

Background: Rubisco activase optimizes photosynthesis in plants, yet the arrangement of subunits is unclear.
Results: The oligomeric state and biological function of Rubisco activase are dependent on protein concentration.
Conclusion: Rubisco activase does not need to be hexameric for full activity.
Significance: Understanding the functioning of Rubisco activase is an important step in determining how it regulates Rubisco.
Keywords: Analytical Ultracentrifugation, Protein Assembly, Protein Complexes, Protein Structure, X-ray Scattering, AAA+ Protein, Rubisco Activase
Abstract
Ribulose-bisphosphate carboxylase/oxygenase (Rubisco) activase uses the energy from ATP hydrolysis to remove tight binding inhibitors from Rubisco, thus playing a key role in regulating photosynthesis in plants. Although several structures have recently added much needed structural information for different Rubisco activase enzymes, the arrangement of these subunits in solution remains unclear. In this study, we use a variety of techniques to show that Rubisco activase forms a wide range of structures in solution, ranging from monomers to much higher order species, and that the distribution of these species is highly dependent on protein concentration. The data support a model in which Rubisco activase forms an open spiraling structure rather than a closed hexameric structure. At protein concentrations of 1 μm, corresponding to the maximal activity of the enzyme, Rubisco activase has an oligomeric state of 2–4 subunits. We propose a model in which Rubisco activase requires at least 1 neighboring subunit for hydrolysis of ATP.
Introduction
Rubisco3 activase maintains maximal photosynthetic rates by using the energy from ATP hydrolysis to release tight binding inhibitors from the active site of Rubisco. Recent studies have confirmed that Rubisco activase is a member of the AAA+ family; however, the exact arrangement of subunits is uncertain. Although the crystal structure of tobacco Rubisco activase has a helical structure, with 6 subunits/turn, it is suggested that the enzyme forms closed hexamers in solution (1). Other members of the AAA+ family have been observed to form either closed hexamers or open spirals in solution (2), and it is unclear from the crystal structure which model is preferred for Rubisco activase.
Rubisco catalyzes the incorporation of inorganic carbon into the biosphere through the carboxylation of the 5-carbon sugar ribulose 1,5-bisphosphate, yet it is hindered by a tendency for inhibitory compounds to bind to the active site, forming a dead-end complex. The substrate, ribulose-P2, itself can act as an inhibitor by binding to uncarbamylated active sites (3, 4). Some plant species, such as tobacco and bean, produce a competitive inhibitor, carboxyarabinitol 1-P, which binds to carbamylated sites and is used as a means to down-regulate Rubisco activity (5, 6). This inhibitor is subsequently degraded by a light-dependent phosphatase (7). In a similar pattern, Rubisco can also accumulate inhibitors during the day through the formation of inhibitory compounds during catalysis that remain bound to the active site and hinder catalysis (8, 9). These inhibitors include pentodiulose-P2 and xylulose-P2, and the in vitro formation of these compounds has been well characterized (10–18).
To accelerate the process of inhibitor release, plants contain Rubisco activase, an AAA+ enzyme that interacts with Rubisco and facilitates the release of inhibitors from the active site in a process that requires energy from ATP hydrolysis (for review, see Refs. 19–22). AAA+ proteins are characterized by a common conserved ATPase domain and have a wide range of functions, including protein degradation, DNA replication, signal transduction, and intracellular transport, but with the common activity of the unfolding of macromolecules (for review, see Ref. 2). These AAA+ proteins usually assemble into complexes that form a ring-shaped structure with a central pore and couple the energy from ATP hydrolysis to changes in conformation. Whereas most AAA+ proteins form hexamers, other members of the AAA+ family have been observed to form spiral assemblies in nature, including clamp-loader and initiator proteins (23, 24). The structure and mechanism of CbbX, a red Rubisco activase, have recently been elucidated, showing that it exists as a closed hexamer that can stack to form long filaments (25).
Despite the presence of high resolution crystal structures, it is still uncertain what structure Rubisco activase adopts in solution. The structures of the substrate recognition domain of creosote Rubisco activase (26) and an N- and C-terminally truncated tobacco Rubisco activase (1) have recently been published, showing that the enzyme has a classical AAA+ protein domain architecture. Previous studies have shown that Rubisco activase has a high degree of polydispersity in its oligomeric forms and can form a variety of species ranging from monomers to large 660-kDa complexes, which would correspond to 16 subunits (1, 27–33). Nanoelectrospray ionization mass spectrometry studies of tobacco Rubisco activase showed polydispersity in its oligomeric forms, with the largest species observed being a hexamer (29). Recent studies using fluorescence fluctuation spectroscopy found that in the presence of ADP, cotton Rubisco activase is predominantly monomeric below 0.5 μm and forms complexes larger than a hexamer above 40 μm (27). Oligomer size is influenced by a range of factors, including polyethylene glycol, the presence of which increases the oligomeric state (31), and nucleotide phosphates. ATP has been observed to increase the molecular mass of spinach and cotton Rubisco activase (28, 30, 32) but has less of an effect on the tobacco enzyme (32).
There is a correlation of protein concentration with ATPase and Rubisco activation activity, showing that the biological function of Rubisco activase is critically linked to the oligomeric state (1, 30, 34). Wang et al. (30) observed that the specific activity of ATP hydrolysis by spinach Rubisco activase increased with higher concentrations of enzyme, with maximum activity reached at 1–2 μm. Lilley and Portis (34) showed that spinach Rubisco activase reached a maximum activity of ADP release, and activation of ribulose-P2 bound Rubisco above 1 μm, with lower activities below this concentration. Activity studies using mixtures of wild-type and Rubisco activase mutants suggested that a minimum of 3–5 subunits are required for Rubisco activation activity (1). However, there have been no studies to date that directly correlate the subunit assembly in solution with biological activity.
