

Abstract
Modifications to the gene encoding human α-synuclein have been linked to the development of Parkinson's disease. The highly conserved structure of α-synuclein suggests a functional interaction with membranes, and several lines of evidence point to a role in vesicle-related processes within nerve terminals. Using recombinant fusions of human α-synuclein, including new genetic tags developed for correlated light microscopy and electron microscopy (the tetracysteine-biarsenical labeling system or the new fluorescent protein for electron microscopy, MiniSOG), we determined the distribution of α-synuclein when overexpressed in primary neurons at supramolecular and cellular scales in three dimensions (3D). We observed specific association of α-synuclein with a large and otherwise poorly characterized membranous organelle system of the presynaptic terminal, as well as with smaller vesicular structures within these boutons. Furthermore, α-synuclein was localized to multiple elements of the protein degradation pathway, including multivesicular bodies in the axons and lysosomes within neuronal cell bodies. Examination of synapses in brains of transgenic mice overexpressing human α-synuclein revealed alterations of the presynaptic endomembrane systems similar to our findings in cell culture. Three-dimensional electron tomographic analysis of enlarged presynaptic terminals in several brain areas revealed that these terminals were filled with membrane-bounded organelles, including tubulovesicular structures similar to what we observed in vitro. We propose that α-synuclein overexpression is associated with hypertrophy of membrane systems of the presynaptic terminal previously shown to have a role in vesicle recycling. Our data support the conclusion that α-synuclein is involved in processes associated with the sorting, channeling, packaging, and transport of synaptic material destined for degradation.
Introduction
Parkinson's disease (PD) is the most common neurodegenerative movement disorder characterized by selective loss of dopaminergic neurons in the substantia nigra. Although PD is typically sporadic (Braak et al., 2003), advances in genomic screening technologies reveal an increasing number of mutations in different genes linked to inherited forms of the disease (Zimprich et al., 2004; Dawson et al., 2010). These heritable defects in specific proteins, while observed only in a small percentage of PD patients, appear to collectively highlight a major cellular housekeeping process relating to the protein degradation pathway. For example, rare dominant mutations in α-synuclein (AS) can cause PD. This protein accumulates in Lewy bodies, which are conglomerates of lipids and proteins found in the surviving neurons of PD patients.
The gene encoding human α-synuclein (SNCA) is associated with autosomal-dominant PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004). Either missense mutation or allele multiplication results in early onset autosomal dominant PD (Singleton et al., 2003; Chartier-Harlin et al., 2004), also implicating AS over-expression in disease pathogenesis. Additionally, misfolded AS protein accumulates abnormally in a host of neurodegenerative disorders, including PD, dementia with Lewy bodies, and multiple system atrophy (for review, see Duda et al., 2000). It is unclear whether pathogenicity results from the presence of AS fibrils and/or oligomers or from a derangement of normal protein function. In mouse models or cell culture systems, AS mutations are associated with protein aggregation and cytotoxic effects, although the mechanisms are unclear (Lee et al., 2002; Pandey et al., 2006).
The highly conserved structure of AS, combined with results from other analytical methods, is consistent with a functional interaction with membranes. Specifically, the N terminus of AS is composed of a series of repeats that encode an extended amphipathic α-helix (George et al., 1995). This domain is stabilized by interactions with phospholipid membranes (Davidson et al., 1998) and fatty acid or detergent micelles (Weinreb et al., 1996). PD-associated point mutations are all localized within this lipid-binding domain. When in the membrane-bound state, the helical conformation of AS favors specific interactions with a variety of cellular proteins, including α –tubulin and septin4, suggesting that AS, once incorporated into membranes, serves as a scaffold for protein assembly (Woods et al., 2007). In vitro, AS exerts pleiotropic effects on membranes, from inhibition of mitochondrial fusion to tubulation of phospholipid vesicles (Varkey et al., 2010).
Consistent with its membrane-binding activity, several studies suggested a role for AS in synaptic vesicle trafficking. Interestingly, it has been reported that AS inhibits SNARE complex assembly via sequestration of arachidonic acid, a fatty acid that stimulates SNARE assembly (Darios et al., 2010), while others (Burré et al., 2010) have reported that AS promotes SNARE assembly via direct interactions with VAMP2. However, the precise sites of AS binding to cellular membranes within neurons remain unknown. With the present study, we address this question using novel genetically encoded probes that allow direct examination of the distribution of AS across scales from the supramolecular complex to the whole cell.
Materials and Methods
Molecular cloning of tetracysteine and MiniSOG constructs.
cDNA encoding wild-type human-α-synuclein was cloned into pCDNA3.1-EGFP-tetracysteine (4Cys) and pCDNA3.1-MiniSOG. The correct constructs were confirmed by sequencing to create pCDNA3.1-AS-EGFP-4Cys and pCDNA3.1-AS-MiniSOG.
Primary cell culture and transfection.
For experiments with 4Cys constructs, primary neurons were prepared from rat hippocampus of embryonic day 18 (BrainBits), and cultured on poly-d-lysine-coated coverslips in Neurobasal medium (Invitrogen) supplemented with B27, 0.5 mm glutamine, and 25 μm glutamate. Cells were transfected with Lipofectamine 2000 (Invitrogen) at 7 days in vitro (DIV) and visualized at 14–17 DIV. For the MiniSOG studies, primary neurons were prepared from cortex of 2-day-old Harlan Sprague Dawley rats and cultured on poly-d-lysine-coated dishes in Neurobasal-A medium supplemented with B27 and l-glutamine. Before plating, neurons were transfected with the Amaxa Nucleofector protocol and imaged at 17–21 DIV.
Western blot analysis.
HEK293 cells transfected with AS-EGFP-4Cys using Lipofectamine 2000 (Invitrogen) were collected after 48 h of expression and lysed with CelLytic M (Sigma). Whole-cell lysates from rat cortical neurons expressing AS-MiniSOG were prepared in SDS buffer containing 4% β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Sigma). Cell extracts were separated by SDS-PAGE and then electrophoretically transferred onto nitrocellulose. Membranes were blocked in 4% nonfat dry milk made in TBST (20 mm Tris, 127 mm NaCl, 0.1% Tween 20) for 1 h and incubated with monoclonal antibodies to the AS C terminus (H3C, (Perrin et al., 2003) or GFP (Invitrogen, anti-GFP 3E6), followed by horseradish peroxidase-linked sheep anti-mouse Ig (GE Healthcare). The membrane was processed for enzyme-linked chemiluminescence and exposed to film as described by the ECL Western Blotting Kit (GE Healthcare).
