

Abstract
Purpose
Ewing sarcoma family tumors (ESFT) are aggressive tumors of putative stem cell origin for which prognostic biomarkers and novel treatments are needed. In several human cancers high expression of the polycomb protein BMI-1 is associated with poor outcome. We have assessed the potential clinical significance of BMI-1 expression level in ESFT.
Experimental Design
BMI-1 expression was assessed in 130 tumors by immunostaining and associations with clinical features and outcome determined. The molecular signatures of BMI-1-low and BMI-1-high tumors were compared using microarrays and differentially activated canonical pathways identified by gene specific enrichment analysis. Automated quantitative analysis (AQUA) of phospho-proteins was used to assess relative levels of pathway activation. Sensitivity to IGF1-R inhibition was determined using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assays.
Results
BMI-1 is over-expressed by the vast majority of ESFT. However, in 20% of cases BMI-1 levels are low to undetectable. Significantly, although clinical presentation and outcome were similar between BMI-1-high and BMI-1-low tumors, whole genome expression array analysis showed marked differences in their respective gene expression profiles. Gene specific enrichment analysis identified that several cancer-associated canonical biologic pathways, including IGF1, mTOR and WNT are significantly down-regulated in BMI-1-low compared to BMI-1-high tumors. Consistent with these in vivo data, the response to IGF1-R inhibition in vitro was diminished in BMI-1-low compared to BMI-1-high ESFT cells.
Conclusion
ESFT that do not over-express BMI-1 represent a novel subclass with a distinct molecular profile and altered activation of and dependence on cancer-associated biologic pathways.
INTRODUCTION
The Ewing sarcoma family of tumors (ESFT) are malignant neoplasms of bone and soft tissue that primarily affect children and young adults (1). Genetically they are identified by expression of EWS-FLI1 or another related gene fusion (1). Although their histogenesis remains elusive, recent studies suggest that ESFT may arise from malignant transformation of mesenchymal and/or neural crest stem cells (2–6). Clinically they are highly aggressive malignancies with a high propensity for relapse and metastasis. Unfortunately, despite aggressive systemic cytotoxic therapy and local control measures, relapse after initial clinical remission is not uncommon and overall survival for patients with relapsed or metastatic ESFT remains less than 20% (7). Given the profound need for novel and less toxic treatments for these patients, great hope is being placed on the development of targeted agents that will successfully inhibit biologic pathways known to contribute to ESFT growth (8). However, knowledge about which tumors are likely to respond to these agents, such as the IGF1-R inhibitors that are currently being tested in clinical trials, is crucial if their efficacy is to be optimized. Therefore, coincident development of biomarkers that can predict response to therapy is essential.
BMI-1 is a member of the polycomb group gene family that promotes self-renewal of normal adult stem cells including neural crest stem cells through epigenetic repression of developmental and senescence pathways (reviewed in Ref (9)). In addition, BMI-1 functions as an oncogene in many human cancers and has been implicated in the self-renewal of tumor initiating cancer stem cells (10–12). Importantly, in at least some human cancers, over-expression of the BMI-1 protein is associated with a worse clinical outcome suggesting that in these tumor types BMI-1 might be useful as a biomarker of aggressive disease (13–18). We previously reported that BMI-1 is over-expressed by ESFT and functions as a growth promoting oncogene in these tumors (19). However, our data also revealed that the absolute level of BMI-1 expression is variable among both primary tumors and cell lines (19). For the current study we have assessed whether BMI-1 might be useful as a predictive biomarker in ESFT. We characterized BMI-1 protein expression in a large cohort of primary ESFT samples and evaluated whether BMI-1 expression levels were correlated with clinical outcome in a group of clinically annotated tumors obtained from patients treated on recent Children’s Oncology Group (COG) clinical trials. In addition, we assessed whether differences in the molecular phenotype of BMI-1-high and BMI-1-low tumors might also be used in the future to predict response to pathway-targeted therapies such as IGF1-R inhibition.
MATERIALS AND METHODS
Sample Accrual
Formalin-fixed paraffin-embedded (FFPE) and fresh-frozen sections were acquired from tumor banks at the COG Biorepository in Columbus, Ohio (Cooperative Human Tissue Network -- CHTN), Childrens Hospital Los Angeles (CHLA) and Memorial Sloan Kettering Cancer Center (MSKCC). Diagnosis of ESFT was confirmed for all specimens by pathologic review at each site. Samples were obtained as single tumor slides or as tissue microarrays (TMA). Serial sections of an ESFT TMA created at the Department of Pathology, University of Michigan were used for validation studies. Clinical outcomes data were obtained from chart review and through the Biostatistical Office of the COG. All human specimens and correlative data were obtained in compliance with HIPAA regulations and following protocol review by institutional review boards in accordance with an assurance filed with and approved by the Department of Health and Human Services. Informed consent for use of tumor samples for research purposes was obtained from each subject or subject’s guardian.
Immunohistochemical Analysis
Four-micron tissue sections were deparaffinized, pretreated with CCI™ (Tris/Borate/EDTA buffer pH 8, Ventana Medical Systems, Inc., Tucson, AZ, USA), and incubated with anti-BMI-1 antibody (1:50, Millipore, Billerica, MA, USA) according to the BenchMark UHC/ISH Staining ModuleR BMI-1-T 1/50 protocol (32 minutes at 42°C). All sections were then treated with iView detection kit (Ventana Medical Systems, Inc.). Adjacent sections were stained with H&E to verify the presence of viable tumor tissue. Individually stained tumor slides were directly visualized and scored by direct light microscopy and photomicrographs acquired via digital camera (DP-11, Olympus, Tokyo, Japan). TMA sections were digitally imaged using Aperio ScanScopeT2 software (Aperio, Vista, CA) at Columbus Children’s Research Institute Biopathology Center.
