

Abstract
α2 adrenergic receptor (α2-AR) agonists have been used as antihypertensive agents, in the management of drug withdrawal, and as sedative analgesics. Since α2-AR agonists also influence the regulation of body temperature, we explored their potential as antipyretic agents. This study delineates the central neural substrate for the inhibition of rat brown adipose tissue (BAT) and shivering thermogenesis by α2-AR agonists. Nanoinjection of the α2-AR agonist clonidine (1.2 nmol) into the rostral raphe pallidus area (rRPa) inhibited BAT sympathetic nerve activity (SNA) and BAT thermogenesis. Subsequent nanoinjection of the α2-AR antagonist idazoxan (6 nmol) into the rRPa reversed the clonidine-evoked inhibition of BAT SNA and BAT thermogenesis. Systemic administration of the α2-AR agonists dexmedetomidine (25 μg/kg, i.v.) and clonidine (100 μg/kg, i.v.) inhibited shivering EMGs, BAT SNA, and BAT thermogenesis, effects that were reversed by nanoinjection of idazoxan (6 nmol) into the rRPa. Dexmedetomidine (100 μg/kg, i.p.) prevented and reversed lipopolysaccharide-evoked (10 μg/kg, i.p.) thermogenesis in free-behaving rats. Cholera toxin subunit b retrograde tracing from rRPa and pseudorabies virus transynaptic retrograde tracing from BAT combined with immunohistochemistry for catecholaminergic biosynthetic enzymes revealed the ventrolateral medulla as the source of catecholaminergic input to the rRPa and demonstrated that these catecholaminergic neurons are synaptically connected to BAT. Photostimulation of ventrolateral medulla neurons expressing the PRSx8-ChR2-mCherry lentiviral vector inhibited BAT SNA via activation of α2-ARs in the rRPa. These results indicate a potent inhibition of BAT and shivering thermogenesis by α2-AR activation in the rRPa, and suggest a therapeutic potential of α2-AR agonists for reducing potentially lethal elevations in body temperature during excessive fever.
Introduction
Regulating body temperature in the face of challenges from the environment and from disease processes is one of the most fundamental homeostatic functions of the CNS. Thermoregulatory circuits in the CNS orchestrate fever, an elevated core body temperature (TCORE) that optimizes host immune responses to infection or inflammation. However, the irreversible protein denaturation ensuing from excessive and sustained elevations in TCORE (hyperpyrexia, “lethal fever”) produces significant cell damage, compromising tissue function and contributing to the high mortality from a variety of rapidly fatal diseases, infections, and brain injuries, including cerebral malaria, meningitis, encephalitis, toxemia, HIV/AIDS, the abuse of amphetamine-based drugs, stroke, and intracranial hemorrhage and traumatic brain injury. Because an enhanced level of metabolic heat production (thermogenesis) in brown adipose tissue (BAT) and skeletal muscle (i.e., shivering) contribute significantly to the elevated TCORE in hyperpyrexia, a rapid and sustained inhibition of centrally driven thermogenesis would improve the prognosis and limit brain and tissue damage. However, few effective approaches exist for the rapid reversal of the extreme elevations in TCORE that are resistant to standard pharmacological antipyretic therapies based on cyclooxygenase inhibition. In particular, the effectiveness of attempts to decrease TCORE by mechanical cooling is limited by the reflex activation of thermogenic and heat conservation mechanisms that combat falls in TCORE. Thus, such mechanical approaches to decreasing TCORE during hyperpyrexia would benefit from an adjunctive therapy to block the physiological responses to external cooling.
The centrally generated elevation in TCORE during fever arises as a consequence of increased thermogenesis and reduced heat loss to the environment (Saper and Breder, 1994), the same combination of thermal effector responses stimulated by central thermoregulatory networks to defend TCORE during a cold challenge (Morrison, 2011). The premotor neurons controlling thermogenic effector activation are primarily within the region of the medullary rostral raphe pallidus area (rRPa) (Morrison et al., 2012) and project to the spinal cord to excite the BAT sympathetic preganglionic neurons and the α-motoneurons contributing to skeletal muscle shivering. Neurons in the ventrolateral medulla (VLM) provide a major inhibitory regulation of the sympathetic outflow to BAT, capable of reversing the increases in BAT heat production elicited during the febrile response (Cao et al., 2010). Catecholaminergic neurons within the VLM project to the RPa (Card et al., 2006), and spinally projecting neurons in the rRPa express α2 adrenergic receptors (α2-ARs) (Guyenet et al., 1994). Thus, the current studies tested the hypothesis that centrally acting α2-AR agonists clonidine and dexmedetomidine will block the activation of thermogenic premotor neurons in the rRPa, and thus could be effective antipyretic agents. We also identified the endogenous adrenergic input to the rRPa and demonstrated its capacity to inhibit thermogenesis via activation of α2-ARs in rRPa. These translational discoveries have wide therapeutic potential for reducing potentially lethal, centrally generated elevations in TCORE, and for the induction of therapeutic hypothermia. Identifying the precise site(s) and mechanism(s) of action of α2-AR agonists to reverse febrile thermogenesis could also contribute to the improved design of such pharmacologic agents by reducing off-target effects.
Materials and Methods
All procedures conform to the regulations detailed in the Guide for the Care and Use of Laboratory Animals (National Research Council, eighth edition) and were approved by the Animal Care and Use Committees of the Oregon Health and Science University or the University of Pittsburgh.
Acute physiology experiments in anesthetized rats
Male Sprague Dawley or Wistar rats (300–450 g; Charles River Laboratories) were anesthetized with isoflurane (2–3% in 100% O2) and the femoral artery and vein were cannulated. Rats were transitioned from isoflurane to urethane (750 mg/kg, i.v.) and α-chloralose (60 mg/kg, i.v.) anesthesia, except for those rats prepared for shivering EMG recordings, which were transitioned to Inactin (50 mg · kg−1 initial dose, 20 mg · kg−1 · h−1 continuous infusion thereafter; Sigma) anesthesia. In all rats, except those that were prepared for shivering EMG recordings, the trachea was cannulated, and the animal was artificially ventilated with 100% O2 (stroke volume, 0.75–1 ml/100 g body weight, 50–70 strokes per minute) and paralyzed with d-tubocurarine (0.6 mg/rat, i.v., supplemented thereafter with 0.3 mg when spontaneous respiratory activity was observed). Animals were placed in a stereotaxic frame with the incisor bar positioned 4 mm below the interaural line. A thermocouple inserted in the rectum was used to measure TCORE, which was maintained at 37.0 ± 0.5°C with a heat lamp and a water-perfused thermal blanket, unless noted otherwise. BAT temperature (TBAT) was recorded using a thermocouple placed in the interscapular BAT. Skin temperature (TSKIN) was recorded using a thermocouple placed on the hindquarter skin.
BAT SNA and thermogenesis.
A sympathetic nerve innervating the right interscapular BAT pad was recorded using bipolar hook electrodes as described previously (Madden and Morrison, 2009). Nerve activity was differentially amplified (10,000 to 50,000 times; CyberAmp 380; Molecular Devices), filtered (1–300 Hz), digitized, and recorded onto a hard drive using Spike2 software (Cambridge Electronic Design). A continuous (4 s bins) measure of BAT SNA amplitude was calculated (Spike2 software) from the autospectra of sequential 4 s segments of BAT SNA as the root mean square value (square root of the total power in the 0.1 to 20 Hz band of the BAT SNA). The control level of BAT SNA was taken as the mean BAT SNA amplitude during a 2 min period of minimum BAT SNA recorded when the rat was in a warm condition (TCORE, >37°C) and basal BAT SNA was absent.
Shivering EMG recordings.
Briefly, for EMG recording, bipolar pin electrodes (∼3 mm separation between poles) were inserted into nuchal, gastrocnemius, and masseter muscles, and the signal was filtered (100–5000 Hz) and amplified (2000×) with a CyberAmp 380 (Molecular Devices). As described previously (Nakamura and Morrison, 2011), EMG amplitude was quantified (Spike2; Cambridge Electronic Design) in sequential 4 s bins as the square root of the total power (root mean square) in the 0–500 Hz band of the autospectra of each 4 s segment of EMG.
Experimental protocols
The α2-AR agonist clonidine (1.2 nmol in 60 nl) was nanoinjected into the rRPa (3 mm caudal to lambda, on the midline, 9.6 to 9.8 mm ventral to dura over the cerebellum) to evaluate whether activation of α2-ARs in the rRPa inhibits BAT SNA and BAT thermogenesis evoked by (1) nanoinjection of prostaglandin E2 (PGE2; 60 ng in 60 nl saline; n = 4) into the medial preoptic area (MPA; 0.2 mm caudal to bregma, 0.6–0.8 mm lateral to bregma, 8.0 mm ventral to dura), or (2) cooling of the rat with a water-perfused thermal blanket (n = 8). The clonidine nanoinjection into the rRPa was followed by a nanoinjection of the α2-AR antagonist, idazoxan (6 nmol in 120 nl) or saline vehicle (120nl) into the same rRPa site. Injection sites were verified by localization of fluorescent polystyrene microspheres (FluoSpheres F8797, F8801, or F8803; Invitrogen) included in the injectate (dilution, 1:200).