Immunocytochemistry.
Cultured neurons were fixed in 4% paraformaldehyde/PBS for 15 min, washed, permeabilized in 0.1% Triton X-100, and blocked in 1% BSA and 2% normal serum. The primary antibodies (rabbit anti-α-synuclein polyclonal, Millipore catalog no. AB5038; mouse monoclonal anti-human α synuclein (LB509), Abcam catalog no. ab27766; mouse monoclonal anti-bassoon, Abcam catalog no. AB82958) were mixed in blocking buffer diluted fivefold in PBS. The secondary antibodies were diluted in the same buffer. Images were acquired using an Olympus FluoView1000 laser-scanning confocal microscope.
Photo-oxidation and EM preparation of transfected neuronal cells.
Transfected neurons plated on glass bottom culture dishes (MatTek) were fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, for 1 h on ice, rinsed 5 times in cold cacodylate buffer, and blocked for 30 min with 10 mm KCN, 20 mm aminotriazole, 50 mm glycine, and 0.01% hydrogen peroxide in cacodylate buffer. For 4Cys photo-oxidation, transfected cells were processed as reported by Gaietta et al. (2002). For MiniSOG photo-oxidation, the transfected neurons were identified using a Leica SPE II inverted confocal microscope. Freshly prepared diaminobenzidine (DAB free base, Sigma catalog no. D8001-10G) in blocking buffer was added to the plate, and neurons were illuminated with 450–490 nm light from a xenon lamp for 3–4 min until a light brown reaction product was observed in place of the green fluorescence of MiniSOG. Neurons were then removed from the microscope, washed in cold cacodylate buffer, and postfixed in 1% osmium tetroxide for 30 min on ice. After several washes in cold double distilled water, cells were either en bloc stained with 2% aqueous uranyl acetate for 1 h to overnight at 4°C, or directly dehydrated in a cold graded ethanol series (20%, 50%, 70%, 90%, 100%) 3 min each on ice, then rinsed once in room temperature with 100% ethanol and embedded in Durcupan ACM resin (Electron Microscopy Sciences). In some samples the poststaining step in uranyl acetate was omitted so that the specific EM signal would be generated only by the passage in osmium to add electron density to the DAB precipitates, which represents the AS labeling. Sections were cut with a diamond knife at a thickness of 70–90 nm for thin sections and 250 nm for thick sections for electron tomography. Thin sections were examined using a JEOL 1200 EX operated at 80 kV.
Electron tomography.
Sections were coated with carbon on both sides, and colloidal gold particles (5 and 10 nm diameter) were deposited on each side to serve as fiducial markers. For reconstruction, double or triple tilt series of images were recorded at regular tilt (angular increments of 2° from −60° to +60° increments) with a JEOL 4000EX intermediate high-voltage electron microscope operated at 400 kV. The specimens were irradiated before initiating a tilt series to limit anisotropic specimen thinning during image collection. Tilt series were recorded using a 4k × 4k custom high resolution slow-scan CCD camera system delivering 25% contrast at Nyquist. Fine alignment of projections and 3D reconstruction were performed using the TxBR reconstruction package (Lawrence et al., 2006). This high resolution tomography reconstruction software was used in conjunction with the IMOD package, where reconstructed image volumes were viewed and objects of interest traced manually and reported as 3D models.
Subjects.
Transgenic (Tg) mice (n = 3) overexpressing human wild-type α-synuclein (PDGF–human α-synuclein Tg mice, line D, from Masliah et al., 2000) and their littermate wild-type controls (n = 3), 10- to 17- month-old males, were used in this study. The mice were housed in an accredited animal care facility (Association for Accreditation of Laboratory Animal Care International) on a 12 h light/dark cycle with food and water provided ad libitum. Animal care was in accordance with the Guide for Care and Use of Laboratory Animals (NIH publication 865-23, Bethesda, MD) and approved by the Institutional Animal Care and Use Committee.
Tissue preparation.
Animals were deeply anesthetized with an intraperitoneal injection of pentobarbital (10 mg/g body weight). Next, mice were perfused transcardially with oxygenated Ringer's solution at 37°C (0.79% NaCl, 0.038% KCl, 0.20% MgCl26H2O, 0.018% NaHPO4, 0.125% NaCHO3, 0.03% CaCl22H2O, 0.20% dextrose, and 0.02% xylocaine) for ∼30 s, followed by 0.1 m PBS, pH 7.4, containing 4% paraformaldehyde and 0.1% glutaraldehyde (37°C) for 10 min. The mouse brain was removed and stored in same fixative for 2 h. The brains were divided into hemispheres, and coronal sections were collected using a Vibratome (VT1000E, Leica Microsystems). The left hemisphere was sectioned into 100-μm-thick sections for conventional EM and processing for serial block face scanning EM (SBEM). The right hemisphere was sectioned into 70-μm-thick sections, and serial sections were placed in cryoprotectant for immunohistochemistry.
Preparation of conventionally fixed and embedded tissue samples.
Vibratome sections were placed in sodium cacodylate-buffered 1% OsO4 for 1 h. After three 5 min rinses with double distilled water, the slices were dehydrated with an ethanol series (50%, 70%, 90%, and 100%), followed by dry acetone (2 times, 5 min each). The slices were infiltrated with a solution of 50% acetone–50% Durcupan ACM epoxy resin (Electron Microscopy Sciences) for 1 h and then with 100% resin (overnight). Next, they were placed into fresh Durcupan and transferred to a rotator for 1 h. The slices were embedded by placing them between two mold release-coated slides and left at 60°C for 48 h.
Preparation of fixed and embedded tissue samples for serial block face scanning EM.
One hundred micrometer-thick vibratomed sections were washed with 0.15 m cacodylate buffer with 2 mm CaCl2. Next, sections were placed in 2% OsO4/1.5% potassium ferrocyanide in buffer for1 h. After three 5 min washes, tissue was placed in filtered 1% thiocarbohydrazide (TCH) solution for 20 min. Sections were rinsed again with ddH2O and then placed in a 2% OsO4 solution for 30 min. After this second osmium step, the sections were rinsed with ddH2O and left in 2% uranyl acetate aqueous solution overnight at 4°C. Next, tissue sections were washed with ddH2O and then dehydrated using an ice-cold graded ethanol series of 70%, 90%, and 100% ethanol for 10 min each. Subsequently, the sections were placed into ice-cold 100% acetone for an additional 10 min, followed by a second 100% acetone step at room temperature for 10 min. The tissue was infiltrated with a solution of 50% acetone–50% Durcupan ACM epoxy resin overnight and then placed into fresh 100% Durcupan for 2 h. Lastly, the sections were embedded using ACLAR and two mold-release coated glass slides and left at 60°C for 48 h.
