

Abstract
Although it is well established that pharmacological inhibitors of classical histone deacetylases (HDACs) are protective in various in vivo models of neurodegenerative disease, the identity of the neurotoxic HDAC(s) that these inhibitors target to exert their protective effects has not been resolved. We find that HDAC3 is a protein with strong neurotoxic activity. Forced expression of HDAC3 induces death of otherwise healthy rat cerebellar granule neurons, whereas shRNA-mediated suppression of its expression protects against low-potassium-induced neuronal death. Forced expression of HDAC3 also promotes the death of rat cortical neurons and hippocampally derived HT22 cells, but has no effect on the viability of primary kidney fibroblasts or the HEK293 and HeLa cell lines. This suggests that the toxic effect of HDAC3 is cell selective and that neurons are sensitive to it. Neurotoxicity by HDAC3 is inhibited by treatment with IGF-1 as well as by the expression of a constitutively active form of Akt, an essential mediator of IGF-1 signaling. Protection against HDAC3-induced neurotoxicity is also achieved by the inhibition of GSK3β, a kinase inhibited by Akt that is widely implicated in the promotion of neurodegeneration in experimental models and in human pathologies. HDAC3 is directly phosphorylated by GSK3β, suggesting that the neuronal death-promoting action of GSK3β could be mediated through HDAC3 phosphorylation. In addition to demonstrating that HDAC3 has neurotoxic effects, our study identifies it as a downstream target of GSK3β.
Introduction
Histone deacetylases (HDACs) are a family of proteins that deacetylate histones as well as a variety of other nuclear, cytoplasmic, and mitochondrial proteins (for review, see Yang and Seto, 2008; Haberland et al., 2009). In mammals, the HDAC family consists of 18 members divided phylogenetically into four classes. Class I HDACs (1, 2, 3, and 8) are ubiquitously expressed and are predominantly nuclear proteins. Class II HDACs are comprised of two subgroups: class IIa HDACs (4, 5, 7, and 9) and class IIb HDACs (6 and 10). Class III HDACs, or sirtuins, consist of seven members (SIRT1–SIRT7). HDAC11 shows structural similarity to both class I and class II HDACs, and is the lone member of class IV. Class I, II, and IV HDACs are Zn+-dependent enzymes collectively referred to as classical HDACs. The sirtuins are structurally unrelated to the classical HDACs and require NAD+ for their activity (Yang and Seto, 2008; Haberland et al., 2009).
Administration of inhibitors against classical HDACs inhibits neuronal loss and improves behavioral outcome in a variety of tissue culture and in vivo models of neurodegenerative disease (for review, see Kazantsev and Thompson, 2008; D'Mello, 2009; Sleiman et al., 2009). Although these results implicate one or more of the HDAC proteins in promoting neuronal death, because the inhibitors used in these studies block the activities of all the classical HDACs efficiently, the identity of the HDAC(s) responsible for neurodegeneration and that is targeted by the inhibitors has been unclear. Somewhat counter intuitively given the protective effects of HDAC inhibitors, analysis of several individual members of the HDAC family, including histone deacetylase-related protein (HDRP, a truncated form of HDAC9), HDAC4, and Sirt 1, protect neurons rather than promote degeneration (Chen et al., 2005; Morrison et al., 2006; Qin et al., 2006; Kim et al., 2007; Majdzadeh et al., 2008). Preliminary results indicate that HDAC7 is also a neuroprotective protein (Ma and D'Mello, 2011). HDAC1 has been reported to be neuroprotective in mouse models of Alzheimer's disease and ischemic stroke (Kim et al., 2008). However, another study found that HDAC1 contributes to neurodegeneration in Huntington's disease by inhibiting the autophagic clearance of mutant huntingtin protein aggregates, leading to their accumulation (Jeong et al., 2009). HDAC6 has also been found to be neuroprotective by different laboratories (Kawaguchi et al., 2003; Iwata et al., 2005; Boyault et al., 2007; Pandey et al., 2007), although a recent study has reported that selective pharmacological inhibition of HDAC6 can promote survival and regeneration of neurons (Rivieccio et al., 2009).
In this report, we identify HDAC3 as a protein with neurotoxic activity. Toxicity by HDAC3 was not observed in non-neuronal cell types that we examined. We show that neurotoxicity by HDAC3 is dependent on GSK3β, a kinase widely implicated in promoting neurodegeneration. Furthermore, neurotoxicity by HDAC3 is inhibited by activation of the IGF-1–Akt signaling pathway, which inhibits GSK3β activity and protects against cell death in various models of neurodegenerative disease.