Stained sections were assigned a BMI-1 score using published criteria (18). In brief, the percentage of positive cells (PP) value (ranging from 0 to 3 for < 5% to > 50% positive cells, respectively) was multiplied by the signal intensity (SI) value (which ranges from 0 for absent to 3 for strongly positive) to generate a composite PPxSI value ranging from 0–9. Composite values of 0–1 are negative; 2–3 are 1+; 4–6 are 2+ and values >6 are scored as 3+.
For automated quantitative analysis (AQUA) of total and phosphorylated protein expression in tumor cells double immunofluorescence staining was performed as previously described (20). Deparaffinized and rehydrated TMA slides were subjected to microwave epitope retrieval in 7.5 mM sodium citrate buffer, pH6 (mTOR and P-mTOR) or 1 mM EDTA buffer, pH8 (IGF1R and P-IGF1R). Slides were washed in TBST (10 mM Tris HCL, pH 8 containing 0.154 M NaCl (TBS), 0.05% Tween-20) then endogenous peroxidase activity blocked with 2.5% hydrogen peroxide in methanol and non-specific binding blocked by a 30 minute incubation in “Background Sniper” (BioCare Medical, Concord, CA). Blocked slides were incubated with a tumor specific antibody, CD99 (Rabbit polyclonal antibody, Ab-27271, Abcam, Cambridge, UK, 1:100 or Mouse monoclonal antibody, DAKO, Carpinteria, CA) overnight at 4°C. CD99-stained slides were washed with TBST then incubated with one of five antibodies: BMI-1 (clone 229F6, 1:400, Millipore, Billerica, MA), mTOR (Ab-51089 Abcam, Cambridge, UK 1:200), P-mTOR (Ab-51044 Abcam, Cambridge, UK 1:100) (for 1 hour at room temperature), IGF1-R (Millipore, clone JBW902, 1:1000) or P-IGF1R (Millipore, 07-841, 1:400) (for 4°C overnight in a humid chamber). Double-stained slides were washed then incubated with a combination of goat anti-rabbit or mouse IgG conjugated to AF555 (Molecular probes, Carlsbad, CA, A21424/A, 1:200) in goat anti-mouse or anti-rabbit Envision+ (DAKO) for 60 minutes at room temperature in a dark humidity tray. Slides were washed in TBST and the target image developed by a CSA reaction of Cy5 labelled tyramide (PerkinElmer, Waltham, MA, 1:50). Finally, slides were stained with 4′,6-diaminodo-2-phenylindole (DAPI) in a non-fading mounting media (ProLong Gold, Molecular probes, Carpinteria, CA) and allowed to dry overnight in a dark dry chamber.
The AQUA system (Software v2.2, HistoRx, New Haven, Connecticut) was used for automated image acquisition and analysis (20). Within each slide, the area of tumor was distinguished from stromal and necrotic areas by creating a tumor specific mask from the CD99 stain, which was visualized from Alexafluor 555 signal. Fluorescence pixel intensity of the target protein/antibody complex was obtained from the Cy5 signal and reported as pixel intensity.
Clinical Correlates Analysis
The association between BMI-1 and age was analyzed via analysis of variance and associations between BMI-1 and other clinico-pathologic characteristics were analyzed via Fisher’s exact test. For survival analysis, event free survival (EFS) was defined as the minimum interval from the date of diagnosis to the date of tumor recurrence, progression, occurrence of a second malignancy, death, or last follow-up. Overall survival (OS) was defined as the interval from the date of diagnosis to the date of death or last follow-up. Estimates of EFS and OS percent were based on the product-limit (Kaplan-Meier) estimate with Greenwood standard errors.(21) The association of EFS and OS with BMI-1 protein expression was tested using the logrank test, either univariately or with stratification based on stage at presentation, age, site, treatment era, and EWS-FLI1 transcript type. Survival analyses were performed using STATA software version 9.2. All reported p-values were two-sided and a p-value < 0.05 was considered significant.
Expression microarrays
Total RNA was isolated from ESFT biopsies using Qiagen miRNA kit (Qiagen, Valencia, CA) and processed for whole genome expression profiling using Affymetrix GeneChip Human Exon 1.0 ST oligonucleotide microarrays according to Affymetrix protocols. Affymetrix cel files from similarly processed ESFT cell line RNA were kindly provided by Dr. T. Triche (CHLA). Signal intensities from core probe sets were quantile normalized by robust multichip averaging and transcript expression determined by median summarization using Partek Genomics Suite software (Partek, St. Louis, MO). Hierarchical clustering using both Ward’s and average linkage methods was performed in Partek. To compare relative levels of pathway activation between BMI-1-high and BMI-1-low tumors 639 gene sets representing canonical pathways were downloaded from the Molecular Signatures Database (MSigDB) and gene set enrichment analysis (GSEA) performed as described (22, 23). In brief, the mean of the t-statistics for the transcripts of each gene set was calculated, and a p value was computed for each gene set by randomly permuting the gene labels 10,000 times. The expected false discovery rate (FDR) for multiple testing was controlled using the Benjamini and Hochberg procedure (24). All reported p-values were two-sided. Statistical computations were performed using STATA software version 9.2 and R version 2.8.1 (http://www.R-project.org).
