

Abstract
Purpose of the review
MHC class I molecules control the repertoire and function of CD8+ T cells and NK cells, and both cell types are involved in transplant rejection. Understanding the regulatory role of MHC class I molecules is important in the design of better therapies. This review articles focuses on molecular aspects of allo-reactive recognition of MHC class I molecules by CD8+ T cells and NK cells, and on the functional activities of CD8+ T cells and NK cells in transplant rejection and tolerance.
Recent findings
Recent TCR/peptide-MHC class I crystal structures, and structural and functional analyses of MHC class I interactions with NK cell inhibitory receptors have revealed new insights into molecular aspects of allo-recognition of MHC class I molecules by CD8 T cells and NK cells. In functional studies, CD8+ T cells and NK cells have been shown to have conditional and model-dependent roles in allograft rejection. NK cells have also been shown to have an unexpected role in tolerance induction in the transplantation setting.
Summary
Both CD8+ and NK cells play diverse roles in graft rejection and tolerance induction. Further understanding of molecular interactions between MHC class I molecules and TCR or NK receptors is important and highly relevant to transplantation.
Keywords: MHC class I, T cell receptor (TCR), Killer Inhibitory Receptors (KIR), transplant, human leukocyte antigens (HLA)
Introduction
MHC class I molecules are expressed by all nucleated cells. Structurally, the MHC class I molecule comprises a heavy chain, a light chain and a short antigenic peptide [1]. Assembly of MHC class I molecules occurs in the endoplasmic reticulum (ER) of cells, and involves a complex machinery of assembly factors, those encoded within the MHC, as well as generic ER chaperones including the lectin chaperones calnexin and calreticulin, and the thiol oxido-reductase ERp57 (reviewed in [2–4]). In normal healthy cells, peptides that assemble with MHC class I molecules are derived from the cells own proteins. During infections, a subset of MHC class I molecules become associated with pathogen-derived peptides, which serves to activate CD8+ T cells against self-MHC/pathogen-peptide combinations. Down-modulation of MHC class I during infection or tumorigenesis also triggers NK cell activation, a mode of immune surveillance that is called “missing-self recognition” [5].
The MHC class I heavy chain locus is polygenic, encoding the HLA-A, HLA-B and HLA-C genes in humans and the H2-K, H2-L and H2-D genes in mice. Each human gene is highly polymorphic. For example, there are over 500 known variants of HLA-B genes, over two hundred variants of HLA-A genes, and over 100 variants of HLA-C genes [6]. Each MHC class I protein binds to a large number of peptides, that share similar primary structural features such as the presence of a particular amino acidic residue at a given position of an octamer or a nonamer sequence [7]. Polymorphic MHC class I variants differ in the structures of their peptide binding grooves, which in turn impacts the specificity of peptide binding [8]. MHC class I polymorphisms have evolved to ensure that immune responses can be generated against a diverse array of antigen peptides. Indeed homozygosity at any locus is associated with negative outcomes during infections [9].
