

Abstract
The plt (pale tremor) mouse carries a null mutation in the Fig4(Sac3) gene that results in tremor, hypopigmentation, spongiform degeneration of the brain, and juvenile lethality. FIG4 is a ubiquitously expressed phosphatidylinositol 3,5-bisphosphate phosphatase that regulates intracellular vesicle trafficking along the endosomal–lysosomal pathway. In humans, the missense mutation FIG4I41T combined with a FIG4 null allele causes Charcot-Marie-Tooth 4J disease, a severe form of peripheral neuropathy. Here we show that Fig4 null mice exhibit a dramatic reduction of myelin in the brain and spinal cord. In the optic nerve, smaller-caliber axons lack myelin sheaths entirely, whereas many large- and intermediate-caliber axons are myelinated but show structural defects at nodes of Ranvier, leading to delayed propagation of action potentials. In the Fig4 null brain and optic nerve, oligodendrocyte (OL) progenitor cells are present at normal abundance and distribution, but the number of myelinating OLs is greatly compromised. The total number of axons in the Fig4 null optic nerve is not reduced. Developmental studies reveal incomplete myelination rather than elevated cell death in the OL linage. Strikingly, there is rescue of CNS myelination and tremor in transgenic mice with neuron-specific expression of Fig4, demonstrating a non-cell-autonomous function of Fig4 in OL maturation and myelin development. In transgenic mice with global overexpression of the human pathogenic FIG4 variant I41T, there is rescue of the myelination defect, suggesting that the CNS of CMT4J patients may be protected from myelin deficiency by expression of the FIG4I41T mutant protein.
Introduction
Proper myelination is required for rapid propagation of action potentials, axonal health, and fiber stability. The importance of myelination in the CNS is demonstrated by a number of hereditary leukodystrophies and multiple sclerosis and in the PNS by Guillain-Barre and demyelinating forms of Charcot-Marie-Tooth disease (CMT) (Gold et al., 2000; Kaye, 2001; Dubourg et al., 2006).
The pale tremor (plt) mouse exhibits many neurological deficits, including severe tremor, neurodegeneration, and defective myelination of the sciatic nerve (Chow et al., 2007). The plt mouse is null for Fig4(Sac3), an evolutionarily conserved phosphatase that regulates intracellular levels of the endolysosome-specific phosphoinositide PI(3,5)P2 (Chow et al., 2007; Poccia and Larijani, 2009). Fibroblasts from Fig4−/− mice accumulate enlarged late endosomes and lysosomes that are reminiscent of the enlarged vacuoles in yeast deficient for Fig4p (Rudge et al., 2004; Chow et al., 2007). In the forebrain of Fig4−/− mice, autophagy intermediates accumulate in astrocytes and, to a lesser extent, in neurons, indicating that PI(3,5)P2 is required for completion of basal autophagy (Ferguson et al., 2009). PI(3,5)P2 was shown recently to bind and activate the endolysosome-specific Ca2+ release channel mucolipin (TRPML1) (Dong et al., 2010). It was suggested that activation of TRPML1 by PI(3,5)P2 could trigger membrane fusion events by regulating juxtaorganellar Ca2+ concentration.
FIG4 is part of a large protein complex that includes the PI(5) kinase FAB1 (PIKFyve/PIP5K3) and the scaffold protein VAC14 (ArPIKFyve) (Jin et al., 2008; Dove et al., 2009). Colocalization of FIG4 phosphatase and its antagonistically acting kinase FAB1 in the same protein complex is thought to facilitate rapid and local interconversion of PI(3) and PI(3,5)P2 (Jin et al., 2008; Botelho, 2009). Dynamic regulation of vesicular PI(3,5)P2 levels is thought to permit precise regulation of vesicle trafficking (Di Paolo and De Camilli, 2006).
Mutations of FIG4 and TRPML1 have been identified in patients with neurodegenerative diseases, including CMT and mucolipidosis type IV (Slaugenhaupt, 2002; Chow et al., 2007). CMT4J is a rare, recessive disorder that accounts for ∼0.2% of CMT (Nicholson et al., 2011). CMT4J patients carry the FIG4 missense allele p.I41T in combination with a null allele (Chow et al., 2007). The I41T mutation impairs interaction with the anchor protein VAC14, resulting in instability of the FIG4 protein (Lenk et al., 2011). CMT4J patients lack upper motor signs and cognitive dysfunction (Zhang et al., 2008; Nicholson et al., 2011), indicating that CNS function is essentially intact. This is in marked contrast to the spongiform degeneration in the brain of Fig4−/− null mice (Chow et al., 2007). Global overexpression of a FIG4I41T transgene in null mice results in phenotypic rescue, demonstrating that the mutant protein retains functional activity in vivo (Lenk et al., 2011).
Defects in PNS myelination have been reported for human CMT4J subjects and mice null for Fig4 (Chow et al., 2007; Zhang et al., 2008). The defective PNS myelination, coupled with the severe action tremor that develops in Fig4−/− mice during the second postnatal week, prompted us to investigate the role of Fig4 in CNS myelination. Here we show that Fig4 function is critical for oligodendrocyte (OL) maturation and normal CNS myelination. Transgenic rescue experiments demonstrate that impaired myelination in Fig4 null mice is secondary to neuronal defects and that Fig4I41T is protective of the defect in CNS myelination.
Materials and Methods
Mouse breeding, transgenic mice, and genotyping.
All mice used in this study were housed and cared for in accordance with National Institutes of Health guidelines, and all research conducted was done with the approval of the University of Michigan and University of Rochester Committees on Use and Care of Animals. plt mice carry a spontaneous null mutation of Fig4 that originated on a mixed-strain background (Chow et al., 2007) and is maintained as a recombinant inbred line designated CB.plt that is now at generation N12 (Lenk et al., 2011). To obtain homozygous plt mice of either sex, Fig4 heterozygous mice (Fig4+/−) were crossed and genotyped as described previously (Chow et al., 2007). The transgenic lines Tg705 and Tg721 express a mouse Fig4I41T cDNA carrying the human pathogenic I41T missense mutation under the transcriptional control of the globally expressed chicken β-actin promoter (Lenk et al., 2011). The two independent transgenic mouse lines, TgN1 and TgN3 (C.J.F., G.M.L., and M.H.M., unpublished observation) express full-length wild-type (WT) Fig4 cDNA under the transcriptional control of the 2.8 kb neuron-specific enolase (NSE) promoter derived from the rat Eno2 gene; this promoter has been widely used for neuron-specific transgene expression (Forss-Petter et al., 1990; Race et al., 1995; Kearney et al., 2001).
Brain membrane and myelin preparation.
Brains from postnatal day 21 (P21) Fig4 WT and mutant mice were dissected, and equal amounts of tissue (2.0 g) were homogenized in 10 ml of ice-cold low-sucrose buffer [0.25 m sucrose, 10 mm HEPES, pH 7.4, 3 mm DTT, 5 mm EDTA, and 200 μl of tissue protease inhibitor mix (Sigma)] using a 15 ml Wheaton Dounce tissue homogenizer cooled on ice. The tissue homogenates were adjusted to 1.4 m sucrose by addition of ice-cold high-sucrose buffer (1.9 m sucrose, 10 mm HEPES, pH 7.4, 3 mm DTT, 5 mm EDTA, and 1:200 protease inhibitor mixture). In a 13 ml centrifuge tube, 7 ml of brain extract were underlaid with 5 ml of ice-cold high-sucrose buffer and spun for 1 h at 75,000 × g (Sorval SW41 rotor). Total brain membranes enriched at the 0.25/1.4 m sucrose interphase were collected, diluted 1:10 in ice-cold 10 mm HEPES, pH7.4, homogenized on ice, and spun for 20 min at 75,000 × g. The pellets were resuspended in 10 mm HEPES, the protein concentration was determined with the Pierce BCA kit, aliquoted, and frozen at −80°C. For the isolation of myelin, brain homogenates were fractioned as described previously (Robak et al., 2009).
Western blot analysis.
Proteins from total brain membranes and myelin fractions were separated by SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare). An equal amount of protein (2 μg) was loaded per lane. Membranes were blocked in 2% milk in Tris-buffered saline with 0.3% Triton X-100 for at least 1 h and incubated with primary antibody overnight at 4°C. Primary antibodies included mouse anti-βIII tubulin (1:1000; Promega), mouse anti-GFAP (1:5000; Sigma), rabbit anti-myelin-associated glycoprotein (MAG) (1:1000; custom made), rat anti-myelin basic protein (MBP) (1:500; Millipore), and rabbit anti-proteolipid protein (PLP) (1:5000; custom made). Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3000; Millipore Bioscience Research Reagents or GE Healthcare). Rabbit anti-MAG and rabbit anti-PLP polyclonal antibodies were generated by immunization of rabbits with either a 6-histidine-tagged fusion protein of the rat l-MAG cytoplasmic portion or cytoplasmic loop of rat PLP. Histidine-tagged fusion proteins were expressed in Escherichia coli using the pTrcHis expression system (Invitrogen), purified over an nickel-nitrilotriacetic acid column, and used for immunization (Venkatesh et al., 2005).
Quantitative reverse transcription-PCR.