In vitro assays of ESFT cell lines
ESFT cells were obtained from CHLA (TC-71, TC-248, A673, A4573) and COG (CHLA-9) cell line repositories (Dr. T. Triche and Dr. C.P. Reynolds, www.cogcell.org, respectively). Cell lines were confirmed to be ESFT by RT-PCR amplification of a type 1 EWS-FLI1 fusion and by analysis of short tandem repeats (kindly performed by Dr. C.P. Reynolds). Cells were maintained in 10% RPMI supplemented with 10% FBS, pen/strep and L-glutamine and RT-PCR and western blot analysis of logarithmic phase cells were performed using standard protocols. PCR primer sequences for BMI-1 and EWS-FLI1 are available on request. To assess response to IGF1-R inhibition, subconfluent cells growing in 96-well plates in 0.5% FBS-supplemented RPMI were treated with increasing concentrations of picropodophyllin (PPP; Enzo Life Sciences, Plymouth Meeting, PA), IMC-A12 or DMSO vehicle. IMC-A12 was provided by ImClone systems through the Cancer Therapy Evaluation Program (CTEP, NCI). Cell viability was assessed 72 hrs post-treatment using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assays (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Madison WI). All experiments were repeated at least three times with a minimum of six wells counted/condition.
RESULTS
BMI-1 is over-expressed by most ESFT
BMI-1 expression was evaluated in 130 FFPE ESFT sections by immunohistochemistry (Table 1). Consistent with our prior studies of ESFT cell lines (19), BMI-1 protein was robustly expressed by most tumors (BMI-1-high; Figure 1A&B). Comparison with 16 normal tissues confirmed that non-malignant cells only rarely express such high levels of BMI-1 (not shown). Of note, however, in 20% of ESFT only weak or no BMI-1 was detected (BMI-1-low; Figure 1C & D).
Table 1.
Clinical and Molecular Features of Primary ESFT Biopsies
| BMI-1 Score | ||||||
|---|---|---|---|---|---|---|
| BMI-1-low | BMI-1-high | |||||
| All Patients | Negative | 1+ | 2+ | 3+ | p-value* | |
| Total Number | 130 | 18 | 6 | 33 | 73 | |
| (%) | (100) | (13.8) | (4.6) | (25.4) | (56.2) | |
| Median age, years (range) | 13 (0, 47) | 13 (5, 47) | 15 (6, 31) | 15 (0, 30) | 13 (0, 46) | 0.76(1) |
| Gender | ||||||
| Male (%) | 84 (65) | 13 | 1 | 25 | 45 | 0.04(2); 0.49(3) |
| Female (%) | 46 (35) | 5 | 5 | 8 | 28 | |
| Stage | ||||||
| Localized (%) | 99 (85) | 11 | 4 | 22 | 62 | 0.43(2) |
| Metastatic (%) | 18 (15) | 4 | 1 | 4 | 9 | |
| Unknown | 13 | 3 | 1 | 7 | 2 | |
| Region | ||||||
| Pelvis (%) | 20 (15) | 4 | 1 | 3 | 12 | 0.56(2) |
| Non-Pelvis (%) | 110 (85) | 14 | 5 | 30 | 61 | |
| Tissue of Origin | ||||||
| Bone | 75 | 12 | 3 | 19 | 41 | 0.72(2) |
| Soft Tissue | 50 | 5 | 2 | 11 | 32 | |
| Unknown | 5 | 1 | 1 | 3 | 0 | |
| Chemotherapy status | ||||||
| Pre-Chemotherapy | 99 | 13 | 3 | 24 | 59 | 0.49(2) |
| Post-chemotherapy | 15 | 3 | 1 | 4 | 7 | |
| Unknown | 16 | 2 | 2 | 5 | 7 | |
| Translocation status | ||||||
| EWS-ETS Fusion + | 70 | 5 | 5 | 18 | 42 | 0.13(2) |
| No fusion detected | 5 | 2 | 0 | 0 | 3 | |
| Unknown | 55 | 11 | 1 | 15 | 28 | |
| p16 genomic status known (N=24) | ||||||
| Wild type | 22 | 3 | 2 | 7 | 10 | 0.52(2) |
| Deleted | 2 | 0 | 1 | 0 | 1 | |
| p53 mutation status known (N=24) | ||||||
| Wild type | 16 | 3 | 1 | 5 | 7 | 0.55(2) |
| Mutant | 8 | 0 | 2 | 2 | 4 | |
p-value from analysis of variance1, 4×2 Fisher’s exact test (unknowns excluded) 2, 2×2 Fisher’s exact test3
Figure 1.

BMI-1 expression in ESFT. Most tumors robustly and diffusely express BMI-1 (A & B: BMI-1-high). In contrast, in nearly 20% of tumors BMI-1 staining is absent or minimal (C & D: BMI-1-low). BMI-1 negative cells in (B) are non-tumor stromal cells (see corresponding H&E section).
BMI-1 has been implicated as a marker of minority populations of tumor-initiating cancer stem cells (12, 25). As shown (Figure 1A & B), we found that in the majority of ESFT all tumor cells are BMI-1-positive. Moreover, comparison of diagnostic biopsies with samples obtained post-neoadjuvant chemotherapy revealed no difference in BMI-1 expression between these two groups (p=0.49). Thus, if a subpopulation of chemo-resistant, tumor-initiating cells exists in ESFT, BMI-1 will not be useful as a distinguishing marker.
Clinical presentation and outcome of BMI-1-high and BMI-1-low tumors are similar
In some cancers BMI-1 is a marker of aggressive disease and worse outcome (13–18, 25). Although patients with metastatic ESFT have the worst outcome, prognostic stratification of patients with localized disease remains a challenge. Previous studies have reported that pelvic disease, older age at presentation and variant EWS-FLI1 fusion type are bad prognostic features (26–28). As shown (Table 1), there was no association between any of these features and BMI-1 expression. In addition, BMI-1-low tumors were equally represented among tumors of bone or soft tissue origin. We next assessed 72 patients with localized disease for whom follow-up data were available. No significant difference in either EFS or OS was observed between BMI-1-high and BMI-1-low tumors (Figure 2). Of the 24 events, 20 were disease progression or relapse, 2 were second malignant neoplasm, and 2 were death as a first event. EFS analyses that censor the non-progression events yield identical conclusions to those presented. Thus, although the small number of BMI-1-low tumors in this cohort limits the statistical power of the analysis, our studies suggest that there is little or no association between BMI-1 expression level and either clinical presentation or outcome in patients with localized disease who are treated on current clinical protocols.
