


Abstract
Synthetic biology has significantly advanced the design of synthetic control devices, gene circuits and networks that can reprogram mammalian cells in a trigger-inducible manner. Prokaryotic helix-turn-helix motifs have become the standard resource to design synthetic mammalian transcription factors that tune chimeric promoters in a small molecule-responsive manner. We have identified a family of Actinomycetes transcriptional repressor proteins showing a tandem TetR-family signature and have used a synthetic biology-inspired approach to reveal the potential control dynamics of these bi-partite regulators. Daisy-chain assembly of well-characterized prokaryotic repressor proteins such as TetR, ScbR, TtgR or VanR and fusion to either the Herpes simplex transactivation domain VP16 or the Krueppel-associated box domain (KRAB) of the human kox-1 gene resulted in synthetic bi- and even tri-partite mammalian transcription factors that could reversibly program their individual chimeric or hybrid promoters for trigger-adjustable transgene expression using tetracycline (TET), γ-butyrolactones, phloretin and vanillic acid. Detailed characterization of the bi-partite ScbR-TetR-VP16 (ST-TA) transcription factor revealed independent control of TET- and γ-butyrolactone-responsive promoters at high and double-pole double-throw (DPDT) relay switch qualities at low intracellular concentrations. Similar to electromagnetically operated mechanical DPDT relay switches that control two electric circuits by a fully isolated low-power signal, TET programs ST-TA to progressively switch from TetR-specific promoter-driven expression of transgene one to ScbR-specific promoter-driven transcription of transgene two while ST-TA flips back to exclusive transgene 1 expression in the absence of the trigger antibiotic. We suggest that natural repressors and activators with tandem TetR-family signatures may also provide independent as well as DPDT-mediated control of two sets of transgenes in bacteria, and that their synthetic transcription-factor analogs may enable the design of compact therapeutic gene circuits for gene and cell-based therapies.
INTRODUCTION
During the past 20 years, bioengineers have constructed an impressive portfolio of basic mammalian transcription-control devices (1–3) that are based on the same design principles as the inaugural gene switch known as the tetracycline- (TET) responsive expression or TET system (4). The TET system’s blueprint consists of a bacterial repressor protein (TetR), managing TET resistance in Escherichia coli, that is fused to the Herpes simplex transactivation domain VP16 to form a synthetic TET-dependent transactivator (tTA; TetR-VP16) or to the Krueppel-associated box (KRAB) domain of the human kox-1 gene to form a TET-dependent transsilencer (tTS; TetR-KRAB (5). The tTS binds to TetR-specific operator sites (tetO) downstream of a simian virus 40 promoter (PSV40) and represses PSV40-driven transgene expression in a TET-responsive manner (5). Likewise, tTA binds to a chimeric TET-responsive promoter (PhCMV*−1), assembled by placing a heptameric tetO7 operator module immediately upstream of a minimal version of the human cytomegalovirus immediate early promoter (PhCMVmin), which controls transgene expression in a TET-responsive manner (4). TET system design variants using identical VP16/PhCMVmin modules, but repressor/operator componentry of different bacteria are responsive to erythromycin (6), tryptophan (7), uric acid (8) as well as γ-butyrolactone (9,10), phloretin (11) and vanillic acid (3,12). All of these mammalian transgene-control systems are compatible with each other and can be used for independent control of different transgenes (6) or functionally interconnected to provide higher-order switching behavior such as regulatory cascades (13–15), epigenetic toggle switches (16) hysteretic circuits (17), band-pass filters (18,19), tunable oscillators (20) and single-cell biocomputers (2). Trigger-controlled transcription-tuning devices have also been successfully used for the design of drug circuits (21), and there are increasing efforts across the synthetic biology community to develop control devices for clinical applications (22–27).
TetR family members are characterized by a conserved N-terminal DNA-binding region (28) forming the DNA-binding helix-turn-helix (HTH) DNA-binding motif that is found in most prokaryotic transcription factors (28,29) and a non-conserved C-terminal domain (28) that is responsible for dimerization and ligand recognition (28). TetR has been successfully mutated to expand its dynamic range and functional space (30–34). For example, a few point mutations can reverse TetR’s DNA-binding function so that the repressor binds instead of dissociates in the presence of the trigger molecule (30,32,35), and engineering of TetR enables the repressor to recognize new trigger molecules (31) or operator sites (36). However, undesired heterodimerization of functionally diversified TetR variants has limited their use for independent control of different transgenes (34). To prevent heterodimerization, either the dimerization domain could be modified (37) or monomeric single-chained TetR variants have been constructed by assembling two TetR subunits head-to-tail via a flexible hinge region (33,34).
Bipartite DNA-binding domains have also been identified in nature. For example, several mammalian differentiation factors such as Oct-1 and Oct-2 contain POU domains with two independent DNA-binding motifs (29,38), members of the Pax and Prd family of developmental transcription factors harbor two-part DNA motifs and the Tc1/mariner superfamily of transposable elements include split DNA-binding modules (39).
Despite their recurrent emergence across different kingdoms suggesting that bipartite DNA-binding proteins play an important role in living systems, most of their regulation dynamics remain elusive. Using a synthetic biology-inspired reverse engineering approach, we designed artificial TetR and GntR family-derived bipartite and tripartite transcription factors, which, besides showing independent regulation of different sets of transgenes, exhibited control dynamics reminiscent of double-pole double-throw (DPDT) relay switches. DPDT relay switches are electromagnetically operated mechanical switches that control one or several electric circuits by a fully isolated ‘low-power’ signal. DPDT relays were extensively used in telephone exchanges and early computers to perform logic operations. Likewise, their genetic counterparts may enable an organism to switch between global metabolic networks and could provide logic operations that interface prosthetic networks with endogenous metabolic circuits in future gene- and cell-based therapies.
MATERIALS AND METHODS
Plasmid design
Table 1 lists all plasmids used in this study and provides detailed information about their construction. All relevant genetic components have been confirmed by sequencing (Microsynth, Balgach, Switzerland).
Table 1.