Total RNA was isolated from fresh whole brain using the TriZOL (Invitrogen) reagent. RNA was quantified using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Five microgram aliquots of total RNA from P21 WT or Fig4−/− mice were treated with amplification-grade DNase I (Invitrogen) and used in first-strand cDNA synthesis (Invitrogen) reaction using oligo-dT primers (Invitrogen). Gene expression was determined using predesigned TaqMan assays for MAG (assay identification number Mm00487538_m1), MBP (assay identification number Mm01262037_m1), PLP1 (assay identification number Mm00456892_m1), PDGFRα (assay identification number Mm00440701_m1), and NG2/CSPG4 (assay identification number Mm00507256_m1) using the Gene Expression Master Mix (Applied Biosystems). Fluorescence was measured on a Step-One Real-Time PCR System (Applied Biosystems) at the Microarray Core at the University of Michigan. The mean CT value for each gene was calculated from quadruplicate measurements of each sample. The mean value for each of the genes was normalized to the mean value obtained for the TATA box binding protein (TAT-bp) transcript (assay identification number Mm00446971_m1) assayed in the same experiment: ΔCT = CT(TAT-bp) − CT(gene). This normalized value (ΔCT) is shown for each biological replicate.
Histochemical and morphometric procedures.
Mice were deeply anesthetized with a mixture of ketamine and xylazine and transcardially perfused with 4% paraformaldehyde. Retina, optic nerve, sciatic nerve, brain, and spinal cord were dissected, postfixed in perfusion solution for 1–2 h, and cryoprotected in 30% sucrose.
For in situ hybridization with riboprobes specific for MAG and NG2 transcripts, neural tissues of perfused mice were embedded in OCT (Sakura Finetek), cryosectioned to 20 μm, and mounted onto Superfrost Plus (Thermo Fisher Scientific) microscope slides. After postfixation in 4% paraformaldehyde solution for 20 min, sections were rinsed, treated with 50 μg/ml proteinase K in PBS containing 5 mm EDTA, incubated with 0.25% acetic anhydride in 1% triethanolamine, and prehybridized for 2 h in hybridization solution (1× Denhardt's solution, 100 μg/ml yeast tRNA, 50% formamide, and 5× SSC) at room temperature. For in situ hybridization, unfixed brain tissue of P21 Fig4+/+, Fig4−/− and Fig4−/−,TgN1 mice were cryosectioned at 25 μm and mounted onto Superfrost Plus (Thermo Fisher Scientific) microscope slides. After postfixation in 4% paraformaldehyde solution for 20 min, sections were rinsed and incubated with 0.25% acetic anhydride in 1% triethanolamine and then prehybridized for 12 h at room temperature. A 2.3 kb fragment of the mouse Fig4 cDNA containing the entire coding region was cloned via HindIII into the pSP72 vector (Promega) in both 5′ → 3′ and 3′ → 5′orientations and linearized with Xba1. Sp6–RNA polymerase was used to generate sense and antisense riboprobes as described previously (Giger et al., 1996).
For double-immunofluorescence labeling, tissues were embedded in OCT and cryosectioned at 25 μm for mounting on slides or 50 μm for free-floating sections. Mounted sections of P7, P19, and P21 mice were blocked with PBS containing 5% horse serum and 0.3% Triton X-100 and incubated with mouse anti-MAG (1:1000; Millipore Bioscience Research Reagents), mouse CC1 (APC, 1:250; Calbiochem), rabbit anti-neurofilament-M (NF-M) (1:200; Millipore), rabbit anti-cleaved caspase-3 (1:250; Promega), rabbit anti-Olig2 (1:500; Millipore), mouse anti-p62 (1:200, SQSTMI; Abrora), rabbit anti-GFAP (1:5000; Millipore Bioscience Research Reagents), and detected with Alexa Fluor-488- or Alexa Fluor-594-conjugated secondary antibody (1:3000; Millipore Bioscience Research Reagents or GE Healthcare). Some sections were counterstained with the nuclear dye Hoechst (1:30,000) for 5 min. Processing of free-floating brain sections (50 μm) was performed as described previously (Giger et al., 2000). Briefly, sections were quenched with 3% H2O2/24% DMSO/73% methanol for 4 h, rinsed, blocked with 10% fetal bovine serum, and incubated in rat anti-MBP (1:500; Millipore). HRP-conjugated antibody (1:1000; Millipore) was used to visualize bound anti-MBP. Stained sections were examined with an Olympus IX71 fluorescence microscope attached to a DP72 camera. For quantification of oligodendrocyte precursor cells (OPCs) per field of view (FOV), pictures of the corpus callosum (648 × 864 μm) were taken at P7, before the onset of myelination. The corpus callosum was identified as low-cell-density area after Hoechst nuclear staining with reagent 33342 (1:30,000). For quantification of TuJ1-positive retinal ganglion cells (RGCs) at P19, pictures (FOV, 500 × 250 μm) were analyzed. The number of Oligo2-expressing (Olig2+) cells within the corpus callosum and the number of TuJ1+ RGCs per FOV was averaged over four consecutive sections. A total of three mice per genotype was analyzed. Only anti-Olig2- or TuJ1-immunolabeled cells that had a Hoechst-labeled nucleus were counted. OPCs per FOV in cerebellar lobules were quantified in a similar manner. The number of Olig2+ cells per FOV (324 × 432 μm) was quantified and averaged over four consecutive sections (n = 3) per genotype.
For analysis of nodes of Ranvier, optic nerves were cut into 10 μm sections on a cryostat and mounted on chromalum-subbed microscope slides. These sections were permeabilized in 0.1 m phosphate buffer, pH 7.4, containing 0.3% Triton X-100 and 10% goat serum (PBTGS). Primary antibodies used were monoclonal anti-pan Na+ channels (Rasband et al., 1999) and polyclonal anti-Caspr (Peles et al., 1997). Secondary antibodies were anti-rabbit or anti-mouse, conjugated to Alexa Fluor-488 or Cy3. All antibodies were diluted in PBTGS. These sections were examined on a Nikon Microphot fluorescence microscope and imaged with a Hamamatsu C4742-95 cooled CCD camera controlled by Image-Pro (Media Cybernetics). Images were analyzed by manually counting, tagging, and classifying node/paranode regions. Each FOV was 68 × 55 μm. Twenty-nine FOVs from two Fig4+/+ mice and 27 FOVs from two Fig4−/− mice were analyzed.
Electron microscopy.
Mice at P10, P15, P21, P90, and P120 were deeply anesthetized with a mixture of ketamine and xylazine and perfused with 3% paraformaldehyde (Electron Microscopy Sciences) and 2.5% glutaraldehyde (Ted Pella) in 0.1 m Sorensen's buffer. Retina, optic nerve, sciatic nerve, brain, and spinal cord were dissected and postfixed at 4°C overnight in perfusion solution, incubated in OsO4 (1% solution in 0.1 m Sorenson's buffer) for 1 h, and embedded in epoxy resin. For light microscopy, 0.5 μm semithin sections were stained with toluidine blue. For transmission electron microscopy (TEM), ultrathin (75 nm) sections were cut at the University of Michigan Imaging Laboratory Core and visualized with a Philips CM-100 microscope. TEM images were analyzed at 10,500–64,000× magnification. NIH ImageJ software was used for quantification of fiber diameter, axon caliber, and percentage of axons myelinated in the optic nerve. To account for the oval shape of many axons in optic nerve cross-sections, diameters were measured twice per axon, once across the shorter distance and once across the longer. The mean of the two values obtained was used. To calculate g-ratios of myelinated axons, diameter of an axon in the optic nerve was determined and divided by the diameter of the same axon including the myelin sheath. A minimum of 300 axons was quantified for each developmental stage and genotype. The percentage of axons myelinated was assessed by counting the number of myelinated axons in a randomly selected FOV divided by the total number of axons in the same field. A total of at least 1500 axons was quantified per genotype at each developmental stage examined. For statistical analysis of the data, Student's t test in Microsoft Excel was used.
Optic nerve conduction recordings.
Age-matched pairs of Fig4 WT and mutant mice at P22–P24 were killed by CO2 inhalation. Both optic nerves from each animal were dissected and placed in oxygenated artificial CSF (ACSF). ACSF contained the following (mm): 125 NaCl, 1.25 NaH2PO4, 25 glucose, 25 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, and 2.5 KCl (saturated with 95% O2/5% CO2). Nerves were incubated in oxygenated ACSF for 45–60 min at room temperature and then transferred to a temperature-controlled recording chamber held at 37 ± 0.4°C. Each end of the nerve was drawn into the tip of a suction pipette electrode. One electrode was connected to a constant-current stimulus isolation unit (WPI) driven by the computer and the other to one input of a differential alternating current amplifier of our own design. The other amplifier input was connected to a pipette that was placed near the recording pipette but not in contact with the nerve. This electrode served to subtract most of the stimulus artifact from the records. Signals were filtered at 10 kHz, sampled at 50–100 kHz, and fed into a data acquisition system. For analysis, single peaks were fitted by a third-order polynomial, and the conduction velocity was taken as the length of the nerve divided by the time-to-peak. The multiple components from myelinated axons were fitted by the sum of three Gaussian curves (Chen et al., 2004). Velocities and amplitudes were then derived from the individual components.
The CABLE program (Hines, 1989; Hines and Shrager, 1991), modified to include an independently specified extracellular layer surrounding each segment (Hines and Shrager, 1991), was used to compute action potentials in model axons with 12 nodes and 11 internodes. Internodal lengths were taken from the data of Murray and Blakemore (1980). Other computational model parameters were as described previously (Shrager, 1993), except that for these mammalian axons the delayed rectifier K+ channels were juxtaparanodal.
Results
Reduction of CNS myelin in the plt mouse null for Fig4
To determine the effect of loss of Fig4 on myelination of CNS axons, brain and spinal cord of P21 WT (Fig4+/+) and plt (Fig4−/−) mice were cryosectioned and stained with anti-MBP, a marker for mature myelin. In coronal sections of the Fig4−/− brain, myelinated fiber tracts are reduced in size compared with age-matched Fig4+/+ controls (Fig. 1). Major brain commissures, such as the corpus callosum and the rostral branch of the anterior commissure, appear smaller and show less MBP immunoreactivity. The lateral ventricles of Fig4−/− mice are greatly enlarged (Fig. 1A--B′). In the cerebellum, myelinated fibers projecting into individual lobules show decreased MBP immunoreactivity (Fig. 1C--D′). Additionally, the thickness of the white matter is substantially reduced in the Fig4−/− spinal cord (Fig. 1E,E′). Axons of the corticospinal tract exhibit a particularly strong reduction in MBP immunoreactivity (Fig. 1F,F′).