MHC class I molecules as ligands for T cell receptors
Through cooperative binding via the CD8 co-receptor, T cell receptors (TCR) of CD8+ T cells are able to engage MHC class I molecules on opposing cells in a manner that is exquisitely specific for the MHC class I-associated peptide (reviewed in [10]). The extracellular regions of the TCR comprise α and β chains that contain immunoglobulin-like constant and variable domains. As with immunoglobulins, variable domains of T cell receptors arise by rearrangements of V, D and J gene segments. Hypervariable regions within the variable domains form the combining sites for associations with MHC-peptide complexes (reviewed in [10]). During their maturation in the thymus, T cells are selected for the abilities of their T cell receptors to recognize self-peptide-MHC combinations through positive selection, but those that are strongly reactive towards self-peptide/MHC combinations are deleted through negative selection. These two selection events help maintain the balance between the ability of T cells to recognize peptides in the context of self MHC, and the ability to maintain self-tolerance. Combined with peripheral tolerance mechanisms, activation of CD8+ T cells by self-MHC/self-peptide combinations is generally suppressed. In a transplantation setting in which MHC allotypes of the donor and recipient are mis-matched, the thymic education process of the recipient does not necessarily eliminate donor MHC/peptide-reactive T cells, or T cells reactive towards donor peptide/recipient-MHC combinations (Figure 1). Thus, allograft-specific peripheral CD8+ T cells can become activated in some allograft settings, given the right contexts, and can mediate immune attack against transplanted organs. A recent study showed that while CD8+ T cells generated from transgenic mice that expressed single peptide-MHC class I complexes (H2-Kb engineered as a single chain with peptides derived from ovalbumin or vesicular stomatitis virus (VSV)) were largely specific for H-2b, reactivity towards H-2d and H-2k was also detectable [11]•. Thus, while a majority of CD8+ T cells selected by single peptide-MHC class I combinations are specific for the selecting MHC class I molecule, CD8+ T cells can arise at a low frequency that are capable of cross-reactivity towards different MHC class I types. It is noteworthy that whereas T cell clones derived from the transgenic mice expressing single peptide-H2-Kb complexes were largely specific for peptides presented by H2-Kb molecules, T cell clones derived from mice expressing H2-Db complexes with a normal repertoire of peptides were cross-reactive towards H2d and H2k at much higher frequencies [11]•. These findings raise the possibility that the frequency of generation of cross-reactive clones is correlated with the diversity of peptides presented by the selecting MHC class I molecule, although additional studies are needed to directly address this possibility.
Figure 1. Recognition of donor-peptide/donor-MHC (A) or donor-peptide/recipient-MHC (B) combinations by allo-reactive T cells of the recipient.
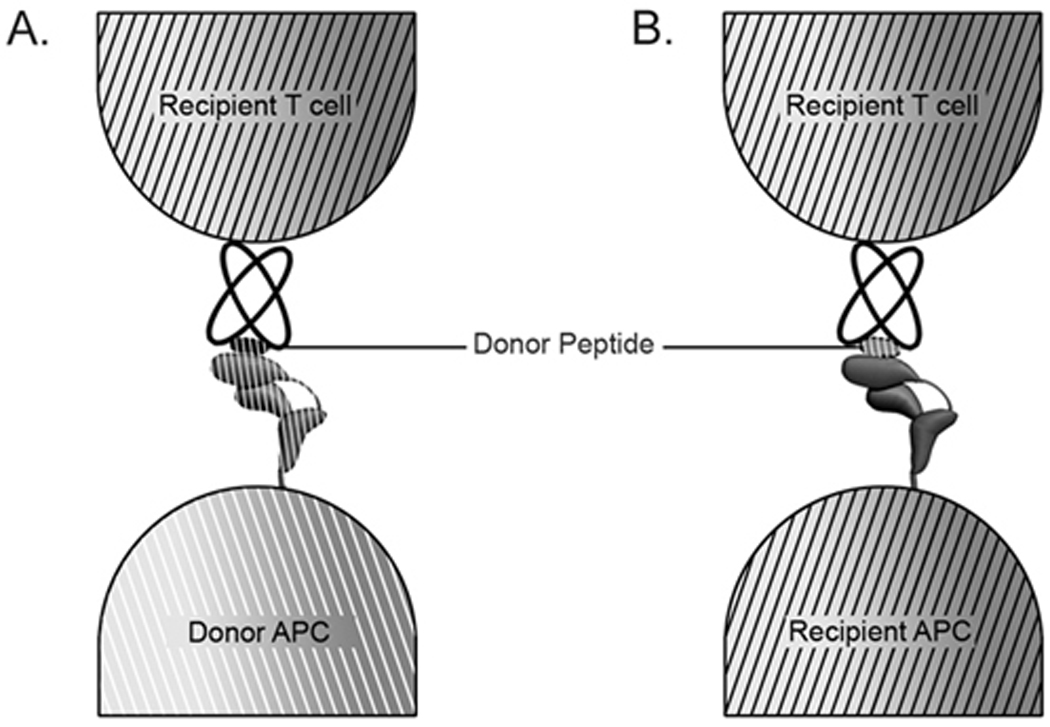
Donor cells can be directly recognized as in A, or alternatively, donor-derived proteins can be cross-presented by recipient antigen presenting cells (APC). Both modes of recognition are relevant to organ transplantation.
