

Background: Oxidative stress increases vascular permeability though caveolin-1 phosphorylation. The exact role of AMPK is unknown.
Results: AMP-dependent kinase (AMPK) inhibits caveolin-1 phosphorylation by stabilizing the interaction between c-Abl and Prdx-1.
Conclusion: AMPK activation inhibits oxidant induced-vascular permeability.
Significance: The present study shows a novel protective role of AMPK in the vascular homeostasis.
Keywords: AMP-activated kinase (AMPK), Caveolae, Caveolin, Endocytosis, Oxidative Stress
Abstract
Caveolin-1 is the primary structural component of endothelial caveolae that is essential for transcellular trafficking of albumin and is also a critical scaffolding protein that regulates the activity of signaling molecules in caveolae. Phosphorylation of caveolin-1 plays a fundamental role in the mechanism of oxidant-induced vascular hyper permeability. However, the regulatory mechanism of caveolin-1 phosphorylation remains unclear. Here we identify a previously unexpected role for AMPK in inhibition of caveolin-1 phosphorylation under oxidative stress. A pharmacological activator of AMPK, 5-amino-4-imidazole carboxamide riboside (AICAR), inhibited oxidative stress-induced phosphorylation of both caveolin-1 and c-Abl, which is the major kinase of caveolin-1, and endocytosis of albumin in human umbilical vein endothelial cell. These effects were abolished by treatment with two specific inhibitors of AICAR, dipyridamole, and 5-iodotubericidin. Consistently, knockdown of the catalytic AMPKα subunit by siRNA abolished the inhibitory effect of AICAR on oxidant-induced phosphorylation of both caveolin-1 and c-Abl. Pretreatment with specific c-Abl inhibitor, imatinib mesylate, and knock down of c-Abl significantly decreased the caveolin-1 phosphorylation after H2O2 exposure and abolished the inhibitory effect of AICAR on the caveolin-1 phosphorylation. Interestingly, knockdown of Prdx-1, an antioxidant enzyme associated with c-Abl, increased phosphorylation of both caveolin-1 and c-Abl and abolished the inhibitory effect of AICAR on the caveolin-1 phosphorylation. Furthermore, co-immunoprecipitation experiment showed that AICAR suppressed the oxidant-induced dissociation between c-Abl and Prdx1. Overall, our results suggest that activation of AMPK inhibits oxidative stress-induced caveolin-1 phosphorylation and endocytosis, and this effect is mediated in part by stabilizing the interaction between c-Abl and Prdx-1.
Introduction
Vascular endothelial permeability plays a pivotal role in regulating many physiological and pathological processes, including inflammation and angiogenesis and in diseases such as cancer, rheumatoid arthritis, atherosclerosis, and diabetes (1). There are two pathways regulating vascular permeability; one paracellular, through inter-endothelial junctions, and the other transcellular, via caveolae-mediated vesicular transport (2). Caveolae, fission of plasma membrane macrodomains enriched with caveolin-1, are particularly abundant in endothelial cells and essential for transport of albumin, albumin-bound ligands, and hormones and for control of tissue oncotic pressure in the normal continuous endothelium (1).
Caveolin-1, an integral membrane protein (20–22 kDa), is the primary structural component of endothelial caveolae and is also a critical scaffolding protein functioning as a “master regulator” of signaling molecules in caveolae (3). Although caveolin-1 and caveolin-2 are abundantly expressed in the endothelium, caveolae formation and transcytosis of albumin are exclusively regulated by caveolin-1 (4–6). Recent studies indicated that phosphorylation of caveolin-1 at Tyr-14 plays a fundamental role in the mechanism of oxidant-induced vascular hyperpermeability (7). However, the regulatory mechanisms of caveolin-1 phosphorylation are obscure.
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that regulates energy homeostasis and metabolic stress (8). AMPK acts as a sensor of cellular energy status and maintains the balance between ATP production and consumption. In mammals, AMPK exists as a heterotrimer with α, β, and γ subunits, each of which is encoded by two or three genes (α1, α2, β1, β2, γ1, γ2, and γ3). The α subunit possesses catalytic activity, whereas the β and γ subunits are regulatory and maintain the stability of the heterotrimer complex. The importance of AMPKα is illustrated by the fact that dual deficiency of AMPKα1 and AMPKα2 is embryonic lethal (9).
Recent evidence suggests that AMPK has a much wider range of functions, including the regulation of cell growth, cell proliferation, cell polarity, and autophagy (10, 11). We have recently demonstrated that activation of AMPK inhibits retinoblastoma cell proliferation, tumor growth, angiogenesis, ocular inflammation, and MMP-9 expression (11–15). Because these functions of AMPK are closely linked to the vascular hyper-permeability induced by oxidative stress, we hypothesized that AMPK has a protective role for vascular permeability. Indeed, recent studies reported that AMPK protects the paracellular pathway by supporting the adherens junction proteins of N-cadherin and VE-cadherin (16). However, the role of AMPK in the transcellular pathway remains unclear. Thus, in the present study we examined the role of AMPK in the transcellular pathway, particularly the effect of AMPK activation on the phosphorylation of caveolin-1 under oxidative stress.
