

The ability to discriminate between like functional groups, so as to transform organic molecules in a “site-selective” or “chemoselective” manner,[1] precludes the requirement of protecting groups and, hence, carries the potential to dramatically enhance synthetic efficiency.[2] Though an exceptionally daunting challenge, systematic efforts toward catalytic methods for the site-selective transformation of polyfunctional molecules have begun to emerge. For example, Miller[3] and Taylor[4] report catalytic methods for the site-selective manipulation of diols and higher polyols. Site-selective metal catalyzed cross-couplings have been catalogued.[5] Further, in what is perhaps the most formidable theatre for site-selectivity, impressive advances in catalytic methods for C-H functionalization have been achieved, as illustrated in the seminal work of Barton,[6a] Murai and Kakiuchi,[6b] and in more recent studies by Davies,[6c] Sanford,[6d] Yu,[6e] Daugulis,[6f] Baran[6g] and others.
Although methods for the site-selective diol oxidation have been reported, including iridium catalyzed methods,[7,8] merged redox-C-C bond construction events involving chemoselective polyol oxidation are unknown.[9] In connection with ongoing studies of C-C bond forming hydrogenation, we recently found that certain iridium and ruthenium complexes catalyze hydrogen exchange between primary alcohols and π-unsaturated reactants to generate organometal-aldehyde pairs that combine to form products of carbonyl addition.[10] In these transformations, primary alcohol reactants are subject to oxidation, yet the secondary alcohol products are not. This fact, and the ability to perform certain transfer hydrogenative couplings in aqueous organic media, suggested unprotected polyols might engage in site-selective carbinol CH-functionalization. Here, we report that the cyclometallated π-allyliridium C,O-benzoate complex derived from (R)- or (S)-SEGPHOS and 4-cyano-3-nitro-benzoic acid catalyzes the redox-triggered allylation[11,12] of unprotected diols and higher polyols with a pronounced kinetic preference for primary alcohol dehydrogenation. In this way, chemo- and stereoselective carbinol CH-allylation of polyols is achieved in the absence of protecting groups, chiral auxiliaries, premetallated reagents and discrete alcohol-to-aldehyde oxidation (Scheme 1).
Scheme 1.
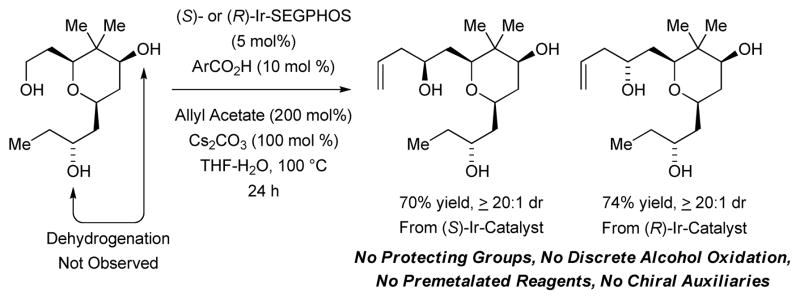
Site selective dehydrogenation enables protecting group-free allylation of diols.
In an initial set of experiments, a series of chromatographically isolated π-allyliridium complexes modified by (S)-SEGPHOS and 4-substituted-3-nitro-C,O-benzoate moieties were assayed in the allylation of diol 1 (Figure 1). The iridium complexes (R)-Ir-b-NO2 and (S)-Ir-a-CN, displayed roughly equal efficiency, with the latter providing the 1,3-syn-diol 2a in 67% yield with complete levels of catalyst-directed diastereoselectivity[13] and without any detectable oxidation of the unprotected secondary alcohol. Similarly, using the enantiomeric catalyst (R)-Ir-CN, diol 1 was transformed to the 1,3-anti-diol 2b in 59% yield without any detectable over-oxidation. Inspired by these results, diols 3, 5, and 7 and were subjected to the enantiomeric catalysts (S)- and (R)-Ir-CN. In each case, the corresponding products of allylation 4a and 4b, 6a and 6b, and 8a and 8b, respectively, were formed in good to excellent yields with exceptional control of diastereoselectivity and without oxidation of the unprotected secondary hydroxyl moieties. Notably, the preparation of 4b in four steps through iterative redox-triggered allylation represents a synthesis of the C11-C18 fragment of the oxopolyene macrolide RK-397, which was previously prepared in 7 steps from the same starting material (Scheme 2).[14] Finally, even the unprotected triol 9 is converted to adducts 10a and 10b using the enantiomeric catalysts (S)- and (R)-Ir-CN, respectively, in good yields and complete levels of catalyst directed diastereoselectivity (Table 1).