Materials and Methods
Materials.
Unless indicated otherwise, all cell culture media and reagents were purchased from Invitrogen and all chemicals [including homocysteic acid (HCA)] were from Sigma-Aldrich. Expression plasmids for HDAC3-Flag, constitutively active Akt, dominant-negative GSK3β (K85A), and control shRNA plasmid (pLK0.1) were purchased from Addgene. HDAC3 shRNA plasmids (TRCN0000039391 and TRCN0000039392) were purchased from Sigma-Aldrich. Flag antibody was from Sigma-Aldrich and CDK5 and GFP antibodies were from Santa Cruz Biotechnology. GSK3β antibody was from BD Transduction. HDAC3 antibody for Western blots was from Sigma-Aldrich and immunoprecipitationss were done with a rabbit polyclonal antibody from Santa Cruz Biotechnology. All radioactive reagents were purchased from PerkinElmer.
Cell culture, transfections, and pharmacological treatments.
Cerebellar granule neurons (CGNs) were cultured from rats as previously described (D'Mello et al., 1993). These cultures were transfected 5 d after plating using the calcium phosphate method as previously described (Pfister et al., 2008). Briefly, the cultures were switched to high-potassium medium (HK; serum-free BMEM supplemented with 25 mm KCl) or low-potassium medium (LK; serum-free BMEM) 8 h after transfection and the viability of the transfected neurons, identified by immunocytochemistry, evaluated 24 h after the switch. Unless mentioned otherwise, IGF-1 and inhibitor treatments were done at the time of transfection. The following concentrations were used for the treatments: IGF1 at 50 ng/ml, SP600125 at 10 μm, Roscovitine at 50 μm, SB415286 at 30 μm, and SB216763 at 5 μm.
Cortical cultures were obtained from embryonic d 18 or 19 rats as described previously (Chen et al., 2008; Majdzadeh et al., 2008; Wang et al., 2010) and transfected on the day after plating as described for CGNs. The neurons were treated with HCA 8 h later by directly adding HCA (to final concentration of 1 mm) to the culture medium, and neuronal viability was assessed 20 h later. Transfection efficiency for both CGNs and cortical neurons was ∼1%.
Kidney fibroblasts were cultured and maintained as described previously (Zhao et al., 2010). HT22, HeLa, and HEK293 cells were maintained in DMEM medium containing 10% FBS. Transfection of these cell types was performed using Lipofectamine 2000 (Invitrogen).
Quantification of cell viability.
Neuronal viability of transfected cells, identified by immunocytochemistry, was quantified by staining with 4′, 6′-diamidino-2-phenylindole hydrochloride (DAPI) as we have previously described (Majdzadeh et al., 2008; Pfister et al., 2008). Cells with condensed and fragmented nuclei were scored as dead. CGN viability results were confirmed by TUNEL staining, performed using the DeadEnd Fluorometric TUNEL System (Promega). Viability of kidney fibroblasts and various cell lines was also quantified by DAPI staining and restricted to successfully transfected cells. Data for all viability experiments were generated from at least three independent experiments performed in duplicate. Statistical analysis was performed using the Student's t test. For comparisons of multiple datasets, one-way ANOVA was performed with Bonferroni's multiple-comparison posttest.
shRNA-mediated suppression.
CGNs were cotransfected with GFP and either one of HDAC3 shRNAs or control plasmid in the ratio 1:6.5 on d 4 after plating. The cultures were switched to HK or LK media after 48 h and cell viability assessed 24 h later. Cortical neurons were transfected 1 d after plating. HCA was added 30 h after transfection and cell viability assessed 20 h later. Suppression of endogenous HDAC3 expression in CGNs was assessed by immunocytochemistry using an HDAC3 antibody. HT22 cells were transfected with either the control or one of the shRNA plasmids for 36 h, after which either call lysates were collected for Western blot analysis or RNA was extracted and reverse transcriptase (RT)-PCR analysis performed.
Expression studies.
RT-PCR and Western blot analysis were performed as previously described (Chen et al., 2008; Majdzadeh et al., 2008). For Western blot analyses, all antibodies were used at 1:1000, except for tubulin antibody, which was used at a 1:2500 dilution.
In vitro kinase assays.