EXPERIMENTAL PROCEDURES
Materials
Antibodies for (p-) caveolin-1, (p-) c-Abl, peroxiredoxin I (Prdx1),2 (p-) AMPK, AMPKα1, AMPKα2, and VE-cadherin were purchased from Cell Signaling Technologies (Beverly, MA). Antibodies for β-actin and p-caveolin-1 (for immunofluorescence) were obtained from Abcam (Cambridge, MA) and R&D Systems (Minneapolis, MN), respectively. Secondary antibodies of Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 647 goat anti-rabbit IgG were purchased from Invitrogen. 5-Amino-4-imidazole carboxamide riboside (AICAR), a pharmacological activator of AMPK, was purchased from Toronto Research Chemicals (Toronto, ON, Canada). Hydrogen peroxide (H2O2), 5-iodotubericidin (IODO), and dipyridamole (DPY) were purchased from Sigma. Imatinib mesylate, c-Abl inhibitor, was purchased from Cayman Chemicals (Ann Arbor, MI). siRNAs targeting c-Abl, AMPKα1, AMPKα2, and Prdx1 and control siRNA were purchased from Thermoscientific (Rockford, IL).
Cell Culture
HUVECs (Lonza, Walkersville, MD) were cultured in endothelial growth medium (Lonza, Walkersville, MD). For all experiments cells were grown at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Experiments were performed on cells below passage 3–6 grown to 80–90% confluence.
Protein Extraction and Western Blotting
Protein extraction and Western blotting were carried out as described previously (15). Densitometric analysis of bands was performed using ImageJ software. Lane-loading differences were normalized by β-actin.
Immunoprecipitation
Immunoprecipitation was performed with the Universal Magnetic Co-IP Kit (Active Motif North America, Carlsbad, CA) according to the manufacturer's instruction.
siRNA
Cells were transfected with siRNAs using a Nucleofection kit (Amaxa Biosystems, Gaithersburg, MD) following the manufacturer's protocol. The medium was changed at 6 h after transfection. The down-regulation of each protein was evaluated at 3 days after nucleofection.
Albumin Endocytosis Assay
After serum starvation overnight, HUVECs were pretreated with AICAR (2 mm) for 2 h and then stimulated with H2O2 (2 mm) for 30 min. We added BSA conjugated with Alexa 555 (50 μg/ml, Invitrogen) in the medium during the experiment. Cells on coverslips were washed 3 times with cold TBS and fixed in 100% methanol at −20 °C for 15 min. Cells were then permeabilized in 0.3% Triton X-100, 0.15% BSA in TBS with 0.05% Tween 20 (TBST) for 15 min at room temperature and blocked with 0.5% skim milk in TBST for 60 min at room temperature. Cells were incubated in p-caveolin-1 antibody diluted 1:200 and VE-cadherin antibody diluted 1:400 overnight at 4 °C and then incubated for 2 h in secondary antibody diluted 1:300. Cells were then rinsed three times in TBST before mounting in Toto3 (Invitrogen). Images were acquired with a confocal microscope, Leica TCS SP2 spectral confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany).
Statistical Analysis
All experiments were repeated a minimum of three times. All data were expressed as the means ± S.D. Statistical differences were analyzed by the unpaired Student's t test. Differences were considered significant at p < 0.05.
RESULTS
AICAR Suppresses Oxidative Stress-induced Phosphorylation of Caveolin-1 and c-Abl
It has already been reported that caveolin-1 is phosphorylated on tyrosine 14 under hyperosmotic shock and oxidative stress (17, 18) and that c-Abl, which is an upstream kinase of caveolin-1, is required for oxidative stress-induced phosphorylation of caveolin-1 (19). To study the effect of oxidative stress on the phosphorylation of caveolin-1 and c-Abl in HUVEC, we exposed HUVEC to H2O2 and determined the phosphorylation by Western blotting. Incubation with H2O2 resulted in the phosphorylation of both caveolin-1 and c-Abl dose-dependently (Fig. 1A). To investigate whether AMPK activation inhibits oxidative stress-induced phosphorylation of caveolin-1 and c-Abl, we pretreated HUVEC with a pharmacological activator of AMPK, AICAR, before H2O2 exposure. As shown in Fig. 1, A, B, and C), AICAR significantly suppressed the phosphorylation of both caveolin-1 and c-Abl. Caveolin-1 is the main component of the caveolae plasma membranes and is involved in receptor-independent endocytosis (2, 20). To determine the effect of H2O2 and AICAR on the endocytosis, we evaluated the amount of fluorescein-conjugated albumin endocytosed by HUVEC. Exposure to H2O2 resulted in the elevation of albumin endocytosis together with caveolin-1 phosphorylation (Fig. 1D). By contrast, pretreatment by AICAR suppressed both endocytosis and caveolin-1 phosphorylation (Fig. 1D).
FIGURE 1.

AICAR suppresses phosphorylation of caveolin-1 and c-Abl, and albumin endocytosis under oxidative stress. A, cells were cultured in AICAR (2 mm) containing medium for 2 h and then stimulated with each different concentration (0, 0.5, 1.0, 2.0 mm) of H2O2 for 30 min. The amounts of p-caveolin-1 and p-c-Abl in HUVEC were then examined by Western blotting. B, densitometry of p-caveolin-1 in A is shown. C, densitometry of p-c-Abl in A. D, shown is an albumin endocytosis assay. a, control (untreated cells). b, H2O2 (2 mm) stimulation for 30 min. c, pretreated with AICAR (2 mm) for 2 h. d, pretreated with AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. BSA was conjugated with Alexa 555 (red), p-caveolin (green), VE-cadherin (blue). Scale bar = 50 μm. A, representative blots are shown. *, p < 0.01.
