caspari T. (2003) Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Gene Dev. 17, 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oron E., Mannervik M., Rencus S., Harari-Steinberg O., Neuman-Silberberg S., Segal D., Chamovitz D. A. (2002) COP9 signalosome subunits 4 and 5 regulate multiple pleiotropic pathways in Drosophila melanogaster. Development 129, 4399–4409 [DOI] [PubMed] [Google Scholar]
- 28. Mundt K. E., Liu C., Carr A. M. (2002) Deletion mutants in COP9/signalosome subunits in fission yeast Schizosaccharomyces pombe display distinct phenotypes. Mol. Biol. Cell 13, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shackleford T. J., Claret F. X. (2010) JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao R., Yeung S. C. J., Chen J., Iwakuma T., Su C. H., Chen B., Qu C., Zhang F., Chen Y. T., Lin Y. L., Lee D. F., Jin F., Zhu R., Shaikenov T., Sarbassov D., Sahin A., Wang H. M., Wang H., Lai C. C., Tsai F. J., Lozano G., Lee M. H. (2011) Subunit 6 of the COP9 signalosome promotes tumorigenesis in mice through stabilization of MDM2 and is upregulated in human cancers. J. Clin. Invest. 121, 851–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Denti S., Fernandez-Sanchez M. E., Rogge L., Bianchi E. (2006) The COP9 signalosome regulates Skp2 levels and proliferation of human cells. J. Biol. Chem. 281, 32188–32196 [DOI] [PubMed] [Google Scholar]
- 32. Yoshida A., Yoneda-Kato N., Kato J. Y. (2013) CSN5 specifically interacts with CDK2 and controls senescence in a cytoplasmic cyclin E-mediated manner. Sci. Rep. 3, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei N., Chamovitz D. A., Deng X. W. (1994) Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell 78, 117–124 [DOI] [PubMed] [Google Scholar]
- 34. Gusmaroli G., Figueroa P., Serino G., Deng X. W. (2007) Role of the MPN subunits in COP9 signalosome assembly and activity, and their regulatory interaction with Arabidopsis Cullin3-based E3 ligases. Plant Cell 19, 564–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oren-Giladi P., Krieger O., Edgar B. A., Chamovitz D. A., Segal D. (2008) Cop9 signalosome subunit 8 (CSN8) is essential for Drosophila development. Genes Cells 13, 221–231 [DOI] [PubMed] [Google Scholar]
- 36. Yang X., Menon S., Lykke-Andersen K., Tsuge T., Xiao D., Wang X., Rodriguez-Suarez R. J., Zhang H., Wei N. (2002) The COP9 signalosome inhibits p27kip1 degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Curr. Biol. 12, 667–672 [DOI] [PubMed] [Google Scholar]
- 37. Pick E., Lau O. S., Tsuge T., Menon S., Tong Y., Dohmae N., Plafker S. M., Deng X. W., Wei N. (2007) Mammalian DET1 regulates Cul4A activity and forms stable complexes with E2 ubiquitin-conjugating enzymes. Mol. Cell Biol. 27, 4708–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cope G. A., Deshaies R. J. (2006) Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC biochemistry 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su H., Huang W., Wang X. (2009) The COP9 signalosome negatively regulates proteasome proteolytic function and is essential to transcription. Int. J. Biochem. Cell Biol. 41, 615–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukumoto A., Tomoda K., Yoneda-Kato N., Nakajima Y., Kato J. Y. (2006) Depletion of Jab1 inhibits proliferation of pancreatic cancer cell lines. FEBS Lett. 580, 5836–5844 [DOI] [PubMed] [Google Scholar]
- 41. Tian L., Peng G., Parant J. M., Leventaki V., Drakos E., Zhang Q., Parker-Thornburg J., Shackleford T. J., Dai H., Lin S. Y., Lozano G., Rassidakis G. Z., Claret F. X. (2010) Essential roles of Jab1 in cell survival, spontaneous DNA damage, and DNA repair. Oncogene 29, 6125–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mori M., Yoneda-Kato N., Yoshida A., Kato J. Y. (2008) Stable form of JAB1 enhances proliferation and maintenance of hematopoietic progenitors. J. Biol. Chem. 283, 29011–29021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peth A., Berndt C., Henke W., Dubiel W. (2007) Downregulation of COP9 signalosome subunits differentially affects the CSN complex and target protein stability. BMC biochemistry 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tomoda K., Kato J. Y., Tatsumi E., Takahashi T., Matsuo Y., Yoneda-Kato N. (2005) The Jab1/COP9 signalosome subcomplex is a downstream mediator of Bcr-Abl kinase activity and facilitates cell-cycle progression. Blood 105, 775–783 [DOI] [PubMed] [Google Scholar]