A crystal structure comparison was recently reported of allo-reactive T cell receptor peptide-MHC class I complexes compared to self-peptide MHC class I complexes, which provide new insights into how TCRs interact with self-MHC-peptide and allo-MHC-peptide complexes [12]•. Comparisons of the crystal structure of the murine 2C TCR in complex with a self MHC-peptide (H2-Kb-dEV8) or an allogeneic MHC-peptide complex (H2-Ld-QL9) revealed that the 2C TCR adopted very similar conformations in the two structures [12]•. However, surprisingly, few contacts were found to be shared between the two structures. Thus, although the overall binding geometry was preserved in the two complexes, the details of the contacts were strikingly different in an allo-complex compared to a self-complex. Greater structural complementarity was observed in the allo-complex 2C-H2-Ld-QL9 compared to the self complex 2C- H2-Kb-dEV8.
In another recent study [13]•, crystal structures were solved of complexes between two TCR clones that were both reactive towards the HLA-B8/FLRGRAYGL complex. The memory response to the HLA-B8/FLRGRAYGL epitope in HLA-B8+ individuals was previously shown to be highly immunodominant, with virtually identical TCR protein sequences being used by CD8+ T cell clones derived from multiple EBV+ individuals [13]•. A large number of clones expressing such a “public” TCR were found to be cross-reactive with HLA-B44 allotypes, and furthermore the “public” TCR sequences were not prevalent in HLA-B8+/HLA-B44+ individuals [13]•. Although CD8+ T cell responses to HLA-B8/FLRGRAYGL were detectable in HLA-B8+/HLA-B44+ individuals, TCR usage was distinct than that in HLA- B8+ individuals (who did not also express HLA-B44). TCR clones derived from HLA- B8+ and HLA-B8+/HLA-B44+ individuals (LC13 and CF34 respectively) demonstrated different modes of interaction towards HLA-B8/FLRGRAYGL, as revealed by crystal structures of each complex. LC13 binding focused towards the peptide carboxy-terminus, whereas CF34 binding focused on the peptide amino-terminus [13]•.
Together, these new structural findings reveal mechanisms of allo-recognition at the molecular level, and also reveal how self-tolerance alters recognition modes of the TCR repertoire.
Role of CD8+ T cells in transplant rejection
Transplantation of MHC incompatible grafts to immune competent recipients triggers the activation of alloreactive T cells, and in the absence of effective immune interventions, the activated T cells mediate robust transplant rejection via several effector mechanisms. It is well recognized that T cells are necessary and sufficient for the initiation of transplant rejection, but graft destruction often involves many other cell types including cells in the innate immune system such as NK cells and macrophages [14]. Thus, the relative significance of various cell types in the rejection response and their complex interactions in conferring tolerance resistance are an interesting and important issue in transplant research. It is generally believed that CD4+ T cells, upon activation by alloantigens, differentiate into functionally different subsets (e.g., Th1, Th2, Th17) that mediate transplant rejection by producing powerful pro-inflammatory cytokines as well as by recruitment of innate inflammatory cells into the graft. In this case, both T cells and innate immune cells contribute to graft damage [15,16]. Activated CD8+ T cells often acquire potent cytolytic activities that can induce graft injury by directly killing target cells. Under some conditions, activated CD8+ T cells can also produce potent inflammatory cytokines (e.g., IL-17) that mediate graft damage [17]. Interestingly, differentiation of activated CD8+ T cells may require CD4+ T cell help, and this appears to be mediated by cytokines produced by activated CD4+ T cells that are needed for CD8+ T cell proliferation or by CD4+ T cell-triggered activation of dendritic cells, which indirectly prime CD8+ T cells to mediate graft injury [18].