AICAR Inhibits H2O2-induced Phosphorylation of Caveolin-1 via Activation of AMPK
It has been reported that AICAR has several effects independent of AMPK pathway (21–24). To determine the effect of AICAR on AMPK phosphorylation in HUVEC, we investigated phosphorylation of AMPK after AICAR administration by Western blotting. As shown in Fig. 2A, AICAR phosphorylated AMPK dose-dependently. Next, we used two different inhibitors of AICAR, DPY and IODO, to exclude the possibility that the inhibitory effect of AICAR on caveolin-1 phosphorylation was caused by mechanisms other than AMPK activation. DPY blocks adenosine transporters and prevents uptake of AICAR into the cells (11, 25). IODO inhibits adenosine kinase in the cell and prevents conversion of AICAR to AICAR monophosphate (ZMP), which activates AMPK (11, 25). Pretreatment with DPY or IODO inhibited AICAR-induced AMPK phosphorylation dose-dependently (Fig. 2, B and F). Furthermore, pretreatment with DPY or IODO before H2O2 exposure significantly restored the inhibitory effect of AICAR on phosphorylation of both caveolin-1 and c-Abl (Fig. 2). These results indicate that ZMP accumulation through both transport and phosphorylation of AICAR is required for the suppression of caveolin-1 phosphorylation, suggesting that AMPK activation is a key process for the inhibitory effect of AICAR.
FIGURE 2.

AICAR inhibits H2O2-induced phosphorylation of caveolin-1 via activation of AMPK. A, cells were treated with each concentration of AICAR for 2 h. B, cells were treated with each concentration of DPY for 1 h and then stimulated with 2 mm AICAR for 2 h. C, cells were treated with 8 μm DPY for 1 h and then stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. D, densitometry of p-caveolin-1 in C is shown. E, densitometry of p-c-Abl in C is shown. F, cells were treated with each concentration of IODO for 1 h and then stimulated with 2 mm AICAR for 2 h. G, cells were treated with 0.4 μm IODO for 1 h and then stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. H, densitometry of p-caveolin-1 in G is shown. I, densitometry of p-c-Abl in G is shown. A[en]E, representative blots are shown. *, p < 0.01.
Both AMPKα1 and -α2 Isoforms Are Required for the Inhibition of Caveolin-1 Phosphorylation under Oxidative Stress
The catalytic subunit of AMPK, AMPKα, has two isoforms (i.e. AMPKα1 and -α2) that show differential tissue-specific expression (8, 9, 15). To determine the role of both isoforms in the inhibitory effect of AMPK on caveolin-1 phosphorylation under oxidative stress, we used RNA interference technology to knock down AMPKα1 or -α2 in HUVEC. Knockdown of either isoform of AMPKα abolished the inhibitory effect of AICAR on H2O2-induced phosphorylation of caveolin-1 and c-Abl (Fig. 3). Knockdown of both AMPK isoforms with two different siRNA oligos showed similar results (Fig. 4). These results suggest that both AMPKα1 and -α2 isoforms are required to inhibit caveolin-1 phosphorylation under oxidative stress.
FIGURE 3.

Both AMPKα1 and -α2 isoforms are required for the inhibition of caveolin-1 phosphorylation under oxidative stress. A and D, the amounts of p-caveolin-1 and p-c-Abl in HUVEC were examined by Western blotting. Cells were transfected with siRNA against AMPKα1 (A) or α2 (D). Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. B, densitometry of p-caveolin-1 in A is shown. C, densitometry of p-c-Abl in A is shown. E, densitometry of p-caveolin-1 in D is shown. F, densitometry of p-c-Abl in D is shown. A and B, representative blots are shown. *, p < 0.01.
FIGURE 4.

AMPK mediates AICAR effects on c-Abl and caveolin1 phosphorylation. A, cells were co-transfected with both AMPKα1 and -α2 siRNAs (two independent oligos) (α1, (PRKAA1)-CCAUACCCUUGAUGAAUUA; α2 (PRKAA2)-CGACUAAGCCCAAAUCUUU). Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. The amounts of p-c-Abl and p-caveolin-1 were examined by Western blotting. B, densitometry of p-c-Abl in A is shown. C, densitometry of p-caveolin-1 in A is shown. D, cells were co-transfected with both AMPKα1 and -α2 siRNAs (two independent oligos: α1′, (PRKAA1)-GCCCAGAGGUAGAUAUAUG, α2′ (PRKAA2)-GAGCAUGUACCUACGUUAU). Three days after transfection, cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. The amounts of p-c-Abl and p-caveolin-1 were examined by Western blotting. E, densitometry of p-c-Abl in D is shown. F, densitometry of p-caveolin-1 in D is shown. A and D, representative blots are shown. *, p < 0.01; NS, not significant.
Inhibitory Effect of AMPK on Caveolin-1 Phosphorylation under Oxidative Stress Is Dependent on c-Abl
Next, to determine the role of c-Abl in the oxidative stress-induced phosphorylation of caveolin-1, we utilized a c-Abl inhibitor, imatinib mesylate (26, 27). As shown in Fig. 5, A, B, and C, imatinib mesylate inhibited H2O2-induced phosphorylation of both caveolin-1 and c-Abl dose- and time-dependently, indicating that c-Abl is an upstream kinase of caveolin-1 in HUVEC. We next investigated the role of c-Abl in the inhibitory effect of AICAR on canveolin-1 phosphorylation by knockdown c-Abl with siRNA. Deletion of c-Abl resulted in the significant decrease in caveolin-1 phosphorylation after H2O2 exposure (Fig. 5, D and E). Furthermore, pretreatment with AICAR before H2O2 exposure did not change caveolin-1 phosphorylation significantly, suggesting that the inhibitory effect of AICAR on caveolin-1 phosphorylation under oxidative stress is dependent on c-Abl (Fig. 5, D and E).