These data have been uploaded in the Cell Centered Database (National Center for Microscopy and Imaging Research, La Jolla, CA) and are available for visualization and download for reanalysis both as full resolution images and 3D movies.
Results
Human α-synuclein associates with tubulovesicular structures in presynaptic terminals
We generated a construct (indicated as AS-EGFP-4Cys in Fig. 1) that directs expression of full-length human AS with C-terminally fused EGFP and a short 4Cys motif (FLNCCPGCCMEP) that mediates binding to biarsenical ligands, such as the red fluorescent ReAsH-EDT2 (for reference, see Gaietta et al., 2002). Protein expression was confirmed by transfection of AS-EGFP-4Cys and the parent vector EGFP-4Cys in HEK293 cells, followed by immunoblot with antibodies to AS and GFP, respectively (Fig. 1B). The constructs, when expressed in primary neurons, demonstrate colocalizing pattern with the presynaptic marker Synapsin I (Fig. 1D) and thus are comparable to what has been established for endogenous AS (Murphy et al., 2000).
Figure 1.

Recombinant AS-EGFP-4Cys is targeted to presynaptic terminals. A, The combinatorial tag formed by EGFP and the 4Cys motif FLNCCPGCCMEP was fused to the carboxyl terminus of human AS. B, Whole-cell lysates from HEK293 cells expressing AS-EGFP-4Cys (lanes 1 and 3) and EGFP-4Cys (lanes 2 and 4) were Western blotted with a monoclonal antibody specific for GFP (lanes 1 and 2) or AS (lanes 3 and 4). C, The labeling obtained with ReAsH-EDT2 is specific as indicated by the colocalization with the GFP signal (displayed in yellow in the merged image). Inset at higher magnification highlights the strong correspondence between ReAsH labeling and GFP fluorescence (arrowheads). Scale bars, 20 μm. D, Colocalization pattern of AS-EGFP-4Cys with the presynaptic marker synapsin I. Scale bar, 20 μm.
Initial attempts at labeling AS proteins with ReAsH using the standard labeling protocol for cell lines (900 nm ReAsH-EDT2 with 12.5 μm ethanedithiol (EDT) for 1 h) produced high background in primary cultures of neurons. To optimize the biarsenical labeling in primary neurons, we ran extensive tests, varying the dye concentration (2.5 μm, 2 μm, 1.8 μm, 1 μm) and the labeling time (30 min, 1 h, 2 h). Using increased dye concentration with decreased incubation time still yielded intense background. Thus, we tried lowering the dye concentration (0.9 μm, 0.45 μm, 0.225 μm), prolonging the incubation time (6 h, 8 h, 10 h, 16 h, 18 h) and increasing the EDT (12.5 μm, 25 μm) to help reduce background staining. Results showed that neurons did not survive well under overnight labeling (16–18 h) using 900 nm ReAsH-EDT2 plus 12.5 μm EDT, but survived well under overnight labeling in the presence of lower concentrations of ReAsH-EDT2 (200–400 nm) with higher concentration of EDT (25 μm). Moreover, 225 nm ReAsH-EDT2 with 25 μm EDT produced the best labeling with low background and strong staining, as indicated by the overlapping of signal with the EGFP fluorescence (displayed as the yellow color in the merged view in Fig. 1C, with the inset representing a view at higher magnification).
One of the advantages of the 4Cys technology using the biarsenical ligand ReAsH is the ability to highly correlate light microscopy and electron microscopy (CLEM) by fluorescence photo-oxidation. When bound to the 4Cys motif, ReAsH becomes fluorescent and upon intense illumination induces the deposition of a DAB reaction product that can be rendered electron dense by a passage in osmium and visualized by EM. Therefore, the EM-visible local precipitate reflects the specific AS labeling. In our experiments ReAsH was indirectly excited through fluorescent resonance energy transfer (FRET) from GFP to avoid nonspecific photo-oxidation of background staining. Figure 2 shows the FRET-mediated photo-oxidation of ReAsH-labeled AS proteins expressed in primary neurons. High concentration of ReAsH/AS staining is observed specifically in membrane-bounded structures, both vesicular and tubular, within the presynaptic terminals.
Figure 2.

FRET-based fluorescent photo-oxidation of ReAsH-labeled AS proteins expressed in neurons specifically labels membranous structures in the presynaptic terminals. A–F, Neurons were transfected with AS-EGFP-4Cys, cultured for 17 days, labeled with ReAsH, and FRET photo-oxidized to avoid nonspecific photo-oxidation of background staining. The EM staining reveals both large tubular membranous structures (arrows) and smaller vesicular structures (arrowheads). C and D are the same terminal, as are E and F. Scale bars, 2 μm (A), 1 μm (B), 200 nm (C–E), 100 nm (F).
MiniSOG tagging illuminates the trafficking and degradation of α-synuclein
Prompted by the success of the 4Cys technology in allowing us to visualize the detailed subcellular tubulovesicular membrane localization of AS within the presynaptic terminals, we turned to the new and more powerful MiniSOG molecular labeling system for CLEM (Shu et al., 2011). This new marker system is more sensitive than the 4Cys probe and is based on a completely new fluorescent protein, “MiniSOG,” which, like the 4Cys and its fluorescent ligands, has been designed to enable labeling for both observations of dynamics in light microscope systems and subsequent high resolution imaging by electron microscopy. It has several significant advantages; the MiniSOG system is a much more powerful singlet oxygen generator, and as a singlet oxygen-based marker system it is at least 20 times more sensitive than the 4Cys probe system. The marker is applicable for imaging both tissue culture systems and animal tissues, whereas the 4Cys system, which requires a second ligand to be applied, proved difficult to be used for proteins expressed in animal models of disease. We added this label to the AS gene (Fig. 3A) and confirmed the expression in rat cortical neuronal cultures by Western blot using an antibody specific to AS (Fig. 3B) and by immunofluorescence (Fig. 3C). The MiniSOG fluorescence (Fig. 3D) overlaps with the AS antibody staining (Fig. 3E) and the presynaptic marker Bassoon (Fig. 3F, merged in G). The greater sensitivity of MiniSOG compared to 4Cys provided better resolution images and revealed more labeling of the components of the protein degradation system (Fig. 4). Since this system is also based on fluorescence photo-oxidation, the darker intensity of the signal observed in the electron micrographs is due to the EM-visible local precipitate of DAB and reflects the specific AS labeling. AS expression appeared in large membranous organelles and smaller vesicular structures within the presynaptic terminal, consistent with the results obtained with the 4Cys probe. In addition we observed staining in the membrane of intraluminar vesicles of multivesicular bodies (MVBs) along the axons and in lysosomes at the cell body of transfected neuronal cells (Fig. 4A). Conversely, the controls, nontransfected neurons that underwent the same photo-oxidation procedure, did not snow this pattern of labeling. Three-dimensional electron microscopic tomography highlighted intense AS staining in extensive membranous elements in presynaptic terminals of overexpressing neurons as well as in the membrane of the vesicles (Fig. 4B). The 3D model obtained from the segmented volume (Fig. 4C) shows an example of such contiguous membranes depicted in three different colors (blue, green, and yellow).