GFP or Flag-HDAC3 was overexpressed in HEK293 cells and cell lysates collected after 24 h. The lysates were incubated with 5 μg of either Flag or GFP antibody and 25 μl of protein A/G PLUS agarose beads (Santa Cruz Biotechnology) overnight. Immunoprecipitates were incubated with 0.5 μg of either active GSK3β, active MEK, or 10 μl of kinase buffer and 5 μl of [γ-32P] ATP mixture for 30 min, after which the reaction was stopped and analyzed by PAGE and autoradiography. When used, GSK3 inhibitor SB415286 was added to the kinase reaction at a concentration of 60 μm. For some experiments, endogenous GSK3β from neurons were immunoprecipitated and used with HDAC3 immunoprecipitated from HEK293 cells, as described above.
32P metabolic labeling.
Metabolic labeling of CGNs was performed as described previously (Chen et al., 2008). Briefly, 60 mm dishes of CGN cultures were treated with phosphate-free HK media for 3 h and then switched to phosphate-free HK, LK, or LK medium supplemented with GSK3 inhibitors (SB415826 at 30 μm and SB216763 at 5 μm) along with 250 μCi/ml [32P] orthophosphate. Three hours after the switch, cell lysates were collected and HDAC3 was immunoprecipitated. The level of HDAC3 phosphorylation was assessed by PAGE and autoradiography.
Results
Elevated HDAC3 is selectively toxic to neurons
CGNs cultured from postnatal rats are healthy in medium containing a depolarizing level of potassium (HK medium) but undergo apoptosis in LK medium. Forced expression of HDAC3 induces a substantial level of neuronal loss in HK medium and causes complete neurodegeneration in LK medium (Fig. 1A,B). Toxicity by HDAC3 was confirmed by TUNEL-staining (Fig. 1B) and active caspase-3 immunocytochemistry (data not shown). Expression of HDAC3 also induces death in otherwise healthy cortical neurons and exacerbates death of these neurons resulting from exposure to HCA, a stimulus that induces oxidative stress (Fig, 1C). In contrast to neurons, HDAC3 has no effect on the viability of primary kidney fibroblasts (Fig. 1C), indicating that toxicity by HDAC3 is cell-selective and that neuronal cells are vulnerable. In support of this conclusion, we find that forced expression of HDAC3 is toxic in the hippocampally derived HT22 cell line, but has no effect on the survival of the HEK293 or HeLa cell lines, both of which are of non-neuronal origin (Fig. 1D). Control experiments confirmed that ectopic HDAC3 was expressed at comparable levels across the different cell types (data not shown).
Figure 1.
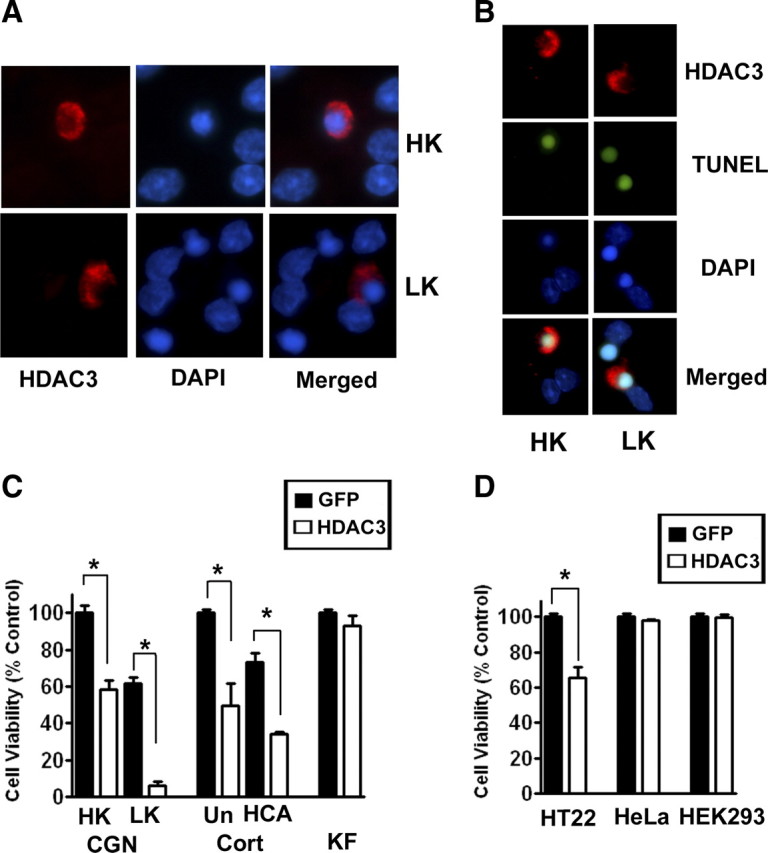
Forced expression of HDAC3 kills neurons selectively. Primary neuronal and non-neuronal cells and cell lines were transfected with plasmids encoding HDAC3-Flag or GFP. Effect of elevated HDAC3 expression on cell viability was evaluated. A, Immunofluorescence images of CGNs transfected with HDAC3-Flag and later switched to HK or LK medium for 24 h. B, TUNEL staining of HDAC3-Flag transfected CGNs after treatment with HK or LK for 24 h. C, Quantification of viability in primary cultures. CGNs, cortical neurons (Cort), and kidney fibroblasts (KF) were transfected with HDAC3 or GFP-expressing plasmids. Eight hours after transfection, CGNs were switched to either HK or LK medium and cortical neurons were either left untreated (Un) or treated with HCA. Kidney fibroblasts were left without additional treatments. D, Quantification of viability in cell lines. HT22, HeLa, and HEK293 cells were transfected with HDAC3 or GFP. Viability was quantified 32 h after transfection and compared with control cultures transfected with GFP. *p < 0.001.