FIGURE 5.

Inhibitory effect of AMPK on caveolin-1 phosphorylation under oxidative stress is dependent on c-Abl. A, cells were treated with 10 or 20 μm imatinib mesylate for 24, 48, or 72 h before stimulation with H2O2 (2 mm) for 30 min. B, densitometry of p-caveolin-1 in A is shown. C, densitometry of p-c-Abl in A is shown. D, cells were transfected with siRNA against c-Abl. Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by stimulation with H2O2 (2 mm) for 30 min. E, densitometry of p-caveolin-1 in B is shown. A and D, representative blots are shown. *, p < 0.01; NS, not significant.
Prdx1 Is Indispensable for the Inhibitory Effect of AMPK on the H2O2-induced Phosphorylation of Caveolin-1
Prdx1, one of the antioxidant enzymes, plays a protective role in cells against oxidative stress. In cytoplasm, Prdx1 exists as a protein complex with c-Abl-SH domain (28–31) and protects c-Abl from phosphorylation (32). Under oxidative stress, oxidant dissociates the protein-protein interaction and phosphorylates liberated c-Abl. To investigate the role of Prdx1 in the inhibitory effect of AICAR on canveolin-1 phosphorylation, we knocked down Prdx1 in HUVEC with siRNA and determined the level of caveolin-1 phosphorylation by Western blotting. As shown in Fig. 6, A, B, and C, knockdown of Prdx1 resulted in increased phosphorylation of both caveolin-1 and c-Abl after H2O2 exposure. Furthermore, lack of Prdx1 abolished the inhibitory effect of AICAR on the H2O2-induced phosphorylation of both caveolin-1 and c-Abl. These results indicate that Prdx1 is indispensable for the inhibitory effect of AMPK on the H2O2-induced phosphorylation of caveolin-1.
FIGURE 6.

AICAR inhibits caveolin-1 phosphorylation under oxidative stress by suppressing the dissociation between Prdx1 and c-Abl. A, cells were transfected with siRNA against Prdx1. Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. The amounts of p-c-Abl, p-caveolin-1 were examined by Western blotting. B, densitometry of p-caveolin-1 in A is shown. C, densitometry of p-c-Abl in A is shown. D, cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. After total cell lysates of each group were collected, the interaction between Prdx1 and c-Abl was examined by immunoprecipitation with anti-Prdx1 antibody. Immunoprecipitates were then subjected to immunoblotting using anti-c-Abl antibody. E, densitometry of p-c-Abl in D is shown. F, cells were transfected with siRNA against AMPKα1 or -α2. Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. After total cell lysates of each group were collected, the interaction between c-Abl and Prdx1 was examined by immunoprecipitation (IP) with anti-Prdx1 antibody. Immunoprecipitates were then subjected to immunoblotting using anti-c-Abl antibody. G, densitometry of c-Abl in F. H, cells were transfected with siRNA against AMPKα1 or -α2. Three days after transfection cells were stimulated with 2 mm AICAR for 2 h followed by H2O2 (2 mm) stimulation for 30 min. After total cell lysates of each group were collected, the interaction between total AMPK and Prdx1 or c-Abl was examined by immunoprecipitation with anti-total AMPK antibody. Immunoprecipitates were then subjected to immunoblotting using anti-c-Abl and Prdx1 antibody. A, D, and F, representative blots are shown. *, p < 0.01; NS, not significant.
AMPK Inhibits Caveolin-1 Phosphorylation under Oxidative Stress by Suppressing the Dissociation between Prdx1 and c-Abl
To investigate the relationship between AMPK and protein interaction of c-Abl and Prdx1, we performed co-immunoprecipitation experiments. As shown in Fig. 6D, oxidative stress resulted in the dissociation between Prdx1 and c-Abl. The dissociation was inhibited by treatment with AICAR before H2O2. In contrast, treatment with IODO before H2O2 and AICAR restored the dissociation, indicating AICAR inhibits the H2O2-induced dissociation between c-Abl and Prdx1. To confirm this, we further conducted co-immunoprecipitation for the cell lysates from HUVEC lacking AMPKα1 or -α2. Deletion of either AMPKα1 or -α2 isoform decreased the inhibitory effect of AICAR on the dissociation between c-Abl and Prdx1 (Fig. 6, F and G). These results indicated that activation of AMPK inhibits caveolin-1 phosphorylation under oxidative stress by suppressing the dissociation between Prdx1 and c-Abl.
AMPK Is Not Detected in the Prdx1·c-Abl Complex
To further investigate the mechanism, we asked if AMPK directly associates with the c-Abl·Prdx1 complex. Co-immunoprecipitation experiments (Fig. 6H) failed to show any direct association. This could be because the association is very weak or because the effects of AMPK on the Prdx1·c-Abl complex are indirect.
DISCUSSION
In the present study we provide evidence that activation of AMPK suppresses both caveolin-1 phosphorylation and endocytosis in HUVEC under oxidative stress and stabilizes Prdx1·c-Abl complex. These results reveal for the first time to the best of our knowledge the suppressive role of AMPK in oxidation-induced caveolin-1 and c-Abl phosphorylation and in stabilization of the Prdx1·c-Abl complex.