Depending on models and the degree of MHC mismatch, there have been complex interactions identified to date between CD4+ and CD8+ T cells in transplant rejection. In fully MHC mismatched organ transplants (MHC class I and class II mismatch), both CD4+ and CD8+ T cells are activated in response to alloantigen stimulation, they conspire with each other to mediate graft destruction. Prevention of transplant rejection in this setting is more difficult and often requires strategies to target both CD4+ and CD8+ T cells. Studies using CD4 and CD8 deficient mice, MHC class I and class II deficient mice, or selective depletion of CD4+ or CD8+ T cells in various transplant models have demonstrated that CD4+ T cells are capable of inducing prompt transplant rejection without CD8+ T cells. However, CD8+ T cells by themselves are potent effector cells in rejection of some allografts (e.g., skin allografts) but much less effective in rejection of other grafts such as the heart allografts [19], suggesting that CD8+ T cells are critical in the rejection response, but their roles can be conditional and model-dependent.
MHC class I molecules as ligands for NK cell receptors
Natural killer cells are lymphocytes that are involved in the innate immune response. Rather than expressing clonotypic antigen receptors as do B cells and T cells, NK cells express a panel of receptors with activating and inhibitory functions [20]. Several of the inhibitory receptors have specificities for self-MHC class I proteins, and engagement of self-MHC by inhibitory receptors of NK cells counters NK cell activation. This is one of the mechanisms by which NK cells ensure self tolerance. Human and mouse inhibitory NK cell receptors, which include the Killer Inhibitory Receptors (KIR) and Ly49 respectively, recognize polymorphic MHC class I determinants, and genes encoding the receptors themselves are also polymorphic [21,22]. The KIR2DL1 receptors bind HLA-C molecules with N77/K80 in their heavy chain sequences, whereas KIR2DL2 and KIR2DL3 receptors bind HLA-C molecules with S77/N80 in their heavy chain sequences [23,24]. Some recent studies indicate that these specificities may not be absolute in the case of KIR2DL2/L3, which can also react with HLA-C molecules containing N77/K80 in their heavy chain sequences [25,26]. HLA-B molecules are classified into either Bw4 or Bw6 serotypes. The KIR3DL1 receptor is specific for the Bw4 serotype [27], although not all HLA-B(Bw4) molecules are equivalently recognized (for example, [28,29]). NK cells have the potential to recognize and destroy allogeneic target cells, when target cells lack expression of MHC class I allotypes that are specific for the KIR expressed by NK cell (Figure 2). This feature of NK cells that has been used in the immunotherapy of leukemias (reviewed in [30,31]). HLA binding specificities of different allelic variants of several KIR receptors remain incompletely or poorly defined, which represents an important area for future investigation, because of the clinical relevance of KIR ligand mis-matching in stem cell transplantation.