Several studies have concluded that Rubisco activase forms hexamers in solution and that this may be the active form. The structure of tobacco Rubisco activase forms a helical arrangement in the crystal structure, with 6 subunits/turn (1), and a hexameric arrangement would be consistent with many AAA+ proteins. Several studies have focused on the effects of mutating the Arg-294 residue of tobacco Rubisco activase on quaternary structure. This residue is located in the sensor 2 region of the enzyme, which is associated with recognition of the Rubisco substrate (35) and is involved with interactions at the intersubunit interface (1). In the presence of the substrate analog ATPγS, R294A was found to form stable complexes with a mass consistent with a hexamer (29), whereas negative stain EM showed that the R294V mutant forms ring structures with 6-fold symmetry in the presence of ATP or ATPγS (1).
Binding of ATP or ATPγS to Rubisco activase triggers a change in protein conformation, as observed by changes in the intrinsic fluorescence, but the effect of nucleotide binding on oligomeric state is less defined. It has been proposed that the increase in intrinsic fluorescence upon binding of ATP or ATPγS to Rubisco activase reflects a change in conformational state associated with Trp-109 and Trp-250 (30, 36). Trp-250 is located adjacent to the hinge region between the large and small AAA+ domains, close to where nucleotides bind, and changes in conformation are likely to involve movement of the small domain relative to the large domain, as occurs in the closely related ClpX and FtsH proteins (37, 38).
It has been suggested that ATP and ADP may increase the oligomerization state of Rubisco activase. Gel filtration studies of spinach Rubisco activase show that the presence of ADP did not affect the molecular size, whereas Rubisco activase incubated in the presence of Mg2+ and ATP or ATPγS was significantly larger (30). Similar studies of the tobacco Rubisco enzyme showed little difference in size between enzyme incubated with ADP or ATP (32). Dynamic light scattering studies of cotton β-activase report that ADP, ATP, or MgCl2 increase the degree of Rubisco activase self-association (33). It has been proposed that Rubisco activase may have functional similarities to actin (34), in that ATP could promote the formation of higher order species.
Within the AAA+ family, there are several instances in which the presence of ATP induces hexamer formation, including ClpA (39), which forms hexameric rings only in the presence of ATP. All initiator complexes, such as Aquifex aeolicus DnaA, require ATP to form a higher order complex from monomers (24).
The assembly pathway of Rubisco activase is poorly characterized, and formation of a hexameric intermediate could involve several different mechanisms. First, stepwise addition of monomers could generate dimeric, trimeric, tetrameric, and pentameric intermediates, which may be similar to isodesmic systems in which the dissociation constant for monomer addition to any species is considered to be the same. This mechanism could lead to the formation of helical units with 6-fold symmetry, such as those observed for the tobacco Rubisco activase crystal structure. It has been suggested that Rubisco activase may also form spiraling arrangements in solution (27, 33). Second, hexamer formation could proceed via a dimeric and tetrameric intermediate, which has been observed for the formation of hexamers by ClpA (39). This AAA+ protease assembles into hexamers via a tetrameric intermediate, and hexamer formation is favored by the presence of ATP. Finally, hexamers could be formed via a trimeric intermediate. Studies utilizing fluorescence fluctuation spectroscopy of cotton Rubisco activase in the presence of Mg·ADP used a model based on a monomer-dimer-tetramer-hexamer system (27).
In the current study we describe experiments aiming to determine the biologically relevant state of Rubisco activase in an effort to better model the mechanism by which Rubisco activase regulates Rubisco. A better understanding of this process will provide insights into how photosynthesis may be optimized and appreciating how plants will adapt to climate change (40).
EXPERIMENTAL PROCEDURES
Expression and Purification
Plasmids encoding tobacco Rubisco activase were a kind gift from Dr. Spencer Whitney (Australian National University). These plasmids contained the rca gene amplified from a tobacco cDNA preparation and cloned into pET28A+ incorporating a C-terminal His7 tag. Protein expression was performed as described previously (41). Protein concentration was determined by A280, and the A280/260 ratio was monitored to ensure that nucleotide was not carried over.
ATP Hydrolysis Measurements
ATP hydrolysis of Rubisco activase was assayed by coupling ADP production to NADH oxidation, as reported previously (36).
Rubisco Reactivation Activity
Rubisco activity was followed by continuous measurement of glycerate 3-P formation, using tobacco ER that was formed as has been described previously (17, 18). Rubisco activase was added to the assay mixture 30 s prior to addition of Rubisco. Rubisco reactivation rates were determined by measuring the Rubisco activity after 3 min relative to the background rate of Rubisco activation.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman Coulter model XL-I analytical ultracentrifuge equipped with UV-visible scanning optics. Reference (380 μl; 20 mm Bistris propane, 20 mm KCl, 0.2 mm EDTA, pH 8.0) and sample (360 μl) solutions were loaded into 12-mm double-sector cells with quartz windows, and the cells were then mounted in an An-60 Ti four-hole rotor. Proteins were centrifuged at various rotor speeds at 20 °C, and radial absorbance data were collected at appropriate wavelengths in continuous mode every 8 min without averaging. Data were fitted to a continuous size-distribution (cs) model using the program SEDFIT (42, 43). The partial specific volume (¯v) of the proteins (0.735 ml g−1), buffer density (1.005 g·ml−1), and buffer viscosity (1.021 cp) were computed using the program SEDNTERP (44). Weight average molecular masses were calculated from weight average sedimentation coefficients and f/f0 values resulting from the fits using SEDFIT (42, 43).
Size-exclusion Chromatography
Gel filtration was carried out at 28 °C using a Superdex 200 10/300 GL column (GE Healthcare). 100 μl of enzyme (18–142 μm) was loaded onto the column and eluted with 20 mm Bistris propane, 20 mm KCl, 0.2 mm EDTA, pH 8.0, at 0.5 ml/min. A Viscotek TDA unit was used to measure the refractive index, low angle and right angle light scattering, and viscosity. BSA was used as a standard to calibrate the instrument.