Our finding of negative control of AMPK on caveolin-1 phosphorylation complements the work by Levine et al. (33) where they showed that siRNA-mediated knockdown of caveolin-1 significantly enhanced AMPK phosphorylation, suggesting that AMPK is negatively regulated by caveolin-1. Thus our work together with the work of Levine et al. (33) suggests a negative feedback loop between AMPK and caveolin-1. This loop has not been described before.
The initial observed effects of AICAR were mediated by AMPK. First, AICAR activated AMPK in HUVEC dose-dependently (Fig. 2A). Second, two different inhibitors of AICAR entry into the cell and activation of AMP, DPY, and IODO suppressed phosphorylation of AMPK and restored the inhibitory effect of AICAR on caveolin-1 phosphorylation (Fig. 2). Finally, knockdown of AMPKα by two different oligo siRNA abolished the inhibitory effect of AICAR on H2O2-induced phosphorylation of caveolin-1 (Fig. 3). These results reveal for the first time to our knowledge the suppressive role of AMPK in the oxidative stress-induced caveolin-1 phosphorylation. Although the possibility of caveolin-1 phosphorylation as a therapeutic target for oxidant-mediated vascular diseases has been indicated, only Wogonin, an O-methylated flavone compound that is found in the plant Scutellaria baicalensis, has been shown to inhibit caveolin-1 phosphorylation in HUVEC under oxidative stress (34). Here, we identified AICAR as a novel chemical inhibitor of caveolin-1, thus adding it to the short list of known inhibitory reagent of caveolin-1 phosphorylation.
Using a specific inhibitor of c-Abl, imatinib mesylate, we showed that c-Abl is upstream of oxidative stress-induced caveolin-1 phosphorylation in HUVEC. Furthermore, by knocking down c-Abl by siRNA, we found that AMPK requires c-Abl for its inhibitory effect on caveolin-1 phosphorylation. These results indicate that AMPK suppresses caveolin-1 phosphorylation by inhibiting c-Abl activation. Although it has been shown that caveolin-1 is a substrate for kinases of the c-Abl and Src families (18, 35–37), the importance of these kinases in caveolin-1 phosphorylation remains unclear. Using fibroblasts derived from c-Abl or Src knock-out mice, Sanguinetti and Mastick (19) showed that c-Abl phosphorylates caveolin-1 independently of Src. In contrast, in pulmonary microvascular endothelial cells, Sun et al. (2, 20, 38) reported that c-Abl activation and subsequent caveolin-1 phosphorylation is in part dependent on Src activity. They further mentioned that the c-Abl pathway becomes more important particularly when higher concentrations of H2O2 are used to stimulate caveolin-1 phosphorylation. In this study imatinib mesylate, a c-Abl inhibitor, and c-Abl siRNA did inhibit H2O2-induced caveolin-1 phosphorylation but failed to abolish the phosphorylation completely (Fig. 5). These results suggest two possibilities; 1) involvement of a c-Abl independent pathway such as Src pathway and 2) increase in the contributing rate of c-Abl dependent pathway on caveolin-1 phosphorylation by a high concentration of H2O2 (2 mm), which we applied to HUVEC in the present study (19, 39, 40).
Recent studies have shown that AMPK plays a protective role in the oxidative stress-induced vascular endothelial dysfunction. However, the precise mechanism by which AMPK protects the endothelium from oxidative stress remains unclear. Ceolotto et al. (41) reported that AMPK protects HUVEC against glucose-induced oxidative stress by suppressing hyperactivity of NAD(P)H oxidase. Deng et al. (42) reported that AMPK protects human aortic endothelium by increasing NO production via endothelial NOS phosphorylation. In contrast to these studies, the present study revealed that AMPK protects the endothelium by inhibiting the dissociation between c-Abl and Prdx1. The activity of c-Abl is mostly inactive in cells and is highly regulated by other proteins, which bind the SH3 domain of c-Abl. Among a number of known SH3 binding proteins, Prdx1 is the principal protein that inhibits c-Abl activation (28–30). Under oxidative stress, reactive oxygen species dissociate c-Abl from Prdx1 and activates c-Abl. Our results using siRNA and co-immunoprecipitation of Prdx1 indicate that AMPK inhibits oxidative stress-induced c-Abl activation by suppressing the dissociation between c-Abl and Prdx1. To our knowledge this is the first report showing the protective role of AMPK in the association between c-Abl and Prdx1. To further investigate the mechanism, we asked if AMPK directly associates with the c-Abl·Prdx1 complex. Co-immunoprecipitation experiments (Fig. 6H) failed to show any direct association. This could be because the association is very weak or because the effects of AMPK on the Prdx1·c-Abl complex are indirect.
The complex of c-Abl·Prdx1 is thought to be a major target of oxidative stress and has been shown to play an important role in the pathogenesis of vascular disease, such as atherosclerosis and diabetes mellitus (43–46) as well as cancer and other aging-related diseases such as osteoporosis (47). Further study using disease-model animal is required to elucidate the significance of protective role of AMPK on c-Abl·Prdx1 complex in these diseases.