Figure 2.
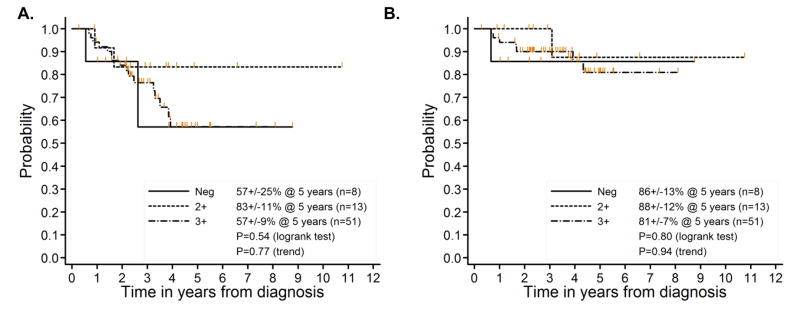
Kaplan-Meier survival analysis of 72 patients with localized disease shows no significant association of BMI-1 expression with outcome. A. Event free survival (EFS) and B. Overall survival (OS).
Gene expression signatures differ between BMI-1-low and BMI-1-high tumors
Silencing of innate p16/RB or p53 tumor suppressor pathways is necessary for tolerance of the EWS-FLI1 oncoprotein in normal cells (29, 30). BMI-1 is known to epigenetically repress p16 and p53 pathways in both normal and tumor cells via transcriptional silencing the CDKN2A locus (31). We therefore reasoned that if BMI-1-high tumors silence p16 and p53 pathways epigenetically, BMI-1-low tumors may be more dependent on secondary genetic mutations. Homozygous deletion of p16 and/or mutation in p53 occurs in 20–25% of primary ESFT (32). To begin to address whether such mutations are more common in BMI-1-low tumors we evaluated 24 tumors for which p53 and p16 status had been previously documented (32). As shown (Table 1), tumors harboring secondary genetic mutations were equally distributed between BMI-1-high (N=18) and BMI-1-low (N=6) groups. Thus, in this small cohort of tumors, we found no association between BMI-1 expression and mutations in p53 or p16. Studies with larger numbers of tumors are necessary but these initial findings suggest that other mechanisms of tumor suppressor inactivation are likely to exist in BMI-1-low tumors.
To more fully define molecular differences between BMI-1-high and BMI-1-low ESFT we performed whole genome expression profiling of five age- and stage-matched tumors from each sub-class (five 3+ tumors versus four negative and one 1+) (GEO submission pending). An EWS-FLI1 fusion was detected by RT-PCR in nine of ten tumors (not shown) and bone and extra-osseous tumors were included in each group. Signal intensity data confirmed significantly lower expression of the BMI-1 transcript in the BMI-1-low tumor cohort (Figure 3A). Unexpectedly, unsupervised principal components analysis of the ten microarrays using all 17,881 core probeset-interrogated transcripts clearly segregated the tumors into two non-overlapping groups (Figure 3B). Thus, despite their common histologic appearance and expression of EWS-ETS fusions, BMI-1-high and BMI-1-low tumors displayed remarkably dissimilar transcriptional profiles. Consistent with this, over 4,000 transcripts were identified as being differentially expressed between the two groups (FDR <0.05). To better assess the potential biologic and clinical significance of these genes we performed gene set enrichment analysis (GSEA) and identified differential activation of 100 different canonical pathways (see methods) (FDR <0.05). The most highly statistically significant of these pathways are shown in Table 2. Importantly, the IGF1, mTOR, ubiquitin-mediated proteolysis and WNT pathways were all identified as being significantly down-regulated in BMI-1-low tumors.
Figure 3.

Gene expression profiling studies demonstrate distinct molecular signatures of BMI-1-low tumors. A. BMI-1 raw signal intensities from 5 BMI-1-low and 5 BMI-1-high primary tumors (arbitrary units). B. Principal components analysis of 10 tumors using all core transcripts segregates BMI-1-low and BMI-1-high tumors into two distinct clusters. Cluster ellipses encompass 2 standard deviations. C. BMI-1-low tumors display down-regulation of genes in the IGF1 pathway relative to BMI-1-high tumors. The fold change in median signal intensity for each gene was calculated for BMI-1-high and BMI-1-low tumors relative to median expression in all 10 tumors. Error bars represent SEM for 5 tumors. D. AQUA analysis reveals variable expression of BMI-1 protein in an independent cohort of primary ESFT. Comparison between the lowest BMI-1 expressors (9 tumors, bottom 15% of 58 cases) and the highest BMI-1 expressors (29 tumors, top 50%) demonstrates that phospho-IGF1R, phospho-mTOR and total mTOR levels are significantly reduced in BMI-1-low tumors.
Table 2.