Figure 1.
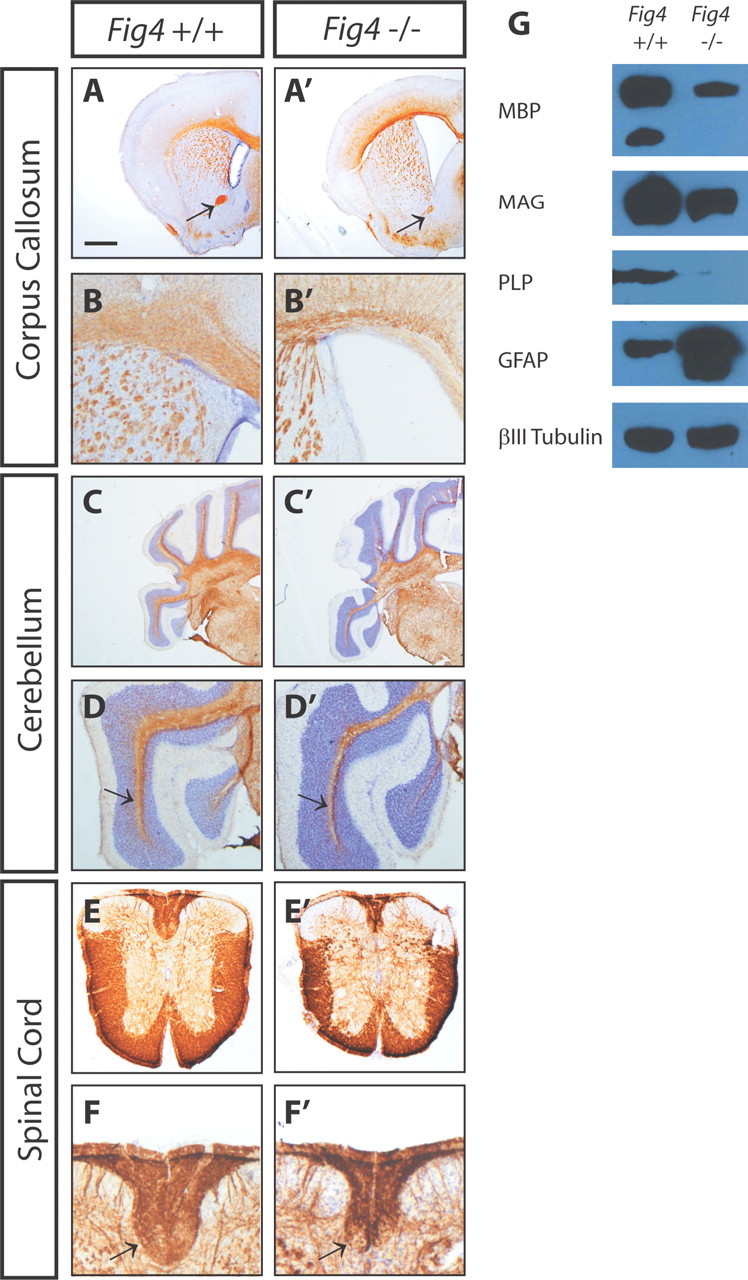
CNS hypomyelination in the Fig4−/− mouse. P21 WT (Fig4+/+) and mutant (Fig4−/−) brain and spinal cord sections were immunostained with anti-MBP (brown) and counterstained with Nissl (purple). A, A′, Coronal sections of forebrain show labeling of the corpus callosum and rostral branch of the anterior commissure (arrow). B, B′, Close-up of forebrain showing thinning of the corpus callosum and enlarged lateral ventricles in Fig4−/− compared with Fig4+/+ mice. C, C′, Coronal sections at the level of cerebellum and brainstem. D, D′, Close-up of cerebellum shows thinning of white matter (arrow) in lobules of Fig4−/− mice. E, E′, Cross-sections of thoracic spinal cord show loss of white matter in Fig4−/− mutants. F, F′, Close-up of dorsal funiculus shows decreased MBP labeling in Fig4−/− compared with Fig4+/+ spinal cord. Arrows point to corticospinal tract. G, Representative immunoblots of equal protein amounts of P21 Fig4+/+ and Fig4−/− brain membranes. A reduction of the myelin-associated proteins MBP, PLP, and MAG is observed in Fig4−/− brain membranes. The most pronounced reduction is observed for PLP. GFAP is greatly increased in Fig4−/− mutants, although no changes in neuron-specific class III β-tubulin are found. Scale bar: A, A′, C, C′, 1 mm; B, B′, D, D′, 0.3 mm; E, E′, 0.5 mm; F, F′, 0.2 mm.
Despite their reduced body size (Chow et al., 2007), the wet weight of brain from Fig4−/− mice (0.38 ± 0.04 g, n = 8) and Fig4+/+ mice (0.39 ± 0.03 g, n = 6) at 1 month of age (28 ± 2.5 d) do not differ. To independently assess the apparent decrease of myelin in Fig4−/− mice, total brain membranes from Fig4+/+ and Fig4−/− mice were isolated and analyzed by immunoblotting with antibodies specific for MBP, PLP, and MAG. Consistent with the immunohistochemical data, immunoblots showed a reduction of MBP in Fig4−/− brain membranes (Fig. 1G). In addition, PLP and MAG are less abundant in the Fig4−/− brain compared with age-matched Fig4+/+ controls (Fig. 1G). Analysis of purified myelin revealed a >50% decrease in total myelin protein, but the protein composition of the Fig4−/− myelin appears indistinguishable from Fig4+/+ controls (data not shown). In agreement with previous observations (Ferguson et al., 2009), GFAP, a marker for astrocytes in the more mature CNS, is elevated in Fig4 null brain, indicative of reactive gliosis. However, there is no apparent change in the neuronal marker class III β-tubulin at P21 (Fig. 1G). Collectively, immunohistochemical and biochemical studies show that loss of Fig4 leads to a pronounced reduction in CNS myelin.
Normal number of axons in optic nerve of Fig4 null mice
Because of its easy accessibility and well-defined anatomy, the optic nerve was chosen for in-depth analysis of defects in CNS myelination. Because Fig4−/− mice die between 1 and 2 months of age (Chow et al., 2007), analysis of the retina and optic nerve was performed at P21, a late developmental stage when myelination of RGC axons is essentially complete (Fig. 2). At 3 weeks of age, the optic nerve diameter of Fig4−/− mutants (249 ± 5 μm, n = 6) is 22% smaller than in Fig4+/+ (319 ± 10, n = 6) mice (p < 0.05). To determine whether the reduced nerve size is a result of retinal defects, we examined retinae of Fig4+/+ and Fig4−/− mice at P19–P21. Toluidine blue staining revealed normal retinal stratification and proper development of the optic nerve head (Fig. 2A,B). The inner plexiform layer (IPL) of Fig4−/− mutants contained vacuole-like structures reminiscent of those in deep layers of the neocortex reported previously (Chow et al., 2007). The penetrance of this phenotype is 100%, but its expressivity varies greatly, ranging from few small (∼20 μm) vacuoles in the IPL (Fig. 2B) to numerous large (∼50 μm) vacuoles (Fig. 2D). At P19, the number of TuJ1-positive RGCs is indistinguishable between the Fig4+/+ (27 ± 5, n = 3) and Fig4−/− (30 ± 4, n = 3) mice per FOV (Fig. 2E,F), and GFAP immunofluorescence appears not significantly altered (Fig. 2G,H). At P19, cell death in the RGC layer, as assessed by staining for activated caspase-3, was not elevated in Fig4−/− mice (data not shown). For an independent assessment of RGC numbers, axons in the optic nerve of Fig4+/+ and Fig4−/− mice were counted. Analysis of P21 optic nerve cross-sections by TEM revealed an average of 72,300 ± 4100 axons per Fig4+/+ optic nerve (n = 3) and 74,200 ± 3700 axons per Fig4−/− optic nerve (n = 3). Morphometric studies revealed a small shift in the distribution profile of axon caliber in Fig4−/− toward smaller calibers (Fig. 2K). This is most likely a reflection of the severe hypomyelination described below, because it has been shown that myelination increases the axon caliber (Yin et al., 1998). Together, these studies show that the number of RGCs in Fig4 WT and mutant mice is comparable.
Figure 2.