Figure 1.
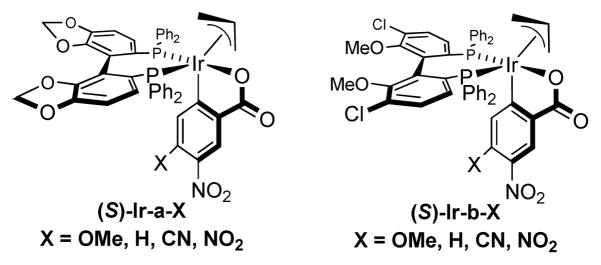
Cyclometallated iridium catalysts for site-selective allylation.
Scheme 2.
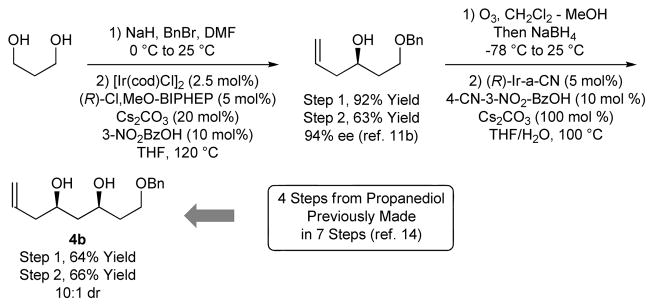
Synthesis of the C11-C18 fragment of RK-397.a
aAs described for Table 1.
Table 1.
Protecting group-free catalyst-directed diastereoselective C-C coupling of chiral diols 1, 3, 5, 7 and 9 with allyl acetate.a
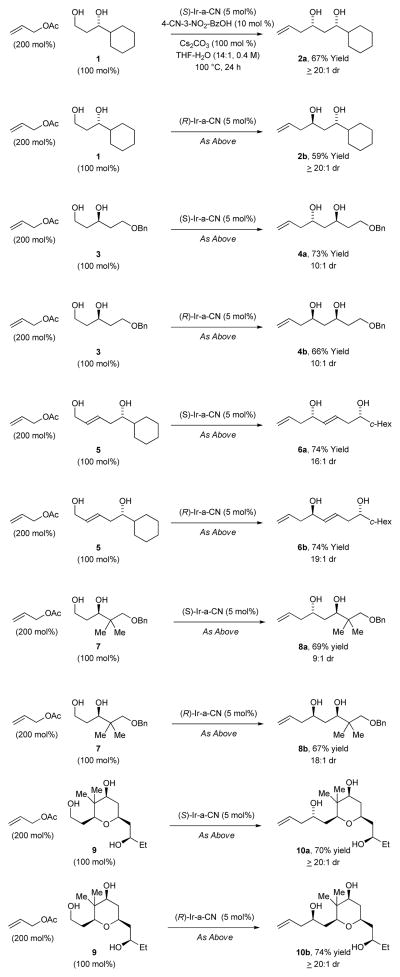
|
Cited yields are of diastereomeric mixtures isolated by silica gel chromatography. Stereoisomeric ratios were determined by 1H NMR analysis of crude reaction mixtures (major diastereomer:sum of two minor diastereomers). See Supporting Information for further experimental details.