Plasmids and oligonucleotides designed and used in this study
| Plasmid | Description | Reference |
|---|---|---|
| pZeoSV2(+) | Constitutive mammalian expression vector encoding the zeocin resistance gene (PhCMV-zeo-pA) | Invitrogen, CA |
| pDA43 | TET-responsive GLuc expression vector (PhCMV*−1-GLuc-pA) | (40) |
| pMF111 | TET-responsive SEAP expression vector (PhCMV*−1-SEAP-pA) | (41) |
| pMF205 | Constitutive tTS expression vector (PSV40-tTS-pA) | (42) |
| pMG10 | Phloretin-responsive SEAP expression vector (PTtgR1-SEAP-pA) | (11) |
| pMG11 | Constitutive TtgA1 expression vector (PSV40-TtgA1-pA) | (11) |
| pMG250 | Constitutive VanA1 expression vector (PSV40-VanA1-pA) | (12) |
| pMG252 | Vanillic acid-responsive SEAP expression vector (P1VanO2-SEAP-pA) | (12) |
| pSAM200 | Constitutive tTA expression vector (PSV40-tTA-pA) | (41) |
| pWW122 | Constitutive SCA expression vector (PSV40-SCA-pA) | (9) |
| pWW124 | SCB1-responsive SEAP expression vector (PSPA-SEAP-pA) | (9) |
| pSEAP2-Control | Constitutive SEAP expression vector (PSV40-SEAP-pA) | Clontech, CA |
| pAS1 | Low-level constitutive ST-TA expression vector (PhCMVmin-ST-TA-pA). PhCMVmin was PCR-amplified from pWW124 using oligonucleotides OAS1 (5′-GCAGCTAGCAGGTCGAGCTCGGTACCCGG-3′) and OAS2 (5′-CAGGCGGCCGCGCTGACTCTAGAGGATCCCCG-3′), restricted with NheI/NotI and cloned into the corresponding sites (NheI/NotI) of pMX1. | This work |
| pMX1 | Constitutive ST-TA expression vector (PSV40-ST-TA-pA). ScbR was PCR-amplified from pWW122 using oligonucleotides OMF1 (5′-GGTCCCGGATCGAATTGCGGCCGCTAATTC-3′) and OMF2 (5′-TGAATTCTACCCGCGCGGCTGTACGCGGA-3′), restricted with EcoRI/NotI and cloned into the corresponding sites (EcoRI/NotI) of pSAM200. | This work |
| pMX5 | Constitutive TS-TA expression vector (PSV40-TetR-Kozak-ScbR-VP16-pA). TetR was PCR-amplified from pSAM200 using oligonucleotides OMF1 (5′-GGTCCCGGATCGAATTGCGGCCGCTAATTC-3′) and OMF2 (5′-TGAATTCTACCCGCGCGGCTGTACGCGGA-3′), restricted with EcoRI/NotI and cloned into the corresponding sites (EcoRI/NotI) of pWW122. | This work |
| pMX6 | Constitutive TV-TA expression vector (PSV40-TetR-VanR-VP16-pA). VanR was PCR-amplified from pMG250 using oligonucleotides OPS213 (5′-GGGTAGAATTCATATGGACATGCCGCGCATAAAGC-3′) and OPS545 (5′-TTTTTCGTACGCGCGCGGCTGTACGCGGAGTCGGCGCGAATGCTCCACGCCGCGCCCAGCGGCGC-3′), restricted with EcoRI/BssHII and cloned into the corresponding sites (EcoRI/BssHII) of pMX10. | This work |
| pMX8 | ST-TA/TS-TA-specific TET- and SCB1-responsive hybrid promoter-driven SEAP expression vector (PST-TA/TS-TA1-SEAP-pA; PST-TA/TS-TA1, tetO-21bp-OPapRI-PhCMVmin). | This work |
| PSPA was PCR-amplified from pWW124 using oligonucleotides OMX26 (5′-TAGACGTCTTTACCACTCC′CTATCAGTGATAGAGAAAAGTGAAAGTCGGCCATTGACAAACCGACCGT-3′) and OMX24 (5′-CTTGAGCACATAGCCTGGACCGTTTCCGTA-3′), restricted with AatII/EcoRI and cloned into the corresponding sites (AatII/EcoRI) of pWW124. | ||
| pMX9 | ST-TA/TS-TA-specific TET- and SCB1-responsive hybrid promoter-driven SEAP expression vector (PST-TA/TS-TA2-SEAP-pA; PST-TA/TS-TA2, tetO-21bp-tetO-21bp-OPapRI-PhCMVmin). | This work |
| PSPA was PCR-amplified from pWW124 using oligonucleotides OMX27 (5′-TAGACGTCCTCTATCAGTGATAGAGAAAAGTGAAAGTCGAGCTCTATCAGTGATAGAGAAAAGTGAAAGTCGGCCATTGACAAACCGACC-3′) and OMX24 (5′-CTTGAGCACATAGCCTGGACCGTTTCCGTA-3′), restricted with AatII/EcoRI and cloned into the corresponding sites (AatII/EcoRI) of pWW124. | ||
| pMX10 | Constitutive TS-TA expression vector (PSV40-TetR-ScbR-VP16-pA). | This work |
| ScbR was PCR-amplified from pMX5 using oligonucleotides OMX10a (5′-AGAATTCGGGGCATGGCCAAGCAGGACCGG-3′) and OMX10b (5′-TTGGCGCGCGGCTGTACGCGGAGTCCTTCC-3′) to remove the Kozak sequence, restricted with EcoRI/BssHII and cloned into the corresponding sites (EcoRI/BssHII) of pMX5. | ||
| pMX25 | Constitutive VT-TA expression vector (PSV40-VanR-TetR-VP16-pA). | This work |
| VanR was PCR-amplified from pMG250 using oligonucleotides OMX12 (5′-CCCGGATCGGAATTGCGGCCGCTAATTCATATGGACATGCCGCGCATAAAGCCGGG-3′) and OMX13 (5′-GCCCCGAATTCTACCCGCGCGGCTGTACGCGGAGTCGGCGCGAATGCTCCACGCCG-3′), restricted with EcoRI/NotI and cloned into the corresponding sites (EcoRI/NotI) of pMX1. | ||
| pMX27 | Constitutive PTS-TA expression vector (PSV40-TtgR-TetR-ScbR-VP16-pA). | This work |
| TtgR-TetR was PCR-amplified from pMX29 using oligonucleotides OMF1 (5′-GGTCCCGGATCGAATTGCGGCCGCTAATTC-3′) and OMX14 (5′-GCCCCAATTGTACCCGCGCGGCTGTACGCGGACCCACTTTCACATTTAAGTTGTT-3′), restricted with NotI/MfeI and cloned into the compatible sites (NotI/EcoRI) of pMX10. | ||
| pMX29 | Constitutive PT-TA expression vector (PSV40-TtgR-TetR-pA). | This work |
| TtgR was PCR-amplified from pMG11 using oligonucleotides OMF1 (5′-GGTCCCGGATCGAATTGCGGCCGCTAATTC-3′) and OMF2 (5′-TGAATTCTACCCGCGCGGCTGTACGCGGA-3′), restricted with EcoRI/NotI and cloned into the corresponding sites (EcoRI/NotI) of pMX1. | ||
| pMX31 | Constitutive SP-TA expression vector (PSV40-ScbR-TtgR-pA); | This work |
| TtgR was PCR-amplified from pMG11 using oligonucleotides OMX28 (5′-GGGTAGAATTCATATGGTCCGTCGAACCAAAGAAGA-3′) and OMX29 (5′-TTTTTCGTACGCGCGCGGCTGTACGCGGATTTGCGCAGAGCCGGGCTCAAGCG-3′), restricted with EcoRI/BssHII and cloned into the corresponding sites (EcoRI/BssHII) of pMX1. | ||
| pMX63 | Constitutive ST-TS expression vector (PSV40-ScbR-TetR-KRAB-pA). | This work |
| KRAB was excised from pMF205 using BssHII/BamHI and ligated into the corresponding sites (BssHII/BamHI) of pMX1. | ||
| pMX64 | Constitutive TS-TS expression vector (PSV40-TetR-ScbR-KRAB-pA). | This work |
| KRAB was excised from pMF205 using BssHII/BamHI and ligated into the corresponding sites (BssHII/BamHI) of pMX10. | ||
| pMX65 | SCB1-inducible SEAP expression vector (PSPS-SEAP-pA; PSPS, PSV40- OPapRI). | This work |
| OPapRI-SEAP was PCR-amplified from pSEAP2-Control using oligonucleotides OMX75 (5′- GTAATAAGCTTGAGGCCATTGACAAACCGACCGTGCCGTTTTTTTCCTGCAGGCCACCATGCTGCTGCTGCTGCTGC TGCTGGG-3′) and OMX24 (5′-CTTGAGCACATAGCCTGGACCGTTTCCGTA-3′), restricted with HindIII/NdeI and cloned into the corresponding sites (HindIII/NdeI) of pSEAP2-Control. | ||
| pMX67 | TET-inducible SEAP expression vector (PtTSON2-SEAP-pA; PtTSON2, PSV40- tetO2). | This work |
| tetO2-SEAP was PCR-amplified from pSEAP2-Control using oligonucleotides OMX77 (5′- GTAATAAGCTTTCTATCAGTGATAGAGAAAAGTGAAAGTCGAGCTCTATCAGTGATAGCCTGCAGGCCACCATGCTGCTGCTGCTGCTGCTGCTGGG-3′) and OMX24 (5′-CTTGAGCACATAGCCTGGACCGTTTCCGTA-3′), restricted with HindIII/NdeI and cloned into the corresponding sites (HindIII/NdeI) of pSEAP2-Control. | ||
| pFS1 | Constitutive TP-TA expression vector (PSV40- TetR-TtgR-pA). | This work |
| TtgR was excised from pMX31 using EcoRI/BssHII and ligated into the corresponding sites (EcoRI/BssHII) of pMX10. |
GLuc, Gaussia princeps luciferase; KRAB, Krueppel-associated box domain of the human kox-1 gene; OPapRI, SCA-specific operator; OTtgR, TtgA1-specific operator; pA, polyadenylation site; PhCMV, human cytomegalovirus immediate early promoter; PhCMV*−1, TET-responsive promoter (tetO7-PhCMVmin); PhCMVmin, minimal version of PhCMV; PSPA, SCB1-responsive promoter (OPapRI-PhCMVmin); PST-TA/TS-TA1, TET- and SCB1-responsive promoter (tetO-21bp-OPapRI-PhCMVmin); PST-TA/TS-TA2, TET- and SCB1-responsive promoter (tetO-21bp-tetO-21bp-OPapRI-PhCMVmin); PSV40, simian virus 40 promoter; PSPS, SCB1-inducible promoter (PSV40-OPapRI); PtTSON2, TET-inducible promoter (PSV40-tetO2; tetO2, tetO-21bp-tetO); PT-TA, hybrid phloretin- and TET-dependent transactivator (TtgR-TetR-VP16); PTS-TA, hybrid phloretin-, TET and SCB1-dependent transactivator (TtgR-TetR-ScbR-VP16); PTtgR1, phloretin-responsive promoter (OTtgR-PhCMVmin); P1VanO2, vanillic acid-responsive promoter (vanO2-PhCMVmin); SCA, SCB1-dependent transactivator (ScbR-VP16); ScbR, Streptomyces coelicolor quorum-sensing receptor; SCB1, S.coelicolor butanolide 1, 2-(1'-hydroxy-6-methylheptyl)-3-(hydroxymethyl)-butanolide; SEAP, human placental secreted alkaline phosphatase; ST-TA, hybrid SCB1- and tetracycline-dependent transactivator (ScbR-TetR-VP16); ST-TS, hybrid SCB1- and TET-dependent transsilencer (ScbR-TetR-KRAB); SP-TA, hybrid SCB1- and phloretin dependent transactivator (ScbR-TtgR-VP16); tetO2, tetO7, tTA-specific operator sequence; TetR, E. coli Tn10-derived repressor of the TET resistance gene; TP-TA, hybrid phloretin- and TET-dependent transactivator (TetR-TtgR-VP16); TS-TA, hybrid TET- and SCB1-dependent transactivator (TetR-ScbR-VP16); TS-TS, hybrid TET- and SCB1-dependent transsilencer (TetR-ScbR-KRAB); tTA, TET-dependent transactivator (TetR-VP16); tTS, TET-dependent transsilencer (TetR-KRAB); TtgA1, phloretin-dependent transactivator (TtgR-VP16); TtgR, repressor of the Pseudomonas putida DOT-T1E ABC multi-drug efflux pump; TV-TA, hybrid TET- and vanillic acid-dependent transactivator (TetR-VanR-VP16); VanA1, vanillic acid-dependent transactivator (VanR-VP16); VanO2, VanR-specific operator; VanR, repressor of the Caulobacter crescentus VanAB gene cluster; VP16, Herpes simplex virus-derived transactivation domain; VT-TA, hybrid vanillic acid- and TET-dependent transactivator (VanR-TetR-VP16); zeo, zeocin resistance gene.