Small Angle X-ray Scattering (SAXS) Measurements
Measurements were performed at the Australian Synchrotron SAXS/WAXS beamline equipped with a Pilatus 1M detector (170 mm ×170 mm, effective pixel size, 172 × 172 μm). The wavelength of the x-rays was 1.0332 Å. Two sample-detector distances were used, 1576 and 7000 mm, which provided a q range of 0.006–0.400 and 0.0015–0.09 Å−1, respectively (where q is the magnitude of the scattering vector, which is related to the scattering angle (2θ) and the wavelength (λ) as follows: q = (4π/λ)sinθ). Data were collected using a 1.5-mm glass capillary at 10 °C under continuous flow in 2-s intervals in buffer containing 20 mm Bistris propane, 20 mm KCl, 0.2 mm EDTA, pH 8.0. Two-dimensional intensity plots were radially averaged, normalized to sample transmission, and background-subtracted.
SAXS Data Analysis
Data sets for both camera lengths were merged manually, and data reduction was performed using the ATSAS package (45, 46). Guinier fits were produced using PRIMUS (47). Indirect Fourier transform was performed using GNOM (48) to yield the function Pr, which gives both the relative probabilities of distances between scattering centers and the maximum dimension of the scattering particle Dmax. Theoretical scattering curves were generated from atomic coordinates (Protein Data Bank ID codes 3T15 and 3ZW6) and compared with experimental scattering curves using CRYSOL (49). Hydrated particle volumes were extracted from the SAXS data using the program AUTOPOROD (45).
Equilibrium Modeling
Structural models of a putative monomer-hexamer equilibrium were reconstructed ab initio from the lowest concentration SAXS data using the program GASBORMX (45). A Rubisco activase monomer was defined as a random gas of 390 dummy residues and the simultaneous refinement of a monomer-hexamer equilibrium initiated by applying P6 symmetry. The program OLIGOMER (46) was used to estimate the volume fractions of the GASBORMX-derived monomer and hexamer at higher concentrations.
RESULTS
To directly correlate the solution structure of Rubisco activase with biological function, we have used a variety of techniques to investigate the quaternary structure of Rubisco activase at different protein concentrations. The results show that the size of the Rubisco activase oligomer in solution is strongly dependent on protein concentration and ranges from dimeric/monomeric at low concentrations (<0.5 μm) up to complexes larger than a hexamer at higher concentrations (>10 μm) (Fig. 1).
FIGURE 1.

Dependence of activity and oligomeric size on protein concentration. Rubisco activation activity (circles, left axis) and specific ATPase activity (blue squares, right axis) was measured for a range of protein concentrations (top panel). Molecular mass was calculated for a range of protein concentrations using SAXS (black squares), static light scattering (SLS) (dotted lines), and AUC (red circles) as described under “Experimental Procedures.”
Biological Activity
ATPase activity and Rubisco reactivation activity of tobacco Rubisco activase were measured at a range of protein concentrations. The specific activity increased from 0.1 to 1.5 μm, (Fig. 1), as has been previously observed for Rubisco activase (30, 34).
Analytical Ultracentrifugation (AUC)
Sedimentation velocity runs were carried out for a series of Rubisco activase concentrations, ranging from 0.6 to 66 μm protomer (Figs. 1 and 2 and Table 1). Poor signal for the AUC experiments was observed in this study below 600 nm, precluding analysis of conditions at low protein concentrations. Work with cotton B-Rubisco activase has shown that it is monomeric at concentrations below 300 nm (27). The data showed that Rubisco activase exists as a heterogeneous mixture in solution at each concentration, with an integrated sedimentation coefficient ranging from 4.6 S at 600 nm to 16.4 S at 67 μm (Table 1). Whereas sedimentation coefficient distributions (cs) have the advantage of not requiring any prior knowledge of the type of system, they are based on equations for noninteracting systems, and the results will be influenced by reactions on the time scale of sedimentation. Appearance of peaks in the cs distributions provide an indication of species in solution; however, for heterogeneous systems with rapid binding kinetics the peaks may occur at positions that do not necessarily reflect the sedimentation coefficient, but are instead governed by the kinetics of interaction (50). It is likely that the different oligomeric forms of Rubisco activase interact with each other over the time frame of the experiment, and integration of cs to provide a weight-average s value is the most accurate way to compare different protein concentrations (51).
FIGURE 2.
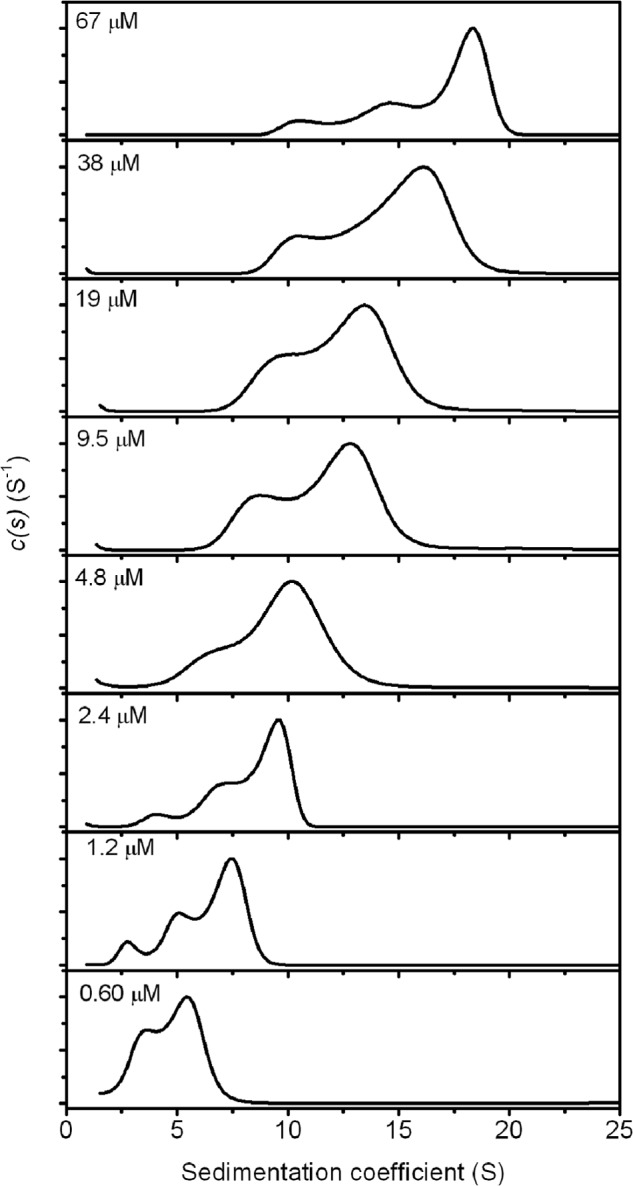
Analytical centrifugation of varying concentrations of tobacco Rubisco activase. Sedimentation velocity experiments of tobacco Rubisco activase were carried out at protein concentrations ranging from 600 nm to 67 μm. Radial absorbance data were collected at different wavelengths depending on the concentration, and cs distributions have been normalized to 1.