The catalytic subunit of AMPK has two isoforms, i.e. AMPKα1 and -α2. Recent studies have shown that these isoforms play a differential role in endothelial functions (16, 48–50). We initially speculated that the α1 isoform of AMPK will be the one to suppress caveolin-1 phosphorylation under oxidative stress, as Creighton et al. (16) reported that AMPKα1 colocalizes with caveolin-1 membranes in pulmonary microvascular endothelial cells. Surprisingly, our results using siRNA toward each specific isoform indicated that both AMPKα1 and -α2 isoforms are required to inhibit caveolin-1 phosphorylation under oxidative stress (Fig. 3). This result is consistent with the previous study showing that AMPKα1 and -α2 isoforms are equally important in the inhibition of lipopolysaccharide-induced endothelial hyperpermeability in pulmonary artery endothelial cells (51). Recently, it was reported that the AMPKβ subunit is not only a scaffold that assembles α and γ subunits but also determines the subcellular localization and substrate specificity of the AMPK heterotrimer complex (25, 52). Thus, further studies understanding the roles of AMPKβ and -γ subunits in the inhibition of caveolin-1 phosphorylation are needed.
In conclusion, we identified AMPK as a novel negative regulator of caveolin-1 phosphorylation in HUVEC under oxidative stress by at least partially stabilizing the Prdx1·c-Alb complex (Fig. 7). This finding along with the finding of negative regulation of AMPK by caveolin-1 by Levine et al. (33) suggest the presence of a negative feedback loop between these two molecules. Because caveolin-1 and AMPK play an important role in major diseases such as cancer, atherosclerosis, diabetes, and inflammation (11–15, 50, 51), our findings might provide insights not only into the regulatory mechanism of caveolin-1 phosphorylation and the function of AMPK but also into therapeutic targets of these diseases.
FIGURE 7.
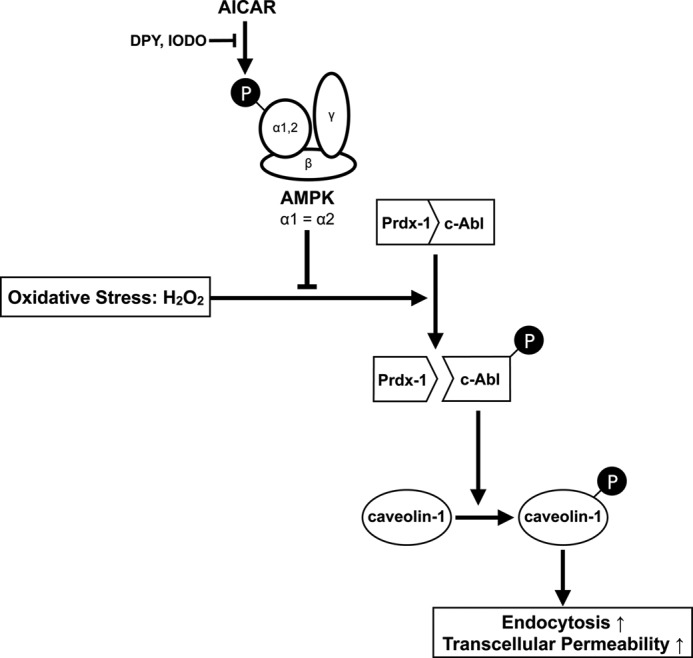
Proposed model for the mechanism by which AMPK suppresses caveolin-1 phosphorylation and endocytosis under oxidative stress. AMPK negatively regulates caveolin-1 phosphorylation by suppressing the dissociation between c-Abl and Prdx-1.
This work was supported, in whole or in part, by National Institutes of Health Grant EY014104 (NEI). This work was also supported by an unrestricted grant to Massachusetts Eye and Ear Infirmary from the Research to Prevent Blindness Foundation (to D. G. V. and J. W. M.), the Lions Eye Research Fund (to D. G. V. and J. W. M.), the Fight for Sight Grant in Aid (to D. G. V.), the Harvard Ophthalmology Department (to D. G. V.), Yeatts Family Fund (to D. G. V. and J. W. M.), and a Bausch and Lomb Japan Vitreoretinal fellowship (to K. T., Y. M., J. S., and M. K.).
- Prdx1
- peroxiredoxin I
- HUVEC
- human umbilical vein endothelial cell(s)
- AICAR
- 5-amino-4-imidazole carboxamide riboside
- IODO
- 5-iodotubericidin
- DPY
- dipyridamole.