Figure 2. Allo-reactive recognition by NK cells.
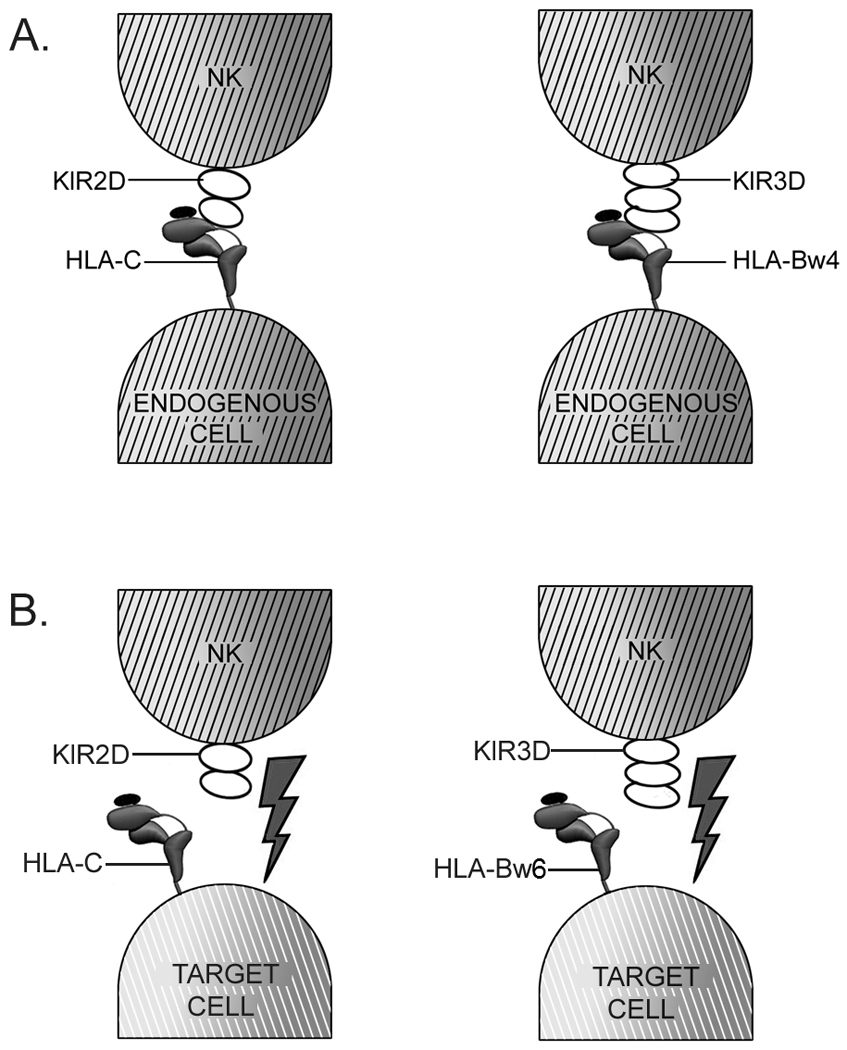
Engagement of HLA-C molecules on target cells by inhibitory KIR2D receptors or of HLA-Bw4 molecules by inhibitory KIR3D receptors inhibits NK cell activation (A). Absence of appropriate KIR-specific HLA-C or HLA-Bw4 ligands on target cells during organ transplantation can lead to blockage of inhibitory signals, and NK cell activation and target cell killing (B), if appropriate activation receptors and their ligands become engaged.
Role of NK cells in transplant rejection
NK cells are frequently found in rejecting allografts, but the exact role of NK cells in regulating the fate of an allograft remains poorly understood. In transplant models, NK cells can respond to the allografts by “missing self” or “missing ligand” recognition [20,32], they possess potent cytolytic function and can also produce copious amounts of inflammatory cytokines such as IFN-γ and TNF-α [32]. Thus, NK cells are well qualified to act as effector cells in graft rejection. Indeed, host NK cells are known effector cells capable of rejecting MHC-incompatible donor stem cells in bone marrow transplant models [33]. Similarly, graft-derived donor NK cells can also destroy MHC mismatched host cancerous cells in leukemia patients who receive bone marrow transplants as a therapy, thus leading to potent graft anti-leukemia effects [34]. In solid organ transplants, the role of NK cells is far more complex. Recent studies have shown that NK cells by themselves are poor effector cells in rejection unless they are deliberately stimulated by IL-15 [35]. However, NK cells contribute significantly to allograft rejection or to chronic vasculopathy in models where CD28 costimulation is absent or T cell activation is inhibited [36,37], but the exact mechanism by which NK cells mediate graft injury remains unclear. Adding to the complexity is the recent demonstration that NK cells can be crucial to the induction of transplant tolerance, as tolerance to islet and skin allografts are difficult to induce in the absence of NK cells [38,39]. We and others reported that host NK cells rapidly kill graft derived donor dendritic cells, thus limiting the direct priming of alloreactive T cells by donor dendritic cells in transplant recipients [38]. Elimination of donor DCs creates a permissive environment for tolerance induction by co-stimulatory blockade. These studies uncover unexpected complexities amongst donor APCs and host NK cells and alloreactive T cells in transplant models, which demand more in-depth studies on the in vivo conditions that govern activation of both innate and adaptive immune cells in transplant settings.