Although elevating HDAC3 levels by ectopic expression kills neurons, the expression of endogenous HDAC3 mRNA or protein is not altered by LK treatment of CGNs (Fig. 2A,B) or in cortical neurons after HCA treatment (data not shown). However, suppression of HDAC3 expression using two separate HDAC3 shRNA constructs protects CGNs and cortical neurons against death resulting from LK or HCA treatment, respectively (Fig. 2C–F). The two HDAC3 shRNAs had no effect on the expression of HDRP and HDAC4, arguing against the possibility that protection was due to a compensatory upregulation of other neuroprotective HDAC proteins (Fig. 2E). These results suggest that HDAC3 plays an essential role in promoting neuronal death.
Figure 2.
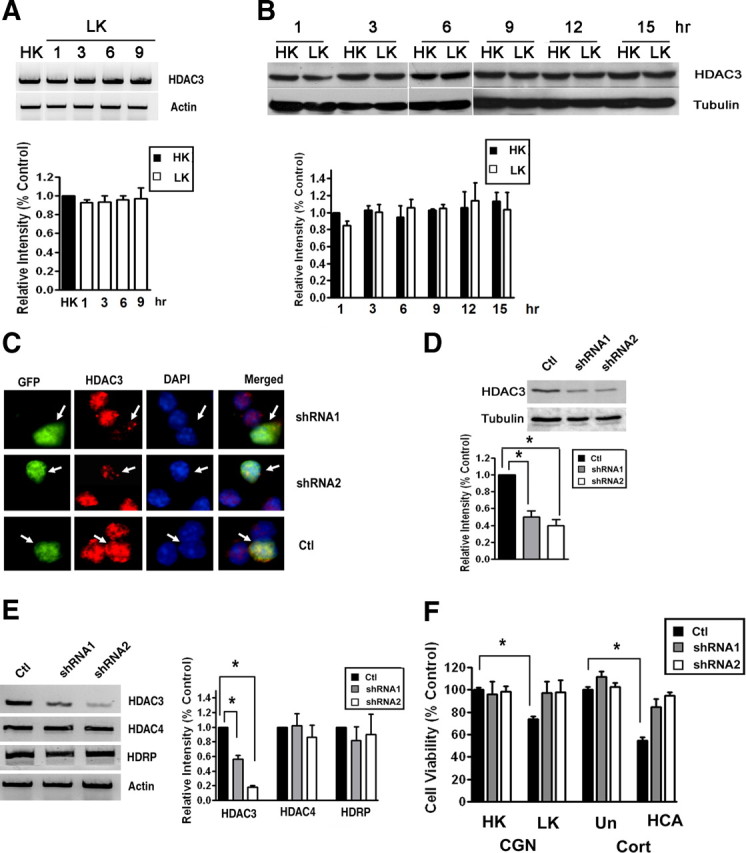
Suppression of HDAC3 expression protects neurons. A, CGNs were treated with LK for 1, 3, 6, or 9 h. Control cultures received HK for 9 h (HK). Total RNA was extracted and RT-PCR performed using specific primers for HDAC3. Actin served as loading control. The lower panel shows the densitometric analysis of the RT-PCR data from three separate experiments. B, CGNs were treated with HK or LK medium for 1, 3, 6, 9, 12, or 15 h. Expression of HDAC3 protein was analyzed by Western blotting. The blot was reprobed with a tubulin antibody. Densitometric analysis of the bands from different experiments was performed and normalized to tubulin. C, CGNs were cotransfected with plasmids expressing GFP with control shRNA (Ctl), shRNA-1, or shRNA-2 for 72 h. The cells were costained with GFP and HDAC3 antibodies and nuclear morphology visualized by DAPI staining. Arrows point to successfully transfected cells (GFP-positive). Neurons receiving either shRNA construct show reduced HDAC3 immunostaining whereas cells cotransfected with the control shRNA show similar HDAC3 staining as untransfected cells. D, E, Lysates from HT22 cells transfected with control shRNA, shRNA-1, or shRNA-2 were analyzed by Western blotting or RT-PCR. D, Western blot results using HDAC3 and tubulin antibodies. Densitometric analysis of the bands was performed and normalized to tubulin. E, Results of RT-PCR analysis of HDAC3, HDAC4, HDRP, and actin (loading control) expression. Also included is densitometric analysis of the data from three different experiments normalized to actin. F, CGNs and cortical neurons (Cort) were transfected with plasmids expressing control shRNA, shRNA-1, or shRNA-2 and cell viability quantified as described in Materials and Methods. After transfection, CGNs were treated with HK or LK for 24 h whereas cortical neurons were either untreated (Un) or treated with HCA for 20 h. Cell viability is normalized to survival in cultures transfected with control shRNA and treated in HK (CGN) or untreated (Cort). *p < 0.05.
IGF-1 suppresses HDAC3-mediated neurotoxicity