Figure 3.
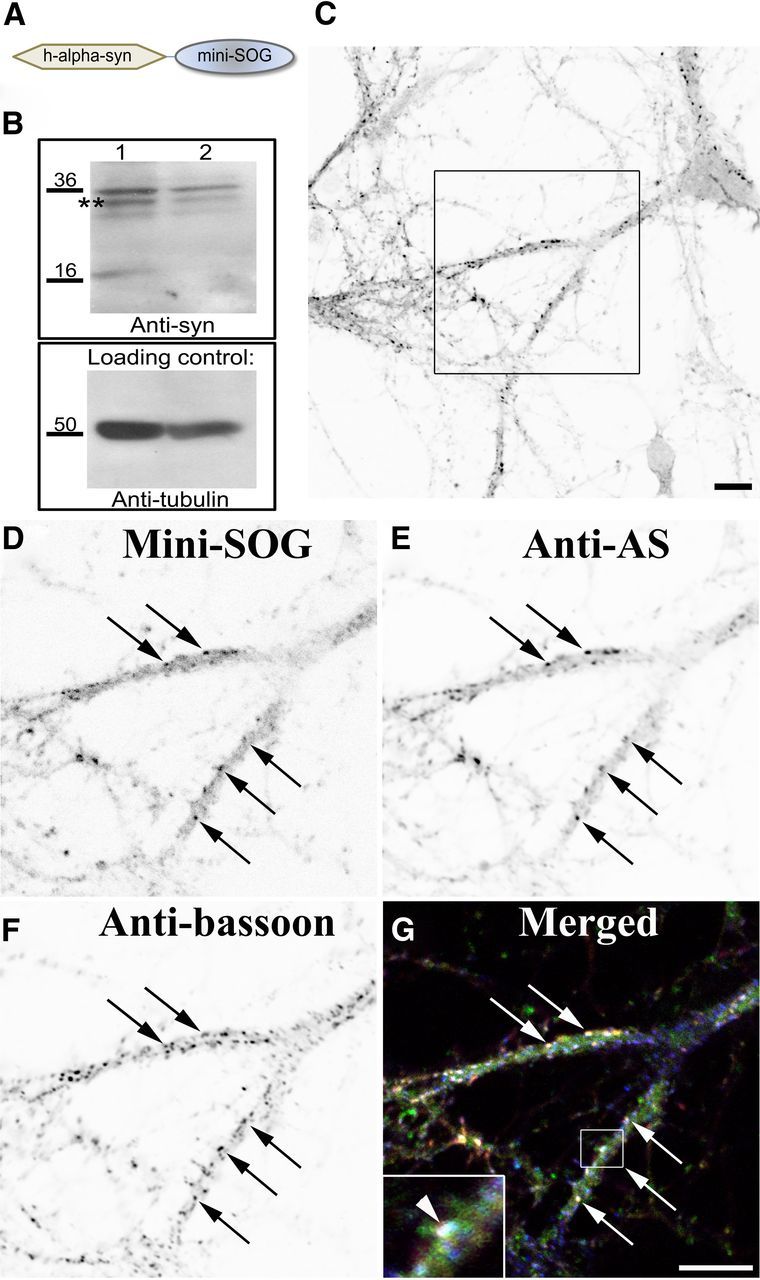
Recombinant AS-MiniSOG localizes in presynaptic terminals. A, The MiniSOG tag was fused to the carboxyl terminus of human α-synuclein. B, Whole-cell lysates from rat cortical neurons expressing the fusion proteins WT-human AS-MiniSOG (lane 1) and A53T-humanAS-MiniSOG (lane 2) were Western blotted with an antibody specific for AS and then stripped and reprobed against anti-α tubulin as a loading control. C, Rat cortical neurons were transfected with human-AS-MiniSOG, cultured for 21 days, and immunostained with an antibody specific to AS. Images represent single scans by confocal microscopy and are displayed with an inverted color table (black is highest signal, white is no signal, i.e., background). Inset in C is shown at higher magnification. D–G, The MiniSOG fluorescence (D, indicated by black arrows) overlaps with the AS signal (E) and the presynaptic marker Bassoon (F), as observed in the merged image (G, white arrows). MiniSOG is displayed in green, AS labeling in red, and Bassoon in blue. White indicates the overlap of all three channels (displayed at higher magnification in the inset in G, white arrowheads). Scale bars, 10 μm.
Figure 4.