TABLE 1.
Parameters calculated from AUC data
| Concentration | Weight-average sedimentation coefficient | f/f0 | Weight-average molecular mass |
|---|---|---|---|
| μm | S | kDa | |
| 0.60 | 4.6 | 1.43 | 78 |
| 1.2 | 6.3 | 1.40 | 121 |
| 2.4 | 8.2 | 1.41 | 182 |
| 4.8 | 9.5 | 1.40 | 225 |
| 9.5 | 11.5 | 1.40 | 300 |
| 19 | 12.2 | 1.35 | 310 |
| 38 | 14.5 | 1.47 | 457 |
| 67 | 16.4 | 1.43 | 528 |
As the protein concentration is increased, the distribution of species becomes broader, suggesting increasing heterogeneity of the oligomeric species. The observation of very large species is consistent with previous studies that have observed supramolecular complexes larger than hexamers at concentrations >10 μm (27). Indeed, the weight-average sedimentation coefficient of the complex at 67 μm (16.4 S) indicates an average molecular mass approaching that of the 550-kDa Rubisco holoenzyme complex (18 S) (52). This corresponds to a Rubisco activase complex larger than a 12-mer.
At the lowest concentration measured (600 nm), the cs distribution shows lower heterogeneity of oligomers than at the higher concentrations. The sedimentation coefficient of a globular protein corresponding to the monomeric molecular mass of Rubisco activase (42 kDa) is estimated at ∼3.5–4.0 S. Thus, the cs distribution suggests that the smallest species present at 600 nm is monomeric and that the distribution comprises mainly monomeric and dimeric forms of the protein.
Static Light Scattering
Rubisco activase samples (loading concentration 18–142 μm) were eluted from a size exclusion chromatography column, and right angle light scattering detectors were used to calculate the molecular mass for each point. As has previously been observed for Rubisco activase, the protein has an asymmetrical elution peak with a trailing edge (Fig. 3). Right angle scattering measurement of the eluting protein showed that the molecular mass was not constant over the peak and that there was a strong correlation between the molecular mass and eluted protein concentration (Fig. 1).
FIGURE 3.

Light scattering analysis of varying concentrations of tobacco Rubisco activase. Different concentrations of tobacco Rubisco activase (18–142 μm) were loaded onto a Superdex 200 10/300 column and eluted at 0.5 ml·min−1. Refractive index (top panel) and right angle scattering were measured and used to calculate the molecular mass (lower panel) as described under “Experimental Procedures.”
At concentrations below 500 nm, the calculated molecular mass corresponded to that of a monomer/dimer, which is in good agreement with results seen by others and with the results determined by analytical ultracentrifugation. Species larger than a hexamer were observed at concentrations above 3 μm, which is also in good agreement with the analytical ultracentrifugation data. There were no distinct phases in the size distribution, suggesting that the mechanism of assembly is continuous, rather than involving discreet intermediate steps.
SAXS
To confirm the results seen for light scattering and analytical ultracentrifugation, SAXS data were collected for a series of different Rubisco activase concentrations (0.6–76 μm) (Fig. 4a). For each concentration, molecular mass and particle volume were calculated and showed that both were strongly correlated with protein concentration (Fig. 1, Table 2). Similar to the AUC and light scattering data, SAXS data indicated that species larger than a hexamer are present at concentrations higher than 3 μm. The parameters extracted from the SAXS data describe an average of multiple species in solution, as shown by AUC, and there is good agreement with the sizes determined using light scattering techniques.
FIGURE 4.

X-ray scattering data of Rubisco activase. a, data were collected for tobacco Rubisco activase for a range (0.6–76 μm) of protein concentrations (top panel). b, data for 2.4 μm Rubisco activase overlaid with the scattering profiles, calculated using CRYSOL, for the open hexamer structure (generated from 3T15), closed hexamer structure (3ZW6) and monomeric structure (3T15). c, distance distribution functions, pr were determined using the indirect Fourier transformation package GNOM (bottom panel).
TABLE 2.
Parameters calculated from SAXS data
| Concentration | Radius of gyrationa | I(0)a | Dmaxb | Porod volumeb | Molecular massb |
|---|---|---|---|---|---|
| μm | Å | nm | Å3 | kDa | |
| 0.60 | 47.3 | 0.0025 | 15.0 | 267,000 | 128 |
| 1.2 | 57.2 | 0.0060 | 19.2 | 437,000 | 154 |
| 2.4 | 62.3 | 0.015 | 20.8 | 315,000 | 193 |
| 4.8 | 59.8 | 0.036 | 21.3 | 406,000 | 231 |
| 9.5 | 72.3 | 0.095 | 24.3 | 544,000 | 305 |
| 19 | 73.6 | 0.229 | 26.0 | 704,000 | 368 |
| 38 | 77.9 | 0.532 | 27.5 | 1,001,000 | 427 |
| 57 | 82.4 | 0.815 | 35.0 | 1,183,000 | 436 |
| 76 | 84.1 | 1.283 | 36.0 | 1,318,000 | 515 |
a Calculated using GNOM.
b Calculated using AUTOPOROD.
The real-space distance distributions, pr generated from the scattering data (Fig. 4c) are positively skewed with a tail at long distances, indicative of elongated structures in solution. The proportion of larger distances increased with increasing protein concentration, consistent with the formation of higher order species.
In an effort to determine whether the recently solved crystal structure of tobacco Rubisco activase accurately reflects the structure in solution, the experimental scattering data were compared with the scattering expected from a closed hexamer, open hexamer, or monomeric model (Fig. 4b). There was some correlation to the hexameric model, but there was insufficient information to distinguish between an open and closed hexamer.