IGF-1, the physiological survival-promoting factor for CGNs (as well as many other neuronal populations) in vivo, also protects cultured CGNs from LK-induced death (D'Mello et al., 1997; Dudek et al., 1997). As shown in Figure 3A, death induced by forced HDAC3 expression is prevented by IGF-1. It is well established that the survival-promoting effect of IGF-1 is mediated through the activation of the PI-3 kinase–Akt signaling pathway (D'Mello et al., 1997; Dudek et al., 1997). Not unexpectedly, the neuroprotective effect of IGF-1 against HDAC3 neurotoxicity is reduced by wortmannin and LY294002, two structurally distinct inhibitors of P1–P3 kinase (Fig. 3B). Additionally, the expression of a constitutively active form of Akt prevents HDAC3-induced neuronal death (Fig. 3A).
Figure 3.

HDAC3-induced neurotoxicity is inhibited by IGF-1 and activation of the PI-3 kinase–Akt signaling pathway. A, CGNs were transfected with HDAC3-Flag alone or in combination with a plasmid expressing HA-tagged constitutively active Akt (CA-Akt-HA). The transfected cultures were then treated with no additives or with IGF-1. B, CGNs were transfected with HDAC3-Flag and then treated with no additives or with IGF-1, IGF-1, and wortmannin, or IGF-1 and LY294002. Eight hours later, the cultures were switched to serum-free medium containing inhibitors, and cell viability assessed 24 h later. C, CGNs were transfected with HDAC3-Flag and treated with HK medium containing either no additives, supplemented with JNK inhibitor (SP600125), CDK inhibitor (roscovitine), GSK3 inhibitor (SB415826 or SB216763), or cotransfected with a dominant-negative form of GSK3β (GSK3β-K85A). Cell viability was assessed 24 h after switching to HK medium. Viability in all cases was normalized to control cultures, which were transfected with GFP and treated with HK medium. *p < 0.01.
Inhibition of GSK3β protects against HDAC3-induced neurotoxicity
An important downstream target of Akt is GSK3β, a kinase implicated in promoting neurodegeneration in experimental models as well as patients (Bhat et al., 2004; Hernández et al., 2009). Akt inhibits GSK3β by directly phosphorylating it (Cross et al., 1995; Shaw et al., 1997). As shown in Figure 3C, treatment with either of two separate but specific inhibitors of GSK3β, SB415826 or SB216763, blocked HDAC3-induced neurotoxicity. Similarly, expression of a dominant-negative form of GSK3β (K85A) substantially reduces HDAC3-mediated neurotoxicity (Fig. 3C). Activation of c-Jun N-terminal kinase (JNK) and cyclin-dependent kinases (CDKs) also plays an important role in promoting neuronal death in a variety of in vitro and in vivo experimental paradigms (Borsello and Forloni, 2007; Greene et al., 2007). However, inhibition of JNK (using SP600125) and CDK (using roscovitine) has no effect on the neurotoxicity by HDAC3 (Fig. 3C). It is noteworthy that, unlike GSK3β, JNK and the CDKs are not targets of Akt. Control experiments showed that both inhibitors blocked LK-induced neuronal death completely (data not shown).