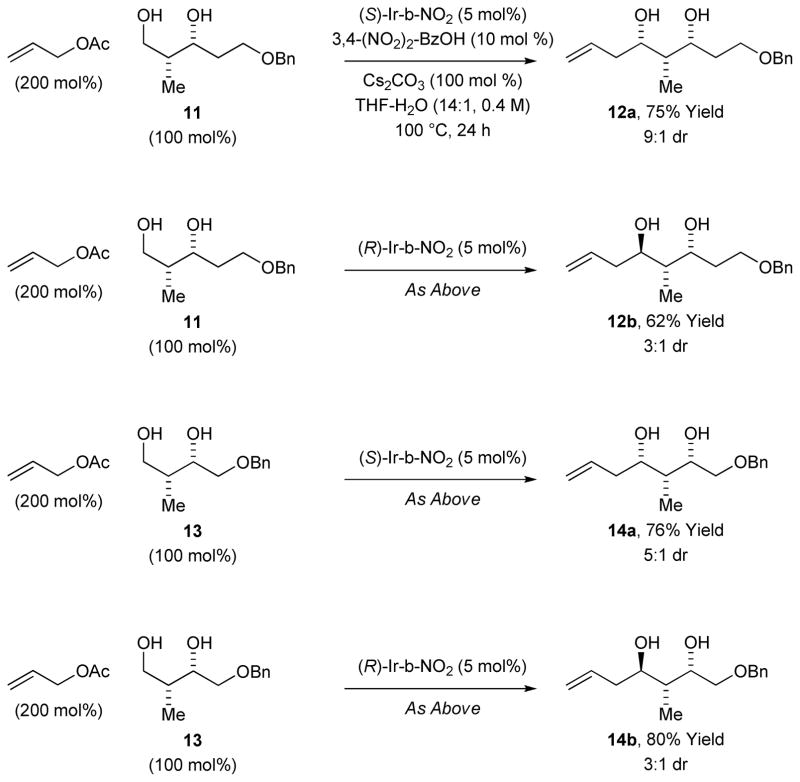
An even more challenging class of substrate is represented by the unprotected chiral β,γ-stereogenic alcohols 11 and 13. Exposure of 1,3-glycol 11 to the catalyst (S)-Ir-NO2 under standard conditions delivers the product of allylation 12a as a 9:1 mixture of diastereomers. In this case, the diastereofacial bias of the catalyst matches the preference of the transient aldehyde for Felkin-Anh addition.[15] In the mismatched case using the enantiomeric catalyst (R)-Ir- NO2, the diastereomeric product 12b is formed as a 3:1 mixture of diastereomers. Similarly, the 1,3-glycol 13 is converted to diastereomeric allylation products 14a (5:1 dr) and 14b (3:1 dr) using (S)-Ir-NO2 and (R)-Ir-NO2, respectively. For the allylation of unprotected chiral β,γ-stereogenic alcohols 11 and 13, the minor diastereomer obtained principally stems from epimerization of the transient α-stereogenic aldehyde (Table 2). Although the levels of catalyst-directed diastereoselectivity are modest, these results are significant in view of the intractability of β-hydroxy-α-substituted aldehydes. Whereas substrate-directed allylation of unprotected β-hydroxyaldehydes is possible with allylindium reagents to provide anti-1,3-diols,[16a] the corresponding 1,3-syn-allylation is only possible with stoichiometric allyltitanium-TADDOL complexes.[16b]
Table 2.
Protecting group-free catalyst-directed diastereoselective C-C coupling of chiral β,γ-stereogenic alcohols 11 and 13 with allyl acetate.a
|
As described for Table 1.
It has been posited, and generally appreciated, that an ideal synthesis would be composed solely of construction events.[9,17] As organic compounds, by their very definition, are compounds composed of carbon and hydrogen, catalytic C-C bond constructions that occur through the addition, transfer or removal of hydrogen potentially provides a direct means of accessing native molecule structure in a highly atom efficient manner. Site-selectivity further enhances efficiency by dispensing with the need to install and remove protecting groups. The site-selective redox-allylations described in this account, which occur by virtue of the strong kinetic preference of the iridium catalyst for primary alcohol dehydrogenation, allow chemo- and stereoselective carbinol CH-allylation in the absence of protecting groups, chiral auxiliaries, premetallated reagents and discrete alcohol-to-aldehyde redox manipulations, thus representing one step toward fulfillment of these ideals.