To establish that systemically administered α2-AR agonists can inhibit febrile thermogenesis via activation of α2-ARs in the rRPa, rats (n = 6) were prepared for recordings of BAT SNA and BAT thermogenesis and received a nanoinjection of PGE2 (60 ng in 60 nl saline) into the MPA. A systemic injection of clonidine (100 μg/kg, i.v.) was followed by a nanoinjection of idazoxan (6 nmol in 120 nl) into the rRPa. A separate group of rats (n = 6) was prepared for recording shivering EMGs and received a nanoinjection of PGE2 (60 ng in 60 nl saline) into the MPA followed by a systemic injection of dexmedetomidine (25 μg/kg, i.v.) and a subsequent nanoinjection of idazoxan (6 nmol in 120 nl) into the rRPa.
Optogenetic experiments.
Male Sprague Dawley rats (n = 6; 350–400 g; Charles River Laboratories) were anesthetized with isoflurane (2–3% in 100% O2) and placed in a stereotaxic frame (incisor bar positioned at −11 mm from interaural zero). Following removal of a portion of the occipital bone, two nanoinjections (200 nl per injection of 3 × 108 lentiviral particles per ml) of a lentiviral vector consisting of eight tandem repeats of the Phox2-binding site and an enhanced version of the photoactivatable cationic channel rhodopsin receptor 2 (H134R) fused to mCherry (PRSx8-ChR2-mCherry) (Abbott et al., 2009) were made into the VLM (0.8 and 1.2 mm rostral, 1.7 mm lateral, and 2.6 mm ventral to calamus scriptorius). After a 14–34 d recovery period to allow for sufficient expression of the lentiviral vector, rats were prepared for BAT SNA recording as in the acute physiology protocol above. The photostimulation sites in the VLM targeted the approximate center of the region of the VLM that we demonstrated previously to inhibit BAT SNA (Cao et al., 2010). Briefly, a fiber optic cable (200 μm diameter; Thorlabs) was stereotaxically positioned in the VLM (1.0–1.5 mm rostral, ±1.7 mm lateral, and 2.3–2.5 mm ventral to calamus scriptorius). A diode-pumped 473 nm laser (model CL-473-075-O; CrystaLaser) was used as the light source. The continuous output of the laser was set at 10 mW by direct light meter (Thorlabs) measurement of the power ouput at the end of the fiber optic cable. Photostimulation of the VLM consisted of 60 s trains of 10 ms light pluses delivered at 5, 10, 20, or 40 Hz. Photostimulation was performed before and after nanoinjection of idazoxan (6 nmol in120 nl) into the rRPa (on the midline, 2.8–3 mm rostral, and 2.8 mm ventral to calamus scriptorius). Rats were then perfused transcardially with saline followed by 4% paraformaldehyde; brains were postfixed for 2–12 h in 4% paraformaldehyde and transferred to a 30% sucrose solution at 4°C for at least 1 d, at which time they were cut on a microtome (35 μm, 1:6 series); and sections were preserved in cryoprotectant at −20°C until processed for immunohistochemistry.
Physiology experiments in free-behaving rats.
Rats were implanted with a temperature transponder (IPTT-300; Bio Medic Data Systems) into the interscapular BAT. During a 1 week recovery period, rats received daily intraperitoneal injections of 0.5 ml sterile saline and were also acclimated for 2 h/d to an environmental chamber maintained at 24°C. On the experimental day, rats received an injection of either sterile saline (0.5 ml, i.p.; n = 5) or dexmedetomidine (100 μg/kg, i.p.; n = 4) and, within 5 min, an injection of lipopolysaccaride (LPS; 10 μg/kg/ml, i.p.). Rats were then placed in the environmental chamber maintained at 24°C. TBAT was assessed before and every 30 min for 3 h following the initial injection. In a separate group, rats received an injection of LPS (10 μg/kg/ml, i.p.) followed 1.5 h later by an injection of either sterile saline (0.5 ml, i.p.; n = 4) or dexmedetomidine (100 μg/kg, i.p.; n = 5). TBAT was assessed every 30 min for 3.5 h beginning just before the injection of LPS.
Anatomy experiments
Cholera toxin subunit b injections and immunohistochemical procedures.
Standard stereotaxic and immunohistochemical procedures were used as described previously (Madden, 2012). Briefly, male Sprague Dawley rats (n = 4, 350–400 g, Charles River Laboratories) were anesthetized with isoflurane (2–3% in 100% O2) and placed in a stereotaxic frame (incisor bar positioned at −4 mm from interaural zero). Via a burr hole in the interparietal bone, cholera toxin subunit b (CTb) conjugated to Alexa Fluor 488 or Alexa Fluor 594 (100–200 nl of 1 mg/ml; Invitrogen) was nanoinjected into the rRPa (3 mm caudal to lambda, on midline, 9.8 mm ventral to dura). One week later, rats were deeply anesthetized (100 mg/kg pentobarbital, i.p.) and perfused transcardially with saline followed by 4% paraformaldehyde; brains were removed, postfixed, saturated in a 30% sucrose solution, and cut on a microtome (35 μm, 1:6 series); and sections were preserved in cryoprotectant at −20°C until processed for immunohistochemistry as follows. Sections were incubated in the primary antibody solution, goat anti-CTb (1:20,000 in antibody dilution solution; List Biological Laboratories, lot #7032A6) and mouse anti-tyrosine hydroxylase (TH; 1:1500; ImmunoStar) or mouse anti-dopamine β hydroxylase (DβH; 1:500; Millipore), for 16 h on a shaker table at room temperature. The tissue was incubated in the secondary antibody solution (donkey anti-mouse-Alexa 488, used at 1:200, A21206, Invitrogen; donkey anti-goat-Alexa 594, used at 1:200, A11058, Invitrogen) for 1 h. The tissue was rinsed in PBS, mounted on slides, and coverslipped using Prolong Gold anti-fade reagent (Invitrogen).
Viral injections and immunohistochemical procedures.
Adult male Sprague Dawley rats (275–350 g; Zivic-Miller Laboratories) were maintained on a 12 h light/dark cycle with ad libitum access to food and water. Rats were housed in a biosafety level 2 facility throughout the experiments. Rats were anesthetized with an intraperitoneal injection of ketamine (60 mg/kg) and xylazine (7 mg/kg). The interscapular BAT was exposed, and 0.1 μl of an attenuated strain of pseudorabies virus [PRV-Bartha; 4 × 108 plaque-forming units (pfu)/ml] was injected into five different sites (total volume, 0.5 μl) using a 1 μl Hamilton syringe. After injections, the skin was closed with wound clips. Different postinoculation times were used to estimate the temporal progression of the virus through the CNS after injection into BAT: 72–98 h. After the appropriate postinoculation time, rats were deeply anesthetized with ketamine (120 mg/kg) and xylazine (14 mg/kg) and perfused transcardially with 0.9% saline followed by paraformaldehyde lysine-periodate fixative. The brains were fixed for 3 h and preserved in 20% sucrose at 4°C. Serial coronal sections (35 μm) were cut with a freezing-stage microtome, collected sequentially in eight sets, and stored in cryoprotectant solution at −20°C before immunohistochemical processing. A dual fluorescence labeling procedure was used to characterize neurons immunoreactive for PRV and DβH or TH. This procedure was similar to our immunohistochemical protocol described previously (Tupone et al., 2011) except that the primary antibodies used in the current study were mouse monoclonal anti-TH (1:2000; Millipore), mouse monoclonal anti-DβH (1:3000; Millipore Bioscience Research Reagents), and a rabbit polyclonal antiserum generated against acetone-inactivated virus (Rb133; 1:2000, kindly supplied by L. Enquist, Princeton University, Princeton, NJ). The secondary antibodies used were Cy3-conjugated donkey anti-rabbit IgG (1:500, red fluorescence to label PRV-infected neurons) and Cy2- conjugated donkey anti-mouse IgG (1:300, green fluorescence to label DβH-containing neurons; both secondary antibodies were from Jackson ImmunoResearch Laboratories). For the longest postinoculation time (98 h; Fig. 5D), to optimize the fluorescence in neurons with robust expression of PRV, fluorescence was reversed, and the secondary antibodies used were Alexa 488 anti-rabbit IgG (1:500, green fluorescence to label PRV-infected neurons) and Alexa 594 anti-mouse IgG (1:500, to label TH-containing neurons; both antibodies were from Invitrogen). Double-labeled, PRV-infected, TH-immunoreactive (TH-ir), or DβH-ir neurons displayed yellow fluorescence.