REFERENCES
- 1. Mehta D., Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 2. Sun Y., Minshall R. D., Hu G. (2011) Role of caveolin-1 in the regulation of pulmonary endothelial permeability. Methods Mol. Biol. 763, 303–317 [DOI] [PubMed] [Google Scholar]
- 3. Minshall R. D., Sessa W. C., Stan R. V., Anderson R. G., Malik A. B. (2003) Caveolin regulation of endothelial function. Am. J. Physiol. Lung Cell Mol. Physiol. 285, L1179–L1183 [DOI] [PubMed] [Google Scholar]
- 4. Komarova Y., Malik A. B. (2010) Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 72, 463–493 [DOI] [PubMed] [Google Scholar]
- 5. Razani B., Engelman J. A., Wang X. B., Schubert W., Zhang X. L., Marks C. B., Macaluso F., Russell R. G., Li M., Pestell R. G., Di Vizio D., Hou H., Jr., Kneitz B., Lagaud G., Christ G. J., Edelmann W., Lisanti M. P. (2001) Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276, 38121–38138 [DOI] [PubMed] [Google Scholar]
- 6. Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F. C., Schedl A., Haller H., Kurzchalia T. V. (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452 [DOI] [PubMed] [Google Scholar]
- 7. Hu G., Minshall R. D. (2009) Regulation of transendothelial permeability by Src kinase. Microvasc. Res. 77, 21–25 [DOI] [PubMed] [Google Scholar]
- 8. Hardie D. G., Hawley S. A. (2001) AMP-activated protein kinase. The energy charge hypothesis revisited. Bioessays 23, 1112–1119 [DOI] [PubMed] [Google Scholar]
- 9. Viollet B., Athea Y., Mounier R., Guigas B., Zarrinpashneh E., Horman S., Lantier L., Hebrard S., Devin-Leclerc J., Beauloye C., Foretz M., Andreelli F., Ventura-Clapier R., Bertrand L. (2009) AMPK. Lessons from transgenic and knockout animals. Front. Biosci. 14, 19–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang W., Guan K. L. (2009) AMP-activated protein kinase and cancer. Acta Physiol (Oxf) 196, 55–63 [DOI] [PubMed] [Google Scholar]
- 11. Theodoropoulou S., Kolovou P. E., Morizane Y., Kayama M., Nicolaou F., Miller J. W., Gragoudas E., Ksander B. R., Vavvas D. G. (2010) Retinoblastoma cells are inhibited by aminoimidazole carboxamide ribonucleotide (AICAR) partially through activation of AMP-dependent kinase. FASEB J. 24, 2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Theodoropoulou S., Brodowska K., Kayama M., Morizane Y., Miller J. W., Gragoudas E. S., Vavvas D. G. (2013) Aminoimidazole carboxamide ribonucleotide (AICAR) inhibits the growth of retinoblastoma in vivo by decreasing angiogenesis and inducing apoptosis. PLoS ONE 8, e52852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki J., Manola A., Murakami Y., Morizane Y., Takeuchi K., Kayama M., Miller J. W., Sobrin L., Vavvas D. G. (2011) Inhibitory effect of aminoimidazole carboxamide ribonucleotide (AICAR) on endotoxin-induced uveitis in rats. Invest. Ophthalmol. Vis. Sci. 52, 6565–6571 [DOI] [PubMed] [Google Scholar]
- 14. Suzuki J., Yoshimura T., Simeonova M., Takeuchi K., Murakami Y., Morizane Y., Miller J. W., Sobrin L., Vavvas D. G. (2012) Aminoimidazole carboxamide ribonucleotide ameliorates experimental autoimmune uveitis. Invest Ophthalmol Vis. Sci. 53, 4158–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morizane Y., Thanos A., Takeuchi K., Murakami Y., Kayama M., Trichonas G., Miller J., Foretz M., Viollet B., Vavvas D. G. (2011) AMP-activated protein kinase suppresses matrix metalloproteinase-9 expression in mouse embryonic fibroblasts. J. Biol. Chem. 286, 16030–16038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Creighton J., Jian M., Sayner S., Alexeyev M., Insel P. A. (2011) Adenosine monophosphate-activated kinase α1 promotes endothelial barrier repair. FASEB J. 25, 3356–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Volonté D., Galbiati F., Pestell R. G., Lisanti M. P. (2001) Cellular stress induces the tyrosine phosphorylation of caveolin-1 (Tyr-14) via activation of p38 mitogen-activated protein kinase and c-Src kinase. Evidence for caveolae, the actin cytoskeleton, and focal adhesions as mechanical sensors of osmotic stress. J. Biol. Chem. 276, 8094–8103 [DOI] [PubMed] [Google Scholar]
- 18. Aoki T., Nomura R., Fujimoto T. (1999) Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp. Cell Res. 253, 629–636 [DOI] [PubMed] [Google Scholar]
- 19. Sanguinetti A. R., Mastick C. C. (2003) c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell. Signal. 15, 289–298 [DOI] [PubMed] [Google Scholar]
- 20. Sun Y., Hu G., Zhang X., Minshall R. D. (2009) Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 105, 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 22. Guigas B., Taleux N., Foretz M., Detaille D., Andreelli F., Viollet B., Hue L. (2007) AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 404, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mukhtar M. H., Payne V. A., Arden C., Harbottle A., Khan S., Lange A. J., Agius L. (2008) Inhibition of glucokinase translocation by AMP-activated protein kinase is associated with phosphorylation of both GKRP and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R766–R774 [DOI] [PubMed] [Google Scholar]
- 24. Foretz M., Hébrard S., Leclerc J., Zarrinpashneh E., Soty M., Mithieux G., Sakamoto K., Andreelli F., Viollet B. (2010) Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 120, 2355–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanz P. (2008) AMP-activated protein kinase. Structure and regulation. Curr. Protein Pept. Sci. 9, 478–492 [DOI] [PubMed] [Google Scholar]
- 26. Müller B. A. (2009) Imatinib and its successors. How modern chemistry has changed drug development. Curr. Pharm. Des. 15, 120–133 [DOI] [PubMed] [Google Scholar]
- 27. Druker B. J. (2008) Translation of the Philadelphia chromosome into therapy for CML. Blood 112, 4808–4817 [DOI] [PubMed] [Google Scholar]