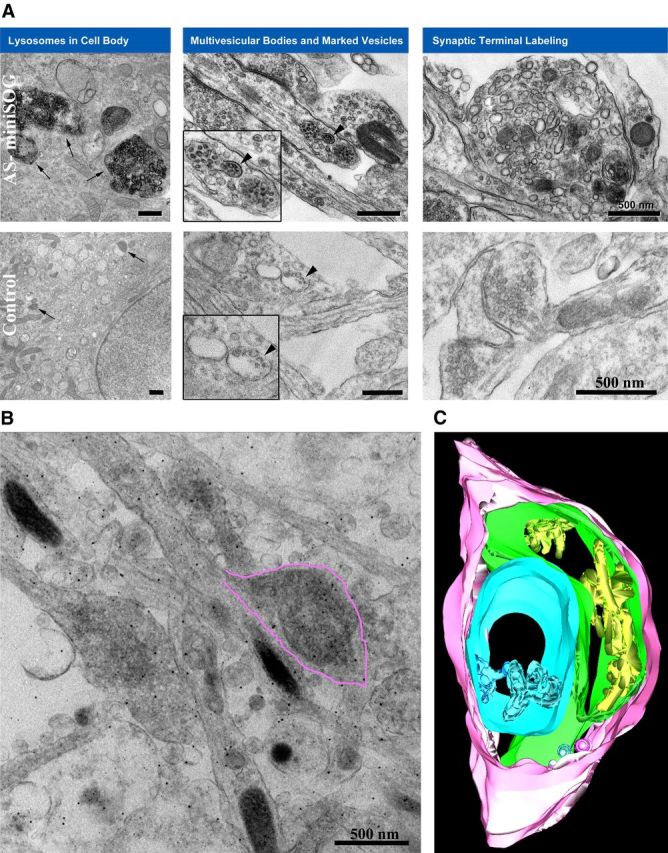
MiniSOG labeling illuminates the trafficking and degradation of AS. A, The electron micrographs show AS-MiniSOG overexpressing neurons processed for photo-oxidation of DAB into an osmiophilic, electron-dense reaction product resolvable by EM. The darker intensity of the signal reflects the specific AS labeling. The cellular distribution of AS (top row) appears in presynaptic terminals, specifically in synaptic vesicle membranes and in larger tubular membranous structures (A, top row, right), similar to what is observed with the 4Cys labeling in Figure 2. In addition, we observed an accumulation in the membrane of intraluminar vesicles of multivesicular bodies (A, top row, middle, black arrowheads, inset at higher magnification), as well as aggregations of the protein at the cell body of transfected neuronal cells (A, top row, left, black arrows). Controls represent nontransfected rat cortical neurons processed in parallel for photo-oxidation and did not snow this pattern of labeling (bottom row, right, normal presynaptic terminal, arrowheads in the middle panel point at a multivesicular body, with inset at higher magnification and arrows in the left panel pointing at lysosomes in a cell body). Scale bars, 500 nm. B, Zero tilt image from tomogram of cultured neurons expressing AS-MiniSOG processed for photo-oxidation. Extensive stained membranous elements are observed in the presynaptic terminal (highlighted in pink to match the 3D model shown in C). C, Three-dimensional model from EM tomogram of same area shows plasma membrane (pink) and contiguous membranes in three different colors (blue, green, and yellow).
Enlarged presynaptic terminals in AS transgenic mice show extensive membranous structures and tubulovesicular architecture
To test whether in vivo AS overexpression is associated with intracellular membrane systems, we analyzed transgenic mice described previously (Masliah et al., 2000; Rockenstein et al., 2002) that overexpress human wild-type AS ∼1.5-fold over the endogenous level. We confirmed the overexpression of AS by immunohistochemistry using a human AS-specific antibody. Strong punctate labeling was observed in the hippocampus and the neocortex of AS transgenic mice (Fig. 5), while no human AS immunoreactivity was observed in nontransgenic controls (littermates). We considered these areas our “hot spots” and analyzed them at high EM resolution using state-of-the-art transmission electron microscopy (TEM), SBEM, and tomography. The combination of these advanced EM tools allowed us to analyze the 3D architecture of several brain areas of the transgenic animals with unprecedented detail but over a large expanse. The large datasets obtained are available for visualization and download from the Cell Centered Database both as full resolution images and 3D movies.
Figure 5.

Immunoreactivity in transgenic mice overexpressing human α-synuclein. Immunohistochemistry using the human AS antibody reveals strong punctate labeling in the hippocampus, both in CA3 (stratum lucidum, SLu) and CA1 (stratum radiatum, Rad), and in the neocortex of transgenic mice. In nontransgenic controls (littermates), no human AS immunoreactivity was observed.
EM analysis in hippocampus CA3 (stratum lucidum) revealed perturbed intracellular membrane systems that appeared as multiple-layered membrane structures in presynaptic terminals of AS Tg mice (Fig. 6), while the non-Tg littermate controls did not show this pattern. These onion-like membrane convolutions were highly represented in the mossy fiber terminals; they often wrapped around presynaptic vesicles within the terminals (Fig. 6A,B), and originated from invaginations of the plasma membrane (Fig. 6C). Interestingly, analysis of hippocampus CA1 (stratum radiatum) revealed the presence of enlarged nerve terminals in AS-Tg mice with extensive membranous networks forming a dense tubulovesicular architecture with contiguous membrane segments stacked on each other (Fig. 7). The non-Tg littermate controls did not show this pattern. Similar atypical synaptic terminals were found in other brain areas, including neocortex and substantia nigra (Fig. 8). In these areas, the enlarged terminals form synapses (evidenced by postsynaptic densities and accumulation of presynaptic vesicles) and presented with extensive endomembrane network that occupied the majority of the terminal volume.
Figure 6.