Bioinformatics
Protein sequence homology was analyzed using BLASTP (http://blast.ncbi.nlm.nih.gov). Structure homology and multiple sequence alignments were scored using the CLUSTAL W program (43) running on the ExPASY web server (http://swissmodel.expasy.org/). Batch secondary structure alignments were performed using the Jpred 3 secondary structure prediction server (44) (http://www.compbio.dundee.ac.uk/jpred).
Cell culture
Human bone marrow stromal cells (hMSCs) immortalized by expression of the human telomerase (hTERT; hMSC-TERT) catalytic subunit (45), as well as human embryonic kidney cells (HEK-293T, ATCC: CRL-11268), and its derivatives were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) (FCS; cat. no. 201F10, lot no. PE01026P, Bioconcept, Allschwil, Switzerland) and 1% (v/v) penicillin/streptomycin solution (Biowest, Nuaillé, France, cat. no. L0022-100, lot no. S09965L0022). All cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2. Viable cell numbers were determined using a Casy® Cell Counter and Analyser Model TT (Roche Diagnostics GmbH, Basel, Switzerland).
Transfections
hMSC-TERT were transfected using an optimized polyethyleneimine (PEI)-based protocol (17). In brief, a transfection solution containing 1.5 µl of PEI (PEI ‘Max’, stock solution 1 mg/ml in ddH2O; Polysciences, Eppelheim, Germany; cat. no. 24765-2) and 0.5 µg of plasmid DNA mixtures was incubated for 15 min at 22°C before it was added dropwise to 1 × 105 hMSC-TERT seeded per well of a 24-well plate 24 h before transfection. HEK-293T were transfected with 0.5 µg of DNA using an optimized calcium phosphate-based protocol. In brief, in each well of a 48-well plate, 25 000 HEK-293T cells were transfected using a DNA-Ca2PO4 precipitate that was prepared by mixing 12.5 µl of the plasmid-containing 0.5 M CaCl2 solution with 12.5 µl of a 2× HEPES-buffered saline (HBS) solution [50 mM HEPES, 280 mM NaCl, 1.5 mM Na2HPO4 (pH 7.1)] for 1 min at 22°C. After 3 h, the cells were washed once with phosphate-buffered saline (PBS) (Dulbecco’s phosphate-buffered saline; Invitrogen, Basel, Switzerland, cat. no. 21600-0069) and cultivated for 48 h in 250 µl of FCS-supplemented DMEM containing different concentrations or combinations of control compounds before reporter protein levels [human placental secreted alkaline phosphatase (SEAP), Gaussia luciferase (GLuc)] were profiled in the culture supernatant. For analysis of the DPDT relay switch expression reversibility, transfected cells were cultivated for 24 h in the presence of TET. Then, the culture medium was collected, the cells were washed twice with 500 µl of fresh medium and grown for another 24 h in fresh medium in the presence or absence of TET before SEAP and GLuc levels were profiled in the culture supernatant.
Stable cell line
The stable HEK-293T-derived cell line HEK-293ST-TA, transgenic for low constitutive PhCMVmin-driven ScbR-TetR-VP16 (ST-TA) expression, was constructed by co-transfection of 800 ng of pAS1 (PhCMVmin-ST-TA-pA) and 100 ng of pZeoSV2(+). After 14 days of selection in DMEM containing 100 μg/ml (w/v) zeocin (Invivogen, San Diego, CA, USA, cat. no. ant-zn-1), resistant cell clones were expanded and screened for functional ST-TA expression by transient co-transfection with pWW124 and pDA43 (ratio 1:1) followed by quantification of SEAP activity in the presence of different concentrations of TET. The best-in-class HEK-293ST-TA cell line was chosen for further experiments.
Quantification of reporter gene expression
SEAP was quantified as described before (40) using an EnVision 2104 multilabel reader (absorbance 405 nm; PerkinElmer, Waltham, MA, USA). GLuc was quantified using the BioLux® GLuc assay (E3300S, New England Biolabs) and the EnVision 2104 multilabel reader (maximum emission 482 nm). Reporter proteins were typically profiled 48 h after transfection. Each error bar is the mean ± standard deviation of the mean of SEAP or GLuc activity measured in triplicate from a representative experiment repeated three times.
Control compounds
TET (Sigma-Aldrich, cat. no. T7660), stock solution 1 mg/ml in ddH2O (41); SCB1 [2-(1′-hydroxy-6-methylheptyl)-3-(hydroxymethyl)-butanolide], stock solution 1 mg/ml in dimethyl sulfoxide (9); Phloretin (Sigma-Aldrich, St. Louis, cat. no. P7912), stock solution 25 mM in dimethyl sulfoxide (11); Vanillic acid (ABCR, Karlsruhe, Germany, cat. no. AB177480), stock solution 200 mM in ethanol (12).
Western blot analysis
Cells were grown in a 10 cm culture dish, detached 72 h after transfection, washed once in phosphate-buffered saline and lysed on ice in 200 µl of lysis buffer [0.14 M NaCl, 0.1 M HEPES (pH 7.4) 1.5% (w/v) Triton X-100 and protease inhibitor cocktail (Sigma-Aldrich; cat. no. P8340)] by sonication for 30 s (Sonopuls mini20, Bandelin electronic GmbH). The cell lysate was centrifuged twice at 14 000 × g for 20 min at 4°C to remove cell debris, and the protein content was quantified using a Bradford assay (BIO-RAD, Hercules, CA, cat. no. 500-0002). In all, 10 µg of crude protein extract was mixed with 5 µl of gel loading buffer [10 ml; 10% SDS; 1.25 ml 0.5 M Tris–HCl (pH 6.8), 3 ml glycerol, 0.2 ml 0.5% (w/v) bromophenol blue, 5.55 ml ddH2O]. The proteins were resolved on a 12% SDS–PAGE and electroblotted (Trans-Blot® SD, Bio-Rad, Reinach, Switzerland) onto a polyvinylidene difluoride (PVDF) membrane (Immobilon®-P, Millipore, Billerica, MA, USA). The membrane was blocked with 5% (w/v) bovine serum albumin (Sigma-Aldrich, cat. no. A9418) diluted in Tris-buffered saline [20 mM Tris, 150 mM NaCl (pH 7.6), 0.1% Tween-20] for 1 h at 22°C and incubated over night at 4°C with a mouse monoclonal anti-TetR antibody (MoBiTec, Göttingen, Germany; cat. no. TET02, lot no. MO2204010). The membrane was then washed three times in Tris-buffered saline and incubated for 1 h with a horseradish peroxidase-coupled sheep anti-mouse immunoglobulin G (GE Healthcare, Switzerland; cat. no. NA931V, lot no. 399402). Likewise, for loading control, the membrane was probed with a rabbit polyclonal anti-α-actin antibody (Sigma, Saint Louis, USA; cat. no A2066, lot no.03044844) and a horseradish peroxidase-coupled sheep anti-rabbit immunoglobulin G (AbD Serotec, Oxford, UK, cat. no. STAR54). ECL-Plus western blot detection reagents (Amersham, Piscataway, NJ, USA; cat. no. RPN2232) were used for chemiluminescence-based signal detection using a Chemilux CCD camera (ImageQuantTM, LAS 400 mini, GE Healthcare, Switzerland).