HDAC3 is a substrate of GSK3β
Given that pharmacological inhibition of GSK3β inhibits HDAC3 neurotoxicity, we explored the possibility that HDAC3 was directly phosphorylated by GSK3β. As shown in Figure 4A, purified GSK3β enzyme does phosphorylate HDAC3 when tested in an in vitro kinase assay. To validate this result in cultured neurons, we performed in vivo metabolic labeling using 32P to label endogenous HDAC3 that was phosphorylated. Although phosphorylation of HDAC3 is barely discernible in HK, it is clearly evident in cultures treated with LK (Fig. 4B). The extent of phosphorylation is markedly reduced by treatment with GSK3β inhibitors. To confirm that the LK-induced phosphorylation in neurons is mediated by GSK3β, we immunoprecipitated GSK3β from CGNs treated with and without the GSK3β inhibitor SB415826, and tested its ability to phosphorylate HDAC3 in vitro. The robust level of phosphorylation seen in LK is abolished in cultures treated with SB415826 (Fig. 4C). In contrast to GSK3β, phosphorylation of HDAC3 was not observed with CDK5, another kinase that is known to regulate neuronal death and implicated in neurodegenerative pathologies (Shelton and Johnson, 2004; Camins et al., 2006).
Figure 4.
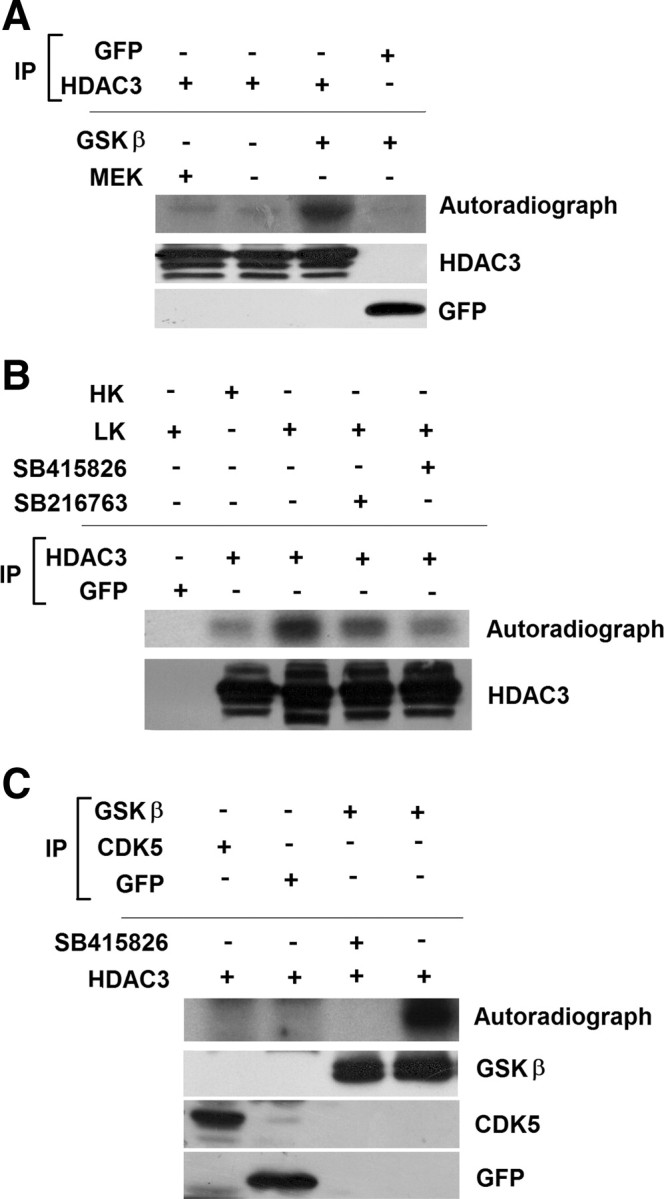
HDAC3 is directly phosphorylated by GSK3β. A, Lysates from HEK293 cells transfected with either GFP or HDAC3 were immunoprecipitated (IP) using either GFP or Flag antibody. The immunoprecipitated proteins were used in an in vitro kinase assay with or without active GSK3β or with active MEK. The reaction mixture was subjected to PAGE and phosphorylation evaluated by autoradiography. Immunoblots show that HDAC3-Flag and GFP were immunoprecipitated in the samples used for the kinase assay. B, CGN cultures were metabolically labeled with [32P] orthophospate in HK, LK, or LK media with GSK3 inhibitors for 3 h. Lysates from the cultures were immunoprecipitated using GFP or HDAC3 antibody and the extent of phosphorylation evaluated by PAGE and autoradiography. The bottom shows that HDAC3 was immunoprecipitated in the samples used for the kinase assay. C, Equal amounts of GSK3β, CDK5, or GFP immunoprecipitated from CGNs treated with LK for 6 h were combined with HDAC3 in an in vitro kinase assays with or without a GSK3 inhibitor as indicated. HDAC3 phosphorylation was evaluated by PAGE of the reaction mixture and autoradiography. Bottom, Similar amounts of GFP, GSK3β, and CDK5 were used in the assay.