In another approach, ab initio models describing a monomer-hexamer equilibrium were reconstructed from the SAXS data using the program GASBORMX. A concentration was chosen that was consistent with the presence of these species, as suggested by the AUC and light scattering experiments (2.4 μm) (Fig. 1). These models provide a very good fit to the experimental data at low concentration (Fig. 5a), neatly describing an equilibrium of 52% monomer and 48% hexamer. Runs were also conducted for equilibria including dimers and tetramers, but these models provided a very poor fit to the data (data not shown).
FIGURE 5.
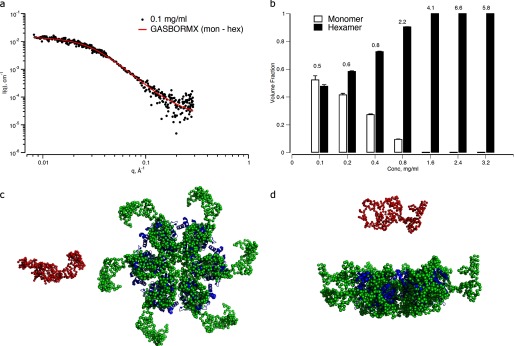
Ab initio monomer-hexamer equilibrium of Rubisco activase generated using GASBORMX. a, fit of the model equilibrium to the SAXS data at 0.1 mg·ml−1 sample concentration. b, volume fractions of monomer and hexamer calculated from the best weighted combinations of the model components at each concentration. The corresponding discrepancy in the fits of the models to the data at each concentration are shown above the bars. Error bars, Errors in the volume fractions shown are those propagated from the solution of linear equations used in OLIGOMER (46). S.D. c, ab initio monomer (red spheres) and hexamer (green spheres) reconstructed from the SAXS data at 2.4 μm (0.1 mg·ml−1). The hexamer model is shown overlaid with the structure of tobacco Rubisco activase (Protein Data Bank ID code 3ZW6). d, rotation of the view in a by 90º in the horizontal.
The ab initio bead model generated using GASBORMX shows good agreement with the ring structure proposed by crystallographic and transmission electron micrograph studies (1), as shown by the overlay of the two models in Fig. 5, but with a smaller inner pore, and an extended domain and an extended outer domain. This extended domain may include the 67-residue N-domain that is not present in the crystal structure, following suggestions that it is flexibly attached (1). Although the ab initio model provides information about the location of the missing domain, there is insufficient resolution to distinguish between closed and open hexamers or whether other intermediates are also present.
Using the program OLIGOMER and the ab initio reconstructed equilibrium models as input, a non-negative linear least squares algorithm was employed to generate weighted combinations of these simple models that best fit the SAXS data at each of the measured concentrations (Fig. 5b). The monomer-hexamer equilibrium describes the data well up to 9.5 μm showing an increase in the volume fraction of hexameric species, with the fit starting to deviate at q > 0.1 Å−1 with concentration. At concentrations >9.5 μm not only does the fit continue to deteriorate at high q, but it is clear that larger oligomeric species begin to contribute to the scattering at low q (data not shown) and that the simple monomer-hexamer model can no longer adequately describe the data.
DISCUSSION
Rubisco Activase May Form an Open, Spiraling Structure
In biology, most proteins form a closed structure with a fixed number of subunits. There are relatively few instances of open structures, in which the size of the complex is determined by the protein concentration. Despite the presence of high resolution crystal structures for tobacco and creosote activase (1, 26), it is still uncertain whether Rubisco activase adopts a structure in which closed hexamers are formed that then stack to form larger assemblies or whether an open hexameric spiral is formed in solution, similar to the packing of the tobacco Rubisco activase crystal structure. Our current data fit a model in which Rubisco activase forms an open, helical, spiral in solution. Over the concentration range measured (0.4–76 μm) there is a continuum of species, rather than appearing to favor units of six, as would be expected for a model in which hexamers stack on top of each other. The composition of oligomeric species is highly dynamic, reequilibrating within a matter of minutes when diluted, suggesting a rapid dissociation/association rate. This may be due to isodesmic polymerization, in which the dissociation constant for monomer addition to any protein species is considered to be identical, independent of the size of the polymer (53).
Other members of the AAA+ family have been observed to form spiral assemblies in nature. The yeast clamp-loader complex adopts an open pentameric spiral structure around the 3′ end of the primer-template junction during DNA replication (23. Another set of AAA+ proteins involved in DNA replication are the initiators that recognize replication origins and recruit replication machinery. Bacterial and archaebacterial initiators are monomeric in solution, but binding of ATP allows A. aeolicus DnaA to transition from a monomeric state into a large oligomeric complex that has an open helix arrangement (24). It has been proposed that these spiral, open ring AAA+ assemblies form the core element of all initiator complexes, and the length of the spiral can vary to accommodate differences in the length of the DNA boxes (24).
Clamp-loader and initiator proteins form distinct clades in the AAA+ family, with the initiator clade being highly divergent in terms of sequence, but having a characteristic α-helix located after strand 2 (54). It is unclear whether Rubisco activase contains the additional α-helix located after strand 2, as the crystal structure of tobacco Rubisco activase is disordered in this region (1); however, there is a short α-helical region that may be similar to that observed for initiator structures. The data in the current study suggest that Rubisco activase may be more likely to adopt a spiraling supramolecular, oligomeric structure similar to the clamp-loader or initiator clades of AAA+ protein, rather than the closed hexameric rings observed for proteases.
Rubisco Activase Does Not Need to Be Hexameric for Full Activity
Formation of higher order structures by Rubisco activase is associated with increased rates of ATP hydrolysis and Rubisco reactivation activity, showing the importance of Rubisco activase oligomeric state in biological function. Our data show that the activity of tobacco Rubisco activase increased with the concentration of Rubisco activase, reaching maximum activity at 1 μm protomer, similar to that seen previously for spinach Rubisco activase (30, 34).