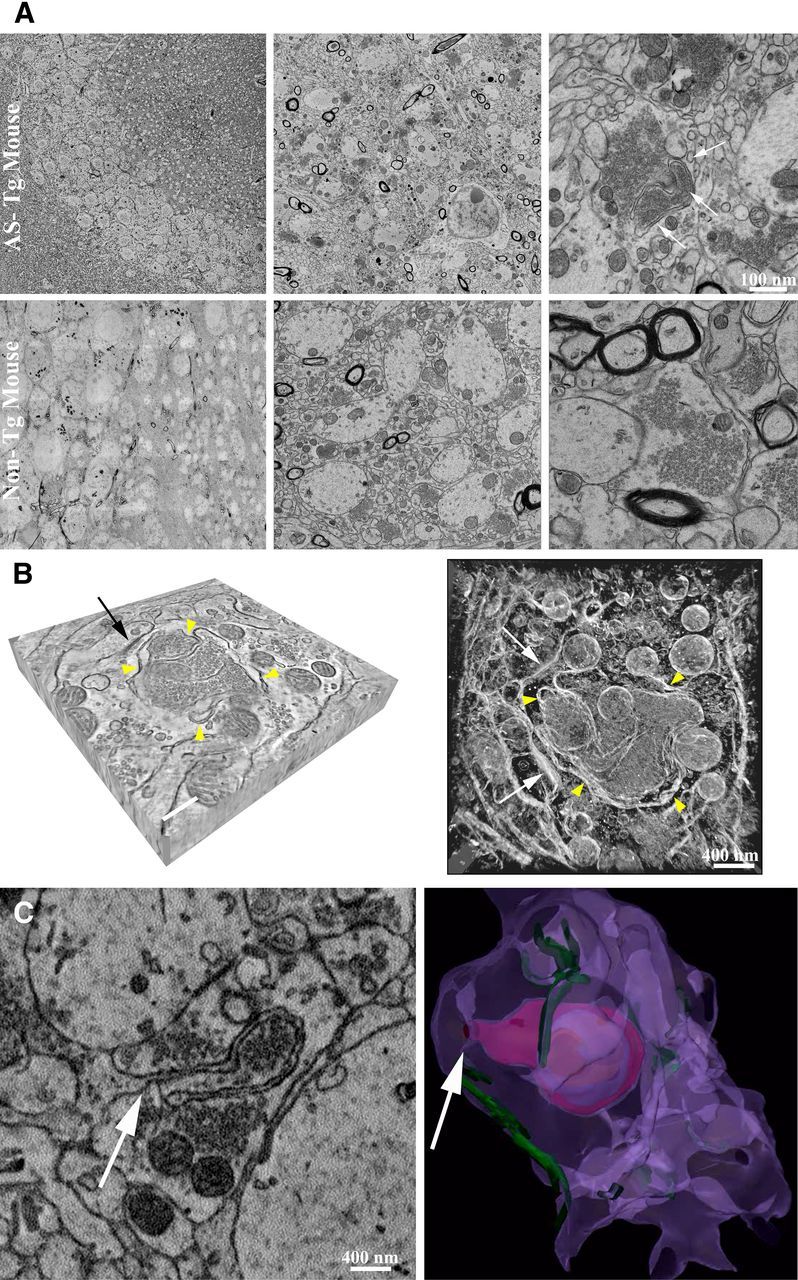
Presynaptic terminals in hippocampus CA3 of AS transgenic mice show convoluted membranous structures. A, EM analysis using TEM in hippocampus CA3 (stratum lucidum) revealed multiple-layered membrane structures in presynaptic terminals of Tg mice overexpressing human AS (top row, white arrows). The non-Tg controls (bottom row) did not show this pattern. B, Analysis of tomographic reconstruction showed that the presynaptic terminal was filled with vesicles and frequent membranous elements around them (left, yellow arrowheads in a tomographic slice). Synapse is indicated by a black arrow. Scale bar, 300 nm. On the right, maximum intensity projection of tomographic reconstruction of the same presynaptic terminal is displayed with an inverted color table (white is highest EM signal, black is no signal). Yellow arrowheads point at membrane convolutions inside the terminal. Two synapses are indicated by the white arrows. C, Using SBEM we observed that the membrane convolutions originate from invaginations of the plasma membrane, as indicated by the white arrow. Three-dimensional volume segmentation of SBEM datasets (image on the right) shows these membrane perturbations inside the complete terminal (plasma membrane is in purple, postsynaptic densities in green, membrane convolutions in magenta, blue, and orange).
Figure 7.

Presynaptic terminals in hippocampus CA1 of AS transgenic mice show extensive tubulovesicular architecture. Comparison of presynaptic nerve terminals found in the hippocampus CA1 (stratum radiatum) in non-Tg (top left, reconstruction in light blue with synapses in yellow) and AS-Tg mice (top right, reconstruction in green with synapses in red). Enlarged nerve terminals of AS-Tg mice show extensive membranous networks (middle row, white arrowheads) and form synapses (evidenced by postsynaptic densities and accumulation of presynaptic vesicles). Both EM analysis using SBEM (top two rows) and tomography (bottom row) revealed large presynaptic nerve terminals filled with tubulovesicular structures similar to what we observed in vitro. Segmentation of contiguous membrane segments from tomographic reconstruction is shown in three different colors (inset in bottom row, right, corresponding to the area indicated by arrowheads).
Figure 8.
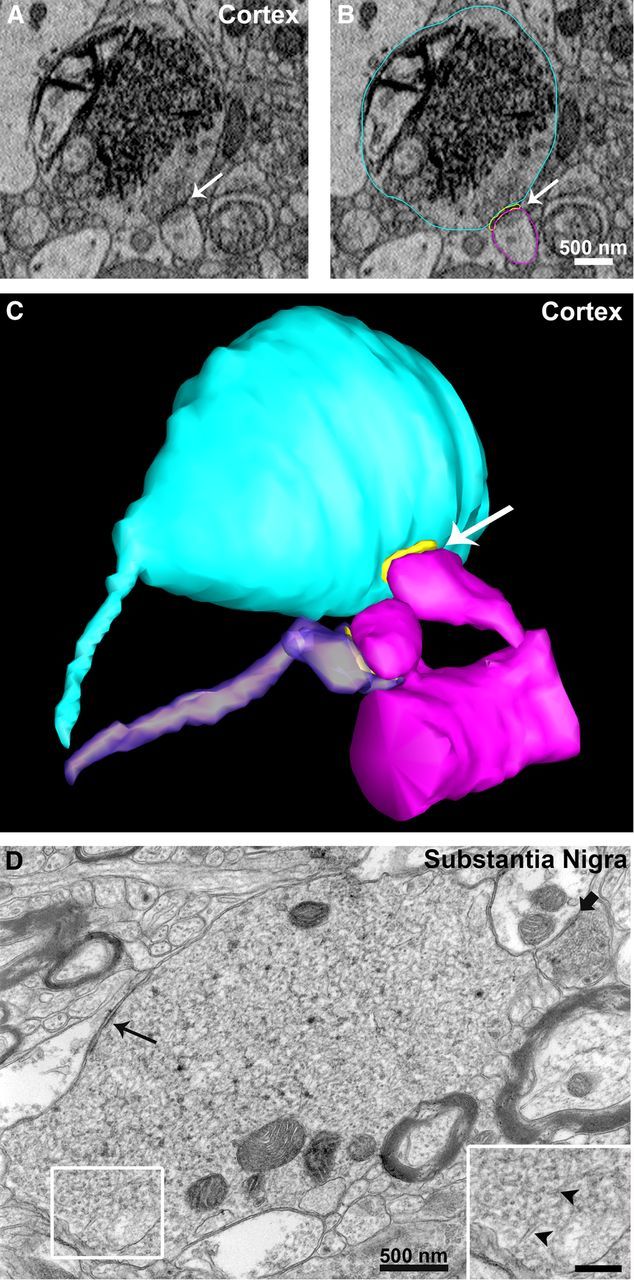
Atypical presynaptic terminals in neocortex and substantia nigra of AS transgenic mice. A–C, Using SBEM we observed, in the neocortex of AS transgenic mice, enlarged nerve terminals massively filled with an endomembrane network formed of tubulovesicular structures similar to those observed in hippocampus CA1. In C, three-dimensional volume segmentation shows the enlarged terminal (light blue) forming a synapse (postsynaptic density is in yellow, indicated by the white arrow) with a dendritic spine (magenta) compared to a normal terminal (purple) forming a synapse with an adjacent spine on the same dendrite. D, Similarly, TEM analysis of thin sections from substantia nigra of AS transgenic mice revealed endomembrane perturbations in presynaptic terminals characterized by extensive tubulovesicular structures (arrowheads in inset at higher magnification, scale bar, 250 nm). Slim black arrow points at a synapse formed by the enlarged terminal, while the shorter black arrow points at a synapse formed by a normal terminal.