Figure 5.
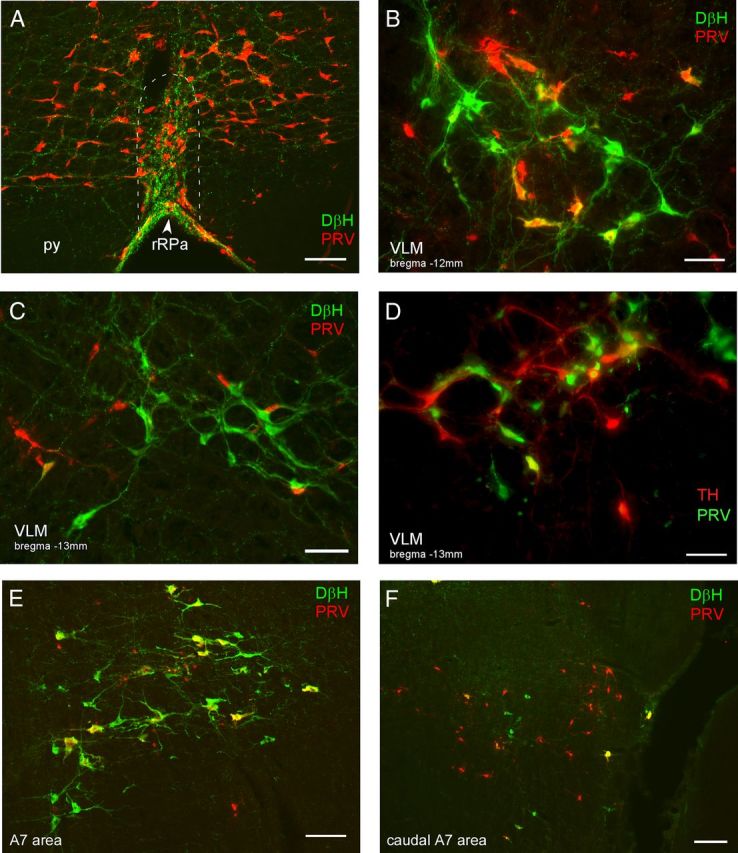
Neurons in the medulla and pons are synaptically connected to BAT. A–F, Photomicrographs are taken from rats that were killed 90 h (A–C, E) or 98 h (D, F) after injection of PRV into BAT. A, PRV-infected neurons (red) and DβH-ir fibers (green) in the rRPa (approximate boundaries indicated by the dashed white line), note the density of varicose DβH-ir fibers in close apposition to the PRV-ir neurons in the rRPa. py, Pyramidal tract. Photomicrographs illustrating PRV-ir (red), DβH-ir (green), and double-labeled (yellow) neurons in the VLM, ∼12.0 mm caudal to bregma (B) and 13.5 mm caudal to bregma (C). Note the double-labeled (yellow) neurons in the rostral VLM (B) but not the caudal VLM (C) at 90 h post-inoculation. D, Photomicrograph illustrating PRV-ir (green), TH-ir (red), and double-labeled (yellow) neurons in the VLM ∼13.3 mm caudal to bregma. Note that caudal VLM neurons become infected at long postinoculation times (98 h). E, A7 neurons of the rostral pons are infected at early postinoculation times, as illustrated by the double-labeled (DβH-ir, PRV-ir, yellow) neurons. F, At later postinoculation times, in the region of the pons containing the caudal aspect of the A7 population (DβH-ir, green neurons) there are many PRV-ir (red) neurons that are not DβH-ir. Scale bars: A, E, 100 μm; B, C, D, 50 μm; F, 200 μm.
Tissue analysis.
The neuroanatomical locations of PRV-infected neurons and TH-ir neurons in the VLM were based on a rat brain atlas (Paxinos and Watson, 2007). PRV-infected neurons (non-TH-ir) and PRV-infected, DβH-ir neurons (dual-labeled neurons) were counted in the VLM in consecutive sections from a 1:8 set of sections (245 μm distance between consecutive sections). To account for the inherent variability in the viral infection of adipose tissue (Cano et al., 2003), cases were grouped based on the total number of PRV-infected neurons in the VLM (intermediate infection, 145–200 neurons infected; late infection, 500–700 neurons infected), and the mean ± SEM was calculated for each group. Photomicrographs of PRV-infected neurons and DβH-ir neurons and fibers were obtained using an image capture system (Simple PCI, version 6.6; Hamamatsu) attached to an Olympus BX51 fluorescence microscope. The photomicrographs were assembled into a plate using Adobe Photoshop to adjust contrast and brightness without altering the original colors.
Data and statistical analyses
BAT SNA, TBAT, TCORE, TSKIN, expired CO2, arterial pressure, and stimulus trigger pulses were digitized (Micro3 1401; Cambridge Electronic Design) and recorded onto a computer hard drive for analysis (Spike2; Cambridge Electronic Design).
All statistics were performed using Systat software (version 10; Cranes Software International). Data are expressed as mean ± SEM. For each variable, statistical comparisons were done between the 30 s period before treatment and the 30 s window at the peak or nadir of the treatment-evoked effect. Statistical significance was assessed using a two-sample t test, or an ANOVA with repeated measures and post hoc testing as appropriate. Results with p < 0.05 were considered significant.
Results
α2-AR activation in the rRPa reverses nonshivering thermogenesis
We tested the hypothesis that α2-AR activation in the rRPa would inhibit the increases in BAT SNA, BAT thermogenesis, and heart rate (HR) evoked during the febrile response to injection of PGE2 into the MPA. As illustrated in Figure 1, A and B, nano-injection of PGE2 (60 pmol) into the MPA increased BAT SNA, TBAT, expired CO2, MAP, and HR. Subsequent nanoinjection of the α2-AR agonist clonidine (1.2 nmol) into the rRPa promptly reversed the PGE2-evoked increase in BAT SNA (t(3) = 2.458, p = 0.046, compared to PGE2-evoked peak), resulting in reductions in the elevated levels of TBAT and expired CO2 (an indirect indicator of metabolic O2 consumption; t(3) = 5.461, p = 0.006). The clonidine-evoked effects were long lasting (in cases where saline or no subsequent treatment was administered, the sympathoinhibitory effect of clonidine persisted for >20 min). Nanoinjection of the α2-AR antagonist idazoxan (6 nmol) into the same rRPa site completely reversed the effects of clonidine (BAT SNA, t(3) = −2.384, p = 0.049; expired CO2, t(3) = −4.825, p = 0.008; Fig. 1A,B).
Figure 1.

Effect of activation of α2-ARs in the rRPa on BAT thermogenesis evoked by PGE2 or cooling. A, C, The stimulation of BAT SNA evoked by nanoinjection of PGE2 into the medial preoptic area (A) or cooling (C) was reversed by nanoinjection of the α2-AR agonist clonidine into the rRPa. Subsequent nanoinjection of the α2-AR antagonist idazoxan into the rRPa restored the PGE2-evoked (A) or cooling-evoked (C) BAT SNA, whereas nanoinjection of saline failed to reverse the effect of clonidine. Changes in TBAT, expired CO2, and HR paralleled those in BAT SNA. However, due to the short time period between injections, the thermogenic increase in TCORE and the thermal inertia of the BAT, TBAT, remained significantly elevated following nanoinjection of clonidine. A, D, Insets, Diagrams of anatomical drawings depict the locations of the centers of the nanoinjection sites. 3V, Third ventricle; ac, anterior commissure; OX, optic chiasm; LPO, lateral preoptic area; py, pyramidal tract. B, The means ± SEM (n = 4) of the time courses of the physiological variable (each point is a 30 s average of the variable value) for the PGE2-evoked responses are shown. *p < 0.05 (significant increases compared with the basal value before PGE2 administration); #p < 0.05 (compared with the peak PGE2-evoked value just before clonidine administration). D, Means ± SEM of the peak BAT SNA during cooling, the nadir within 10 min of nanoinjection of clonidine into the rRPa, and the peak within 10 min of subsequent injection of idazoxan (n = 4) or saline into the rRPa (n = 4). *p < 0.05 (compared with cooling evoked peak); #p < 0.05 (compared with clonidine-evoked nadir).