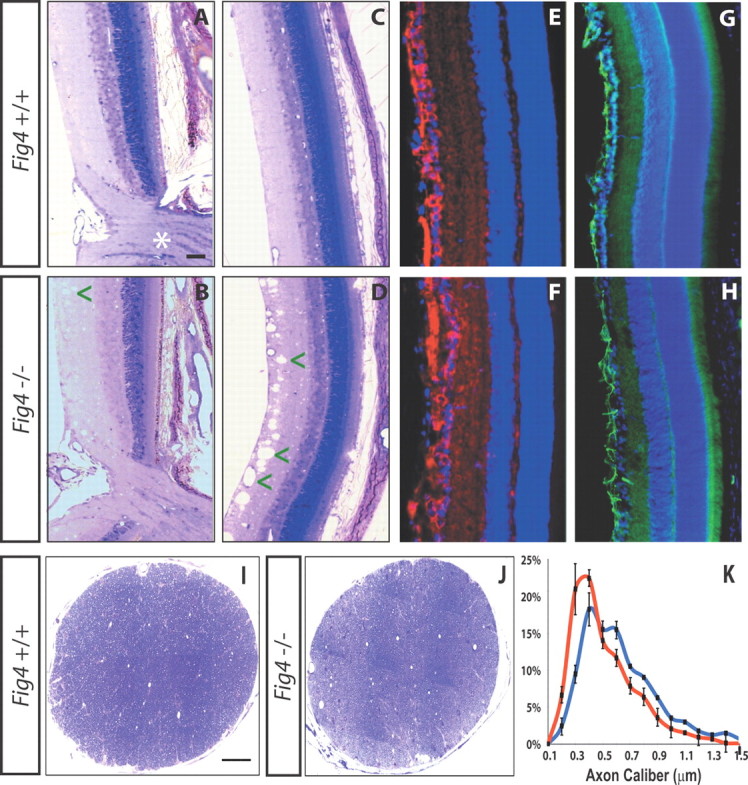
No evidence for loss of RGCs or optic nerve axons in Fig4−/− mice. A–D, Toluidine blue labeling of P21 WT (Fig4+/+) and mutant (Fig4−/−) retina. No major defects in retinal lamination or the optic nerve head (asterisk) are observed in Fig4−/−mutants. In Fig4−/− mice, the inner retina shows small-diameter (arrowhead in B) and sometimes large-diameter (arrowheads in D) vacuoles. E, F, P19 retinae labeled with TuJ1 to visualize RGCs (red) and counterstained with the Hoechst nuclear dye 33342 (blue). G, H, P19 retinae labeled with anti-GFAP (green) and counterstained with a nuclear dye (blue). Scale bars: A–H, 50 μm. I, J, Low-magnification images of optic nerve cross-sections of P21 Fig4+/+ and Fig4−/− mice stained with toluidine blue. Scale bars: I, J, 50 μm. K, Morphometric assessment of axon caliber distribution in P21 optic nerve of Fig4+/+ (blue graph, n = 4) and Fig4−/− (red graph, n = 4) mice. Measurements of axon diameter were made from electron microscopy images and revealed a shift toward smaller-sized axons in Fig4−/− mutant mice.
Loss of Fig4 causes severe hypomyelination of the optic nerve
Next, we examined whether a reduction in myelination contributes to the decreased optic nerve diameter in Fig4−/− mice. As shown in Figure 3A–C, the fraction of myelinated axons in the optic nerve is reduced in a manner proportional to the Fig4 gene dosage (Fig. 3A–C). In Fig4+/+ mice at P21, 94 ± 2% of axons are myelinated. In Fig4+/− mice, 50 ± 2% and, in Fig4−/− mice, 27 ± 2% of axons in the optic nerve are myelinated (Fig. 3F). Thus, loss of one or both copies of the Fig4 gene leads to a significant decrease in myelinated axons. The deficit in Fig4+/− heterozygotes is a developmental delay that is overcome by P90 (Fig. 3D). This is the first demonstration of neural impairment in Fig4+/− heterozygotes, with implications for a potential role of Fig4+/− heterozygosity in amyotrophic lateral sclerosis (ALS) patients (Chow et al., 2009).
Figure 3.
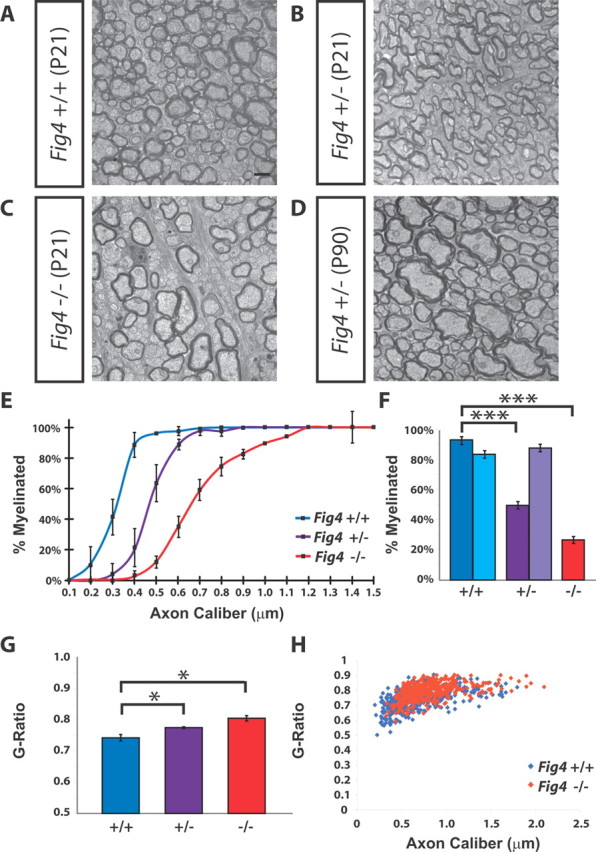
Small-caliber axons are not myelinated in the Fig4−/− optic nerve. A–C, Electron microscopy images of optic nerve cross-sections of P21 Fig4+/+, Fig4+/−, and Fig4−/− mice shows a Fig4 gene dosage-dependent decrease in the percentile of myelinated axons. D, Image of optic nerve cross-section of Fig4+/− at P90. E, Graph showing the percentage of myelinated axons at P21 as a function of axon caliber within each genotype. Fifteen different size groups, ranging from 0.1 to 1.5 μm in axon caliber, were used to subdivide optic nerve axons. Three mice for each genotype (Fig4+/+, Fig4+/−, and Fig4−/−) were included in the analysis. Differences in the percentile of myelination between Fig4+/+ and Fig4−/− are not significant for axons <0.2 μm; significant for axons between 0.2 and 0.3 μm (p < 0.05), highly significant for axons between 0.3 and 1.0 μm (p < 0.007), and not significant for axons between 1.1 and 1.5 μm in diameter. Results are presented as the mean ± SEM; Student's t test. F, Quantification of percentile of myelinated axons in the optic nerve at P21 [dark blue (Fig4+/+), dark purple (Fig4+/−), and red (Fig4−/−)] and at P90 [light blue (Fig4+/+) and light purple (Fig4+/−)]. At P21, 94 ± 2% (Fig4+/+, n = 4 mice), 50 ± 2% (Fig4+/−, n = 4), and 27 ± 2% (Fig4−/−, n = 3 mice) of axons are myelinated. At P90, 84 ± 2% (Fig4+/+, n = 3 mice) and 88 ± 2% (Fig4+/−, n = 4 mice) of axons are myelinated. Results are presented as the mean ± SEM; ***p < 0.001, Student's t test. G, Quantification of g-ratio for all myelinated axons in P21 Fig4+/+, Fig4+/−, and Fig4−/− optic nerve. Results are presented as the mean ± SEM; *p < 0.05 (n = 4 mice for each genotype). H, Scatter plot showing the distribution of g-ratios of myelinated axons as a function of axon caliber at P21 for Fig4+/+ (blue) and Fig4−/− (red) optic nerves (n = 4). Scale bars: A–D, 1 μm.
There is a striking axon size dependence in the myelin deficit of Fig4 mice. In Figure 3E, the percentage of myelinated axons is plotted as a function of axon diameter. In Fig4+/+ mice, virtually all optic nerve axons >0.4 μm are myelinated at P21. However, in Fig4−/− mice, the majority of axons <0.6 μm lack myelin entirely, and even some large-diameter axons (>1 μm) are only partially myelinated. This latter point is addressed in more detail below. Loss of only one copy of Fig4 leads to an intermediate phenotype. Compared with WT controls, Fig4+/− optic nerve axons with a caliber between 0.3 and 0.7 μm show decreased myelination (Fig. 3E). This indicates that, in P21 optic nerve, Fig4 is haploinsufficient for proper myelination. The reduced size of the optic nerve diameter in Fig4 null mutants nicely correlates with the observed reduction of myelinated axons.
To further analyze the myelination deficits in the optic nerve, we determined g-ratios (the ratio of the inner axonal diameter to the total fiber diameter) and myelin periodicity (the thickness of the myelin wraps). In Fig4+/+ mice, the g-ratio of myelinated axons is 0.74 ± 0.010. There is a Fig4-dependent increase in the g-ratio, with significantly higher values in heterozygous (Fig4+/−, 0.78 ± 0.003) and null (Fig4−/−, 0.81 ± 0.01) mice, indicative of less myelin (Fig. 3F). Plotting the g-ratio of myelinated axons as a function of axon caliber shows that the g-ratio in Fig4−/− mutants is slightly shifted toward higher values compared with Fig4+/+ controls (Fig. 3G). The shift is observed independently of the caliber of the myelinated axons. Consistent with this observation, analysis of high-magnification TEM images shows a small but significant difference in the periodicity of compact myelin: 12.6 ± 0.1 nm in Fig4+/+ and 11.8 ± 0.2 nm in Fig4−/− mice (p = 0.02).