Discussion
In the current study we investigated, at high resolution by 3D electron microscopy, the effects of human wild-type AS overexpression both in vitro and in vivo that recapitulates the clinical circumstances of increased expression of α-synuclein associated with the pathogenesis of sporadic as well as familial PD (Braak et al., 2003; Singleton et al., 2003; Chartier-Harlin et al., 2004). The successful application of both the 4Cys and MiniSOG probes allowed us to precisely determine the intracellular membrane systems with which AS associates when overexpressed in cultured neurons. Based on our observations, AS binds to membranes and directly affects membrane dynamics, suggesting a dysfunctional interaction of overexpressed AS with intracellular membranes. Similarly, we confirmed accumulation of AS in membrane systems of presynaptic terminals in vivo, and showed for the first time at high resolution the existence of profound perturbations of the membrane architecture of presynaptic terminals in several brain areas, including hippocampus, substantia nigra, and neocortex of transgenic mice, overexpressing human wild-type AS. These observations suggest a correlation between structural abnormalities in nerve terminals and synaptic dysfunction as the substrate for neurodegeneration in PD. Consistent with our identification of alterations to a presynaptic endomembrane system associated with AS overexpression, recent reports have suggested that similar overexpression of AS inhibits neurotransmitter release, particularly modulating the size and dynamics of synaptic vesicle pools. Nemani et al. (2010) reported a reduction in synaptic vesicle clustering and the size of the recycling pool in transgenic mice expressing AS ∼3-fold over endogenous AS. Recently, Scott and Roy (2012) reported that excessive AS inhibits intersynaptic vesicle trafficking, influencing the overall size of recycling pools at synapses and their release probability. Thus, it is conceivable that the changes in membrane architecture of the presynaptic terminals we observed in our study might perturb the normal distribution of synaptic vesicles and alterations in vesicle recycling machinery, therefore compromising delivery of vesicles to release sites and causing impairment of neurotransmitter release. Conversely, deletion of all three members of the synuclein family (α, β, and γ) has been reported to have a dramatic effect on synapse structure and decreased presynaptic terminal size both in the CA1 and CA3 subfields of the hippocampus (Greten-Harrison et al., 2010), suggesting a possible function of synucleins in modulating the flow of the synaptic membrane. This is in line with our observation that AS overexpression is associated with hypertrophy of a complex endomembrane system of the presynaptic terminal.
Recombinant AS is described as having preferential affinity for negatively charged lipids, such as phosphatidylserine (Davidson et al., 1998) or 50% phosphatidylglycerol/50% phosphatidylethanolamine (Zakharov et al., 2007). AS was shown to have a stabilizing effect on lipid packing in small vesicles (Nuscher et al., 2004; Kamp and Beyer, 2006) and in regulating the number of presynaptic vesicles (Murphy et al., 2000), which suggests that this could be a physiological function of AS. Overall, AS membrane interactions appear to be physiologically important, and the lipid composition of the cellular membranes may affect these interactions in vivo, which could explain the various membrane architectures we observed in different brain areas/neuronal cell types in AS transgenic mice.
Our observations also support a role of AS in synaptic membrane biogenesis and vesicle turnover. In fact, the most striking association of AS is with a complex network or stack of tubules similar to structures previously determined to play a role in recycling of synaptic proteins and production of synaptic vesicles (Heuser and Reese, 1973; Lenzi et al., 2002). The labeling of these core membrane structures of the nerve terminal and nearby MVBs, as well as labeling of interlinking vesicles, argues strongly for a role of this presynaptic membranous organelle system and AS in retrieval and delivery of presynaptic constituents to the cell's lysosomal machinery for degradation and thus implicates perturbations in this pathway in the pathogenesis of PD and other synucleinopathies.
In addition, the accumulation of AS in the membrane of intraluminar vesicles of MVBs suggests the possibility of an exosome-associated release of AS as a plausible mechanism of propagation of AS pathology. The notion that AS can be transferred from cell to cell comes from observations in PD patients that underwent transplantation as part of the disease treatment and showed host-to-graft transmission of the pathology more than a decade after transplantation. Similar findings were observed also in vitro and in vivo in transgenic mice (Danzer et al., 2009; Desplats et al., 2009; Hansen et al., 2011; Volpicelli-Daley et al., 2011; Luk et al., 2012). However, the mechanism of secretion of α-synuclein remains unclear. Possible routes have been proposed (Vekrellis et al., 2011; Hansen and Li, 2012), one of them being an exosome-mediated secretion (Danzer et al., 2012). Our novel observations would therefore support this hypothesis. Exosomes are typically expelled in the extracellular space upon fusion of the MVB to the plasma membrane, suggesting exosomal release as a way of secreting membrane proteins meant to be discarded or to be passed to other cells. In the CNS, a role of exosomes has been associated with secretion of other proteins such as prion and amyloid precursor proteins (Fevrier et al., 2005; Rajendran et al., 2006). Also, movement of MVBs to synapses, fusion to the plasma membrane, and exosome secretion have been linked to synaptic plasticity as a way of local redistribution of surface receptors at synapses undergoing plastic changes (for review, see Smalheiser, 2007; Chivet et al., 2012). Although the neuron-to-neuron secretion of α-synuclein at the synapse has been recently proposed in vivo (Luk et al., 2012), additional studies are needed to understand the precise pathways involved in AS transcellular spreading.