Skin cooling also increased BAT SNA. The BAT SNA response to environmental cooling was inhibited by nanoinjection of clonidine (1.2 nmol) into rRPa (t(3) = 7.628, p = 0.005 and t(3) = 2.417, p = 0.047 for the groups receiving subsequent idazoxan and saline, respectively; Fig. 1C,D). Subsequent nanoinjection of idazoxan (6 nmol) into the same rRPa site resulted in a small transient (∼2–3 min) decrease in residual BAT SNA followed by a complete reversal of the clonidine-evoked sympathoinhibition (Fig. 1C,D; t(3) = −9.196, p = 0.003, compared to clonidine-evoked nadir). Nanoinjection of saline into the rRPa failed to reverse the clonidine-evoked inhibition of BAT SNA (Fig. 1D; saline rRPa compared to cooling evoked, t(3) = 2.512, p = 0.043). Under warm conditions when BAT SNA was absent, injection of idazoxan alone did not increase BAT SNA (data not shown).
Nanoinjection of idazoxan in rRPa reverses the inhibition of both nonshivering and shivering thermogenesis evoked by intravenous α2-AR agonists
To assess whether systemically administered α2-AR agonists can inhibit febrile BAT thermogenesis, we determined the effect of an intravenous injection of clonidine on BAT thermogenesis evoked by nanoinjection of PGE2 in MPA (Fig. 2). Nanoinjection of PGE2 (60 pmol) into the MPA increased BAT SNA, TBAT, expired CO2, MAP, and HR (Fig. 2A,B). Subsequent injection of clonidine (25 μg/kg, i.v.) promptly and completely reversed the increases in BAT SNA (t(5) = 3.586, p = 0.016), TBAT (t(5) = 5.085, p = 0.004), expired CO2 (t(5) = 4.392, p = 0.007), and HR (t(5) = 10.078, p < 0.001; Fig. 2A,B). Intravenous clonidine also significantly elevated MAP above the level attained following PGE2 in MPA (t(5) = −4.895, p = 0.004). The increase in MAP is attributable to peripheral vascular effects of the bolus administration of α2-AR agonists (van Zwieten, 1980) and is avoided clinically by slowly infusing highly selective α2-AR agonists (Flower and Hellings, 2012; Lam et al., 2012). To identify the site of action for the antipyretic effect of systemically administered α2-AR agonists, we determined whether nanoinjection of idazoxan in the rRPa would reverse the antipyretic effect of intravenous clonidine. Nanoinjection of idazoxan in rRPa reversed the decreases in BAT SNA (t(5)= −4.192, p = 0.009), TBAT (t(5) = −5.315, p = 0.003), CO2 (t(5) = −5.590, p = 0.003), and HR (t(5) = −10.786, p < 0.001) evoked by intravenous injection of clonidine (Fig. 2A,B).
Figure 2.

Effect of systemic administration of an α2-AR agonist and subsequent blockade of α2-ARs specifically in the the rRPa on BAT thermogenesis evoked by PGE2. A, Nanoinjection of PGE2 into the MPA increased BAT SNA, TBAT, expired CO2, mean arterial pressure (MAP), and HR. Subsequent intravenous administration of clonidine reversed the increases in BAT SNA, TBAT, expired CO2, and HR, whereas MAP was increased further. Nanoinjection of the α2-AR antagonist idazoxan into rRPa restored the PGE2-evoked BAT SNA, TBAT, expired CO2, and HR. Inset, Diagrams of anatomical drawings depict the locations of the centers of the nanoinjection sites. 3V, Third ventricle; ac, anterior commissure; LPO, lateral preoptic area; OX, optic chiasm; py, pyramidal tract. B, Means ± SEM (n = 6) of the time courses of the physiological variables (each point is a 30 s average of the variable value). *p < 0.05 (significant increases compared with the basal value before PGE2 administration or cooling); #p < 0.05 (compared with the peak PGE2- or cooling-evoked value just before clonidine administration).
To determine whether systemically administered α2-AR agonists also inhibit febrile shivering thermogenesis, dexmedetomidine (25 μg/kg, i.v.) was injected during episodes of shivering evoked by nanoinjection of PGE2 in MPA (Fig. 3). Systemic administration of dexmedetomidine promptly and completely inhibited shivering EMGs recorded from the nuchal (t(4) = 3.356, p = 0.028), gastrocnemius (t(3) = 3.242, p = 0.048), and masseter (t(4) = 10.983, p < 0.001) muscles and reversed the PGE2-evoked increase in TCORE (t(4) = 2.236, p = 0.045; Fig. 3A,B). The dexmedetomidine-induced inhibition of shivering was reversed by nanoinjection of idazoxan in the rRPa (nuchal, t(4) = −2.415, p = 0.037; gastrocnemius, t(3) = −2.151, p = 0.06; masseter, t(4) = −10.058, p < 0.001; Fig. 3A,B).
Figure 3.

Effect of systemic administration of an α2-AR agonist and subsequent blockade of α2-ARs specifically in the rRPa on shivering thermogenesis evoked by PGE2. A, Nanoinjection of PGE2 into the MPA increased shivering EMG activity in the nuchal, gastrocnemius, and masseter muscles and increased TCORE. Subsequent intravenous injection of dexmedetomidine (DEX) completely inhibited all EMG activity and decreased TCORE. Nanoinjection of the α2-AR antagonist idazoxan into the rRPa reversed the DEX-induced inhibition of shivering EMG activity in all recorded muscles. Inset, Diagrams of anatomical drawings depict the locations of the centers of the nanoinjection sites. ac, Anterior commissure; LPO, lateral preoptic area; OX, optic chiasm; py, pyramidal tract. B, Means ± SEM (n = 6) of the time courses of the physiological variables (each point is a 30 s average of the variable value). *p < 0.05 (significant increases compared with the basal value before PGE2 administration); #p < 0.05 (compared with the peak PGE2-evoked value just before DEX administration).
Source of the catecholaminergic input to rRPa
There is a dense catecholaminergic innervation of the rRPa, as indicated by the many fibers and axonal swellings within this area that are immunoreactive for the catecholamine biosynthetic enzymes DβH (Figs. 4A, 5A). To determine which catecholaminergic neurons innervate the rRPa and thereby provide potential endogenous sources of catecholamines to activate the thermogenesis inhibiting α2-ARs in this region, we injected the retrograde tracer CTb into the rRPa (Fig. 4B,C) and assessed the presence of CTb in neurons that were immunoreactive for DβH, TH, or phenylethanolamine N-methyltransferase (PNMT). The extent of the CTb injection sites encompassed the entire rRPa and also extended into the raphe magnus (RMg) as well as around the medial edges of the pyramidal tracts (Fig. 4B,C). Double-labeled, DβH-ir, and CTb-ir neurons (Fig. 4D–G) were found in the VLM (Fig. 4H), extending caudally from ∼0.5 mm caudal to the facial nucleus for ∼2 mm, but concentrated in the region between 12.5 to 13.5 mm caudal to bregma (Paxinos and Watson, 2007). This region corresponds largely to the caudal C1 cell population (Card et al., 2006; Schreihofer and Sved, 2011), and indeed many CTb-labeled neurons in the VLM were immunoreactive for PNMT (data not shown). Based on the difference in the number of DβH-ir versus PNMT-ir neurons in the VLM, it is likely that some rostral A1 neurons were also retrogradely labeled from the rRPa. In addition, there were many CTb-labeled neurons in the VLM that were not immunoreactive for DβH or TH (Fig. 4I). There were also many CTb-labeled neurons in the rostral pons near the medial paralemniscal nucleus in close proximity to the A7 catecholaminergic cell population; however, none of these neurons were immunoreactive for either DβH or TH (Fig. 4J–M). In addition, very few A2 neurons (fewer than five cells per rat) or A5 cells (fewer than five cells per rat) and no A6 cells contained CTb.
Figure 4.