RESULTS
Mce3R belongs to a new family of Actinomycetes repressors
Mycobacterium tuberculosis harbors four homologous mce operons with similar genetic arrangement (46,47) that have been suggested to manage the pathogen’s lipid import and metabolism (48,49). The mce3 gene cluster is negatively controlled by Mce3R (Rv1963c), a TetR family transcriptional regulator that contains a unique double TetR-family signature consisting of two DNA-binding HTH motifs (50,51). The weak homology between the tandem TetR units suggests that Mce3R might have resulted from a fusion of two diverse TetR repressors rather than representing an evolutionary snapshot of a standard gene duplication-diversification process. The function, control characteristics and tuning dynamics of this tandem TetR–TetR fusion motif as well as the question whether Mce3R could possibly be responsive to two distinct trigger molecules and address different target genes for differential control of metabolic activities remain elusive. Multiple alignment analysis of Mce3R’s secondary structure revealed that 204 other putative Actinomycetes repressor proteins share such a double TetR-family signature, suggesting that Mce3R is a prototype of a larger family of repressor fusion proteins with common control characteristics (Figure 1A).
Figure 1.

Tandem TetR-family scheme. (A) Multiple alignment analysis of Mce3R’s predicted alpha helical secondary structure reveals a double TetR-family signature consisting of typical DNA-binding HTH and ligand-binding domain (LBD) domains that is present in over 200 other putative Actinomycetes repressor proteins. Therefore, Mce3R may be a prototype of a larger family of natural repressor fusion proteins sharing specific transcription-control characteristics. (B) Synthetic bipartite TetR-family repressor-derived mammalian transcription factors with dual input–output control capacity. Bipartite TetR-family repressor-derived mammalian transactivator variants assembled by fusing the TET- and γ-butyrolactone-dependent repressor proteins (TetR, ScbR) to the Herpex simplex virus-derived transactivation domain (VP16). Corresponding target promoters contain specific operator sites (tetO7, OPapR1) immediately 5′ of a minimal version of the human cytomegalovirus immediate early promoter (PhCMVmin) and control expression of the human placental SEAP gene. (C) Interaction diagram of individual and tandem transactivator components with corresponding expression units.
Design of synthetic bipartite mammalian transactivators
Using Mce3R’s unique architecture as a blueprint, we have used a synthetic biology-inspired reverse engineering approach to design tandem TetR-family repressor-derived fusion proteins with dual input–output control capacity. Therefore, two validated TetR-family repressor proteins such as the TET-responsive repressor TetR [T; (4)] and the γ-butyrolactone-adjustable repressor ScbR [S; (9)] were fused and linked to a Herpes simplex virus-derived VP16 transactivation domain to form chimeric dual-input transcription factors that could individually bind and program transcription of their specific pairs of target promoters such as the TET [PhCMV*−1; (4)] or SCB1 [PSPA; (9)] -responsive promoters (Figure 1B and C). Indeed, in human cells, the synthetic dual TetR-family transactivator ST-TA (pMX1) was able to induce its cognate target promoters PSPA and PhCMV*−1 (PSPA-SEAP, pWW124; PhCMV*−1-SEAP, pMF111) and provide similar SEAP production characteristics compared with the individual transactivators ScbR-VP16 (SCA; pWW122) and TetR-VP16 (tTA; pSAM200) (Figure 2A). Also, ST-TA retained the control capacity of their isogenic individual counterparts and remained responsive to TET and the butyrolactone SCB1 (Figure 2B), which confirms that, even in a scaffold configuration, TetR family repressor proteins individually act as fully functional DNA-binding domains, and that they can share a common transactivation domain to trigger expression of different transgenes. Thus, the Mce3R-mimetic ST-TA pioneers a new class of composite control proteins that could adjust their individual target promoters in response to specific molecular inputs. As the relative position of TetR family members within the scaffold transactivator may have an effect on its control capacity, we have reversed the position of ScbR and TetR, which resulted in TS-TA (TetR-ScbR-VP16; pMX10) (Figure 1B). However, TS-TA could only marginally trigger SEAP expression from its individual target promoters PhCMV*−1 (pMF111) and PSPA (pWW124) (Figure 2A).
Figure 2.

Performance of bipartite TetR-family transcription factors. (A) Regulation characteristics of the ScbR-TetR-VP16 (ST-TA) and TetR-ScbR-VP16 (TS-TA) transactivators. HEK-293T cells were co-transfected with ST-TA (pMX1) or TS-TA (pMX10) and either PhCMV*−1- (pMF111) or PSPA- (pWW124) driven SEAP expression vectors. Isogenic cultures expressing tTA (pSAM200) or SCA (pWW122) instead of ST-TA (pMX1) were used as controls. Cells were grown for 48 h in the presence or absence of the trigger molecules TET or the γ-butyrolactone (SCB1) before the SEAP levels in the culture supernatant were quantified. (B) TET- and γ-butyrolactone- (SCB1) adjustable ST-TA-driven transgene expression. HEK-293T cells were transfected with either pMX1/pWW124 or pMX1/pMF111 and cultivated for 48 h in the presence of different concentrations of TET or SCB1 before SEAP was scored in the culture supernatant. HEK-293T transfected with either tTA (pSAM200) or SCA (pWW122) instead of ST-TA (pMX1) were used as controls. (C) Synthetic bipartite TetR-family repressor-derived mammalian transsilencers. Tandem TetR-family repressor-derived mammalian transsilencer variants assembled by fusing the TET- and γ-butyrolactone-dependent repressor proteins (TetR, ScbR) to the KRAB domain of the human kox-1 gene. Corresponding target promoters contain specific operator sites (tetO2, OPapR1) immediately 3′ of the constitutive simian virus 40 promoter (PSV40) and control expression of the human placental SEAP. (D) Performance of the ScbR-TetR-KRAB (ST-TS) and TetR-ScbR-KRAB (TS-TS) transsilencers. HEK-293T cells were co-transfected with ST-TS (pMX63) or TS-TS (pMX64) and either PSPS- (pMX65) or PtTSON2- (pMX67) driven SEAP expression vectors and grown for 48 h in the presence (1 µg/ml) or absence of the trigger molecules TET or the γ-butyrolactone (SCB1) before SEAP levels were scored in the culture supernatant. Isogenic cultures transfected with either pMX65 or pMX67, but no transsilencer-encoding plasmids were used as controls.
Design of synthetic tandem transsilencers
To design tandem transcription factors with ON-type switch characteristics, we replaced the VP16 transactivation domains of ST-TA and TS-TA with the KRAB domain of the human kox-1 gene, which resulted in the transsilencer (TS) variants ST-TS (ScbR-TetR-KRAB; pMX63) and TS-TS (TetR-ScbR-KRAB; pMX65) (Figure 2C). In the absence of any trigger compounds, ST-TS bound to specific operator modules (OpapRI, tetO2) and silenced the corresponding chimeric target promoters PSPS (PSV40-OPapRI-SEAP-pA; pMX65) and PtTSON2 (PSV40-tetO2-SEAP-pA; pMX67). In the presence of either SCB1 or TET, ST-TS was released from PSPS and PtTSON2, and constitutive SEAP expression was restored (Figure 2D). TS-TS failed to control both promoters, suggesting that not all dual-TetR architectures are fully functional (data not shown).