Gene-specific enrichment analysis identifies differentially expressed canonical pathways in BMI-1-low ESFT (FDR<0.01 for all listed pathways)
| A. Down-regulated in BMI-1-Low |
| CELL_CYCLE_KEGG |
| FASPATHWAY |
| HSA00280_VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION |
| HSA00970_AMINOACYL_TRNA_BIOSYNTHESIS |
| HSA04110_CELL_CYCLE |
| HSA04120_UBIQUITIN_MEDIATED_PROTEOLYSIS |
| IGF1PATHWAY |
| MRNA_PROCESSING_REACTOME |
| MTORPATHWAY |
| PROTEASOMEPATHWAY |
| RNA_TRANSCRIPTION_REACTOME |
| HSA00240_PYRIMIDINE_METABOLISM |
| HSA00563_GLYCOSYLPHOSPHATIDYLINOSITOL_ANCHOR_BIOSYNTHESIS |
| HSA03050_PROTEASOME |
| HSA04130_SNARE_INTERACTIONS_IN_VESICULAR_TRANSPORT |
| KREBS_TCA_CYCLE |
| VALINE_LEUCINE_AND_ISOLEUCINE_DEGRADATION |
| P35ALZHEIMERSPATHWAY |
| WNTPATHWAY |
| HSA03022_BASAL_TRANSCRIPTION_FACTORS |
| MAPKPATHWAY |
| PROPANOATE_METABOLISM |
| B. Up-regulated in BMI-1-Low |
| GPCRDB_CLASS_A_RHODOPSIN_LIKE |
| HSA01430_CELL_COMMUNICATION |
| HSA04020_CALCIUM_SIGNALING_PATHWAY |
| HSA04060_CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION |
| HSA04080_NEUROACTIVE_LIGAND_RECEPTOR_INTERACTION |
| HSA04512_ECM_RECEPTOR_INTERACTION |
| HSA04514_CELL_ADHESION_MOLECULES |
| HSA04640_HEMATOPOIETIC_CELL_LINEAGE |
| HSA04940_TYPE_I_DIABETES_MELLITUS |
| HSA04950_MATURITY_ONSET_DIABETES_OF_THE_YOUNG |
| HSA05217_BASAL_CELL_CARCINOMA |
| PEPTIDE_GPCRS |
| STRIATED_MUSCLE_CONTRACTION |
| GPCRDB_OTHER |
| HSA00590_ARACHIDONIC_ACID_METABOLISM |
| HSA04610_COMPLEMENT_AND_COAGULATION_CASCADES |
Levels of IGF1R and mTOR phosphorylation are significantly reduced in BMI-1-low tumors
Next, we sought to confirm that differences in gene expression were truly reflective of differential levels of pathway activation. To achieve this we used quantitative immunohistochemistry tools (20) to analyze levels of BMI-1 as well as total and phosphoryated IGF1-R and mTOR proteins in an independent cohort of ESFT samples obtained from newly diagnosed patients at the University of Michigan. Consistent with non-fluorescent staining protocols (Figure 1), AQUA analysis of 58 ESFT revealed a wide range of BMI-1 expression (range 15 – 860 fluorescent intensity units, median signal=142). In order to quantify relative levels of kinase pathway activation in BMI-1-low and BMI-1-high tumors we compared tumors with the lowest BMI-1 signal (bottom 15%, median BMI-1 signal=24) to tumors with the highest signal (top 50%, median BMI-1 signal=416). As shown (Figure 3D), consistent with gene expression data, levels of IGF1-R protein were equivalent between BMI-1-high and BMI-1-low tumors. However, the relative levels of IGF1-R phosphorylation as well as mTOR phosphorylation were significantly reduced in BMI-1-low tumors (Figure 3D). Thus, both gene expression and protein array data suggest that BMI-1-low tumors represent a distinct molecular sub-class of ESFT that may be less dependent on IGF1-R, mTOR and other canonical cancer-associated pathways for growth and survival. As such, they may also be less responsive to pathway-targeted agents currently in development and in early phase clinical trials.
ESFT cells with low expression of BMI-1 are less sensitive to IGF1-R inhibition
IGF1-R inhibitors are currently being evaluated as novel therapeutic agents in ESFT (8). As discussed above, molecular profiling studies revealed that the IGF1 pathway is one of the most significantly down-regulated pathways in BMI-1-low ESFT and levels of IGF1-R phosphorylation and its downstream effector mTOR are reduced in these tumors (Figure 3C & D). In light of these data we hypothesized that BMI-1-low ESFT might be less sensitive to the effects of IGF1-R inhibition. To test this hypothesis we first needed to identify representative cell lines for in vitro assays. We previously showed that BMI-1 is over-expressed by ESFT cell lines in vitro (19). To identify candidate BMI-1-low cell lines we performed hierarchical clustering of whole genome expression profiles obtained from the ten aforementioned primary tumors and 19 ESFT cell lines. Interestingly, only the TC-248 cell line clustered with the BMI-1-low tumors (Figure 4A). Although no other BMI-1-low cell lines were identified, among BMI-1-high cell lines transcript expression was found to vary from very high (e.g. TC-32, TC-71, A673) to more moderate (e.g. CHLA-9, TC-466, TTC-487) levels. Reduced expression of BMI-1 in TC-248 cells was confirmed by western blot (Figure 4B) and expression of a type 1 EWS-FLI1 fusion confirmed by RT-PCR (Figure 4C). Consistent with our hypothesis, TC-248 was significantly less sensitive than BMI-1-high cell lines to the growth inhibitory effect of picropodophyllin (PPP), a selective small molecule inhibitor of IGF1-R (33)(Figure 4D). Similarly, TC-248 was relatively insensitive to the IGF1-R inhibitory antibody IMC-A12 (Figure 4E). These in vitro studies of ESFT cell lines support our studies of primary human tumors in vivo and suggest that patients whose tumors express low levels of BMI-1 may be less sensitive to IGF1-R-targeted therapy. Prospective testing of patient samples is now required to clinically test this novel hypothesis.
Figure 4.

A. Hierarchical clustering of 19 ESFT cell lines and 10 tumors identifies TC-248 as a BMI-1-low cell line. B. Western blot confirms low expression of BMI-1 protein in TC-248 compared to 2 BMI-1-high cell lines. C. RT-PCR confirms expression of a type 1 EWS-FLI1 fusion in TC-248 cells. D. Treatment with an IGF1-R-specific small molecule inhibitor picropodophyllin (PPP) results in growth inhibition of BMI-1-high cells after 72 hours. In contrast, TC-248 cells are significantly less sensitive to PPP (p<0.0001 compared to each of 4 BMI-1-high cell lines at 500 nM dose). E. Growth of TC-248 cells is not significantly inhibited by exposure to the anti-IGF1-R-targeted antibody IMC-A12.