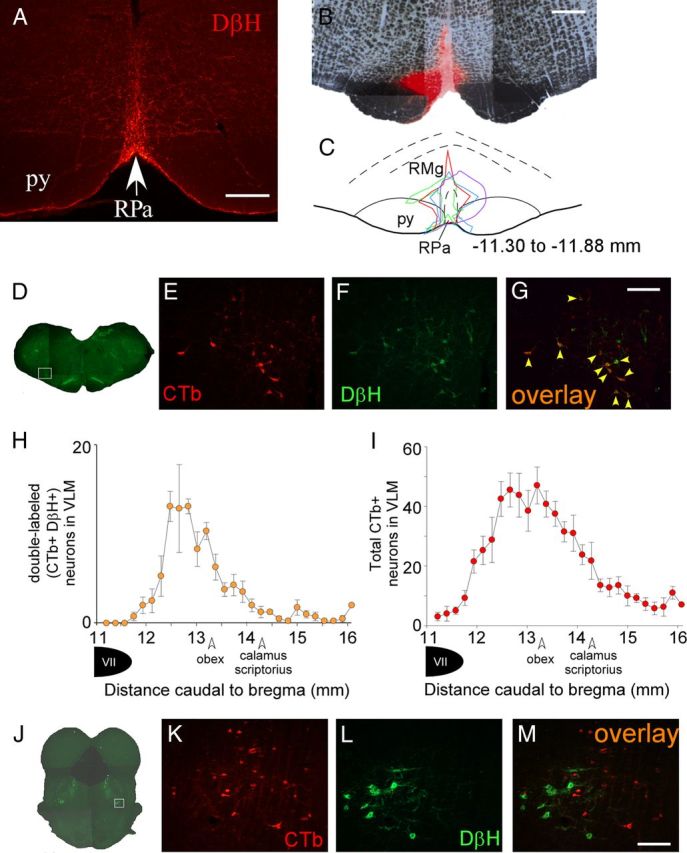
The caudal C1 area of the VLM provides the principal source of catecholaminergic input to the rRPa. A, Photomicrograph of DβH-ir highly varicose fibers in the rRPa. B, Representative example of a CTb injection site in the rRPa. C, Diagram drawing of the four CTb injection sites, each encompassing the entirety of the rRPa. Numbers represent the approximate distance caudal to bregma (Paxinos and Watson, 2007). D, Composite photomicrograph of a brainstem section representative of those in which CTb labeling was found in DβH-containing neurons in the caudal C1 region of the VLM; the white box depicts the area that is presented at higher magnification in E–G. CTb-ir neurons (red), and DβH-ir neurons (green) in the VLM and pons are illustrated in E, F, K, and L. G, Double-labeled (CTb-ir, DβH-ir) neurons (orange, indicated by yellow arrowheads) in the VLM. H, Counts (mean ± SEM, n = 4) of double-labeled neurons in the VLM with respect to the distance from bregma. I, Total number of CTb-ir neurons (mean ± SEM, n = 4) in the VLM. J, Composite photomicrograph of a pontine section through the region containing the A7 catecholaminergic cell population. The white box depicts the area presented at higher magnification in K–M. Note that there are no double labeled neurons in the pons (M). Scale bars: A, 250 μm; B, 500 μm; G (for E–G), M (for K–M), 200 μm.
Catecholaminergic neurons in the VLM are synaptically connected to BAT
To define the anatomical locations of catecholaminergic neurons in the VLM that are synaptically connected to BAT, we injected PRV into BAT and assessed the presence of PRV in catecholaminergic neurons in the VLM at a series of postinoculation times following PRV injection. As reported previously (Cano et al., 2003), neurons in the RPa are infected at early infection times, consistent with their infection arising from the direct spinal projection of the BAT sympathetic premotor neurons in rRPa (Morrison et al., 2012). The PRV-infected neurons in the rRPa are found in a dense network of highly varicose DβH-ir fibers (Fig. 5A). A few DβH-ir neurons in the rostral VLM (RVLM) are infected by PRV at early infection times. Many DβH-ir neurons in the rostral VLM are infected by PRV at intermediate and late infection times (Figs. 5B, 6A,B). At later infection times (Fig. 5D) double-labeled, PRV-ir, and DβH-ir neurons appear more caudally in the region of the VLM between 12.5 and 13.5 mm caudal to bregma, corresponding to the area where DβH-ir neurons are labeled following injection of CTb in rRPa (compare Figs. 4H, 6B). The timing of the PRV infection of these VLM neurons is consistent with their infection arising from their axonal projections to the RPa, or to other brain regions that are infected earlier. In addition to the DβH-ir neurons in the VLM, there were many infected neurons in the VLM that were not DβH-ir (Fig. 5B–D). As with the CTb-labeled projection from the VLM to the rRPa, the noncatecholaminergic population of VLM neurons comprised the majority of PRV-labeled cells within this area and was intermingled with the DβH-ir, PRV-infected population.
Figure 6.

A, B, Counts (mean ± SEM) of PRV-only (white circles) and PRV-ir, DβH-ir (black squares) neurons throughout the VLM (from 11 to 15 mm caudal to bregma) at intermediate (A; n = 2) and late infection times (B; n = 3). Note the progression of infection: at the intermediate infection time, very few catecholaminergic neurons are infected with PRV in the caudal VLM (∼12.75–13.5 mm caudal to bregma), whereas at the late infection time, many catecholaminergic neurons of this region begin to become infected with PRV.
At intermediate infection times there were many A7 neurons labeled with PRV (Fig. 5E), consistent with their infection arising from a direct spinal projection. At later infection times, some PRV-infected neurons lacking DβH immunoreactivity appeared medial to the A7 neurons. At the caudal level of the A7 cell population, there were PRV-infected neurons lacking DβH immunoreactivity lateral to the A7 cell population (Fig. 5F) in an area where noncatecholaminergic, CTb-labeled neurons were observed following injection of CTb in rRPa (compare Figs. 4M, 5F).
Optogenetic activation of VLM neurons inhibits BAT thermogenesis
To determine whether activation of catecholaminergic neurons in the VLM inhibits BAT SNA and BAT thermogenesis via activation of α2-ARs in the rRPa, we used an optogenetic approach to preferentially activate catecholaminergic neurons within the VLM, and we determined whether blockade of α2-ARs in the rRPa would prevent the photostimulation-evoked inhibition of BAT SNA and BAT thermogenesis. Injection of the PRSx8-ChR2-mCherry lentivirus into the VLM produced mCherry expression in neurons (Fig. 7B), most of which (53 ± 7%; n = 6) were immunoreactive for DβH (Fig. 7C). PRSx8-ChR2-mCherry expression was robust and, in addition to expression in the cell soma, was found in fibers and terminals of infected neurons, with an especially dense expression in fibers and axonal swellings in the rRPa (Fig. 7D). Photostimulation of the right VLM (60 s; 5–40 Hz; 10 ms pulses; ∼10 mW), the side containing ChR2-expressing neurons, abruptly inhibited cold-evoked BAT SNA and decreased TBAT in a frequency-dependent manner, with the 40 Hz stimulation completely inhibiting BAT SNA (Fig. 7E,F). This BAT sympathoinhibition was attenuated by nanoinjection of idazoxan into the rRPa (two-way repeated measures ANOVA, drug effect, F(1,6) = 12.497, p = 0.012; frequency effect, F(3,18) = 19.868, p < 0.001; no significant interaction effect, F(3,18) = 2.329, p = 0.114; Fig. 7G–I). The photostimulation-evoked decreases in TBAT were modest and did not differ significantly between the trials before and after nanoinjection of idazoxan (Fig. 7J), likely due to the short duration of photostimulation and the thermal inertia of BAT. Photostimulation of the left, uninfected VLM had no effect.
Figure 7.
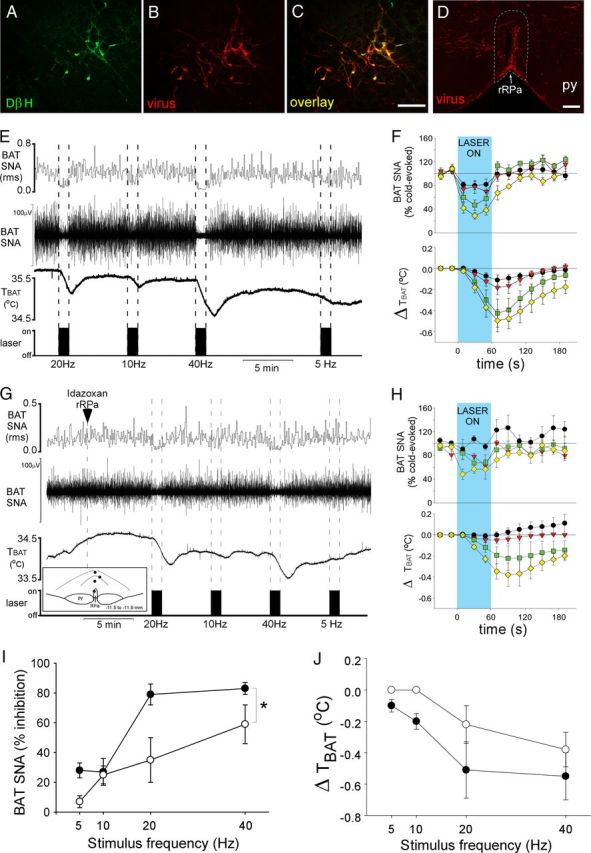
Photostimulation of VLM neurons inhibits BAT SNA via activation of α2-adrenergic receptors in the rRPa. A–C, Photomicrographs of the same area of the VLM, illustrating neurons expressing DβH (green), PRSx8-ChR2-mCherry lentivirus (red), and both DβH and lentivirus (yellow). D, Photomicrograph of the rRPa (approximate borders are denoted by the white dotted line) containing highly varicose fibers expressing the lentivirus (red). E, G, BAT SNA and TBAT during photostimulation of the VLM at several stimulus frequencies before (E) and after nanoinjection of idazoxan (G) into the rRPa. G, Inset, Diagram depicts the centers of the injection sites. F, H, The group data (mean ± SEM; n = 4) of the time course of changes in BAT SNA and TBAT evoked by photostimulation (blue shaded region) at 5 Hz (black circles), 10 Hz (red triangles), 20 Hz (green squares), and 40 Hz (yellow diamonds) stimulation frequencies before (F) and after nanoinjection of idazoxan (H) in the rRPa. I, J, The photostimulation frequency response curve of the percentage of inhibition of BAT SNA (I) and the maximal change in TBAT (J) before (filled circles) and after nanoinjection of idazoxan (open circles) in rRPa. The asterisk indicates two-way repeated measures ANOVA: drug effect, p < 0.05; frequency effect, p < 0.001; and no significant interaction effect, p = 0.114. py, Pyramidal tract. Scale bars: 100 μm.