Supplementary Material
Footnotes
Acknowledgment is made to the Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) for partial financial support. The Cancer Prevention Research Institute of Texas (CPRIT) is acknowledged for Postdoctoral Fellowship support (D.C.S.). Dr. Taichiro Touge of Takasago is thanked for the generous donation of SEGPHOS ligands.
References
- 1.The term chemoselectivity broadly refers to the ability to selectively act upon like or unlike functional groups: Trost BM. Science. 1983;219:245–250. doi: 10.1126/science.219.4582.245. The term site-selectivity will be used in the more specific context of discrimination between like functional groups en route to constitutionally isomeric products.
- 2.Young IS, Baran PS. Nature Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 3.a) Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869–8875. [Google Scholar]; b) Lewis CA, Sculimbrene BR, Xu Y, Miller SJ. Org Lett. 2005;7:3021–3023. doi: 10.1021/ol051061a. [DOI] [PubMed] [Google Scholar]; c) Lewis CA, Miller SJ. Angew Chem. 2006;118:5744–5747. [Google Scholar]; Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]; d) Lewis CA, Merkel J, Miller SJ. Bioorg Med Chem Lett. 2008;18:6007–6011. doi: 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lewis CA, Longcore KE, Miller SJ, Wender PA. J Nat Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Fowler BS, Laemmerhold KM, Miller SJ. J Am Chem Soc. 2012;134:9755–9761. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Lee D, Taylor MS. J Am Chem Soc. 2011;133:3724–3727. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]; b) Chan L, Taylor MS. Org Lett. 2011;13:3090–3093. doi: 10.1021/ol200990e. [DOI] [PubMed] [Google Scholar]; c) Gouliaras C, Lee D, Chan L, Taylor MS. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; d) Lee D, Williamson CL, Chan L, Taylor MS. J Am Chem Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- 5.For reviews on site-selective metal catalyzed cross-coupling of polyhalogenated molecules, see: Fairlamb IJS. Chem Soc Rev. 2007;36:1036–1045. doi: 10.1039/b611177g.Joucla L, Djakovitch L. Adv Synth Catal. 2009;351:673–714.Wang JR, Manabe K. Synthesis. 2009:1405–1427.Rossi R, Bellina F, Lessi M. Adv Synth Catal. 2012;354:1181–1255.
- 6.For reviews on site-selective metal catalyzed C-H functionalization, see: Kakiuchi F, Murai S. Top Organomet Chem. 1999;3:47–79.Barton DHR. Tetrahedron. 1998;54:5805–5817.Davies HML, Morton D. Chem Soc Rev. 2011;40:1857–1869. doi: 10.1039/c0cs00217h.Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f.Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g.Daugulis O. Top Curr Chem. 2010;292:57–84. doi: 10.1007/128_2009_10.Newhouse T, Baran PS. Angew Chem. 2011;123:3422–3435.Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368.
- 7.a) Lin Y, Zhu X, Zhou YJ. Organomet Chem. 1992;429:269–274. [Google Scholar]; b) Suzuki T, Morita K, Tsuchida M, Hiroi K. Org Lett. 2002;4:2361–2363. doi: 10.1021/ol026091h. [DOI] [PubMed] [Google Scholar]; c) Suzuki T, Yamada T, Watanabe K, Katoh T. Bioorg Med Chem Lett. 2005;15:2583–2585. doi: 10.1016/j.bmcl.2005.03.043. [DOI] [PubMed] [Google Scholar]; d) Suzuki T, Morita K, Ikemiyagi H, Watanabe K, Hiroi K, Katoh T. Heterocycles. 2006;69:457–461. [Google Scholar]; e) Königsmann M, Donati N, Stein D, Schönberg H, Harmer J, Sreekanth A, Grützmacher H. Angew Chem. 2007;119:3637–3640. doi: 10.1002/anie.200605170. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2007;46:3567–3570. doi: 10.1002/anie.200605170. [DOI] [PubMed] [Google Scholar]; f) Oger C, Brinkmann Y, Bouazzaoui S, Durand T, Galano JM. Org Lett. 2008;10:5087–5090. doi: 10.1021/ol802104z. [DOI] [PubMed] [Google Scholar]; g) Arita S, Koike T, Kayaki Y, Ikariya T. Chem Asian J. 2008;3:1479–1485. doi: 10.1002/asia.200800137. [DOI] [PubMed] [Google Scholar]; h) Kosaka M, Sekiguchi S, Naito J, Uemura M, Kuwahara S, Watanabe M, Harada N, Hiroi K. Chirality. 2005;17:218–232. doi: 10.1002/chir.20156. [DOI] [PubMed] [Google Scholar]; i) Suzuki T, Morita K, Matsuo Y, Hiroi K. Tetrahedron Lett. 2003;44:2003–2006. [Google Scholar]
- 8.For an excellent review of iridium-catalyzed oxidation, see: Suzuki T. Chem Rev. 2011;111:1825–1845. doi: 10.1021/cr100378r.