- 45. Tanguy G., Drévillon L., Arous N., Hasnain A., Hinzpeter A., Fritsch J., Goossens M., Fanen P. (2008) CSN5 binds to misfolded CFTR and promotes its degradation. Biochim. Biophys. Acta 1783, 1189–1199 [DOI] [PubMed] [Google Scholar]
- 46. Yan J., Walz K., Nakamura H., Carattini-Rivera S., Zhao Q., Vogel H., Wei N., Justice M. J., Bradley A., Lupski J. R. (2003) COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell Biol. 23, 6798–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei N., Deng X. W. (2003) The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19, 261–286 [DOI] [PubMed] [Google Scholar]
- 48. Schwechheimer C., Isono E. (2010) The COP9 signalosome and its role in plant development. Eur. J. Cell Biol. 89, 157–162 [DOI] [PubMed] [Google Scholar]
- 49. Adler A. S., Littlepage L. E., Lin M., Kawahara T. L. A., Wong D. J., Werb Z., Chang H. Y. (2008) CSN5 isopeptidase activity links COP9 signalosome activation to breast cancer progression. Cancer Res. 68, 506–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dohmann E. M. N., Levesque M. P., De Veylder L., Reichardt I., Jürgens G., Schmid M., Schwechheimer C. (2008) The Arabidopsis COP9 signalosome is essential for G2 phase progression and genomic stability. Development 135, 2013–2022 [DOI] [PubMed] [Google Scholar]
- 51. Massagué J. (2004) G1 cell-cycle control and cancer. Nature 432, 298–306 [DOI] [PubMed] [Google Scholar]
- 52. Lange C., Calegari F. (2010) Cdks and cyclins link G1 length and differentiation of embryonic, neural, and hematopoietic stem cells. Cell Cycle 9, 1893–1900 [DOI] [PubMed] [Google Scholar]
- 53. Salomoni P., Calegari F. (2010) Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol. 20, 233–243 [DOI] [PubMed] [Google Scholar]
- 54. Orford K. W., Scadden D. T. (2008) Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 9, 115–128 [DOI] [PubMed] [Google Scholar]
- 55. Lange C., Huttner W. B., Calegari F. (2009) Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5, 320–331 [DOI] [PubMed] [Google Scholar]
- 56. Lim S., Kaldis P. (2012) Loss of Cdk2 and Cdk4 induces a switch from proliferation to differentiation in neural stem cells. Stem Cells 30, 1509–1520 [DOI] [PubMed] [Google Scholar]
- 57. Wei N., Tsuge T., Serino G., Dohmae N., Takio K., Matsui M., Deng X. W. (1998) The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr. Biol. 8, 919–922 [DOI] [PubMed] [Google Scholar]
- 58. Serino G., Su H., Peng Z., Tsuge T., Wei N., Gu H., Deng X. W. (2003) Characterization of the last subunit of the Arabidopsis COP9 signalosome: implications for the overall structure and origin of the complex. Plant Cell 15, 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Freilich S., Oron E., Kapp Y., Nevo-Caspi Y., Orgad S., Segal D., Chamovitz D. A. (1999) The COP9 signalosome is essential for development of Drosophila melanogaster. Curr. Biol. 9, 1187–1190 [DOI] [PubMed] [Google Scholar]
- 60. Rosel D., Kimmel A. R. (2006) The COP9 signalosome regulates cell proliferation of Dictyostelium discoideum. Eur. J. Cell Biol. 85, 1023–1034 [DOI] [PubMed] [Google Scholar]
- 61. Busch S., Schwier E. U., Nahlik K., Bayram O., Helmstaedt K., Draht O. W., Krappmann S., Valerius O., Lipscomb W. N., Braus G. H. (2007) An eight-subunit COP9 signalosome with an intact JAMM motif is required for fungal fruit body formation. Proc. Natl. Acad. Sci. U.S.A. 104, 8089–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]