Based on our observations, we hypothesized a working model where intracellular components associated with protein degradation are decorated by overexpressed AS (Fig. 9). The protein is transported from the cell body to the nerve terminals by axonal transport in a soluble, disorganized form and associates with membranes in the presynaptic terminal where synaptic vesicle recycling occurs, causing profound membrane perturbations. AS accumulates in MVBs, which are primary lysosomes without hydrolases, possibly perturbing the normal function of MVBs to degrade proteins. These primary lysosomes are trafficked to the cell body by retrograde transport machinery, where they presumably become full-fledged lysosomes containing AS. Overexpressed AS is seen in all elements of this protein degradation pathway, suggesting that this system represents the main target in PD. In PD patients, besides the dramatic loss of dopaminergic neurons, the overall rate of neuronal death is very low, showing slow progression over many years. These observations suggest a mechanism distinct from classical apoptosis and are in line with our hypothesis that the PD pathogenesis is rooted in disruption of the complicated intracellular machinery responsible for protein degradation. Many of the other PD-associated mutant proteins lead to visible changes in the function or structure of the protein degradation pathway, reinforcing the notion that PD develops due to disturbances to or overburdening of subcellular mechanisms responsible for protein degradation.
Figure 9.

Overexpressed AS highlights the intracellular system associated with protein degradation. Soluble AS protein is transported from the cell body to nerve terminals by axonal transport and associates with intracellular membranes in the presynaptic terminal, causing profound membrane perturbations. Consequently, accumulation of AS in components of the protein degradation system such as MVBs and lysosomes might interfere with their normal function to degrade proteins.
Finally, despite considerable research efforts, knowledge of the physiological function of AS is still lacking, and this represents a puzzling question in the field that requires urgent investigation to understand how deviations in its normal activity may play a role in the various forms of neurodegeneration.
Our work supports the idea that a primary role of both the alpha and gamma synucleins (Nguyen et al., 2011) is to associate with subcellular components early in the sorting process, thereby highlighting their involvement in the crucial steps of membrane structure modification and protein collection for movement into the first stages of the degradation process. It appears that the synucleins accompany these products through their journey to proteolysis, also becoming irreversibly altered along the way.
Notes
Supplemental material for this article is available at http://ccdb.ucsd.edu/index.shtm. The microscopy data have been uploaded in the Cell Centered Database (CCDB) and are available for visualization and download for reanalysis both as full resolution images and 3D movies. The CCDB is a web accessible database for high resolution 2D, 3D, and 4D data from light and electron microscopy, including correlated imaging. This material has not been peer reviewed.
Footnotes
This project involved use of facilities supported by grants from the NIH National Center for Research Resources (5P41RR004050-24) and the National Institute of General Medical Sciences (8 P41 GM103412-24) from the National Institutes of Health to M.H.E. It also benefitted from support to R.Y. Tsien and M.H.E. (R01 GM086197-05) for development of new probes for CLEM. The Parkinson's-specific work was supported by awards from the Branfman Family Foundation (M.H.E., J.M.G.) and a subcontract to M.H.E. from the Institute for Systems Biology as part of the Luxembourg Center for Systems Biomedicine research program on neurodegenerative disease. Dr. E. Masliah was supported by NIH Grants AG184440 and AG022074. We thank Dr. R.Y. Tsien for helpful discussions; Dr. Guido Gaietta, Tom Deerinck, Edward Rockenstein, and Wendy Woods for outstanding technical assistance, and Dr. Andrew Noske for assistance with illustrations. This work is also linked to activities of a consortium of researchers working with the Luxembourg Center for Systems Biomedicine on Parkinson's disease.
References
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chivet M, Hemming F, Pernet-Gallay K, Fraboulet S, Sadoul R. Emerging role of neuronal exosomes in the central nervous system. Front Physiol. 2012;3:145. doi: 10.3389/fphys.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem. 2009;111:192–203. doi: 10.1111/j.1471-4159.2009.06324.x. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Ruipérez V, López I, Villanueva J, Gutierrez LM, Davletov B. Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 2010;11:528–533. doi: 10.1038/embor.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Février B, Vilette D, Laude H, Raposo G. Exosomes: a bubble ride for prions? Traffic. 2005;6:10–17. doi: 10.1111/j.1600-0854.2004.00247.x. [DOI] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. Alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C, Li JY. Beyond alpha-synuclein transfer: pathology propagation in Parkinson's disease. Trends Mol Med. 2012;18:248–255. doi: 10.1016/j.molmed.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F, Beyer K. Binding of alpha-synuclein affects the lipid packing in bilayers of small vesicles. J Biol Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lawrence A, Bouwer JC, Perkins G, Ellisman MH. Transform-based backprojection for volume reconstruction of large format electron microscope tilt series. J Struct Biol. 2006;154:144–167. doi: 10.1016/j.jsb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi D, Crum J, Ellisman MH, Roberts WM. Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron. 2002;36:649–659. doi: 10.1016/s0896-6273(02)01025-5. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Murphy D, Rueter S, Trojanowski JQ, Lee V. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente-Soriano FJ, Yang Z, Davis CH, Bedont JL, Son JL, Wei JO, Buchman VL, Zack DJ, Vidal-Sanz M, Ellisman MH, Marsh-Armstrong N. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A. 2011;108:1176–1181. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuscher B, Kamp F, Mehnert T, Odoy S, Haass C, Kahle PJ, Beyer K. Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J Biol Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- Pandey N, Schmidt RE, Galvin JE. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp Neurol. 2006;197:515–520. doi: 10.1016/j.expneurol.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, Ye L, George JM. Epitope mapping and specificity of the anti-alpha-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett. 2003;349:133–135. doi: 10.1016/s0304-3940(03)00781-x. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Scott D, Roy S. Alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smalheiser NR. Exosomal transfer of proteins and RNAs at synapses in the nervous system. Biol Direct. 2007;2:35. doi: 10.1186/1745-6150-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–1025. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Woods W, Boettcher JM, Zhou DH, Kloepper KD, Hartman KL, Ladror DT, Qi Z, Rienstra CM, George JM. Conformation-specific binding of alpha-synuclein to novel protein partners detected by phage display and NMR spectroscopy. J Biol Chem. 2007;282:34555–34567. doi: 10.1074/jbc.M705283200. [DOI] [PubMed] [Google Scholar]
- Zakharov SD, Hulleman JD, Dutseva EA, Antonenko YN, Rochet JC, Cramer WA. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry. 2007;46:14369–14379. doi: 10.1021/bi701275p. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]