Discussion
Although it is well established that pharmacological inhibitors of classical HDACs are protective in various cell culture and in vivo models of neurodegenerative disease, the identity of the neurotoxic HDAC(s) targeted by these inhibitors to exert their protective effects is less clear. In this study, we show that HDAC3 has strong neurotoxic activity and thus represents a potential target of HDAC inhibitors found to be protective in models of neurodegenerative disease. Although a more comprehensive study is required, our limited analysis suggests that the toxic activity of HDAC3 is selective for primary neurons and cell lines of neuronal origin. Indeed, whereas primary CGNs, cortical neurons, and HT22 cells are sensitive, the viability of primary kidney cells and the HEK293 and HeLa cell lines is not affected by elevated HDAC3. Our conclusion that HDAC3 is likely to be involved in neurodegenerative disorders is supported by the finding that suppression of Hda-3 (a nematode homolog of HDAC-3) expression suppresses polyQ-huntingtin toxicity in a C. elegans model of Huntington's disease (HD), whereas neuronal expression of hda-3 restored toxicity by polyQ-huntingtin (Bates et al., 2006). Another laboratory has reported that a pharmacological inhibitor selective for HDAC3 reverses silencing of the frataxin gene in lymphocytes isolated from patients with Frederich's ataxia and is efficacious in a mouse model of HD (Rai et al., 2008; Thomas et al., 2008).
Although our studies demonstrate that HDAC3 is a potent stimulator of neuronal death when overexpressed, CGNs and cortical neurons express HDAC3 normally. Moreover, its expression is not increased when these neurons are induced to die. Yet the suppression of endogenous HDAC3 expression protects neurons from death. This suggests that under normal conditions, the apoptotic activity of HDAC3 is kept in check by survival-promoting signaling molecules. We propose that in neurons primed to die, the levels or activities of these survival-promoting molecules are reduced, permitting HDAC3 to induce neuronal death. Our results suggest that Akt is such a survival-promoting signaling molecule. Consistent with this conclusion is our finding that expression of an active form of Akt or treatment with IGF-1, a physiological activator of Akt, protects against the neurotoxic effect of HDAC3.
A well established target of Akt is GSK3β. Inhibition of GSK3β either pharmacologically or caused by expressing a dominant-negative form of the kinase protects against HDAC3-mediated neurotoxicity. Activation of GSK3β has been implicated in the pathogenesis of different neurodegenerative diseases, and the inhibition of this kinase has been considered a therapeutic approach in the treatment of these disorders (Bhat et al., 2004; Hernández et al., 2009). Despite the long-standing acceptance that GSK3β plays a key role in promoting neuronal death, little is known about what its substrates are in the context of neurodegeneration. We show that HDAC3 is directly phosphorylated by GSK3β, suggesting that the neurodegeneration-promoting effect of GSK3β could be mediated through the phosphorylation of HDAC3. The specific residues within HDAC3 that are phosphorylated by GSK3β remain to be delineated (sequence analysis of HDAC3 reveal three consensus sites for GSK3β phosphorylation). More work is also needed to determine the downstream mechanism by which phosphorylation of HDAC3 promotes neurotoxicity. An obvious possibility is that GSK3β-mediated phosphorylation stimulates the deacetylase activity of HDAC3, resulting in the deacetylation of specific proteins that regulate neuronal death.