- 9.Baran PS, Hoffmann RW, Burns NZ. Angew Chem. 2009;121:2896–2910. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 10.For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem. 2008;121:36–48.Angew Chem Int Ed. 2008;48:34–46.Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim Acta. 2008;41:95–104.Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278–7287. doi: 10.1039/b917243m.Bower JF, Krische MJ. Top Organomet Chem. 2011;43:107–138. doi: 10.1007/978-3-642-15334-1_5.Hassan A, Krische MJ. Org Proc Res Dev. 2011;15:1236–1242. doi: 10.1021/op200195m.
- 11.a) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340–6341. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891–14899. doi: 10.1021/ja805722e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514–2520. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350–2354. doi: 10.1021/jo200068q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For selected reviews on enantioselective carbonyl allylation, see: Ramachandran PV. Aldrichim Acta. 2002;35:23–35.Denmark SE, Fu J. Chem Rev. 2003;103:2763–2794. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463–472.Marek I, Sklute G. Chem Commun. 2007:1683–1691. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644–1655.Lachance H, Hall DG. Org React. 2008;73:1–544.Yus M, Gonzalez-Gomez JC, Foubelo F. Chem Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474.
- 13.For selected examples of catalyst-directed diastereoselectivity, see: Minami N, Ko SS, Kishi Y. J Am Chem Soc. 1982;104:1109–1111.Ko SY, Lee AWM, Masamune S, Reed LA, Sharpless KB, III, Walker FJ. Science. 1983:949–951. doi: 10.1126/science.220.4600.949.Kobayashi S, Ohtsubo A, Mukaiyama T. Chem Lett. 1991:831–834.Hammadi A, Nuzillard JM, Poulin JC, Kagan HB. Tetrahedron: Asymmetry. 1992;3:1247–1262.Doyle MP, Kalinin AV, Ene DG. J Am Chem Soc. 1996;118:8837–8846.Trost BM, Calkins TL, Oertelt C, Zambrano J. Tetrahedron Lett. 1998;39:1713–1716.Balskus EP, Jacobsen EN. Science. 2007;317:1736–1740. doi: 10.1126/science.1146939.Han SB, Kong JR, Krische MJ. Org Lett. 2008;10:4133–4135. doi: 10.1021/ol8018874.
- 14.Fu F, Loh TP. Tetrahedron Lett. 2009;50:3530–3533. [Google Scholar]
- 15.Mengel A, Reiser O. Chem Rev. 1999;99:1191–1223. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 16.The allylation of unprotected β-hydroxyaldehydes using allylindium reagents provides anti-1,3-diols. The corresponding 1,3-syn-allylation of unprotected β-hydroxyaldehydes is only possible using chirally modified allyltitanium-TADDOL reagents: Paquette LA, Mitzel TM. J Am Chem Soc. 1996;118:1931–1937.BouzBouz S, Cossy J. Org Lett. 2000;2:501–504. doi: 10.1021/ol991349y.
- 17.“The ideal synthesis creates a complex skeleton… in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality.” Hendrickson JB. J Am Chem Soc. 1975;97:5784–5800.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.