For each of the methods used in the present study, the data indicate that tobacco Rubisco activase species larger than a hexamer are present at concentrations higher than 3 μm, that species smaller than a trimer are observed at concentrations lower than 1 μm. This is in good agreement with fluorescence fluctuation spectroscopy studies of cotton Rubisco activase (27), which found that monomer is the dominating species <0.5 μm and hexamers formed at concentrations >4 μm. At 1 μm, where Rubisco activase has full activity, light scattering gives an average molecular mass of ∼120 kDa, and analytical ultracentrifugation gives a weight-average sedimentation coefficient of 6.5 S, corresponding to a molecular mass of 121 kDa, suggesting that the main species in solution are 2–4 subunits.
Together, these data support the hypothesis that Rubisco activase needs to form an oligomer with 2–4 subunits to catalyze hydrolysis of ATP and activation of Rubisco. Studies that involved titrating mixtures of wild-type and Rubisco activase mutants that blocked oligomerization also suggested that a minimum of 3–5 subunits are required for Rubisco activation activity (1). This is in contrast to other AAA+ proteins, which require at least 6 subunits for activity.
The formation of an open, helical oligomer by Rubisco activase obviates any requirement for the minimum unit for activity to be a hexamer. Studies of the ClpX protein suggested that the most likely mechanism for ATP hydrolysis is a probabilistic model, rather than a concerted model in which simultaneous binding and hydrolysis of ATP occurs in all modules, or a sequential model in which ATP hydrolysis in one subunit is followed sequentially by hydrolysis in the neighboring subunit (55). If this is the case for Rubisco activase, the enzyme could behave similarly, even as an open ring, as there is no need for symmetry. Given that the nucleotide binding pocket is located between 2 subunits, with the arginine finger from 1 subunit interacting with nucleotide bound in the neighboring subunit (56), it is reasonable to assume that the probability of ATP hydrolysis in a specific subunit will depend on the structural constraints of a neighboring subunit, which may or may not have nucleotide bound.
Thus, we propose a model in which ATP hydrolysis occurs at a faster rate when a subunit has neighboring subunits, with a dimer having some activity, and larger species having higher specific activity. The size of the oligomer is determined by protein concentration and forms open spirals, but may include closed hexamer structures, such as those observed for mutant variants of the Arg-294 residue (1, 29).
Mechanism of Rubisco Activation
Activation of Rubisco must involve some form of conformational change in Rubisco activase that triggers opening of the Rubisco active site and release of inhibitors. Nearly all AAA+ proteins act through the central pore region, whether by destabilizing DNA or through translocation of a polypeptide chain through the central pore. CbbX, a red-type Rubisco activase, has recently been characterized and shown to function by transiently pulling the C-terminal domain of the Rubisco large subunit into the central pore (25). Despite functional similarities, there are several key differences between CbbX and higher plant Rubisco activase enzymes, including regulation by ribulose-P2, and differences in the central pore region (1, 25).
Our low resolution dummy-residue models generated from the SAXS data suggest that the central pore of tobacco Rubisco activase may be smaller than in the crystal structure (Fig. 5), which is consistent with a role proposed for three loop segments (residues 141–145, 177–191, and 232–237) that are not resolved in the crystal structure, but face the inner solvent channel (1). Modeling of the solution scattering data also suggests that the N-terminal domain that is missing from the crystal structure may be located on the periphery of the ring structure.
Specificity of Rubisco activase for Rubisco is mediated through the sensor 2′ domain (35), a region that is located on the outside of the spiral rather than in the central pore. It has been speculated that this region interacts with the Rubisco enzyme via hydrophobic patches on an extended helix of Rubisco activase (26). As such, the initial association of Rubisco activase with Rubisco must be via the outside of the assembly. Residues on the surface of the Rubisco large subunit that are important for Rubisco activase interactions have also been identified (57, 58). These residues are located near the loop 6 region that closes over the Rubisco active site during catalysis, and it has been postulated that an interaction with Rubisco activase may trigger subtle changes in the N-terminal domain of Rubisco that are responsible for the release of inhibitors (58). It is also important to appreciate that the site of initial recognition may differ from the site at which activation occurs.
One of the implications of our proposed model is that species as small as 2–4 subunits may be able to activate Rubisco. Indeed, other studies using titrations of mutant enzymes have suggested that the minimum oligomeric size for Rubisco activation activity is 3–5 subunits, and Rubisco activase concentrations of as low as 0.5 μm are sufficient to show full Rubisco activational activity (34). At these concentrations, others have also shown that the main oligomeric species are no larger than a trimer (27). The mechanism for this activation, however, remain elusive, pending more detailed structural information and a better understanding of the conformational changes associated with ATP hydrolysis.
In planta, the holoenzyme concentration of Rubisco is ∼500 μm, and the concentration of the Rubisco activase protomer is thought to be 170–500 μm (19, 59). These concentrations would favor Rubisco activase to form a higher order species; however, it may be the case that there are many smaller species associated with the large number of Rubisco enzymes, rather than a few larger species.
With an increased appreciation that Rubisco activase forms very dynamic structures in solution, we hope to be able to better understand how it is that this enzyme can optimize photosynthesis. Remaining questions include the role of nucleotide binding in formation of higher species, the conformational changes that are triggered by the binding of ATP, and the mechanism by which Rubisco activase interacts with Rubisco to trigger opening of the Rubisco active site.
Acknowledgments
Parts of this research were undertaken at the SAXS beamlines of the Australian Synchrotron, Victoria, Australia. We give special thanks to Nigel Kirby at the SAXS beamline. Travel to the Australian Synchrotron was supported by the New Zealand Synchrotron Group. We thank Prof. Juliet Gerrard and Prof. George Lorimer for active discussion and a critical reading of the manuscript. We thank Assistant Prof. Spencer Whitney for the Rubisco activase plasmid and for providing laboratory facilities after the Canterbury earthquake.