DISCUSSION
New therapies for ESFT are needed to both overcome resistant disease and to reduce the short- and long-term side effects associated with current cytotoxic regimens (34). Recent pre-clinical data has generated great enthusiasm for biologic pathway-targeted agents, including IGF1-R antagonists (8). In this study, we have identified a distinct subclass of ESFT that is characterized by low expression of BMI-1. These tumors represent up to 20% of cases and are molecularly distinct from the more common BMI-1-high tumors. Importantly, genetic profiling studies revealed that numerous cancer-associated pathways, including the IGF1 and mTOR pathways, are relatively less active in this subset of ESFT. In support of this observation, we showed, in an independent cohort of ESFT, that the levels of IGF1-R and mTOR phosphorylation are significantly reduced in BMI-1-low compared to BMI-1-high tumors. Moreover, preliminary in vitro studies confirmed that that the growth inhibitory effects of IGF1-R inhibition are significantly diminished in BMI-1-low cells. These studies suggest that BMI-1 might be useful as a predictive biomarker of response to IGF1-R targeted agents in vivo.
Recent work from the Pediatric Preclinical Testing Program (PPTP) found that the in vivo efficacy of IMC-A12 therapy in ESFT xenografts was not predicted by the extent of in vitro response (35). Moreover, only one of five ESFT xenografts displayed a significant response to IMC-A12 as a single agent (35). Analysis of Affymetrix gene expression array data provided by PPTP (https://sharedoc.nchri.org/PPTP/default.aspx) shows that the ESFT xenograft that displayed the lone in vivo response to IMC-A12, EW-5, also expressed the highest level of BMI-1 (absolute as well as normalized signal intensities, not shown). Moreover, EW-5 xenografts showed complete regression when treated with combined therapy targeting IGF1-R and mTOR (36). Finally, we have found that the level of BMI-1 in TC-71 cells grown in vitro is significantly higher than that expressed by TC-71 xenografts (JH Hsu, ER Lawlor, unpublished data). We speculate that this difference in BMI-1 expression may contribute to the unexpected and disappointing differences in response that have been observed between IMC-A12-treated TC-71 cells in vitro and in vivo (35). Together these data lend pre-clinical in vivo support to our hypothesis that targeted therapies against IGF1-R and mTOR will be most effective in ESFT with the highest levels of BMI-1.
In some tumors BMI-1 over-expression is a feature of tumor-initiating cells and since normal somatic stem cells also express high levels of BMI-1 it has been proposed that BMI-1 might be a universal marker of tumor-initiating cancer stem cells (10–12). Our studies show that, in BMI-1-high ESFT, all tumor cells robustly express the protein. Thus, if a discrete population of tumorigenic stem cells exists in ESFT, BMI-1 will not be useful as discriminating marker. Alternately, if BMI-1 is a marker of cells with tumor-initiating potential then all cells within BMI-1-high tumors would be predicted to possess tumor-initiating capability. This latter possibility is supported by the observation that, in the absence of systemic chemotherapy, ESFT are destined to recur at distant sites indicating that circulating tumor cells with tumor-initiating potential are universally present in ESFT patients at the time of diagnosis. Further studies with large numbers of prospectively acquired patient samples are required to definitively address this question and to determine if there is a relationship between BMI-1 and CD133, a recently reported marker of putative cancer stem cells in ESFT (37).
Given the role of BMI-1 in p16-RB and p53 pathway suppression in other tumor types we hypothesized that mutations in these pathways might substitute for BMI-1 over-expression in the BMI-1-low tumors. Analysis of a limited subset of cases did not reveal any significant increase in p16 deletion or p53 mutation among BMI-1-low tumors. Although these are the two most common secondary mutations in ESFT multiple alternate mechanisms of p16-RB and p53 pathway inactivation exist, including p16 promoter hyper-methylation and MDM2 amplification (1). However, the absence of BMI-1-mediated repression of CDKN2A in ESFT cell lines (19) suggests that other p16-independent functions of BMI-1 also contribute to its function as an oncogene in this family of tumors. Indeed, we have found that BMI-1 promotes ESFT tumorigenicity through modulation of cell adhesion (19). Significantly, in the current study GSEA analysis of biologic pathways identified differential up-regulation of cell adhesion molecules in BMI-1-low tumors (Table 2B). Moreover, recent studies of neural stem cells and brain tumors also link BMI-1 to regulation of cell:cell and cell:matrix adhesion pathways (38-40). These data suggest that deregulation of cell adhesion pathways, either through BMI-1-dependent (in BMI-1-high tumors) or BMI-1-independent (in BMI-1-low tumors) mechanisms is essential for ESFT tumorigenesis. Further mechanistic studies are now required to test this hypothesis and to fully elucidate the relationship between BMI-1, cell adhesion and ESFT pathogenesis.
With the exception of metastatic disease, no clinical features allow prognostic stratification of ESFT patients. High expression of BMI-1 has been associated with an unfavorable prognosis in numerous malignancies (13, 16–18, 25). In contrast, in malignant melanoma loss of BMI-1 expression is associated with disease progression (41), and in breast cancer conflicting reports have demonstrated associations of BMI-1 with either aggressive (14, 15) or more favorable disease (42, 43). We identified no clinical or pathologic features that could distinguish BMI-1-high from BMI-low ESFT and patients treated on current clinical protocols fared equally well irrespective of BMI-1 expression level. Despite their clinical similarities, however, gene expression profiling studies showed that BMI-1-high and BMI-1-low tumors are markedly different at a molecular level. Significantly, many of these differences in gene expression are reflected in marked disparities in biologic pathway activation, including several pathways that are the focus of novel targeted agents currently in development. Whether these inherent biologic differences are reflective of alternate cellular origins and/or alternate paths to malignant transformation remains to be determined and will require study in in vitro and in vivo models of ESFT initiation. Nevertheless, these findings have profound implications for the design and interpretation of pre-clinical and clinical trials designed to test biologic pathway-targeted agents in ESFT. The cell lines used in most pre-clinical studies of ESFT are representative of the more common BMI-1-high tumors (e.g. TC-71, A673, TC-32, A4573, 6647, CHLA-9). In contrast, the sole BMI-1-low cell line that we have identified, TC-248, is rarely studied (44). Our findings suggest that current pre-clinical models of drugs that target canonical biologic pathways are likely to be most informative for patients with BMI-1-high tumors and less predictive of response in the subset of patients who present with BMI-1-low ESFT. In addition, we propose that the existence of this novel molecular subclass will be worthy of consideration when evaluating treatment responses in clinical trials that test these agents.