Intravenous injection of dexmedetomidine prevents/reverses LPS-evoked fever
To assess whether systemic administration of an α2-AR agonist was capable of preventing or reversing fever in a free-behaving rat, an intraperitoneal injection of dexmedetomidine or saline was given to rats either before or 1.5 h after an intraperitoneal injection of LPS. Following saline administration, an injection of LPS increased TBAT (Fig. 8A). The increase in TBAT evoked by LPS was prevented for >3 h by pretreatment with dexmedetomidine (two-way repeated measures ANOVA, drug effect, saline vs dexmedetomidine, F(1,7) = 77.514, p < 0.001; time effect, F(6,42) = 6.808, p < 0.001; interaction effect, F(6,42) = 9.684, p < 0.001). In fact, TBAT decreased by nearly 3°C following pretreatment with dexmedetomidine (Fig. 8A). Similarly, when dexmedetomidine was injected 1.5 h after administration of LPS, the hyperthermia evoked by LPS was reversed, and indeed TBAT decreased by ∼2.5°C (falling ∼2.0°C below the basal, pre-LPS temperature; Fig. 8B; two-way repeated measures ANOVA, drug effect, saline vs dexmedetomidine, F(1,7) = 20.372, p = 0.003; time effect, F(7,49) = 11.434, p < 0.001; interaction effect, F(7,49) = 15.824, p < 0.001).
Figure 8.

Systemic administration of the α2-adrenergic receptor agonist dexmedetomidine (100 μg/kg) prevents and reverses LPS (10 μg/kg)-evoked thermogenesis. Symbols represent the group means ± SEM of TBAT just before administration of LPS, at the time of LPS administration (time 0), and at 30 min intervals following injection of LPS. A, The change in TBAT following intraperitoneal injection of LPS preceded by an intraperitoneal injection of saline (open circles; n = 5) or dexmedetomidine (filled circles; n = 4). The asterisk indicates two-way repeated measures ANOVA: drug effect, p < 0.001; time effect, p < 0.001; and interaction effect, p < 0.001. B, The increase in TBAT following an intraperitoneal injection of LPS is reversed 1.5 h later by an intraperitoneal injection of dexmedetomidine (filled circles; n = 5) but not saline (open circles; n = 4). The asterisk indicates two-way repeated measures ANOVA: drug effect, p < 0.005; time effect, p < 0.001; interaction effect, p < 0.001.
Discussion
In the present study, we demonstrate that systemic administration of α2-AR agonists reverses LPS- and central PGE2-evoked fevers and can induce a modest hypothermia in normal ambient temperatures. These effects arise from a potent inhibition of the metabolic heat production in BAT and in skeletal muscle during shivering due to an α2-AR-mediated inhibition of the populations of premotor neurons in the rRPa that are responsible for the descending excitatory drives to spinal BAT sympathetic preganglionic neurons and to spinal α-motoneurons. The catecholaminergic neurons providing the endogenous sources of adrenergic agonist for the α2-AR in the rRPa are located in the region of the caudal C1/rostral A1 catecholaminergic cell groups in the VLM. These findings explicate a novel mechanism for the inhibitory regulation of metabolic heat production and support a novel pharmacological approach to the control of excessive or neurogenic fevers that are resistant to commonly prescribed, cyclooxygenase inhibition-based antipyretic therapies.
α2-AR agonists cause a modest hypothermia in the rodent (Millan et al., 2000; Lähdesmäki et al., 2003), attenuate febrile responses in rabbits (Szreder, 1997) and horses (Kendall et al., 2010), and reduce human shivering in clinical hypothermic settings (Weant et al., 2010; Logan et al., 2011). Early studies to identify the site and mechanism underlying the hypothermic effects of α2-AR agonists focused on the thermoregulatory circuits in the preoptic area (Mallick and Alam, 1992; Quan et al., 1992; Romanovsky et al., 1993; Millan et al., 2000). However, the results were inconclusive, with other studies reporting hyperthermia (Romanovsky et al., 1993; Feleder et al., 2004, 2007) or no change in body temperature (Osaka, 2009) following preoptic injection of an α2-AR agonist. Our demonstration that rRPa injection of an α2-AR antagonist reversed the inhibitions of BAT thermogenesis and shivering evoked by systemic administration of α2-AR agonists strongly supports the view that the hypothermic and antipyretic effects of α2-AR agonists are mediated by activation of α2-ARs in the rRPa. The localization of α2-ARs on spinally projecting neurons in the RPa (Guyenet et al., 1994) and the essential role played by spinally projecting premotor neurons in the rRPa in controlling thermoregulatory and febrile activations of BAT thermogenesis and shivering (Nakamura and Morrison, 2011; Morrison et al., 2012) are consistent with a model in which α2-AR agonists binding to inhibitory α2-ARs on thermogenic premotor neurons in rRPa reduces their responses to excitatory inputs, including those from the dorsomedial hypothalamus, which are essential for the activation of thermogenic premotor neurons in rRPa controlling metabolic heat production during cold exposure and fever (Madden and Morrison, 2004; Nakamura and Morrison, 2007, 2011).
Potent inhibition of BAT SNA and BAT thermogenesis can be elicited by activation of neurons over an ∼3 mm rostrocaudal extent of the VLM that extends from the facial nucleus to the obex (Cao et al., 2010). The present demonstrations that neurons in the caudal C1/rostral A1 region of the VLM provide the catecholaminergic input to the rRPa and that α2-AR activation in the rRPa inhibits BAT SNA and BAT thermogenesis suggest that activation of α2-ARs in the rRPa by catecholamines released from these VLM neurons contributes significantly to the potent inhibition of BAT SNA evoked from this region of the VLM. However, because idazoxan injections into the rRPa did not completely block the inhibition of BAT SNA evoked from the caudal C1/rostral A1 region of the VLM, a nonadrenergic input from the VLM to the rRPa could contribute, since retrograde tracing revealed a significant noncatecholaminergic input from the caudal C1/rostral A1 region of the VLM to the rRPa and catecholaminergic neurons in the VLM may release other neurotransmitters (Stornetta et al., 1999, 2002; Abbott et al., 2012). VLM neurons may contribute to the inhibition of BAT SNA via an indirect pathway. In this regard, following lentivirus injection into the VLM, we observed PRSx8-ChR2-mCherry-expressing fibers in brain regions including the median preoptic area, paraventricular hypothalamus, and nucleus tractus solitarius, from which inhibitions of BAT SNA can also be elicited (Nakamura and Morrison, 2008; Madden and Morrison, 2009; Cao et al., 2010).
Although catecholaminergic neurons in the caudal C1/rostral A1 region of the VLM (12.5 to 13.5 mm caudal to bregma) project strongly to the rRPa (Fig. 4H), we found very few C1 cells in the RVLM (∼11.75 to 12.25 mm caudal to bregma) that were retrogradely labeled from the rRPa. These results are consistent with the report that the rostral levels of RPa are devoid of EGFP-expressing axon terminals following virally mediated EGFP expression selectively in the rostral C1 catecholaminergic neurons of the RVLM (Card et al., 2006). Thus, if the C1 cells in the RVLM participate in the inhibition of BAT thermogenesis elicited from this most rostral level of the VLM (Cao et al., 2010), we postulate that this effect would be indirect, rather than through a direct input to the rRPa.