Current strategies to block recognition by CD8+ T cells and NK cells
Given a key role of T cells in transplant rejection, various strategies have been designed and tested to target alloreactive T cells in transplant models, and such strategies include blocking initial TCR signals critical to T cell activation (e.g. calcineurin inhibitors), broad depletion of T cells including CD8+ T cells, antibody-mediated blockade of CD8 molecule, T cell co-stimulatory blockade, and blocking growth factor signals required for T cell proliferation [40]. These strategies, either used alone or in various combinations, have demonstrated efficacy in prolonging transplant survival. However, in transplant models where rejection is mediated by both CD4+ and CD8+ T cells, the relative contribution of CD4+ versus CD8+ T cells to graft rejection remains an interesting issue and it is often difficult to ascertain how much of the therapeutic effect is due to specific inhibition of CD8+ T cells, as CD4+ and CD8+ T cells often use overlapping signals and mechanisms for activation and proliferation. In experimental models in which rejection is mediated solely by CD8+ T cells, blocking growth factor signaling by rapamycin or targeting critical co-stimulatory molecules using blocking antibodies and recombinant fusion proteins can inhibit CD8+ T cell activation and induce allograft tolerance [19]. In contrast, antibody-mediated depletion of CD8+ T cells only prolongs graft survival, but fails to induce tolerance [41]. Thus, different protocols are not therapeutically equivalent in blocking CD8+ T cell mediated rejection.
Despite recent interests in NK cells in transplant models, we have a limited understanding on the role of NK cells in transplant rejection and tolerance induction. Thus, therapeutic protocols that selectively and specifically target NK cells for the induction of transplant tolerance are still lacking. Given the fact that NK cells are phenotypically and functionally diverse, harnessing the role of NK cells for the purpose of transplant tolerance is unlikely to be straightforward. In addition, they are involved in both graft rejection and tolerance induction, therapeutic strategies that can uncouple the pro-inflammatory and anti-inflammatory effects of NK cells in transplant settings are a clinically important issue. Clearly, given the broad effect of NK cells in transplant models, global depletion of NK cells using depleting Abs is not ideal and may induce unwanted side effects. As the impact of NK cells on transplant outcomes is compelling, there is a need in developing NK specific protocols as adjunctive therapies to promote transplant tolerance.
Problems associated with current therapies
Current immunosuppressive protocols, while effective in prolonging transplant survival, are fraught with significant sides effects including infections, malignancy, and metabolic disorders, and sometimes these side effects can be life threatening. Furthermore, most transplants are eventually lost to chronic rejection despite life long immunosuppression. In fact, in spite of drastic improvement in short-term graft survival, current immunosuppressive drugs have made little improvement in long-term graft acceptance [42]. In addition, most of the drugs used by transplant patients are associated with significant toxicities. These problems provide compelling reasons for the development of better and more specific tolerance induction strategies. Unfortunately, we have limited understanding on the underlying mechanisms that mediate tolerance resistance, but certain evidence suggests that memory T cells, especially memory CD8+ T cells are resistant to tolerance induction [43]. Interestingly, NK cells have been recently shown to acquire “memory” features that are traditionally ascribed to T cells [44,45]••, and the exact role of memory NK cells in transplant models, especially in tolerance induction is completely unknown. In addition, the impact of memory T cells and NK cells on the induction of active immune regulation and how the commonly used immunosuppressive drugs would affect this process remain incompletely defined. These areas should deserve more attention in transplant research in the future.
Conclusions
MHC class I molecules are selecting and presenting ligands regulating various aspects of CD8+ T cell and NK cell development, maturation and function. Both CD8+ T cells and NK cells are involved in the allograft response, but complex mechanisms determine the participation of these cell types, and the resulting functional outcomes. Recent biochemical and structural studies have revealed significant insights into receptor-ligand interaction relevant to allo-recognition by both CD8+ T cells and NK cells. However, much remains to be understood about impacts of KIR polymorphisms upon recognition of different HLA class I ligands. With the discovery of the functional heterogeneity of CD8+ T cells and NK cells, it is also important to obtain a better understanding of the involvement of different cellular subsets in transplant rejection vs. tolerance maintenance.