Footnotes
- Rubisco
- ribulose-bisphosphate carboxylase/oxygenase
- AAA+ proteins
- ATPases associated with diverse cellular activities
- ATPγS
- adenosine 5′-3-O-(thio)triphosphate
- AUC
- analytical ultracentrifugation
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1. Stotz M., Mueller-Cajar O., Ciniawsky S., Wendler P., Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Structure of green-type Rubisco activase from tobacco. Nat. Struct. Mol. Biol. 18, 1366–1370 [DOI] [PubMed] [Google Scholar]
- 2. Ogura T., Wilkinson A. J. (2001) AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6, 575–597 [DOI] [PubMed] [Google Scholar]
- 3. Pon N. G., Rabin B. R., Calvin M. (1963) Mechanism of the carboxydismutase reaction. I. The effect of preliminary incubation of substrates, metal ion and enzyme on activity. Biochem. Z. 338, 7–19 [PubMed] [Google Scholar]
- 4. Laing W. A., Christeller J. T. (1976) A model for the kinetics of activation and catalysis of ribulose-1,5-bisphosphate carboxylase. Biochem. J. 159, 563–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gutteridge S., Parry M. A., Burton S., Keys A. J., Mudd A., Feeney J., Servaites J. C., Pierce J. (1986) A nocturnal inhibitor of carboxylation in leaves. Nature 324, 274–276 [Google Scholar]
- 6. Berry J. A., Lorimer G. H., Pierce J., Seemann J. R., Meek J., Freas S. (1987) Isolation, identification, and synthesis of 2-carboxyarabinitol 1-phosphate, a diurnal regulator of ribulose-bisphosphate carboxylase activity. Proc. Natl. Acad. Sci. U.S.A. 84, 734–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andralojc P. J., Madgwick P. J., Tao Y., Keys A., Ward J. L., Beale M. H., Loveland J. E., Jackson P. J., Willis A. C., Gutteridge S., Parry M. A. (2012) 2-Carboxy-d-arabinitol 1-phosphate (CA1P) phosphatase: evidence for a wider role in plant Rubisco regulation. Biochem. J. 442, 733–742 [DOI] [PubMed] [Google Scholar]
- 8. Keys A. J., Major I., Parry M. A. (1995) Is there another player in the game of Rubisco regulation? J. Exp. Bot. 46, 1245–1251 [Google Scholar]
- 9. Paul M. J., Andralojc P. J., Banks F. M., Parry M. A. J., Knight J. S., Gray J. C., Keys A. J. (1996) Altered Rubisco activity and amounts of a daytime tight-binding inhibitor in transgenic tobacco expressing limiting amounts of phosphoribulokinase. J. Exp. Bot. 47, 1963–1966 [Google Scholar]
- 10. Edmondson D. L., Badger M. R., Andrews T. J. (1990) A kinetic characterization of slow inactivation of ribulose-bisphosphate carboxylase during catalysis. Plant Physiol. 93, 1376–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edmondson D. L., Badger M. R., Andrews T. J. (1990) Slow inactivation of ribulose-bisphosphate carboxylase during catalysis is not due to decarbamylation of the catalytic site. Plant Physiol. 93, 1383–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edmondson D. L., Badger M. R., Andrews T. J. (1990) Slow inactivation of ribulose-bisphosphate carboxylase during catalysis is caused by accumulation of a slow, tight-binding inhibitor at the catalytic site. Plant Physiol. 93, 1390–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Edmondson D. L., Kane H. J., Andrews T. J. (1990) Substrate isomerization inhibits ribulose-bisphosphate carboxylase-oxygenase during catalysis. FEBS Lett. 260, 62–66 [Google Scholar]
- 14. Zhu G., Jensen R. G. (1991) Fallover of ribulose-1,5-bisphosphate carboxylase/oxygenase activity: decarbamylation of catalytic sites depends on pH. Plant Physiol. 97, 1354–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu G., Jensen R. G. (1991) Xylulose 1,5-bisphosphate synthesized by ribulose-1,5-bisphosphate carboxylase/oxygenase during catalysis binds to decarbamylated enzyme. Plant Physiol. 97, 1348–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu G. H., Bohnert H. J., Jensen R. G., Wildner G. F. (1998) Formation of the tight-binding inhibitor, 3-ketoarabinitol-1,5-bisphosphate by ribulose-1,5-bisphosphate carboxylase/oxygenase is O2-dependent. Photosynth. Res. 55, 67–74 [Google Scholar]
- 17. Pearce F. G. (2006) Catalytic by-product formation and ligand binding by ribulose bisphosphate carboxylases from different phylogenies Biochem. J. 399, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pearce F. G., Andrews T. J. (2003) The relationship between side reactions and slow inhibition of ribulose-bisphosphate carboxylase revealed by a loop 6 mutant of the tobacco enzyme. J. Biol. Chem. 278, 32526–32536 [DOI] [PubMed] [Google Scholar]
- 19. Portis A. R. (2003) Rubisco activase: Rubisco's catalytic chaperone. Photosynth. Res. 75, 11–27 [DOI] [PubMed] [Google Scholar]
- 20. Salvucci M. E., Ogren W. L. (1996) The mechanism of Rubisco activase: insights from studies of the properties and structure of the enzyme. Photosynth. Res. 47, 1–11 [DOI] [PubMed] [Google Scholar]
- 21. Portis A. R., Jr., Salvucci M. E. (2002) The discovery of Rubisco activase: yet another story of serendipity. Photosynth. Res. 73, 257–264 [DOI] [PubMed] [Google Scholar]
- 22. Portis A. R., Jr., Li C.., Wang D., Salvucci M. E. (2008) Regulation of Rubisco activase and its interaction with Rubisco. J. Exp. Bot. 59, 1597–1604 [DOI] [PubMed] [Google Scholar]
- 23. Bowman G. D., O'Donnell M., Kuriyan J. (2004) Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429, 724–730 [DOI] [PubMed] [Google Scholar]
- 24. Erzberger J. P., Mott M. L., Berger J. M. (2006) Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat. Struct. Mol. Biol. 13, 676–683 [DOI] [PubMed] [Google Scholar]
- 25. Mueller-Cajar O., Stotz M., Wendler P., Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Structure and function of the AAA+ protein CbbX, a red-type Rubisco activase. Nature 479, 194–199 [DOI] [PubMed] [Google Scholar]