Loss of Fig4 causes CNS dysmyelination
Hypomyelination of the optic nerve in Fig4−/− mice could be caused by either loss of myelin sheaths of axons that were myelinated at an earlier time point (demyelination) or a developmental failure of axons to undergo myelination (dysmyelination). To distinguish between these, we analyzed the time course of optic nerve myelination. At P10, few myelinated axons are found in the optic nerve of Fig4+/+ (7 ± 2%) and Fig4−/− (2 ± 1%) mice (Fig. 4). Even at this early developmental stage, a significant reduction (p = 0.02) in the percentage of myelinated axons was noticed (Fig. 4A,A′,D). Although progressively more axons become myelinated at P15 and P21 in Fig4 WT and mutant mice, at each developmental stage, the proportion of myelinated axons in Fig4−/− optic nerve is dramatically reduced (Fig. 4B--D). The average diameter of myelinated axons is comparable for Fig4+/+ and Fig4−/− mice at P10 and P15 but is slightly larger for Fig4−/− at P21 (Fig. 4E). This is likely a reflection of the preferential myelination of large-caliber axons in the mutant mice (Fig. 3E). Moreover, g-ratios at P10 and P15 are similar for Fig4 WT and mutants but are increased in Fig4−/− mice at P21 (Figs. 3G, 4F). Based on these developmental studies, we conclude that, in Fig4 null mutants, a large proportion of axons fail to undergo myelination. Thus, the severe hypomyelination phenotype observed at P21 (Fig. 3) is primarily a reflection of axon dysmyelination rather than demyelination. A hallmark of remyelination is the presence of uniformly thin myelin sheaths associated with large- and small-caliber axons, coupled with the presence of myelin debris. We found neither of these signs of axon remyelination in the optic nerve of Fig4−/− mice. Although remyelination is observed in the sciatic nerve of Fig4−/− mice (Lenk et al., 2011), it does not occur in the optic nerve. Collectively, our studies suggest that developmental failure to myelinate small- and intermediate-sized axons is the primary reason for the Fig4−/− CNS hypomyelination phenotype.
Figure 4.
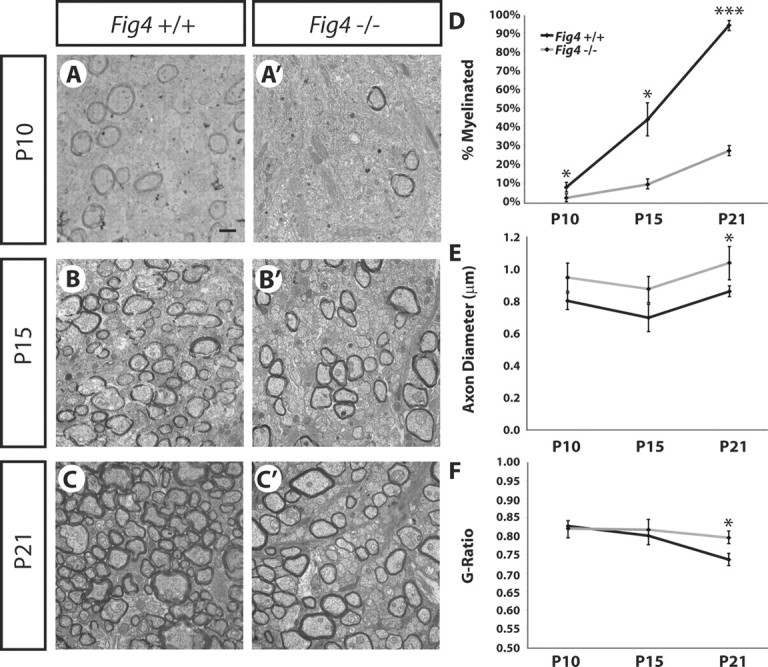
Fig4−/− mice exhibit severe dysmyelination of the optic nerve. Developmental time study of axon myelination. Ultrastructural images of optic nerve cross-sections from Fig4+/+and Fig4−/− mice at P10 (A, A′), P15 (B, B′), and P21 (C, C′) are shown. Scale bar, 1 μm. D, Quantification of percentage of myelinated axons in Fig4+/+and Fig4−/− optic nerve at P10 (n = 3), P15 (n = 3), and P21 (n = 4). E, Quantification of average diameter of myelinated axons in Fig4+/+and Fig4−/− optic nerve at P10 (n = 3), P15 (n = 3), and P21 (n = 4). F, Fiber g-ratios at different developmental stages were determined and are shown as a function of age at P10 (n = 3), P15 (n = 3), and P21 (n = 4). Only myelinated fibers were included to determine g-ratios. D–F, Results are presented as the mean ± SEM; *p < 0.05, ***p < 0.001, Student's t test.
Abnormal nodes of Ranvier in Fig4−/− optic nerve
To examine whether loss of Fig4 influences nodal or paranodal organization, optic nerve sections were immunostained to localize pan Na+ channels and the paranodal axonal protein Caspr at nodes of Ranvier (Menegoz et al., 1997; Peles et al., 1997). Figure 5A–F shows representative images. Figure 5, A and B, is from a Fig4+/+ optic nerve and illustrates the largest and smallest myelinated fibers seen, respectively. In the latter, the diameter at the node is ≤0.5 μm. Figure 5C–F are from Fig4−/− nerves. In Figure 5C, the nodes within these large axons of Fig4−/− mice appear normal. However, there were many heminodes with focal Na+ channel label in these sections (Fig. 5D), indicating segmental absence of myelin. Because myelin debris was not seen, this is likely a developmental dysmyelination, not a demyelination. Furthermore, other abnormalities appeared. In Figure 5E, the paranodes are retracted, and Na+ channels now populate the much wider gap. In Figure 5F, there is a heminode in which Na+ channels again occupy a wide region, away from the original nodal gap. Additionally, small myelinated axons were observed much less frequently in Fig4−/− mutants than in Fig4+/+. These results are quantified in the bar graphs in Figure 5G–I. The number of nodes is reduced by 50% in mutant optic nerves (Fig. 5G). In Figure 5H, the combined data for all myelin abnormalities seen in Figure 5D–F (heminodes, retracted paranodes, and broad Na+ channel immunoreactivity) are shown. There were clearly many more dysmyelinated sites in the Fig4−/− mutants than in Fig4+/+. The graph in Figure 5I plots the fraction of nodes for axons that were ≤0.5 μm in diameter. Although 32% of nodes in Fig4+/+ were in this range, only ∼7% of Fig4−/− nodes were small. This is consistent with the measurements in electron micrographs, in which the percentage of myelinated axons ≤0.5 μm in diameter with normal nodal structures was 35% in Fig4+/+ and 8% in Fig4−/−. It is thus clear that there is a size dependence to the dysmyelination seen in the CNS of Fig4−/− mutant mice. Larger fibers in the Fig4−/− optic nerve have an increased incidence of paranodal and nodal irregularities, although many smaller axons lack myelin altogether.
Figure 5.

Disorganized nodes of Ranvier and paranodes in the Fig4−/− optic nerve. A–F, Optic nerve sections immunostained for Na+ channels (red) and Caspr (green) to visualize nodes and paranodes, respectively. Fig4+/+ (A, B) and Fig4−/− (C–F) optic nerve at P21. A, C, Normal nodal structure in large-diameter axons is found in both genotypes. B, A thin myelinated axon, common in Fig4+/+ but rare in Fig4−/− mice. Abnormal structure in Fig4−/− mutants included focal heminodes (D), retracted paranodes (E), and heminodes with broad regions of Na+ channel label (F). Bar graphs in G–I plot means ± SEM. G, The number of nodes per FOV (68 × 55 μm) is decreased in Fig4−/− mice. H, The fraction of nodes that fit into at least one of the categories shown in D–F is much greater in Fig4−/− mice. I, Plot of the fraction of nodes present on axons with a diameter ≤0.5 μm. FOVs analyzed: Fig4+/+, n = 29; Fig4−/−, n = 27. ***p < 0.001, Student's t test. Scale bars: A–F, 10 μm.
Increased number of slow conducting axons in the optic nerve of Fig4−/− mice
To assess potential physiological alterations in optic nerve function, compound action potentials (CAPs) were recorded from Fig4+/+ and Fig4−/− nerves at P22–P24. In acutely isolated Fig4+/+ nerve (Fig. 6A), there was a complex first peak, usually with three components (labeled 1–3). Peaks 1–3 are not fully resolved in these short nerves, and a very small slow component (peak 4) is observed. Peaks 1–3 are likely obtained from myelinated axons and peak 4 from the small fraction of unmyelinated axons found in optic nerves at this age. In Fig4−/− nerves, there was a corresponding fast signal, but peak 3 was very small or absent (Fig. 6B) and the slow component was prominent, propagating at ∼0.5 m/s, corresponding to peak 4 in Fig4+/+ optic nerve. The complex fast signal was analyzed by fitting the data to the sum of three Gaussians (Chen et al., 2004). In Figure 6, C (Fig4+/+) and D (Fig4−/−), the sum of the Gaussian curves is shown in red. In this Fig4−/− trace, only peaks 1 and 2 are present. The peak 3 fit had 0 amplitude in 40% of the Fig4−/− nerves tested. A graph of velocities is shown in Figure 6E. In both Fig4+/+ and Fig4−/− optic nerve, peaks 1–3 are all >1 m/s and thus likely myelinated. Peak 4 is ∼0.5 m/s and therefore likely unmyelinated. Additionally, the fastest component (peak 1) is significantly slower in Fig4−/− compared with Fig4+/+. The difference in velocity is smaller in peak 2 and is not seen in peak 3. This analysis, of course, only looks at Fig4−/− peak 3 when it is present and ignores its amplitude. The overall amplitude of the signal from myelinated axons in Fig4−/− mutants dropped dramatically to ∼23% of the Fig4+/+ value. The amplitude of the unmyelinated peak 4 rose in Fig4−/− to a value several-fold that in Fig4+/+ (Fig. 6A,B). Absolute values of amplitudes have limited usefulness in extracellular recording, although ratios of peaks within records are highly informative. In Figure 6F, we plotted the ratio of the amplitude of peak 4 to that of the overall 1-2-3 complex. The very large difference, combined with the results shown in Figure 3, is consistent with the idea that small fibers that are myelinated in Fig4+/+ (peak 3) are unmyelinated in Fig4−/− and appear now in peak 4.
Figure 6.