Acknowledgements
We thank Yesung Park for assistance with the figures. Research in the authors laboratories are supported by grants from the National Institutes of Health (AI044115 and AI066131 (to MR)), and R01 AI057409 (XCL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 2.Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 3.Raghavan M, Del Cid N, Rizvi SM, Peters LR. MHC class I assembly: out and about. Trends Immunol. 2008;29:436–443. doi: 10.1016/j.it.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic. 2009;10:1745–1752. doi: 10.1111/j.1600-0854.2009.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ljunggren HG, Karre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 6.Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens. 2003;61:403–407. doi: 10.1034/j.1399-0039.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- 7.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 8.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 9.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O'Brien SJ. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 10.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Primeau TM, Myers N, Rohrs HW, Gross ML, Lybarger L, Hansen TH, Connolly JM. A single peptide-MHC complex positively selects a diverse and specific CD8 T cell repertoire. Science. 2009;326:871–874. doi: 10.1126/science.1177627. This paper elucidates features of CD8+ T cells selected by single-peptide MHC class I complexes.
- 12.Colf LA, Bankovich AJ, Hanick NA, Bowerman NA, Jones LL, Kranz DM, Garcia KC. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. These structural studies compare alloreactive and endogenously-reactive recognition of peptide-MHC complexes by a single TCR
- 13.Gras S, Burrows SR, Kjer-Nielsen L, Clements CS, Liu YC, Sullivan LC, Bell MJ, Brooks AG, Purcell AW, McCluskey J, et al. The shaping of T cell receptor recognition by self-tolerance. Immunity. 2009;30:193–203. doi: 10.1016/j.immuni.2008.11.011. These structural studies illustrate how self-tolerance induces alterations to TCR recognition features.
- 14.Alegre ML, Florquin S, Goldman M. Cellular mechanisms underlying acute graft rejection: time for reassessment. Curr Opin Immunol. 2007;19:563–568. doi: 10.1016/j.coi.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 15.Langrehr JM, White DA, Hoffman RA, Simmons RL. Macrophages produce nitric oxide at allograft sites. Ann Surg. 1993;218:159–166. doi: 10.1097/00000658-199308000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall BM. Cells mediating allograft rejection. Transplantation. 1991;51:1141–1151. doi: 10.1097/00007890-199106000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol. 2008;181:3906–3914. doi: 10.4049/jimmunol.181.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trambley J, Bingaman AW, Lin A, Elwood ET, Waitze SY, Ha J, Durham MM, Corbascio M, Cowan SR, Pearson TC, et al. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J Clin Invest. 1999;104:1715–1722. doi: 10.1172/JCI8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vu MD, Amanullah F, Li Y, Demirci G, Sayegh MH, Li XC. Different costimulatory and growth factor requirements for CD4+ and CD8+ T cell-mediated rejection. J Immunol. 2004;173:214–221. doi: 10.4049/jimmunol.173.1.214. [DOI] [PubMed] [Google Scholar]
- 20.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 21.Middleton D, Gonzelez F. The extensive polymorphism of KIR genes. Immunology. 129:8–19. doi: 10.1111/j.1365-2567.2009.03208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217–251. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- 23.Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405:537–543. doi: 10.1038/35014520. [DOI] [PubMed] [Google Scholar]
- 24.Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol. 2001;2:452–460. doi: 10.1038/87766. [DOI] [PubMed] [Google Scholar]
- 25.Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, Parham P. Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J Immunol. 2008;180:3969–3979. doi: 10.4049/jimmunol.180.6.3969. [DOI] [PubMed] [Google Scholar]