Transactivator scaffolding as a general design concept
The principle of designing multi-input/output control devices by assembling different TetR-familiy members in a single protein scaffold could be validated by the construction of PT-TA (TtgR-TetR-VP16; pMX29) and TP-TA (TetR-TtgR-VP16; pFS1), two chimeric transcription factors combining the phloretin [P; (9)] and the TET [T; (4)] -dependent repressors with a single C-terminal VP16 transactivation domain (Figure 3A). Akin to ST-TA, both PT-TA and TP-TA were able to retain the interference-free control of individual PTtgR1- (phloretin-responsive promoter) and PhCMV*−1-driven transgene expression in response to the trigger molecules phloretin and TET, respectively (Figure 3B). This suggests that the design of functional transactivators with a double TetR-family signature could represent a universal principle. By way of preliminary example, we also daisy chained three independent TetR-family repressors (TetR, ScbR and TtgR) to generate the three-in-one transactivator PTS-TA (TtgR-TetR-ScbR-VP16; pMX27) (Figure 3A) that could control all individual target promoters (PhCMV*−1, PSPA, PTtgR1) in response to the specific trigger molecules TET, SCB1 and phloretin, respectively (Figure 3C).
Figure 3.

Characterization of synthetic bi- and tri-partite TetR-family transactivators. (A) Bi- and tri-partite TetR-family repressor-derived mammalian transactivator variants assembled by fusing the TET-, phloretin- and γ-butyrolactone-dependent repressor proteins (TetR, TtgR, ScbR) to the Herpex simplex virus-derived transactivation domain (VP16). All multi-partite transactivator-encoding expression units are driven by the constitutive simian virus 40 promoter (PSV40) and contain a polyadenylation signal (pA). Corresponding target promoters contain specific operator sites (tetO7, OTtgR, OPapR1) immediately 5′ of a minimal version of the human cytomegalovirus immediate early promoter (PhCMVmin) and control expression of the human placental SEAP. (B) Regulation performance of the TtgR-TetR-VP16 (PT-TA) and TetR-TtgR-VP16 (TP-TA) transactivators. HEK-293T cells were co-transfected with PT-TA (pMX29) or TP-TA (pFS1) and either PhCMV*−1- (pMF111) or PTtgR1- (pMG10) driven SEAP expression vectors. Isogenic cultures expressing tTA (pSAM200) or TtgA1 (pMG11) instead of PT-TA (pMX29) or TP-TA (pFS1) were used as controls. Cells were grown for 48 h in the presence or absence of the trigger molecules TET (1 µg/ml) or phloretin (PLT, 0.2 µg/ml) before SEAP levels were scored in the culture supernatant. (C) Regulation performance of the tripartite TtgR-TetR-ScbR-VP16 transactivator (PTS-TA). HEK-293T cells were co-transfected with PTS-TA (pMX27) and either PhCMV*−1- (pMF111), PSPA- (pWW124) or PTtgR1- (pMG10) driven SEAP expression vectors. Isogenic cultures expressing tTA (pSAM200), SCA (pWW122) or TtgA1 (pMG11) instead of PTS-TA (pMX27) were used as controls. Cells were grown for 48 h in the presence or absence of the trigger molecules TET (1 µg/ml), γ-butyrolactone (SCB1, 1 µg/ml) or the phloretin (PLT, 0.2 µg/ml) before SEAP levels were profiled in the culture supernatant.
Transactivator scaffolding is not limited to TetR-family members
To evaluate whether this scaffold standard could be extended beyond the TetR repressor family, we also included the GntR-type transcriptional repressor VanR that controls lignin metabolism of Caulobacter crescentus in response to vanillic acid (11,52). In contrast to the TetR family whose DNA-binding motif contains two alpha-helical structures, VanR’s binding capacity is based on a winged HTH motif that contains three alpha helices (53). We have constructed two VanR-containing dual-control devices by sequentially permutating VanR and TetR: TV-TA (TetR-VanR-VP16; pMX6) and VT-TA (VanR-TetR-VP16; pMX25) (Figure 4A). When co-transfecting either of the dual-control transactivator variants with either VanR- (P1VanO2; pMG252) or TetR (PhCMV*−1; pMF111)-specific reporter constructs into human cells, only VT-TA was able to independently transactivate (Figure 4B) and control (Figure 4C) PhCMV*−1 (pMF111) as well as P1VanO2 (pMG252)-driven transgene expression in response to TET and vanillic acid, respectively. This demonstrates that the concept of fusing prokaryotic repressor proteins of different families to achieve differential small molecule-responsive transgene expression in mammalian cells works in principle, but that there remain some unknown design rules that may limit functionality and predictability.
Figure 4.

Bi-partite TetR/GntR-family repressor-derived mammalian transactivators with dual input–output control capacity. (A) Bi-partite TetR/GntR-family repressor-derived mammalian transactivator variants assembled by fusing the TET- and vanillic acid-responsive repressor proteins (TetR, VanR) to the Herpex simplex virus-derived transactivation domain (VP16). The bi-partite transactivator-encoding expression units are driven by the constitutive simian virus 40 promoter (PSV40) and contain a polyadenylation signal (pA). Corresponding target promoters contain domain-specific operator sites VanO2 and tetO7 immediately 5′ of a minimal version of the human cytomegalovirus immediate early promoter (PhCMVmin) that controls expression of the human placental SEAP. (B) Regulation performance of the TetR-VanR-VP16 (TV-TA) and VanR-TetR-VP16 (VT-TA) transactivators. HEK-293T cells were co-transfected with TV-TA (pMX6) or VT-TA (pMX25) and either PhCMV*−1- (pMF111) or P1VanO2- (pMG252) driven SEAP expression vectors. Isogenic cultures expressing tTA (pSAM200) or VanA1 (pMG250) instead of TV-TA (pMX6) or VT-TA (pMX25) were used as controls. Cells were grown for 48 h before SEAP levels were quantified in the culture supernatant. (C) Trigger responsiveness of the VanR-TetR-VP16 (VT-TA) transactivator. HEK-293T cells were co-transfected with VT-TA (pMX25) and either PhCMV*−1- (pMF111) or P1VanO2- (pMG252) driven SEAP expression vectors and grown for 48 h in the presence and absence of the trigger molecules vanillic acid (VAC, 17 µg/ml) or TET (1 µg/ml) before SEAP levels were scored in the culture supernatant.
Bipartite transactivators show independent two-transgene control characteristics at high intracellular concentrations
With ST-TA’s trigger-controlled fine-tuning of its individual target promoters PSPA and PhCMV*−1 established (Figure 2B), we also assessed whether this bipartite transactivator could differentially adjust two distinct transgenes in response to independent molecular inputs. We have therefore co-transfected HEK-293T cells with high amounts of the ST-TA-encoding plasmid (pMX1) relative to PSPA-driven SEAP (pWW124) and PhCMV*−1-driven GLuc (pDA43) expression vectors (ratio 20:1:1). At high intracellular ST-TA concentrations, the composite transactivator was able to simultaneously control its specific target promoters and independently fine-tune SEAP and GLuc expression in a SCB1 and TET-responsive manner (Figure 5A). As neither SCB1 affected GLuc expression nor TET influenced SEAP expression, ST-TA manages independent interference-free two-input/two-output transcription fine tuning in a most compact format.
Figure 5.

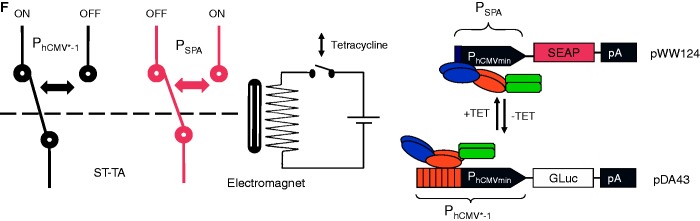
Two-gene control dynamics of the ST-TA transactivator. (A) Independent control of two different transgenes. HEK-293T cells were co-transfected with the ST-TA-encoding plasmid pMX1, the PSPA-driven SEAP- (pWW124) and PhCMV*−1-driven GLuc- (pDA43) expression vectors at a high transactivator-to-reporter ratio (20:1:1) and grown for 48 h in culture medium containing increasing concentrations of TET or γ-butyrolactone (SCB1) before SEAP and GLuc levels were quantified in the culture supernatant. (B) DPDT relay switch characteristics.