- 26. Henderson J. N., Kuriata A. M., Fromme R., Salvucci M. E., Wachter R. M. (2011) Atomic resolution x-ray structure of the substrate recognition domain of higher plant ribulose-bisphosphate carboxylase/oxygenase (Rubisco) activase. J. Biol. Chem. 286, 35683–35688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chakraborty M., Kuriata A. M., Nathan Henderson J., Salvucci M. E., Wachter R. M., Levitus M. (2012) Protein oligomerization monitored by fluorescence fluctuation spectroscopy: self-assembly of Rubisco activase. Biophys. J. 103, 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barta C., Dunkle A. M., Wachter R. M., Salvucci M. E. (2010) Structural changes associated with the acute thermal instability of Rubisco activase. Arch. Biochem. Biophys. 499, 17–25 [DOI] [PubMed] [Google Scholar]
- 29. Blayney M. J., Whitney S. M., Beck J. L. (2011) NanoESI mass spectrometry of Rubisco and Rubisco activase structures and their interactions with nucleotides and sugar phosphates. J. Am. Soc. Mass Spectrom. 22, 1588–1601 [DOI] [PubMed] [Google Scholar]
- 30. Wang Z. Y., Ramage R. T., Portis A. R. (1993) Mg2+ and ATP or adenosine 5′-[γ-thio]-triphosphate (ATPγS) enhances intrinsic fluorescence and induces aggregation which increases the activity of spinach Rubisco activase. Biochim. Biophys. Acta 1202, 47–55 [DOI] [PubMed] [Google Scholar]
- 31. Salvucci M. E. (1992) Subunit interactions of Rubisco activase: polyethylene glycol promotes self-association, stimulates ATPase and activation activities, and enhances interactions with Rubisco. Arch. Biochem. Biophys. 298, 688–696 [DOI] [PubMed] [Google Scholar]
- 32. Li C., Wang D., Portis A. R., Jr. (2006) Identification of critical arginine residues in the functioning of Rubisco activase. Arch. Biochem. Biophys. 450, 176–182 [DOI] [PubMed] [Google Scholar]
- 33. Henderson J. N., Hazra S., Dunkle A. M., Salvucci M. E., Wachter R. M. (2013) Biophysical characterization of higher plant Rubisco activase. Biochim. Biophys. Acta 1834, 87–97 [DOI] [PubMed] [Google Scholar]
- 34. Lilley R. M., Portis A. R., Jr. (1997) ATP hydrolysis activity and polymerization state of ribulose-1,5-bisphosphate carboxylase oxygenase activase: do the effects of Mg2+, K+, and activase concentrations indicate a functional similarity to actin? Plant Physiol. 114, 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li C., Salvucci M. E., Portis A. R., Jr. (2005) Two residues of Rubisco activase involved in recognition of the Rubisco substrate. J. Biol. Chem. 280, 24864–24869 [DOI] [PubMed] [Google Scholar]
- 36. Wang D., Portis A. R., Jr. (2006) Two conserved tryptophan residues are responsible for intrinsic fluorescence enhancement in Rubisco activase upon ATP binding. Photosynth. Res. 88, 185–193 [DOI] [PubMed] [Google Scholar]
- 37. Baker T. A., Sauer R. T. (2012) ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 1823, 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langklotz S., Baumann U., Narberhaus F. (2012) Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta 1823, 40–48 [DOI] [PubMed] [Google Scholar]
- 39. Kress W., Mutschler H., Weber-Ban E. (2007) Assembly pathway of an AAA+ protein: tracking ClpA and ClpAP complex formation in real time. Biochemistry 46, 6183–6193 [DOI] [PubMed] [Google Scholar]
- 40. Sage R. F., Way D. A., Kubien D. S. (2008) Rubisco, Rubisco activase, and global climate change. J. Exp. Bot. 59, 1581–1595 [DOI] [PubMed] [Google Scholar]
- 41. Baker R. T., Catanzariti A. M., Karunasekara Y., Soboleva T. A., Sharwood R., Whitney S., Board P. G. (2005) Using deubiquitylating enzymes as research tools. Methods Enzymol. 398, 540–554 [DOI] [PubMed] [Google Scholar]
- 42. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laue T. M., Shah D. B., Ridgeway T. M., Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Protein Science (Harding S. E., Rowe A. J., Horton J. C., eds), pp. 90–125, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 45. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 48. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 49. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 50. Dam J., Velikovsky C. A., Mariuzza R. A., Urbanke C., Schuck P. (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions cs. Biophys. J. 89, 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dam J., Schuck P. (2004) Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 384, 185–212 [DOI] [PubMed] [Google Scholar]
- 52. Gurevitz M., Somerville C. R., McIntosh L. (1985) Pathway of assembly of ribulose-bisphosphate carboxylase oxygenase from Anabaena-7120 expressed in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 82, 6546–6550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Frieden C. (2007) Protein aggregation processes: in search of the mechanism. Protein Sci. 16, 2334–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iyer L. M., Leipe D. D., Koonin E. V., Aravind L. (2004) Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 [DOI] [PubMed] [Google Scholar]
- 55. Martin A., Baker T. A., Sauer R. T. (2005) Rebuilt AAA plus motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 56. Wendler P., Ciniawsky S., Kock M., Kube S. (2012) Structure and function of the AAA plus nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 [DOI] [PubMed] [Google Scholar]
- 57. Larson E. M., O'Brien C. M., Zhu G., Spreitzer R. J., Portis A. R., Jr. (1997) Specificity for activase is changed by a Pro-89 to Arg substitution in the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase. J. Biol. Chem. 272, 17033–17037 [DOI] [PubMed] [Google Scholar]
- 58. Ott C. M., Smith B. D., Portis A. R., Jr., Spreitzer R. J. (2000) Activase region on chloroplast ribulose-1,5-bisphosphate carboxylase/oxygenase: nonconservative substitution in the large subunit alters species specificity of protein interaction. J. Biol. Chem. 275, 26241–26244 [DOI] [PubMed] [Google Scholar]
- 59. Robinson S. P., Streusand V. J., Chatfield J. M., Portis A. R. (1988) Purification and assay of Rubisco activase from leaves. Plant Physiol. 88, 1008–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]