- 26.Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, Romeo E, Cognet C, Martinetti M, Maccario R, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood. 2009;113:3119–3129. doi: 10.1182/blood-2008-06-164103. [DOI] [PubMed] [Google Scholar]
- 27.Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181:1133–1144. doi: 10.1084/jem.181.3.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foley BA, De Santis D, Van Beelen E, Lathbury LJ, Christiansen FT, Witt CS. The reactivity of Bw4+ HLA-B and HLA-A alleles with KIR3DL1: implications for patient and donor suitability for haploidentical stem cell transplantations. Blood. 2008;112:435–443. doi: 10.1182/blood-2008-01-132902. [DOI] [PubMed] [Google Scholar]
- 29.O'Connor GM, Guinan KJ, Cunningham RT, Middleton D, Parham P, Gardiner CM. Functional polymorphism of the KIR3DL1/S1 receptor on human NK cells. J Immunol. 2007;178:235–241. doi: 10.4049/jimmunol.178.1.235. [DOI] [PubMed] [Google Scholar]
- 30.Velardi A, Ruggeri L, Mancusi A, Aversa F, Christiansen FT. Natural killer cell allorecognition of missing self in allogeneic hematopoietic transplantation: a tool for immunotherapy of leukemia. Curr Opin Immunol. 2009;21:525–530. doi: 10.1016/j.coi.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Moretta A, Pende D, Locatelli F, Moretta L. Activating and inhibitory killer immunoglobulin-like receptors (KIR) in haploidentical haemopoietic stem cell transplantation to cure high-risk leukaemias. Clin Exp Immunol. 2009;157:325–331. doi: 10.1111/j.1365-2249.2009.03983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 33.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 34.Ruggeri L, Mancusi A, Perruccio K, Burchielli E, Martelli MF, Velardi A. Natural killer cell alloreactivity for leukemia therapy. J Immunother. 2005;28:175–182. doi: 10.1097/01.cji.0000161395.88959.1f. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer A, Edtinger K, Li XC. The innate natural killer cells in transplant rejection and tolerance induction. Curr Opin Organ Transplant. 2008;13:339–343. doi: 10.1097/MOT.0b013e3283061115. [DOI] [PubMed] [Google Scholar]
- 36.McNerney ME, Lee KM, Zhou P, Molinero L, Mashayekhi M, Guzior D, Sattar H, Kuppireddi S, Wang CR, Kumar V, et al. Role of natural killer cell subsets in cardiac allograft rejection. Am J Transplant. 2006;6:505–513. doi: 10.1111/j.1600-6143.2005.01226.x. [DOI] [PubMed] [Google Scholar]
- 37.Uehara S, Chase CM, Kitchens WH, Rose HS, Colvin RB, Russell PS, Madsen JC. NK cells can trigger allograft vasculopathy: the role of hybrid resistance in solid organ allografts. J Immunol. 2005;175:3424–3430. doi: 10.4049/jimmunol.175.5.3424. [DOI] [PubMed] [Google Scholar]
- 38.Yu G, Xu X, Vu MD, Kilpatrick ED, Li XC. NK cells promote transplant tolerance by killing donor antigen-presenting cells. J Exp Med. 2006;203:1851–1858. doi: 10.1084/jem.20060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beilke JN, Kuhl NR, Van Kaer L, Gill RG. NK cells promote islet allograft tolerance via a perforin-dependent mechanism. Nat Med. 2005;11:1059–1065. doi: 10.1038/nm1296. [DOI] [PubMed] [Google Scholar]
- 40.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 41.Minamimura K, Sato K, Yagita H, Tanaka T, Arii S, Maki T. Strategies to induce marked prolongation of secondary skin allograft survival in alloantigen-primed mice. Am J Transplant. 2008;8:761–772. doi: 10.1111/j.1600-6143.2007.02143.x. [DOI] [PubMed] [Google Scholar]
- 42.Sayegh MH, Carpenter CB. Transplantation 50 years later--progress, challenges, and promises. N Engl J Med. 2004;351:2761–2766. doi: 10.1056/NEJMon043418. [DOI] [PubMed] [Google Scholar]
- 43.Valujskikh A, Li XC. Frontiers in nephrology: T cell memory as a barrier to transplant tolerance. J Am Soc Nephrol. 2007;18:2252–2261. doi: 10.1681/ASN.2007020151. [DOI] [PubMed] [Google Scholar]
- 44.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A. 2009;106:1915–1919. doi: 10.1073/pnas.0813192106. These reports identify adaptive features of NK cells that are traditionally ascribed to T cells, thus providing new knowledge on modes of function of NK cells in immune responses.