In summary, we have identified a distinct sub-group of ESFT that do not express high levels of BMI-1. These tumors represent up to 20% of cases and have markedly different molecular profiles compared to the more common BMI-1 over-expressing tumors. Importantly, the relative activation of numerous cancer-associated biologic pathways is diminished in BMI-1-low tumors indicating that patients with these tumors may be less likely to respond to novel pathway-targeted agents.
Statement of Translational Relevance.
The development of biologically targeted therapies holds tremendous promise for cancer patients. However, optimal efficacy of these approaches will only be realized if it can be determined which patients are most likely to respond and which tumors are likely to be resistant. The development of biomarkers that predict treatment response is, therefore, absolutely critical if novel biologic agents are to be successfully translated into clinical practice. In the current study we have identified a subset of Ewing sarcoma family tumors (ESFT) that do not over-express BMI-1. These tumors display relative down-regulation of the IGF1 pathway and are less sensitive to the growth inhibitory effects of IGF1-R inhibition. Together these data demonstrate that within ESFT there exists a molecular subclass, defined by absence of BMI-1 over-expression, that are less likely to respond to IGF1-R inhibitors. Evaluation of BMI-1 as a potential predictive biomarker of response to IGF1-R targeted agents is warranted.
Acknowledgments
Research support: NIH SPECS grant 1U01CA11475-05 (ERL, RS, TJT), NIH COG Chair’s grant U10 CA98543-07 (MK, RBW), NIH SDC grant U10 CA98413 (MK), Human Specimen Banking U24 CA114766, NIH T32 CA09659 (JvD) and the My Brother Joey & TJ Martell Foundations (ERL)
The authors thank the CHLA Biorepository and COG Biopathology Center staff for their exceptional support and technical expertise; patients and families for donation of tumor tissues; Betty Schaub and Long Hung for technical assistance; Dr. Michele Wing and members of the Lawlor lab for helpful discussion. We dedicate this work to the memory of Dr. Steven Qualman whose tireless efforts on behalf of children with cancer made this work possible.
References
- 1.Burchill SA. Ewing’s sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol. 2003;56:96–102. doi: 10.1136/jcp.56.2.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA, Ilaria RL., Jr Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1-dependent, ewing sarcoma-like tumors. Cancer Res. 2005;65:8698–705. doi: 10.1158/0008-5472.CAN-05-1704. [DOI] [PubMed] [Google Scholar]
- 3.Riggi N, Cironi L, Provero P, et al. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005;65:11459–68. doi: 10.1158/0008-5472.CAN-05-1696. [DOI] [PubMed] [Google Scholar]
- 4.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11:421–9. doi: 10.1016/j.ccr.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 5.Meltzer PS. Is Ewing’s sarcoma a stem cell tumor? Cell Stem Cell. 2007;1:13–5. doi: 10.1016/j.stem.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Coles EG, Lawlor ER, Bronner-Fraser M. EWS-FLI1 causes neuroepithelial defects and abrogates emigration of neural crest stem cells. Stem Cells. 2008;26:2237–44. doi: 10.1634/stemcells.2008-0133. [DOI] [PubMed] [Google Scholar]
- 7.Scotlandi K, Remondini D, Castellani G, et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol. 2009;27:2209–16. doi: 10.1200/JCO.2008.19.2542. [DOI] [PubMed] [Google Scholar]
- 8.Seddon BM, Whelan JS. Emerging chemotherapeutic strategies and the role of treatment stratification in Ewing sarcoma. Paediatr Drugs. 2008;10:93–105. doi: 10.2165/00148581-200810020-00004. [DOI] [PubMed] [Google Scholar]
- 9.Gil J, Bernard D, Peters G. Role of polycomb group proteins in stem cell self-renewal and cancer. DNA Cell Biol. 2005;24:117–25. doi: 10.1089/dna.2005.24.117. [DOI] [PubMed] [Google Scholar]
- 10.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowdhury M, Mihara K, Yasunaga S, Ohtaki M, Takihara Y, Kimura A. Expression of Polycomb-group (PcG) protein BMI-1 predicts prognosis in patients with acute myeloid leukemia. Leukemia. 2007;21:1116–22. doi: 10.1038/sj.leu.2404623. [DOI] [PubMed] [Google Scholar]
- 14.Feng Y, Song L, Guo B, et al. Expression and significance of Bmi-1 in breast cancer. Chinese journal of cancer. 2007;26:154–7. [PubMed] [Google Scholar]
- 15.Kim JH, Yoon SY, Jeong SH, et al. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast. 2004;13:383–8. doi: 10.1016/j.breast.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Liu JH, Song LB, Zhang X, et al. Bmi-1 expression predicts prognosis for patients with gastric carcinoma. J Surg Oncol. 2008;97:267–72. doi: 10.1002/jso.20934. [DOI] [PubMed] [Google Scholar]
- 17.van Galen JC, Muris JJ, Oudejans JJ, et al. Expression of the polycomb-group gene BMI1 is related to an unfavourable prognosis in primary nodal DLBCL. Journal of Clinical Pathology. 2007;60:167–72. doi: 10.1136/jcp.2006.038752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Pan K, Zhang HK, et al. Increased polycomb-group oncogene Bmi-1 expression correlates with poor prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:535–41. doi: 10.1007/s00432-007-0316-8. [DOI] [PubMed] [Google Scholar]