Although GSK3β is activated in neurons primed to die (Chin et al., 2005), and we show that GSK3β phosphorylates HDAC3 when active, and that the inhibition of GSK3β protects against HDAC3 toxicity, we have not found a strict correlation between the total levels of Akt or GSK3β or their activity (evaluated indirectly by looking at phosphorylation status) and vulnerability to HDAC3 toxicity in the cell types used in this study. This suggests that activation of GSK3β is by itself not sufficient to explain HDAC3-induced toxicity, pointing to the involvement of other cell-specific components and mechanisms. For example, the accessibility of HDAC3 to GSK3β could be regulated differently in different cell types. It is also possible that the toxic activity of HDAC3 is dependent on the participation of another molecule that is expressed selectively in neurons, or that a molecule capable of blocking the toxic activity HDAC3 could be missing in neurons.
In summary, our study has produced three important findings. First, we have identified HDAC3 as a protein with neurotoxic activity. Although protection by HDAC inhibitors in experimental models of neurodegeneration has pointed to the existence of neurotoxic proteins within the HDAC family, conclusive identification of such a protein has been elusive. Second, we provide evidence suggesting that IGF-1, a trophic factor for many neuronal populations in vivo, promotes neuronal survival at least in part by suppressing the neurotoxic activity of HDAC3. Finally, we show that HDAC3 is a substrate of GSK3β. Although the involvement of GSK3β in promoting neurodegeneration both in experimental models and human pathologies is well accepted, the downstream mechanism by which this kinase promotes neuronal death is poorly understood. We suggest that HDAC3 represents a downstream effector of GSK3β neurotoxicity.
Footnotes
This work was supported by grants from the National Institute of Neurological Diseases and Stroke (NS40408 and NS058462) to S.R.D. We thank Brandi Tompkins for her assistance with some of the transfection experiments.
References
- Bates EA, Victor M, Jones AK, Shi Y, Hart AC. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J Neurosci. 2006;26:2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13:1875–1886. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc'h C, Matthias P, Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camins A, Verdaguer E, Folch J, Canudas AM, Pallàs M. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect. 2006;19:453–460. doi: 10.1358/dnp.2006.19.8.1043961. [DOI] [PubMed] [Google Scholar]
- Chen HM, Wang L, D'Mello SR. A chemical compound commonly used to inhibit PKR, {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one}, protects neurons by inhibiting cyclin-dependent kinase. Eur J Neurosci. 2008;28:2003–2016. doi: 10.1111/j.1460-9568.2008.06491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- Chin PC, Majdzadeh N, D'Mello SR. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res Mol Brain Res. 2005;137:193–201. doi: 10.1016/j.molbrainres.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- D'Mello SR. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009;22:513–524. doi: 10.1358/dnp.2009.9.1428871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor 1 and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta. 2007;1772:392–401. doi: 10.1016/j.bbadis.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández F, Nido JD, Avila J, Villanueva N. GSK3 inhibitors and disease. Mini Rev Med Chem. 2009;9:1024–1029. doi: 10.2174/138955709788922647. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, D'Mello Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem. 2011 doi: 10.1074/jbc.M110.146860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D'Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D'Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D'Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, Geschwind DH, Gottesfeld JM, Pandolfo M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivieccio MA, Brochier C, Willis DE, Walker BA, D'Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci U S A. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416:307–311. doi: 10.1016/s0014-5793(97)01235-0. [DOI] [PubMed] [Google Scholar]
- Shelton SB, Johnson GV. Cyclin-dependent kinase-5 in neurodegeneration. J Neurochem. 2004;88:1313–1326. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA, Coppola G, Desplats PA, Tang B, Soragni E, Burnett R, Gao F, Fitzgerald KM, Borok JF, Herman D, Geschwind DH, Gottesfeld JM. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ankati H, Akubathini SK, Balderamos M, Storey CA, Patel AV, Price V, Kretzschmar D, Biehl ER, D'Mello SR. Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J Neurosci Res. 2010;88:1970–1984. doi: 10.1002/jnr.22352. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Ippolito G, Wang L, Price V, Kim MH, Cornwell G, Fulenchek S, Breen GA, Goux WJ, D'Mello SR. Neuron-selective toxicity of tau peptide in a cell culture model of neurodegenerative tauopathy: essential role for aggregation in neurotoxicity. J Neurosci Res. 2010;88:3399–3413. doi: 10.1002/jnr.22485. [DOI] [PubMed] [Google Scholar]