HEK-293T cells were co-transfected with the ST-TA-encoding plasmid pMX1, the PSPA-driven SEAP- (pWW124) and PhCMV*−1-driven GLuc- (pDA43) expression vectors at a low transactivator-to-reporter ratio (1:12:12) and grown for 48 h in culture medium containing increasing concentrations of TET before SEAP and GLuc levels were quantified in the culture supernatant. (C) HEK-293T cells were co-transfected with the tTA- and SCA- expression vectors (pSAM200, pWW122) and corresponding reporter plasmids pDA43 (PhCMV*−1-GLuc) and pWW124 (PSPA-SEAP) (ratio 1:1:5:5), grown for 48 h in culture medium containing increasing concentrations of TET before expressed SEAP and GLuc levels were quantified in the culture supernatant. (D) DPDT relay switch characteristics of hMSC-TERT cells. hMSC-TERT cells were co-transfected with the ST-TA-encoding plasmid pMX1, the PSPA-driven SEAP- (pWW124) and PhCMV*−1-driven GLuc- (pDA43) expression vectors in a low transactivator-to-reporter ratio (1:12:12) and grown for 48 h in culture medium containing increasing concentrations of TET before SEAP and GLuc levels were quantified in the culture supernatant. (E) Reversibility of ST-TA-mediated DPDT relay switch characteristics. HEK-293T cells were co-transfected with the ST-TA-encoding plasmid pMX1, the PSPA-driven SEAP- (pWW124) and PhCMV*−1-driven GLuc- (pDA43) expression vectors in a low transactivator-to-reporter ratio (1:12:12) and grown for 24 h in the presence (1 µg/ml) or absence of TET and SEAP, and GLuc levels were profiled in the culture supernatant. The same cells were then washed and incubated with fresh medium and grown for another 24 h in the presence (TET, 1 µg/ml) or absence of TET (TET to no TET), before SEAP and GLuc levels were profiled in the culture supernatant. (F) Schematic of the electric and genetic DPDT relay switch.
Composite transactivators show DPDT relay switch characteristics at low intracellular concentrations
When co-transfecting HEK-293T cells with lower amounts of ST-TA-encoding plasmid (pMX1) relative to PSPA-driven SEAP (pWW124) and PhCMV*−1-driven GLuc (pDA43) expression vectors (ratio 1:12:12), ST-TA triggered maximum GLuc levels, whereas SEAP expression remained low. Following addition of increasing concentrations of TET, ST-TA is gradually released from PhCMV*−1, leading to progressive shut down of GLuc expression and switches to exclusive binding of PSPA, which then results in dose-dependent induction of SEAP production (Figure 5B). This TET-induced switch from GLuc to SEAP expression was an exclusive characteristic of the bi-partite ST-TA, as control experiments with individual transactivators (SCA, tTA) resulted in GLuc shut down but not in SEAP induction after addition of TET (Figure 5C). The DPDT relay switch characteristics could be confirmed in telomerase-immortalized hMSC-TERT using an identical experimental set-up (Figure 5D). At lower ST-TA concentration (ratio 1:12:12), the DPDT relay switch is reversible as removal of TET switches ST-TA back to binding of PhCMV*−1, which results in shut down of SEAP expression and concomitant induction of GLuc (Figure 5E and F).
The TET-controlled change over from PhCMV*−1 to PSPA and switch back from PSPA to PhCMV*−1 thereby modulating expression of two different transgenes represents the genetic version of a DPDT relay switch. DPDT relays consist of an electromagnet that, on activation by an electric current (TET), simultaneously switches two moving plates (two poles; TetR and ScbR of ST-TA) that may engage either of two different sets of fixed contacts (PhCMV*−1 and PSPA) to switch one contact from ON-to-OFF (GLuc) and the other one from OFF-to-ON (SEAP) (the double throw) (Figure 5F). Although the electric DPDT relay is limited to digital switching, its genetic counterpart shows additional dimmer switch quality at intermediate inducer concentrations when GLuc expression is gradually shut down while SEAP production is progressively turned on and vice versa (Figure 5B and D).
To validate the DPDT relay switching characteristics at low composite transactivator levels, we produced a stable ST-TA-transgenic cell line HEK-293TST-TA in which ST-TA is driven by leaky transcription of the minimal version of the human cytomegalovirus immediate early promoter (PhCMVmin; pAS1) (Figure 6A) and therefore expresses the tandem transactivator at low constitutive levels (Figure 6B). Co-transfection of HEK-293TST-TA with pDA43 and pWW124 confirmed the circuit’s DPDT relay switching capacity (Figure 6C). To explore functional compatibility of the composite transactivator (ST-TA) and the individual transactivators (tTA, SCA) in the same cell using a DPDT experimental set-up, we co-transfected HEK-293TST-TA with the tTA (pSAM200) and SCA (pWW122) expression vectors as well as with the DPDT reporter plasmids pDA43 (PhCMV*−1-SEAP) and pWW124 (PSPA-SEAP) and profiled reporter protein production before and after addition of TET (Figure 6D), showing that the presence of tTA does not interfere with ST-TA’s switch from PSPA-GLuc to PhCMV*−1-SEAP, as GLuc levels decrease and SEAP levels increase when the culture is switched from −TET to +TET. Likewise, SCA does not interfere with ST-TA’s switch from PSPA-GLuc to PhCMV*−1-SEAP, as GLuc levels decrease and SEAP levels remain constant when the culture is switched from −TET to +TET (Figure 6D). This is remarkable and shows that composite and individual transactivators can in principle operate side-by-side in an interference-free manner in the same cell.
Figure 6.

(A) ST-TA driven by a minimal version of the human cytomegalovirus immediate early promoter. (B) Western blot-based analysis of ST-TA expression levels. Lanes left to right: ST-TA (pMX1) transiently transfected into HEK-293T; HEK-293TST-TA stably expressing ST-TA at low levels; tTA (pSAM200) transiently transfected into HEK-293T; non-transfected HEK-293T cells used as control. (C) HEK-293TST-TA cells were co-transfected with the PSPA-driven SEAP (pWW124) and PhCMV*−1-driven GLuc (pDA43) expression vectors (plasmid ratio 1:1) and grown for 24 h in culture medium containing increasing concentrations of TET before SEAP levels, and GLuc were profiled in the culture supernatant. (D) Compatibility of the DPDT relay with tTA and SCA. HEK-293TST-TA cells were co-transfected with the PSPA-driven SEAP (pWW124) and PhCMV*−1-driven GLuc (pDA43) expression plasmids (ratio 13:20:20), and either the tTA (pSAM200) or SCA (pWW122) expression vectors, and grown for 48 h in the presence or absence of 1 µg/ml TET before SEAP and GLuc levels were profiled in the culture supernatant.
Design of hybrid promoters for bipartite transactivators
To further study the control behavior of tandem TetR-family transcription factors such as ST-TA and TS-TA, we designed hybrid promoter variants that combined one (PST-TA/TS-TA1; tetO-OPapRI-PhCMVmin; pMX8) or two (PST-TA/TS-TA2; tetO-tetO-OPapRI-PhCMVmin; pMX9) TetR-specific binding sites (tetO) with a single ScbR-specific binding site (OPapRI), all separated by two helical turns, with a minimal version of the human cytomegalovirus immediate early promoter (Figure 7A). Although ST-TA was able to activate individual (PhCMV*−1 and PSPA) (Figure 2A) as well as both hybrid promoter versions (PST-TA/TS-TA1, PST-TA/TS-TA2) (Figure 7B), TS-TA exclusively controlled the hybrid promoters (Figures 2A and 7B). As expected, owing to the twin tetO, PST-TA/TS-TA2 always provided higher maximum expression levels when fully induced (Figure 7B). Addition of TET partially reduced SEAP levels to intermediate high-level expression, as TS-TA’s ScbR component continues to bind and activate the hybrid promoter (PST-TA/TS-TA2). Likewise, administration of SCB1 reduces SEAP expression to intermediate low-level expression as TS-TA’s TetR module remains bound to the hybrid promoter. Only in the presence of both trigger molecules, SEAP expression is shut down (Figure 7C). Overall, SCB1 seems to be a more efficient release trigger for TS-TA than TET. Therefore, dual TetR-family transactivators can be programmed by different trigger molecules to provide distinct transcription activities from hybrid promoters and mediate discrete transgene expression levels (Figure 7C). Interestingly, TS-TA, which failed to address single promoters (Figure 2A), was fully operational in combination with hybrid promoters, suggesting that tandem transactivators and hybrid promoters could be matched for optimal regulation performance. These findings suggest that natural tandem TetR-family transcription factors may enable DPDT relay switch characteristics with mutually exclusive graded expression profiles for different sets of target genes driven by individual target promoters while enabling discrete inducer programmed expression levels for hybrid promoter configurations.