- 19.Douglas D, Hsu JH, Hung L, et al. BMI-1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008;68:6507–15. doi: 10.1158/0008-5472.CAN-07-6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCabe A, Dolled-Filhart M, Camp RL, Rimm DL. Automated quantitative analysis (AQUA) of in situ protein expression, antibody concentration, and prognosis. J Natl Cancer Inst. 2005;97:1808–15. doi: 10.1093/jnci/dji427. [DOI] [PubMed] [Google Scholar]
- 21.Cox DR, Oakes D. Analysis of survival data. CRC Press; 1984. [Google Scholar]
- 22.Song S, Black MA. Microarray-based gene set analysis: a comparison of current methods. BMC Bioinformatics. 2008;9:502. doi: 10.1186/1471-2105-9-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tian L, Greenberg SA, Kong SW, Altschuler J, Kohane IS, Park PJ. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci U S A. 2005;102:13544–9. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. CONTROLLING THE FALSE DISCOVERY RATE - A PRACTICAL AND POWERFUL APPROACH TO MULTIPLE TESTING. JOURNAL OF THE ROYAL STATISTICAL SOCIETY SERIES B-METHODOLOGICAL. 1995;57:289–300. [Google Scholar]
- 25.Hayry V, Tynninen O, Haapasalo HK, et al. Stem cell protein BMI-1 is an independent marker for poor prognosis in oligodendroglial tumours. Neuropathol Appl Neurobiol. 2008 doi: 10.1111/j.1365-2990.2008.00949.x. [DOI] [PubMed] [Google Scholar]
- 26.Jenkin RD, Al-Fawaz I, Al-Shabanah M, et al. Localised Ewing sarcoma/PNET of bone--prognostic factors and international data comparison. Med Pediatr Oncol. 2002;39:586–93. doi: 10.1002/mpo.10212. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Galindo C, Liu T, Krasin MJ, et al. Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Cancer. 2007;110:375–84. doi: 10.1002/cncr.22821. [DOI] [PubMed] [Google Scholar]
- 28.de Alava E, Kawai A, Healey JH, et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol. 1998;16:1248–55. doi: 10.1200/JCO.1998.16.4.1248. [DOI] [PubMed] [Google Scholar]
- 29.Deneen B, Denny CT. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 2001;20:6731–41. doi: 10.1038/sj.onc.1204875. [DOI] [PubMed] [Google Scholar]
- 30.Lessnick SL, Dacwag CS, Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer Cell. 2002;1:393–401. doi: 10.1016/s1535-6108(02)00056-9. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 32.Huang HY, Illei PB, Zhao Z, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005;23:548–58. doi: 10.1200/JCO.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 33.Girnita A, All-Ericsson C, Economou MA, et al. The insulin-like growth factor-I receptor inhibitor picropodophyllin causes tumor regression and attenuates mechanisms involved in invasion of uveal melanoma cells. Acta Ophthalmol. 2008;86 doi: 10.1111/j.1755-3768.2008.01183.x. [DOI] [PubMed] [Google Scholar]; Thesis. 4:26–34. [Google Scholar]
- 34.Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572–82. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 35.Houghton PJ, Morton CL, Gorlick R, et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2010;54:921–6. doi: 10.1002/pbc.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurmasheva RT, Dudkin L, Billups C, Debelenko LV, Morton CL, Houghton PJ. The insulin-like growth factor-1 receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGF and synergizes with rapamycin in models of childhood sarcoma. Cancer Res. 2009;69:7662–71. doi: 10.1158/0008-5472.CAN-09-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suva ML, Riggi N, Stehle JC, et al. Identification of Cancer Stem Cells in Ewing’s Sarcoma. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-2242. [DOI] [PubMed] [Google Scholar]
- 38.Bruggeman SW, Hulsman D, Tanger E, et al. Bmi1 controls tumor development in an ink4a/arf-independent manner in a mouse model for glioma. Cancer Cell. 2007;12:328–41. doi: 10.1016/j.ccr.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 39.Bruggeman SW, Hulsman D, van Lohuizen M. Bmi1 deficient neural stem cells have increased integrin dependent adhesion to self-secreted matrix. Biochim Biophys Acta. 2009;1790:351–60. doi: 10.1016/j.bbagen.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Wiederschain D, Chen L, Johnson B, et al. Contribution of polycomb homologues Bmi-1 and Mel-18 to medulloblastoma pathogenesis. Mol Cell Biol. 2007;27:4968–79. doi: 10.1128/MCB.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachmann IM, Puntervoll HE, Otte AP, Akslen LA. Loss of BMI-1 expression is associated with clinical progress of malignant melanoma. Mod Pathol. 2008;21:583–90. doi: 10.1038/modpathol.2008.17. [DOI] [PubMed] [Google Scholar]
- 42.Choi YJ, Choi YL, Cho EY, et al. Expression of Bmi-1 protein in tumor tissues is associated with favorable prognosis in breast cancer patients. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-9909-4. [DOI] [PubMed] [Google Scholar]
- 43.Pietersen AM, Horlings HM, Hauptmann M, et al. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008;10:R109. doi: 10.1186/bcr2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitsiades N, Poulaki V, Kotoula V, Leone A, Tsokos M. Fas ligand is present in tumors of the Ewing’s sarcoma family and is cleaved into a soluble form by a metalloproteinase. Am J Pathol. 1998;153:1947–56. doi: 10.1016/S0002-9440(10)65708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]