Catecholaminergic neurons in the RVLM (i.e., rostral C1 neurons), as well as neurons in rRPa, are among the first brainstem PRV-ir neurons at early postinoculation times, reflecting their infection via their spinal projections and their roles as premotor neurons (Cano et al., 2003). The dense network of highly varicose DβH fibers surrounding the PRV-ir neurons in the rRPa is consistent with a direct catecholaminergic input to BAT sympathetic premotor neurons. The source of this catecholaminergic input to BAT-projecting neurons in rRPa is suggested by the PRV infection of TH-ir neurons in the caudal C1 region of the VLM at later postinoculation times, consistent with these neurons becoming infected via their projections to premotor regions, such as the rRPa, that were infected at the earlier postinoculation times. The localization of CTb retrograde labeling of catecholaminergic neurons in the caudal C1 region of the VLM following CTb injections in rRPa indicates that at least some of the PRV-ir and TH-ir neurons in the caudal C1 region of the VLM became infected specifically via their projection to BAT premotor neurons in the rRPa. Our findings with PRV transsynaptic retrograde tracing from BAT provide further support for the division of the VLM into regions that can inhibit BAT thermogenesis via distinct pathways, with only neurons in the caudal C1 region providing a direct catecholaminergic input to the rRPa. The functions of these distinct inhibitory pathways and the physiological stimuli through which they may be differentially activated remain to be determined.
Our observation that none of the rRPa-projecting neurons in the A7 region of the rostral pons contain DβH or TH is inconsistent with a report that A7 noradrenergic neurons were retrogradely labeled following injection of CTb into the rostral medullary raphe (Yoshida et al., 2009). This discrepancy is most likely explained by differences in the CTb injection sites. Since the CTb injections in the study by Yoshida et al. (2009) were relatively large and not specifically targeted to the rRPa, with many injections including the RMg and pyramidal tract and some injections completely excluding the rRPa, the labeling of A7 neurons may be due to their projections into areas outside of rRPa. Since the current study specifically targeted the rRPa, our CTb injections were nearly completely confined to the rRPa.
Clonidine, dexmedetomidine, and idazoxan have affinities for both α2-ARs and imidazoline receptors (Ernsberger et al., 1987, 1990; Savola and Savola, 1996). Although systemic administration of a specific imidazoline receptor agonist decreases body temperature (Thorn et al., 2012), it is unlikely that the inhibitions of thermogenesis observed in the current studies are mediated by activation of imidazoline receptors in the rRPa because (1) α2-ARs are expressed in spinally projecting neurons of the RPa (Guyenet et al., 1994), (2) the hypothermic actions of α2-AR agonists are attenuated in mice lacking α2-ARs (Hunter et al., 1997), and (3) the imidazoline binding site has not been localized in the rRPa (Brüning et al., 1987; Ernsberger et al., 1995; Ruggiero et al., 1995).
Potential physiological roles for the α2-AR-mediated inhibition of the metabolic heat production and energy consumption by BAT thermogenesis and shivering are suggested by the observations that catecholaminergic neurons of the VLM are activated by hypoxia (Hirooka et al., 1997) and by glucoprivation (Ritter et al., 1998), two stimuli that inhibit BAT SNA and BAT thermogenesis (Madden and Morrison, 2005; Madden, 2012). Additionally, a catecholaminergic inhibition of thermogenesis may contribute to the inability of DβH knock-out mice to initiate torpor (Swoap and Weinshenker, 2008). The physiological role(s) of BAT sympathoinhibitory mechanisms in the VLM and the role of α2-AR activation in the rRPa in specific physiological responses remain to be investigated.
In conclusion, α2-AR activation in the rRPa elicits a potent inhibition of both febrile-evoked and cold-evoked BAT thermogenesis and shivering. Neurons in the caudal C1 region of the VLM provide the endogenous catecholaminergic input activating α2-AR in the rRPa. These discoveries have wide and immediate therapeutic potential for reducing potentially lethal, centrally generated elevations in body temperature, and for complementing existing approaches to the effective and rapid induction of therapeutic hypothermia. Furthermore, by better defining the precise mechanisms of the thermolytic properties of α2-AR agonists, this study provides a foundation for the improved design of agents targeting these pathways.
Footnotes
This work was supported by NIH Grants NS040987 (S.F.M.) and DK-082558 (C.J.M.), an American Heart Association Scientist Development Grant (C.J.M.), and National Science Foundation Grant NSF-0918867 (G.C.). We are grateful to Dr. Patrice Guyenet and Dr. Ruth Stornetta for providing the plasmid containing the lentiviral construct, and to Dr. Lynn Enquist of Princeton University for the gift of PRV virus and antiserum. We acknowledge Rubing Xing for histological and immunohistochemical assistance and Jane Igoe for contributions to this work.
The authors declare no competing financial interests.
References
- Abbott SB, Stornetta RL, Socolovsky CS, West GH, Guyenet PG. Photostimulation of channelrhodopsin-2 expressing ventrolateral medullary neurons increases sympathetic nerve activity and blood pressure in rats. J Physiol. 2009;587:5613–5631. doi: 10.1113/jphysiol.2009.177535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Kanbar R, Bochorishvili G, Coates MB, Stornetta RL, Guyenet PG. C1 neurons excite locus coeruleus and A5 noradrenergic neurons along with sympathetic outflow in rats. J Physiol. 2012;590:2897–2915. doi: 10.1113/jphysiol.2012.232157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning G, Kaulen P, Baumgarten HG. Quantitative autoradiographic localization of alpha 2-antagonist binding sites in rat brain using [3H]idazoxan. Neurosci Lett. 1987;83:333–337. doi: 10.1016/0304-3940(87)90110-8. [DOI] [PubMed] [Google Scholar]
- Cano G, Passerin AM, Schiltz JC, Card JP, Morrison SF, Sved AF. Anatomical substrates for the central control of sympathetic outflow to interscapular adipose tissue during cold exposure. J Comp Neurol. 2003;460:303–326. doi: 10.1002/cne.10643. [DOI] [PubMed] [Google Scholar]
- Cao WH, Madden CJ, Morrison SF. Inhibition of brown adipose tissue thermogenesis by neurons in the ventrolateral medulla and in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol. 2010;299:R277–R290. doi: 10.1152/ajpregu.00039.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, Sved JC, Craig B, Raizada M, Vazquez J, Sved AF. Efferent projections of rat rostroventrolateral medulla C1 catecholamine neurons: Implications for the central control of cardiovascular regulation. J Comp Neurol. 2006;499:840–859. doi: 10.1002/cne.21140. [DOI] [PubMed] [Google Scholar]
- Ernsberger P, Meeley MP, Mann JJ, Reis DJ. Clonidine binds to imidazole binding sites as well as alpha 2-adrenoceptors in the ventrolateral medulla. Eur J Pharmacol. 1987;134:1–13. doi: 10.1016/0014-2999(87)90125-7. [DOI] [PubMed] [Google Scholar]
- Ernsberger P, Giuliano R, Willette RN, Reis DJ. Role of imidazole receptors in the vasodepressor response to clonidine analogs in the rostral ventrolateral medulla. J Pharmacol Exp Ther. 1990;253:408–418. [PubMed] [Google Scholar]
- Ernsberger P, Graves ME, Graff LM, Zakieh N, Nguyen P, Collins LA, Westbrooks KL, Johnson GG. I1-imidazoline receptors. Definition, characterization, distribution, and transmembrane signaling. Ann N Y Acad Sci. 1995;763:22–42. doi: 10.1111/j.1749-6632.1995.tb32388.x. [DOI] [PubMed] [Google Scholar]
- Feleder C, Perlik V, Blatteis CM. Preoptic alpha 1- and alpha 2-noradrenergic agonists induce, respectively, PGE2-independent and PGE2-dependent hyperthermic responses in guinea pigs. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1156–R1166. doi: 10.1152/ajpregu.00486.2003. [DOI] [PubMed] [Google Scholar]
- Feleder C, Perlik V, Blatteis CM. Preoptic norepinephrine mediates the febrile response of guinea pigs to lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1135–R1143. doi: 10.1152/ajpregu.00067.2007. [DOI] [PubMed] [Google Scholar]
- Flower O, Hellings S. Sedation in traumatic brain injury. Emerg Med Int. 2012 doi: 10.1155/2012/637171. Advance online publication. Retrieved November 7, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Riley T, Norton FR, Rosin DL, Lynch KR. Alpha 2A-adrenergic receptors are present in lower brainstem catecholaminergic and serotonergic neurons innervating spinal cord. Brain Res. 1994;638:285–294. doi: 10.1016/0006-8993(94)90661-0. [DOI] [PubMed] [Google Scholar]