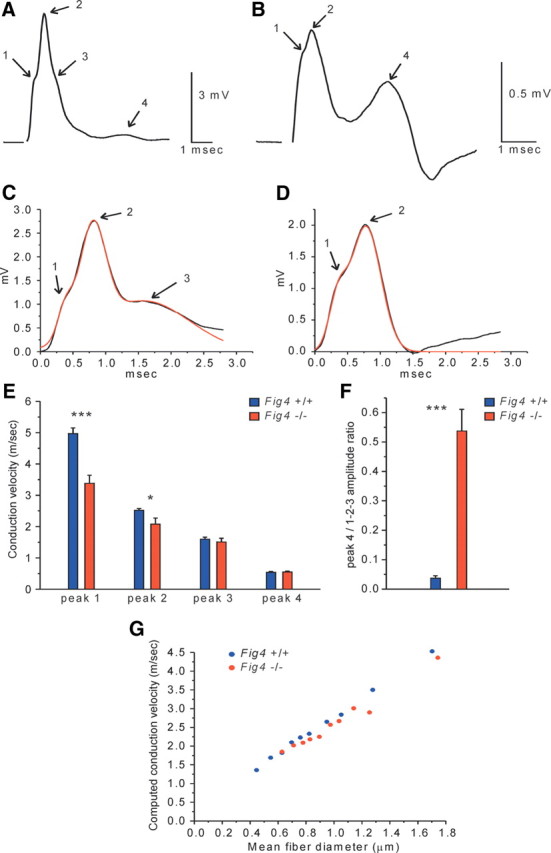
Larger population of slow conducting axons in the optic nerve of Fig4−/− mice. A, CAP recorded from a Fig4+/+optic nerve at P22–P24. Peaks 1–3 reflect contributions from myelinated axons. There is only a small signal from slow unmyelinated axons (peak 4). B, CAP recorded from an optic nerve of an age-matched Fig4−/− mouse. In contrast to Fig4+/+ optic nerve, a very large slow component (peak 4) is seen in mutants. The component likely representing thinner myelinated axons (peak 3) is small or absent in mutants. C, Peaks 1–3 of a Fig4+/+ nerve (black line) are fitted by the sum of three Gaussians (red line). D, Optic nerve from a Fig4−/− mutant in which the fit requires only peaks 1 and 2 (red line). E, Conduction velocities of peaks 1–3 (from Gaussian fits) and peak 4 in Fig4+/+ and Fig4−/− optic nerves. F, The ratio of the amplitude of peak 4 to the maximum amplitude of the myelinated complex (peaks 1–3). Fig4+/+, n = 5 animals (9 nerves), Fig4−/−, n = 5 animals (10 nerves). Error bars indicate SEM; *p < 0.05, ***p < 0.001, Student's t test. G, Predicted conduction velocity computed from measured g-ratios. g-ratio data from electron micrographs of Fig4+/+ fibers (n = 399) and Fig4−/− fibers (n = 400) were sorted by diameter into 10 groups of each genotype, and the mean values from each group were used in calculations. Action potentials were calculated at nodes 5–8 at 37°C. The graph thus represents predicted velocities based on the measured g-ratios, i.e., myelin thickness. Note that CAP velocities in Fig4−/− nerve (red dots) deviate from the linear plot and are decreased compared with WT control (blue dots). The model does not include any nodal/paranodal abnormalities.
As an additional test of the relation between function and structure, we computed predicted action potential conduction based on g-ratios measured from electron micrographs (Hines and Shrager, 1991; Shrager, 1993). For Fig4−/− mice, we found significant deviations from the predicted linear relationship between conduction velocity and fiber diameter (Fig. 6G). However, the measured velocity decrease in Fig4−/− mice exceeded that predicted from the thinner myelin alone, and it is thus likely that the nodal abnormalities shown in Figure 5 also contribute to this functional deficit. Conduction is often blocked at heminodes (Shrager and Rubinstein, 1990), and this is likely to be responsible for the large decrease in amplitude of the component of the CAP attributed to fast myelinated axons.
Markedly fewer myelinating OLs in Fig4 null mice
Thus far, our analysis of Fig4 mice revealed a profound CNS myelination defect that results in faulty nerve conduction. To assess the underlying cellular mechanisms, we assessed whether Fig4 dysmyelination is a reflection of a decreased number of myelinating OLs. P21 cross-sections and longitudinal sections of optic nerves of Fig4+/+ and Fig4−/− mice were subjected to in situ hybridization with a riboprobe specific for MAG mRNA to visualize postmitotic myelinating OLs (Fig. 7A–B′). In Fig4+/+ nerves, the MAG+ OLs exhibit longitudinally ordered arrangement parallel to axons. In Fig4−/− nerves, fewer MAG+ OLs are detected, and the cells stand in isolation and do not show the chain-like organization typically observed in the Fig4+/+ optic nerve. Quantification of MAG+ OLs in P21 optic nerve tissue sections revealed that the number of myelinating OLs is reduced by 66 ± 6% (Fig. 7C). The decrease in myelinating OLs fits nicely with the biochemical data showing a marked reduction of the myelin proteins MBP, PLP, and MAG in Fig4−/− brain at P21 (Fig. 1G). As in the optic nerve, the corpus callosum of 3-week-old Fig4−/− mice contains significantly fewer MAG+ OLs compared with age-matched controls (data not shown). Consistent with a reduced number of myelinating OLs, qRT-PCR on Fig4−/− brain RNA revealed a significant decrease in abundance of MAG, MBP, and PLP transcripts at P21 (Fig. 7D–F). In white matter, the transcription factor Olig2 predominantly labels nuclei in the OL lineage, including premyelinating and myelinating OLs (Dimou et al., 2008). At P21, the optic nerve of Fig4−/− mutant mice shows a 60 ± 4% decrease in Olig2+ cells (Fig. 7G,G′,I). Together with the reduced MBP immunoreactivity in multiple brain regions and the spinal cord (Fig. 1), these data demonstrate that loss of Fig4 results in a significant reduction of myelinating OLs throughout the CNS.
Figure 7.

Reduction of myelinating OLs in Fig4−/− mice. P21 optic nerve sections of WT (Fig4+/+) and mutant (Fig4−/−) mice were stained with markers for the OL lineage. A–B′, MAG in situ hybridization on optic nerve cross-sections and longitudinal sections. C, Quantification of MAG+ cells per optic nerve cross-sections at P21 in Fig4+/+ (n = 4) and Fig4−/− (n = 4) mice. D–F, qRT-PCR on P21 whole-brain RNA normalized to the TATA box binding protein Tbp shows a decrease in copy number of MAG (p = 0.0029), MBP (p = 0.0017), and PLP (p < 0.0001) in Fig4−/− (n = 8) compared with Fig4+/+ (n = 7) mice. G, G′, Olig2 (green) immunostaining of P21 optic nerve cross-sections counterstained with Hoechst nuclear dye 33342 (blue). I, Quantification of the mean number of Olig2+ cells in optic nerve of Fig4+/+ (116 ± 24) and Fig4−/− (40 ± 5) mice (n = 3). J–L, At P21, OPCs are not decreased in Fig4−/− optic nerve or whole brain compared with Fig4+/+ mice. J, qRT-PCR for PDGFRα of Fig4+/+ (n = 4) and Fig4−/− (n = 4) brain revealed no significant difference (p = 0.74). H, H′, NG2 in situ hybridization on P21 optic nerve cross-sections. K, Quantification of NG2+ cells per optic nerve cross-section at P21 in Fig4+/+ and Fig4−/− mice (n = 2 mice per genotype). L, qRT-PCR for NG2 (Cspg4) of Fig4+/+ (n = 4) and Fig4−/− (n = 4) of RNA isolated from 3-week-old brains revealed no significant difference (p = 0.96). Results are presented as the mean ± SEM; *p < 0.05, Student's t test. Scale bar: A, A′, 50 μm; B, B′, 100 μm; G, G′, 50 μm; H, H′, 80 μm.
Normal generation and distribution of OPCs in Fig4−/− mice
Failure in the OL lineage to produce sufficient numbers of myelinating MAG+ OLs may be a reflection of insufficient numbers of OPCs, impaired cell migration, arrest in OL maturation, or cell death (Barres and Raff, 1994; Bradl and Lassmann, 2010). To examine these possibilities, OPCs were labeled at different developmental stages. At P21, in situ hybridization with a riboprobe specific for the OPC marker chondroitin sulfate proteoglycan NG2 did not reveal any reduction in NG2+ cells in the optic nerve (Fig. 7H,H′,K). To independently assess expression of early OL lineage markers, we performed qRT-PCR on total RNA isolated from P21 brain. Consistent with our histochemical experiments, transcripts for PDGFRα and NG2 in Fig4 WT and mutant brains are present at comparable levels (Fig. 7J,L). Collectively, these data suggest that generation, migration, and viability of OPCs is essentially normal in Fig4−/− mice.