Figure 7.

Regulation characteristics of ST-TA and TS-TA-specific hybrid promoters. (A) Schematic of ST-TA- and TS-TA-specific promoter variants combining one (PST-TA/TS-TA1) or two (PST-TA/TS-TA2) tetOs (orange) with a single ScbR-specific binding site (OPapRI, blue), all separated by two helical turns, with a minimal version of the human cytomegalovirus immediate early promoter. (B) Maximum ST-TA- and TS-TA-dependent induction of the hybrid promoters in the absence of any control molecules. HEK-293T cells were co-transfected with either the SCA- (pWW122), tTA- (pSAM200), ST-TA- (pMX1) or TS-TA- (pMX10) encoding plasmids in combination with either PST-TA/TS-TA1 (pMX8, gray) or PST-TA/TS-TA2 (pMX9, black) and cultivated for 48 h before SEAP levels were quantified in the culture supernatant. (C) TET- and γ-butyrolactone (SCB1)-regulated TS-TA (pMX10)-mediated transactivation of the pMX9-encoded hybrid promoter enables programming of discrete SEAP expression levels in response to a specific combination of inhibitory concentrations (1 µg/ml) of TET and SCB1. HEK-293T cells were co-transfected with pMX10 (TS-TA) and pMX9 (PST-TA/TS-TA2) and cultivated in medium containing different combinations of trigger compounds. HEK-293T cells exclusively transfected with pMX9 (PST-TA/TS-TA2) were used as control. Cells were grown for 48 h before SEAP levels were quantified in the culture supernatant.
DISCUSSION
Synthetic biology is an engineering-driven approach to assemble modular biological parts in a rational and predictable manner to design devices, systems and organisms with novel and useful functions (53). Here, we have defined a novel scaffolding architecture for serial assembly of trigger-inducible DNA-binding proteins that could program expression from individual or hybrid target promoters in a small molecule-responsive manner. Scaffold transcription factors or control proteins with multiple DNA-binding domains seem to be an evolution-proven design found to control lipid import and metabolism in bacteria [Mce3R; (50,51)] as well as differentiation in mammalian cells [POU domain-containing proteins such as Oct-1, Oct-2; (29)]. However, the functionality and control dynamics of these bi-partite transcription factors remain largely elusive. Intuitively, bi-partite transcription factors can address two different types of promoters or single promoters containing different operator sites and so program expression of two different sets of genes or (co-)modulate transcription of a single target gene in a specific manner. Indeed, synthetic mammalian tandem transcription factors, assembled by fusing up to three different prokaryotic DNA-binding proteins of the TetR and GntR types to a single transactivation domain, enabled trigger-controlled expression of three different expression units. Permutation of individual DNA-binding proteins within a daisy-chain transcription factor modulated its regulation performance. Overall, multi-partite transcription factors not only retain the individual DNA-binding capacities of their components but also the responsiveness to their specific trigger molecules. As individual repressors and target promoters provide independent transcription control in a simpler fashion, multi-partite transcription-control proteins may not have evolved to exclusively modulate multiple genes.
One additional characteristic of multi-partite transcription factors was discovered when evaluating their performance on hybrid promoters containing matching operator sites. The daisy-chain transcription factors could control hybrid target promoters with discrete expression profiles; full expression in the absence of trigger molecules when both TetR-moieties are bound, intermediate high-level expression in the presence of one inducer molecule when only one TetR component is bound, intermediate low-level expression when the other repressor domain remains bound in the presence of the second trigger compound and repression in the presence of both control molecules, which abolishes binding of the entire bi-partite transcription factor to the hybrid promoter. Similar discrete expression profiles have previously been exclusively achieved by synthetic cascades in which three independent transcription-control systems were serially linked (14,55).
A second feature that bi-partite transcription factors exhibited when expressed at low levels and when addressing two separate promoters was the DPDT relay switch characteristics. The genetic DPDT relay switch enabled reversible and graded mutually exclusive expression swapping between different transgenes, as the daisy-chain transactivator first binds with higher affinity to one promoter and gradually flips over to the second promoter, as increasing concentrations of the trigger compound dose dependently release the bi-partite transcription factor from the original promoter. Like their electric counterparts, genetic DPDT relay switches may enable organisms to gradually and reversibly switch between two distinct metabolic networks using a single pair of input molecules.
Overall, we suggest that multi-partite transcription factors may have evolved to (i) independently fine tune expression of different sets of target genes, (ii) enable discrete expression levels when programming hybrid promoters containing multiple operators and (iii) provide DPDT relay switching of independent metabolic circuits. Owing to the high modularity, orthogonality and interoperability of DNA-binding proteins the multi-partite transactivator architecture will provide a novel addition to the synthetic biology toolbox and may help design complex mammalian designer circuits with unprecedented design, complexity and logic operation capabilities.
FUNDING
European Research Council (ERC) advanced grant (ProNet). Funding for open access charge: ETH Zurich.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Franziska Stolz for technical assistance as well as providing plasmid pFS1 and Joel Busset for critical comments on the manuscript.
REFERENCES
- 1.Jakobus K, Wend S, Weber W. Synthetic mammalian gene networks as a blueprint for the design of interactive biohybrid materials. Chem. Soc. Rev. 2012;41:1000–1018. doi: 10.1039/c1cs15176b. [DOI] [PubMed] [Google Scholar]
- 2.Auslander S, Auslander D, Muller M, Wieland M, Fussenegger M. Programmable single-cell mammalian biocomputers. Nature. 2012;487:123–127. doi: 10.1038/nature11149. [DOI] [PubMed] [Google Scholar]
- 3.Auslander S, Fussenegger M. From gene switches to mammalian designer cells: present and future prospects. Trends Biotechnol. 2013;31:155–168. doi: 10.1016/j.tibtech.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deuschle U, Meyer WK, Thiesen HJ. Tetracycline-reversible silencing of eukaryotic promoters. Mol. Cell. Biol. 1995;15:1907–1914. doi: 10.1128/mcb.15.4.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weber W, Fux C, Daoud-el Baba M, Keller B, Weber CC, Kramer BP, Heinzen C, Aubel D, Bailey JE, Fussenegger M. Macrolide-based transgene control in mammalian cells and mice. Nat. Biotechnol. 2002;20:901–907. doi: 10.1038/nbt731. [DOI] [PubMed] [Google Scholar]
- 7.Bacchus W, Lang M, El-Baba MD, Weber W, Stelling J, Fussenegger M. Synthetic two-way communication between mammalian cells. Nat. Biotechnol. 2012;30:991–996. doi: 10.1038/nbt.2351. [DOI] [PubMed] [Google Scholar]
- 8.Kemmer C, Gitzinger M, Daoud-El Baba M, Djonov V, Stelling J, Fussenegger M. Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Nat. Biotechnol. 2010;28:355–360. doi: 10.1038/nbt.1617. [DOI] [PubMed] [Google Scholar]
- 9.Weber W, Schoenmakers R, Spielmann M, El-Baba MD, Folcher M, Keller B, Weber CC, Link N, van de Wetering P, Heinzen C, et al. Streptomyces-derived quorum-sensing systems engineered for adjustable transgene expression in mammalian cells and mice. Nucleic Acids Res. 2003;31:e71. doi: 10.1093/nar/gng071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folcher M, Gaillard H, Nguyen LT, Nguyen KT, Lacroix P, Bamas-Jacques N, Rinkel M, Thompson CJ. Pleiotropic functions of a Streptomyces pristinaespiralis autoregulator receptor in development, antibiotic biosynthesis, and expression of a superoxide dismutase. J. Biol. Chem. 2001;276:44297–44306. doi: 10.1074/jbc.M101109200. [DOI] [PubMed] [Google Scholar]