- Hirooka Y, Polson JW, Potts PD, Dampney RA. Hypoxia-induced Fos expression in neurons projecting to the pressor region in the rostral ventrolateral medulla. Neuroscience. 1997;80:1209–1224. doi: 10.1016/s0306-4522(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Hunter JC, Fontana DJ, Hedley LR, Jasper JR, Lewis R, Link RE, Secchi R, Sutton J, Eglen RM. Assessment of the role of alpha2-adrenoceptor subtypes in the antinociceptive, sedative and hypothermic action of dexmedetomidine in transgenic mice. Br J Pharmacol. 1997;122:1339–1344. doi: 10.1038/sj.bjp.0701520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall A, Mosley C, Bröjer J. Tachypnea and antipyresis in febrile horses after sedation with alpha-agonists. J Vet Intern Med. 2010;24:1008–1011. doi: 10.1111/j.1939-1676.2010.0528.x. [DOI] [PubMed] [Google Scholar]
- Lähdesmäki J, Sallinen J, MacDonald E, Sirviö J, Scheinin M. Alpha2-adrenergic drug effects on brain monoamines, locomotion, and body temperature are largely abolished in mice lacking the alpha2A-adrenoceptor subtype. Neuropharmacology. 2003;44:882–892. doi: 10.1016/s0028-3908(03)00080-7. [DOI] [PubMed] [Google Scholar]
- Lam F, Ransom C, Gossett JM, Kelkhoff A, Seib PM, Schmitz ML, Bryant JC, Frazier EA, Gupta P. Safety and efficacy of dexmedetomidine in children with heart failure. Pediatr Cardiol. 2012 doi: 10.1007/s00246-012-0546-7. Advance online publication. Retrieved November 7, 2012. [DOI] [PubMed] [Google Scholar]
- Logan A, Sangkachand P, Funk M. Optimal management of shivering during therapeutic hypothermia after cardiac arrest. Crit Care Nurse. 2011;31:e18–e30. doi: 10.4037/ccn2011618. [DOI] [PubMed] [Google Scholar]
- Madden CJ. Glucoprivation in the ventrolateral medulla decreases brown adipose tissue sympathetic nerve activity by decreasing the activity of neurons in raphe pallidus. Am J Physiol Regul Integr Comp Physiol. 2012;302:R224–R232. doi: 10.1152/ajpregu.00449.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden CJ, Morrison SF. Excitatory amino acid receptors in the dorsomedial hypothalamus mediate prostaglandin-evoked thermogenesis in brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2004;286:R320–R325. doi: 10.1152/ajpregu.00515.2003. [DOI] [PubMed] [Google Scholar]
- Madden CJ, Morrison SF. Hypoxic activation of arterial chemoreceptors inhibits sympathetic outflow to brown adipose tissue in rats. J Physiol. 2005;566:559–573. doi: 10.1113/jphysiol.2005.086322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden CJ, Morrison SF. Neurons in the paraventricular nucleus of the hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2009;296:R831–R843. doi: 10.1152/ajpregu.91007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick BN, Alam MN. Different types of norepinephrinergic receptors are involved in preoptic area mediated independent modulation of sleep-wakefulness and body temperature. Brain Res. 1992;591:8–19. doi: 10.1016/0006-8993(92)90972-c. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Dekeyne A, Newman-Tancredi A, Cussac D, Audinot V, Milligan G, Duqueyroix D, Girardon S, Mullot J, Boutin JA, Nicolas JP, Renouard-Try A, Lacoste JM, Cordi A. S18616, a highly potent, spiroimidazoline agonist at alpha(2)-adrenoceptors: I. Receptor profile, antinociceptive and hypothermic actions in comparison with dexmedetomidine and clonidine. J Pharmacol Exp Ther. 2000;295:1192–1205. [PubMed] [Google Scholar]
- Morrison SF. 2010 Carl Ludwig Distinguished Lectureship of the APS Neural Control and Autonomic Regulation Section: Central neural pathways for thermoregulatory cold defense. J Appl Physiol. 2011;110:1137–1149. doi: 10.1152/japplphysiol.01227.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF, Madden CJ, Tupone D. Central control of brown adipose tissue thermogenesis. Front Endocrinol. 2012;3:1–19. doi: 10.3389/fendo.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Morrison SF. Central efferent pathways mediating skin cooling-evoked sympathetic thermogenesis in brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2007;292:R127–R136. doi: 10.1152/ajpregu.00427.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Morrison SF. Preoptic mechanism for cold-defensive responses to skin cooling. J Physiol. 2008;586:2611–2620. doi: 10.1113/jphysiol.2008.152686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Morrison SF. Central efferent pathways for cold-defensive and febrile shivering. J Physiol. 2011;589:3641–3658. doi: 10.1113/jphysiol.2011.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka T. Heat loss responses and blockade of prostaglandin E2-induced thermogenesis elicited by alpha1-adrenergic activation in the rostromedial preoptic area. Neuroscience. 2009;162:1420–1428. doi: 10.1016/j.neuroscience.2009.05.030. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. Sydney: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Quan N, Xin L, Ungar AL, Blatteis CM. Preoptic norepinephrine-induced hypothermia is mediated by alpha 2-adrenoceptors. Am J Physiol. 1992;262:R407–R411. doi: 10.1152/ajpregu.1992.262.3.R407. [DOI] [PubMed] [Google Scholar]
- Ritter S, Llewellyn-Smith I, Dinh TT. Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-D-glucose induced metabolic challenge. Brain Res. 1998;805:41–54. doi: 10.1016/s0006-8993(98)00655-6. [DOI] [PubMed] [Google Scholar]
- Romanovsky AA, Shido O, Ungar AL, Blatteis CM. Genesis of biphasic thermal response to intrapreoptically microinjected clonidine. Brain Res Bull. 1993;31:509–513. doi: 10.1016/0361-9230(93)90117-t. [DOI] [PubMed] [Google Scholar]
- Ruggiero DA, Regunathan S, Wang H, Milner TA, Reis DJ. Distribution of imidazoline receptor binding protein in the central nervous system. Ann N Y Acad Sci. 1995;763:208–221. doi: 10.1111/j.1749-6632.1995.tb32408.x. [DOI] [PubMed] [Google Scholar]
- Saper CB, Breder CD. The neurologic basis of fever. N Engl J Med. 1994;330:1880–1886. doi: 10.1056/NEJM199406303302609. [DOI] [PubMed] [Google Scholar]
- Savola MK, Savola JM. [3H]dexmedetomidine, an alpha 2-adrenoceptor agonist, detects a novel imidazole binding site in adult rat spinal cord. Eur J Pharmacol. 1996;306:315–323. doi: 10.1016/0014-2999(96)00224-5. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Sved AF. The ventrolateral medulla and sympathetic regulation of arterial pressure. In: Llewellyn-Smith I, Verberne AJ, editors. Central regulation of autonomic functions. Ed 2. New York: Oxford UP; 2011. pp. 78–97. [Google Scholar]
- Stornetta RL, Akey PJ, Guyenet PG. Location and electrophysiological characterization of rostral medullary adrenergic neurons that contain neuropeptide Y mRNA in rat medulla. J Comp Neurol. 1999;415:482–500. doi: 10.1002/(sici)1096-9861(19991227)415:4<482::aid-cne5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Sevigny CP, Guyenet PG. Vesicular glutamate transporter DNPI/VGLUT2 mRNA is present in C1 and several other groups of brainstem catecholaminergic neurons. J Comp Neurol. 2002;444:191–206. doi: 10.1002/cne.10141. [DOI] [PubMed] [Google Scholar]
- Swoap SJ, Weinshenker D. Norepinephrine controls both torpor initiation and emergence via distinct mechanisms in the mouse. PLoS One. 2008;3:e4038. doi: 10.1371/journal.pone.0004038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szreder Z. Do cardiovascular mechanisms participate in thermoregulatory activity of alpha 2-adrenoceptor agonists and antagonists in rabbits? Ann N Y Acad Sci. 1997;813:512–525. doi: 10.1111/j.1749-6632.1997.tb51741.x. [DOI] [PubMed] [Google Scholar]
- Thorn DA, An XF, Zhang Y, Pigini M, Li JX. Characterization of the hypothermic effects of imidazoline I(2) receptor agonists in rats. Br J Pharmacol. 2012;166:1936–1945. doi: 10.1111/j.1476-5381.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupone D, Madden CJ, Cano G, Morrison SF. An orexinergic projection from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue thermogenesis. J Neurosci. 2011;31:15944–15955. doi: 10.1523/JNEUROSCI.3909-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zwieten PA. Pharmacology of centrally acting hypotensive drugs. Br J Clin Pharmacol. 1980;10(Suppl 1):13S–20S. doi: 10.1111/j.1365-2125.1980.tb04899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weant KA, Martin JE, Humphries RL, Cook AM. Pharmacologic options for reducing the shivering response to therapeutic hypothermia. Pharmacotherapy. 2010;30:830–841. doi: 10.1592/phco.30.8.830. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Li X, Cano G, Lazarus M, Saper CB. Parallel preoptic pathways for thermoregulation. J Neurosci. 2009;29:11954–11964. doi: 10.1523/JNEUROSCI.2643-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]