Cell death in the OL lineage is not elevated in Fig4−/− mice
At P7 in the mouse, fibers in the brainstem, pons, and base of the cerebellum are immunopositive for MAG. At this age, MAG or MBP immunoreactivity in not yet detected in the corpus callosum, cerebellar lobules, or optic nerve, indicating that myelination in these structures has not yet started (Fig. 8 and data not shown). At P7, the base of the cerebellum harbors numerous MAG+ OLs that overlap with NF-M+ fibers (Fig. 8A–A″). Littermate Fig4−/− pups show very few MAG+ OLs at the base of the cerebellum (Fig. 8B--B″). The reduction in myelination in Fig4−/− is likely not secondary to axonal loss, because NF-M staining was comparable between Fig4 WT and mutant pups (Fig. 8A′,B′). Before onset of CNS myelination, anti-Olig2 staining allows visualization of premyelinating OLs. At P7, the density of Olig2+ cells in cerebellar lobules (Fig. 8C--D″) and the corpus callosum (Fig. 8G–H″) are comparable between Fig4+/+ and Fig4−/− pups, indicating that premyelinating OLs are not reduced in number and show normal CNS distribution. Furthermore, cell death appears not to be substantially increased in the OL lineage. Anti-activated caspase-3 labels few cells in P7 brainstem sections independent of Fig4 genotype. To assess whether cell death in the OL lineage is increased in Fig4−/− pups, P7 Fig4+/+ and Fig4−/− brains were stained with anti-activated caspase-3 and CC1, an antibody that recognizes the cell body of postmitotic OLs. In mutant pups, the number of CC1-positive cells in the pons and brainstem is reduced by 46 ± 9% (n = 3). Double-immunofluorescence labeling revealed no overlap between CC1 and activated caspase-3-labeled cells in either genotype (Fig. 8E–F″). This suggests that, at P7, cell death of postmitotic OLs is not increased in Fig4−/− mice (Fig. 8E–F″). p62/SQSTM1 binds polyubiquitinated protein substrates destined for autophagic degradation. We previously showed that p62 accumulates in astrocytes and to a lesser extent in neurons of Fig4−/− brains, suggesting that autophagy is impaired in these cells (Ferguson et al., 2009). Here we expand on these observations by showing that p62 does not accumulate in Olig2+ cells in the corpus callosum of P7 Fig4−/− pups (Fig. 8G–H′). Together, these observations indicate that OPCs are present at normal numbers and distribution in Fig4−/− mutants. Coupled with the finding that fewer postmitotic OLs are present in the CNS and cell death is not increased, this suggests that loss of Fig4 causes an arrest in OL maturation.
Figure 8.

OPCs are not significantly reduced in the Fig4−/− mice. Sagittal brain sections (25 μm) of P7 WT (Fig4+/+) and mutant (Fig4−/−) pups. A–B″, Double-immunofluorescence labeling of the cerebellum with anti-MAG (green) and anti-NF-M (red). C–D″, P7 cerebellar lobules labeled with anti-Olig2 (green) and Hoechst for nuclear staining (blue). The number of Olig2+ cells per FOV in cerebellar lobules of Fig4+/+ (44 ± 7, n = 3) is not significantly different from Fig4−/−mice (39 ± 6, n = 3). E–F″, Pons of P7 pups stained with CC1 antibody to visualize postmitotic OLs (green) and double stained with anti-activated caspase-3 (red) to assess cell death. Few cells with activated caspase-3 were detected in the pons, and these cells did not overlap with CC1+ OLs. Note that the number of CC1-labeled cells in the Fig4−/− pons is decreased by 46 ± 9% compared with age-matched WT controls. p < 0.01, Student's t test. G–H″, At P7, before the onset of forebrain myelination, the number of Olig2+ cells (red) within the corpus callosum per FOV is not significantly different between Fig4+/+ (167 ± 15, n = 3) and Fig4−/− (152 ± 10, n = 3) mice. At this age, many p62-positive cells (green) are observed in the corpus callosum of Fig4−/− but not Fig4+/+pups. Double immunofluorescence revealed no overlap between p62- and Olig2-positive cells in the corpus callosum. Sections were counterstained with Hoechst nuclear dye 33342 (blue). Cell counts are presented as the mean ± SEM. Scale bars: A–B″, C–D″, G–H″, 60 μm; E–F″, 30 μm.
Neuron-specific expression of transgenic Fig4 is sufficient to rescue the CNS myelination defects in Fig4 null mutants
Fig4 is broadly expressed in the developing and adult nervous system. To determine the cell specificity of Fig4 expression required for rescue of myelination, we examined transgenic mice that express WT Fig4 under the transcriptional control of the NSE promoter (Forss-Petter et al., 1990). Two independent transgenic lines were established, TgN1 and TgN3. Both lines were crossed onto a Fig4 null background to generate Fig4−/−,TgN1 and Fig4−/−,TgN3 mice.
Remarkably, analysis of optic nerve myelination of Fig4−/−,TgN1 and Fig4−/−,TgN3 lines revealed that neuron-specific expression of Fig4 is sufficient to rescue the myelination defects and tremor observed in Fig4−/− mice (Fig. 9A–C″ and data not shown). The fraction of myelinated axons in the optic nerve increases from 27 ± 2% in Fig4−/− mice to 85 ± 12% (Fig4−/−,TgN1) and 89 ± 5% (Fig4−/−,TgN3), which is comparable with WT (90 ± 2%) mice (Fig. 9D). Thus, in two independent mouse lines, neuron-specific expression of Fig4 is sufficient to rescue the hypomyelination defects observed in Fig4 null mutants. As an independent indicator of the extent of myelination in Fig4−/−,TgN1 brain, we analyzed the abundance of MBP. Anti-MBP Western blot analysis of P25 brain lysates demonstrated correction of the MBP deficiency in Fig4−/− mice (Fig. 9E). The level of MBP protein in Fig4−/−,TgN1 mice is comparable with WT mice and nicely correlates with the rescue of the optic nerve dysmyelination. Based on the greatly increased fraction of myelinated axons in the optic nerve and the increased abundance of MBP in the brain, we conclude that neuronal Fig4 is sufficient to rescue the pronounced dysmyelination phenotype observed in Fig4−/− mutants. Thus, arrest of OPC maturation and myelin development in Fig4 null mutants is secondary to neuronal defects, demonstrating a non-cell-autonomous function of Fig4 for proper OL development. The effect is not simply mediated by neuronal survival, because there is no reduction of the total number of RGCs in the retina or axons in the optic nerve of Fig4−/− mice (Fig. 2E,F,K).
Figure 9.
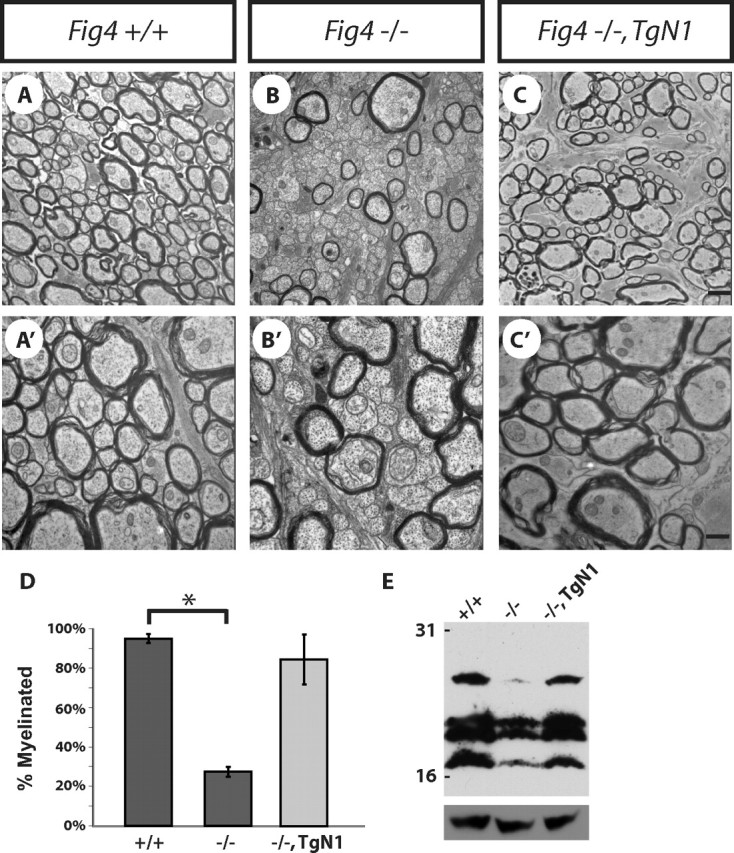
Neuron-specific expression of Fig4 is sufficient to rescue the Fig4−/− myelination defect in vivo. Ultrastructural analysis of optic nerve cross-sections of Fig4+/+ (A, A′) and Fig4−/− (B, B′) mice and transgenic Fig4−/−,TgN1 mice (C, C′). D, Quantification of the fraction of myelinated optic nerve axons revealed greatly enhanced myelination in Fig4−/−,TgN1 mice (85 ± 12%) compared with Fig4−/− mice (27 ± 2%). The percentile of myelinated axons in the Fig4−/−,TgN1 optic nerve is similar to Fig4+/+ mice (92 ± 2%). E, Western blot analysis of whole-brain extracts of P25 Fig4+/+ (lane 1), Fig4−/− (lane 2), and Fig4−/−,TgN1 (lane 3) mice shows that neuronal expression of WT Fig4 on a Fig4 null background is sufficient to restore MBP expression to WT levels. Anti-MBP recognizes four bands between ∼17 and 27 kDa. Anti-GAPDH is shown as loading control. Results are presented as the mean ± SEM; *p < 0.01, Student's t test. Scale bar: A–C, 1 μm; A′–C′, 0.3 μm.
To confirm the lack of expression of the NSE promoter Fig4 transgenes in the OL lineage, brain sections of Fig4−/−,TgN1 mice at P21 were subjected to in situ hybridization with an antisense riboprobe specific for Fig4 transcripts (Fig. 10). In WT (Fig4+/+) mice, weak staining was observed throughout the brain, with somewhat stronger labeling in the caudate–putamen and principal neurons of the hippocampus (Fig. 10A and data not shown). No signal was detected in Fig4−/− mice (Fig. 10C). In Fig4−/−,TgN1 mice, there was robust labeling of large cells in the neocortex and CA1 pyramidal neurons (Fig. 10B) but few labeled cells in the corpus callosum (Fig. 10D,E). To determine whether the labeled cells in the corpus collosum belong to the OL lineage, sections were double stained with anti-Olig2. Fewer than 5% of the Olig2+ cells were also labeled with the Fig4 antisense riboprobe (Fig. 9D–E″). Thus, expression of the transgene in OLs cannot account for the rescue of myelination described above.
Figure 10.