- 11.Gitzinger M, Kemmer C, El-Baba MD, Weber W, Fussenegger M. Controlling transgene expression in subcutaneous implants using a skin lotion containing the apple metabolite phloretin. Proc. Natl Acad. Sci. USA. 2009;106:10638–10643. doi: 10.1073/pnas.0901501106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gitzinger M, Kemmer C, Fluri DA, El-Baba MD, Weber W, Fussenegger M. The food additive vanillic acid controls transgene expression in mammalian cells and mice. Nucleic Acids Res. 2012;40:e37. doi: 10.1093/nar/gkr1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehra S, Charaniya S, Takano E, Hu WS. A bistable gene switch for antibiotic biosynthesis: the butyrolactone regulon in Streptomyces coelicolor. PLoS One. 2008;3:e2724. doi: 10.1371/journal.pone.0002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer BP, Weber W, Fussenegger M. Artificial regulatory networks and cascades for discrete multilevel transgene control in mammalian cells. Biotechnol. Bioeng. 2003;83:810–820. doi: 10.1002/bit.10731. [DOI] [PubMed] [Google Scholar]
- 15.Deans TL, Cantor CR, Collins JJ. A tunable genetic switch based on RNAi and repressor proteins for regulating gene expression in mammalian cells. Cell. 2007;130:363–372. doi: 10.1016/j.cell.2007.05.045. [DOI] [PubMed] [Google Scholar]
- 16.Kramer BP, Viretta AU, Daoud-El-Baba M, Aubel D, Weber W, Fussenegger M. An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol. 2004;22:867–870. doi: 10.1038/nbt980. [DOI] [PubMed] [Google Scholar]
- 17.Wieland M, Auslander D, Fussenegger M. Engineering of ribozyme-based riboswitches for mammalian cells. Methods. 2012;56:351–357. doi: 10.1016/j.ymeth.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Auslander D, Fussenegger M. Optogenetic therapeutic cell implants. Gastroenterology. 2012;143:301–306. doi: 10.1053/j.gastro.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 19.Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R. A synthetic multicellular system for programmed pattern formation. Nature. 2005;434:1130–1134. doi: 10.1038/nature03461. [DOI] [PubMed] [Google Scholar]
- 20.Kampf MM, Engesser R, Busacker M, Horner M, Karlsson M, Zurbriggen MD, Fussenegger M, Timmer J, Weber W. Rewiring and dosing of systems modules as a design approach for synthetic mammalian signaling networks. Mol. Biosyst. 2012;8:1824–1832. doi: 10.1039/c2mb05509k. [DOI] [PubMed] [Google Scholar]
- 21.Grushkin D. The new drug circuit. Nat. Med. 2012;18:1452–1454. doi: 10.1038/nm1012-1452. [DOI] [PubMed] [Google Scholar]
- 22.Folcher M, Fussenegger M. Synthetic biology advancing clinical applications. Curr. Opin. Chem. Biol. 2012;16:345–354. doi: 10.1016/j.cbpa.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Ruder WC, Lu T, Collins JJ. Synthetic biology moving into the clinic. Science. 2011;333:1248–1252. doi: 10.1126/science.1206843. [DOI] [PubMed] [Google Scholar]
- 24.Weber W, Fussenegger M. Emerging biomedical applications of synthetic biology. Nat. Rev. Genet. 2012; 13:21–35. doi: 10.1038/nrg3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YY, Smolke CD. From DNA to targeted therapeutics: bringing synthetic biology to the clinic. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002944. 106ps142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perkel JM. Streamlined engineering for synthetic biology. Nat. Methods. 2013;10:39–42. [Google Scholar]
- 27.Khalil AS, Lu TK, Bashor CJ, Ramirez CL, Pyenson NC, Joung JK, Collins JJ. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 2012;150:647–658. doi: 10.1016/j.cell.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, Gallegos MT, Brennan R, Tobes R. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol. Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Kamionka A, Bogdanska-Urbaniak J, Scholz O, Hillen W. Two mutations in the tetracycline repressor change the inducer anhydrotetracycline to a corepressor. Nucleic Acids Res. 2004;32:842–847. doi: 10.1093/nar/gkh200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scholz O, Kostner M, Reich M, Gastiger S, Hillen W. Teaching TetR to recognize a new inducer. J. Mol. Biol. 2003;329:217–227. doi: 10.1016/s0022-2836(03)00427-3. [DOI] [PubMed] [Google Scholar]
- 32.Resch M, Striegl H, Henssler EM, Sevvana M, Egerer-Sieber C, Schiltz E, Hillen W, Muller YA. A protein functional leap: how a single mutation reverses the function of the transcription regulator TetR. Nucleic Acids Res. 2008;36:4390–4401. doi: 10.1093/nar/gkn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamionka A, Majewski M, Roth K, Bertram R, Kraft C, Hillen W. Induction of single chain tetracycline repressor requires the binding of two inducers. Nucleic Acids Res. 2006;34:3834–3841. doi: 10.1093/nar/gkl316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krueger C, Berens C, Schmidt A, Schnappinger D, Hillen W. Single-chain Tet transregulators. Nucleic Acids Res. 2003;31:3050–3056. doi: 10.1093/nar/gkg421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 36.Krueger M, Scholz O, Wisshak S, Hillen W. Engineered Tet repressors with recognition specificity for the tetO-4C5G operator variant. Gene. 2007;404:93–100. doi: 10.1016/j.gene.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Baron U, Schnappinger D, Helbl V, Gossen M, Hillen W, Bujard H. Generation of conditional mutants in higher eukaryotes by switching between the expression of two genes. Proc. Natl Acad. Sci. USA. 1999;96:1013–1018. doi: 10.1073/pnas.96.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herr W, Cleary MA. The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev. 1995;9:1679–1693. doi: 10.1101/gad.9.14.1679. [DOI] [PubMed] [Google Scholar]
- 39.Vos JC, Plasterk RH. Tc1 transposase of Caenorhabditis elegans is an endonuclease with a bipartite DNA binding domain. EMBO J. 1994;13:6125–6132. doi: 10.1002/j.1460-2075.1994.tb06959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muller M, Auslander S, Auslander D, Kemmer C, Fussenegger M. A novel reporter system for bacterial and mammalian cells based on the non-ribosomal peptide indigoidine. Metab. Eng. 2012;14:325–335. doi: 10.1016/j.ymben.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Fussenegger M, Mazur X, Bailey JE. A novel cytostatic process enhances the productivity of Chinese hamster ovary cells. Biotechnol. Bioeng. 1997;55:927–939. doi: 10.1002/(SICI)1097-0290(19970920)55:6<927::AID-BIT10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 42.Malphettes L, Fussenegger M. Macrolide- and tetracycline-adjustable siRNA-mediated gene silencing in mammalian cells using polymerase II-dependent promoter derivatives. Biotechnol. Bioeng. 2004;88:417–425. doi: 10.1002/bit.20230. [DOI] [PubMed] [Google Scholar]
- 43.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kassem M, Abdallah BM, Yu Z, Ditzel N, Burns JS. The use of hTERT-immortalized cells in tissue engineering. Cytotechnology. 2004;45:39–46. doi: 10.1007/s10616-004-5124-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 47.Zhang F, Xie JP. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol. Cell Biochem. 2011;352:1–10. doi: 10.1007/s11010-011-0733-5. [DOI] [PubMed] [Google Scholar]
- 48.Kendall SL, Withers M, Soffair CN, Moreland NJ, Gurcha S, Sidders B, Frita R, Ten Bokum A, Besra GS, Lott JS, et al. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol. Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl Acad. Sci. USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santangelo MP, Goldstein J, Alito A, Gioffre A, Caimi K, Zabal O, Zumarraga M, Romano MI, Cataldi AA, Bigi F. Negative transcriptional regulation of the mce3 operon in Mycobacterium tuberculosis. Microbiology. 2002;148:2997–3006. doi: 10.1099/00221287-148-10-2997. [DOI] [PubMed] [Google Scholar]
- 51.de la Paz Santangelo M, Klepp L, Nunez-Garcia J, Blanco FC, Soria M, Garcia-Pelayo MC, Bianco MV, Cataldi AA, Golby P, Jackson M, et al. Mce3R, a TetR-type transcriptional repressor, controls the expression of a regulon involved in lipid metabolism in Mycobacterium tuberculosis. Microbiology. 2009;155:2245–2255. doi: 10.1099/mic.0.027086-0. [DOI] [PubMed] [Google Scholar]
- 52.Thanbichler M, Iniesta AA, Shapiro L. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 2007;35:e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang YM, Jeng WY, Ko TP, Yeh YJ, Chen CK, Wang AH. Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl Acad. Sci. USA. 2010;107:8617–8622. doi: 10.1073/pnas.0913302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khalil AS, Collins JJ. Synthetic biology: applications come of age. Nat. Rev. Genet. 2010;11:367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kramer BP, Fischer M, Fussenegger M. Semi-synthetic mammalian gene regulatory networks. Metab. Eng. 2005;7:241–250. doi: 10.1016/j.ymben.2005.02.005. [DOI] [PubMed] [Google Scholar]