- 28. Wen S. T., Van Etten R. A. (1997) The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 11, 2456–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prospéri M. T., Ferbus D., Rouillard D., Goubin G. (1998) The pag gene product, a physiological inhibitor of c-abl tyrosine kinase, is overexpressed in cells entering S phase and by contact with agents inducing oxidative stress. FEBS Lett. 423, 39–44 [DOI] [PubMed] [Google Scholar]
- 30. Cao J., Schulte J., Knight A., Leslie N. R., Zagozdzon A., Bronson R., Manevich Y., Beeson C., Neumann C. A. (2009) Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 28, 1505–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morell M., Espargaró A., Avilés F. X., Ventura S. (2007) Detection of transient protein-protein interactions by bimolecular fluorescence complementation. The Abl-SH3 case. Proteomics 7, 1023–1036 [DOI] [PubMed] [Google Scholar]
- 32. Wong C. M., Zhou Y., Ng R. W., Kung Hf H. F., Jin D. Y. (2002) Cooperation of yeast peroxiredoxins Tsa1p and Tsa2p in the cellular defense against oxidative and nitrosative stress. J. Biol. Chem. 277, 5385–5394 [DOI] [PubMed] [Google Scholar]
- 33. Levine Y. C., Li G. K., Michel T. (2007) Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J. Biol. Chem. 282, 20351–20364 [DOI] [PubMed] [Google Scholar]
- 34. Wang F., Song X., Zhou M., Wei L., Dai Q., Li Z., Lu N., Guo Q. (2013) Wogonin inhibits H2O2-induced vascular permeability through suppressing the phosphorylation of caveolin-1. Toxicology 305, 10–19 [DOI] [PubMed] [Google Scholar]
- 35. Rothberg K. G., Heuser J. E., Donzell W. C., Ying Y. S., Glenney J. R., Anderson R. G. (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68, 673–682 [DOI] [PubMed] [Google Scholar]
- 36. Glenney J. R., Jr., Soppet D. (1992) Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 89, 10517–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ko Y. G., Liu P., Pathak R. K., Craig L. C., Anderson R. G. (1998) Early effects of pp60(v-src) kinase activation on caveolae. J. Cell. Biochem. 71, 524–535 [PubMed] [Google Scholar]
- 38. Sun S. W., Zu X. Y., Tuo Q. H., Chen L. X., Lei X. Y., Li K., Tang C. K., Liao D. F. (2010) Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol. Sin. 31, 1336–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar S., Mishra N., Raina D., Saxena S., Kufe D. (2003) Abrogation of the cell death response to oxidative stress by the c-Abl tyrosine kinase inhibitor STI571. Mol. Pharmacol. 63, 276–282 [DOI] [PubMed] [Google Scholar]
- 40. Cao H., Courchesne W. E., Mastick C. C. (2002) A phosphotyrosine-dependent protein interaction screen reveals a role for phosphorylation of caveolin-1 on tyrosine 14. Recruitment of C-terminal Src kinase. J. Biol. Chem. 277, 8771–8774 [DOI] [PubMed] [Google Scholar]
- 41. Ceolotto G., Gallo A., Papparella I., Franco L., Murphy E., Iori E., Pagnin E., Fadini G. P., Albiero M., Semplicini A., Avogaro A. (2007) Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 27, 2627–2633 [DOI] [PubMed] [Google Scholar]
- 42. Deng G., Long Y., Yu Y. R., Li M. R. (2010) Adiponectin directly improves endothelial dysfunction in obese rats through the AMPK-eNOS Pathway. Int. J. Obes. (Lond.) 34, 165–171 [DOI] [PubMed] [Google Scholar]
- 43. Lassila M., Allen T. J., Cao Z., Thallas V., Jandeleit-Dahm K. A., Candido R., Cooper M. E. (2004) Imatinib attenuates diabetes-associated atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 935–942 [DOI] [PubMed] [Google Scholar]
- 44. Hägerkvist R., Makeeva N., Elliman S., Welsh N. (2006) Imatinib mesylate (Gleevec) protects against streptozotocin-induced diabetes and islet cell death in vitro. Cell Biol. Int. 30, 1013–1017 [DOI] [PubMed] [Google Scholar]
- 45. Hägerkvist R., Sandler S., Mokhtari D., Welsh N. (2007) Amelioration of diabetes by imatinib mesylate (Gleevec). Role of β-cell NF-κB activation and anti-apoptotic preconditioning. FASEB J. 21, 618–628 [DOI] [PubMed] [Google Scholar]
- 46. Louvet C., Szot G. L., Lang J., Lee M. R., Martinier N., Bollag G., Zhu S., Weiss A., Bluestone J. A. (2008) Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 18895–18900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jeyabalan J., Shah M., Viollet B., Chenu C. (2012) AMP-activated protein kinase pathway and bone metabolism. J. Endocrinol. 212, 277–290 [DOI] [PubMed] [Google Scholar]
- 48. Gayard M., Guilluy C., Rousselle A., Viollet B., Henrion D., Pacaud P., Loirand G., Rolli-Derkinderen M. (2011) AMPK α1-induced RhoA phosphorylation mediates vasoprotective effect of estradiol. Arterioscler. Thromb. Vasc. Biol. 31, 2634–2642 [DOI] [PubMed] [Google Scholar]
- 49. Goirand F., Solar M., Athea Y., Viollet B., Mateo P., Fortin D., Leclerc J., Hoerter J., Ventura-Clapier R., Garnier A. (2007) Activation of AMP kinase α1 subunit induces aortic vasorelaxation in mice. J. Physiol. 581, 1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bess E., Fisslthaler B., Frömel T., Fleming I. (2011) Nitric oxide-induced activation of the AMP-activated protein kinase α2 subunit attenuates IκB kinase activity and inflammatory responses in endothelial cells. PLoS ONE 6, e20848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xing J., Wang Q., Coughlan K., Viollet B., Moriasi C., Zou M. H. (2013) Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 182, 1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warden S. M., Richardson C., O'Donnell J., Jr., Stapleton D., Kemp B. E., Witters L. A. (2001) Post-translational modifications of the β-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem. J. 354, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]