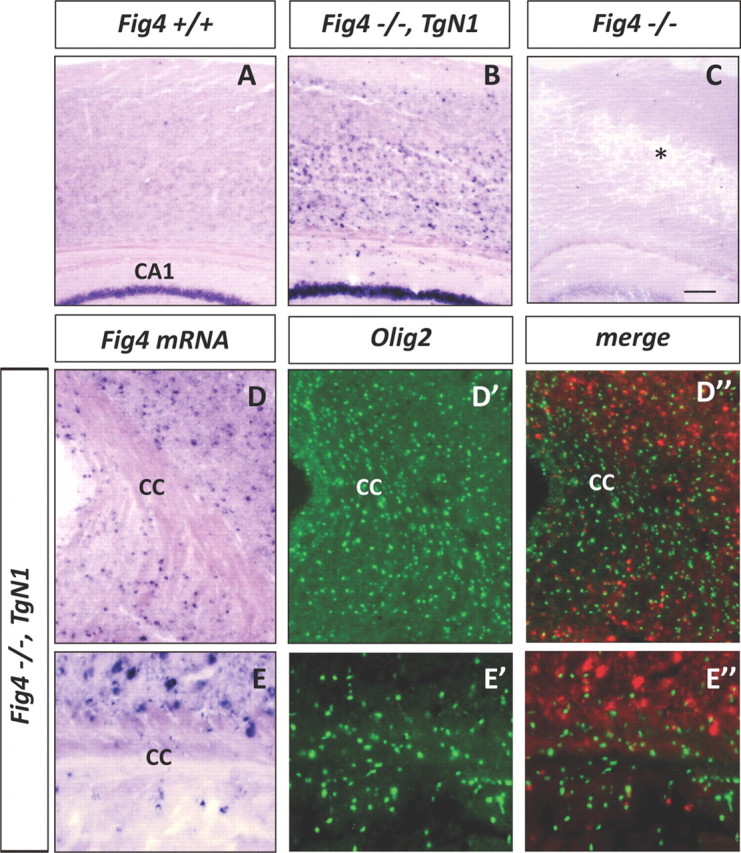
Fig4−/−,TgN1 transgenic mice express Fig4 in neurons but not OLs. A–C, Cross-sections of P21 neocortex of WT mice (Fig4+/+), NSE-transgenic mice crossed onto a Fig4−/− background (Fig4−/−,TgN1), and Fig4 null (Fig4−/−) mice were hybridized with a digoxigenin-labeled cRNA probe specific for Fig4. In Fig4+/+ sections, there is weak labeling in the neocortex and robust staining of hippocampal CA1 pyramidal neurons. In the Fig4−/−,TgN1 mice, there are many strongly labeled cells in the neocortex and intense labeling of CA1 pyramidal neurons. In Fig4−/− null mice, there is degeneration in the neocortex (asterisk) and no hybridization signal. D–D″, Low-magnification images of the corpus callosum (cc) of Fig4−/−,TgN1 brain stained for Fig4 transcript and Olig2 protein. Note in D″, showing merged Fig4 and Olig2 labeling, the Fig4 hybridization signal is shown in red. E–E″, Higher magnification of the corpus callosum demonstrates primarily non-overlapping labeling of Olig2 and Fig4 in Fig4−/−,TgN1 brain. Scale bar: A–C, 100 μm; D–D″, 64 μm; E–E″, 20 μm.
Overexpression of the human variant FIG4I41T rescues CNS hypomyelination of Fig4−/− mice at P21
Unlike Fig4 null mice, CMT4J patients contain residual FIG4 activity from the I41T allele and are therefore less severely affected than the null mice (Lenk et al., 2011). To study the function of the human CMT4J mutation FIG4I41T in CNS myelination in vivo, we examined optic nerve myelination in transgenic mice with ubiquitous, global expression of the I41T cDNA from the chicken β-actin promoter (Lenk et al., 2011). Expression of this human pathogenic variant at two times the normal transcript level (line 705) or five times the normal transcript level (line 721) (corresponding to 4 or 10% of normal protein levels) rescued lethality, brain spongiform degeneration, and sciatic nerve myelination of Fig4 null mice (Lenk et al., 2011). Ultrastructural analysis of P21 optic nerve from these two transgenic lines revealed dose-dependent rescue of the hypomyelination phenotype as well. As shown in Figure 11, in Fig4−/− mice, only 27 ± 2% of optic nerve axons are myelinated. The fraction of myelinated axons is increased to 34 ± 1% in Fig4−/−,Tg705 mice and 78 ± 1% in Fig4−/−,Tg721 mice. In Fig4+/+ controls, 94 ± 4% of optic nerve axons are myelinated (Fig. 11A--E). The average g-ratio of myelinated axons was 0.81 in Fig4−/− mice and was corrected to 0.75 in Fig4−/−,Tg721 mice, a value indistinguishable from Fig4+/+ controls (0.74) (Fig. 11F). Importantly, small-caliber axons that are dysmyelinated in Fig4 null mutants become myelinated in a Fig4I41T dose-dependent manner (Fig. 11G). The rescue of CNS myelination defects in Fig4I41T mice was accompanied by rescue of the tremor phenotype. Collectively, these data demonstrate that transgenic expression of the disease allele Fig4I41T on a Fig4−/− background rescues defects in CNS myelination observed at P21 and may explain the lack of CNS abnormalities in CMT4J patients expressing this mutation.
Figure 11.
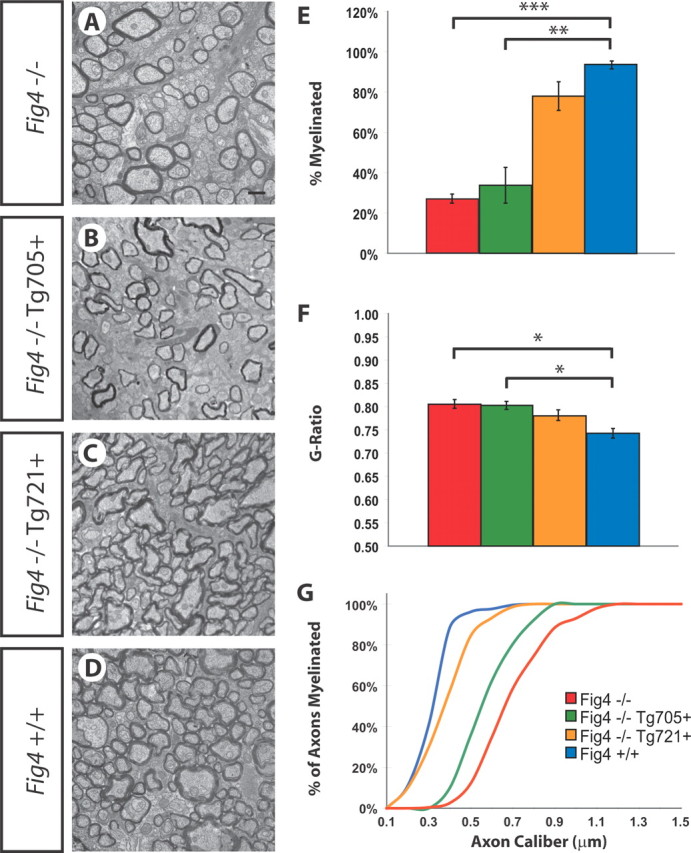
Dose-dependent rescue of Fig4−/− dysmyelination phenotype by overexpression of the human pathogenic variant Fig4I41T. Ultrastructural images of P21 optic nerve section of Fig4 null mice (Fig4−/−) (A), transgenic mice expressing low levels of Fig4I41T (line 705) on a Fig4 null background (Fig4−/−,Tg705) (B), transgenic mice expressing high levels of Fig4I41T (line 721) on a Fig4 null background (Fig4−/−,Tg721) (C), and WT (Fig4+/+) (D) mice. E, Quantification of percentage of myelinated axons for Fig4−/− (red, n = 4), Fig4−/−,Tg705 (green, n = 3), Fig4−/−,Tg721 (yellow, n = 4), and Fig4+/+ (blue, n = 4) optic nerve. F, g-ratio of myelinated axons in P21 Fig4−/− (n = 4), Fig4−/−,Tg705 (n = 3), Fig4−/−,Tg721 (n = 4), and Fig4−/− (n = 4) optic nerve. G, Percentile of myelinated optic nerve axons as a function of axon diameter. The curve for Fig4−/− mice (red, n = 4) shows a shift toward the right compared with Fig4+/+ (blue, n = 4) control mice. Expression of a low dose of I41T, Fig4−/−,Tg705 (green, n = 4) and a high dose of I41T, Fig4−/−,Tg721 (yellow, n = 3) reveals a I41T gene-dosage-dependent shift toward Fig4+/+ (blue graph). Results are presented as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, Student's t test. Scale bars: A–D, 1 μm.
Long-term effect of early dysmyelination in Fig4 null mice
At P21, Fig4−/− mice exhibit pronounced CNS dysmyelination (Fig. 3). Because the null mice rarely survive beyond 6 weeks of age, it is not possible to test them for later completion of myelination, for example, at P90 when myelination in Fig4+/+ and Fig4+/− is complete (Fig. 3). At P21, the severity of optic nerve dysmyeliation is comparable in Fig4 null mice and Fig4−/−,Tg705 transgenic mice (Fig. 11). Because Fig4−/−,Tg705 transgenic mice do survive to P90, we were able to determine whether they could recover from the dysmyelination that was observed at P21. At P90, Fig4−/−,Tg705 mice continue to exhibit severe hypomyelination of the optic nerve, with only 44 ± 2% of axons myelinated (Fig. 12). The low expression of the I41T mutant protein in line 705 is thus insufficient for completion of myelination. We conclude that loss of Fig4 function does not simply result in a temporary delay of optic nerve myelination but is necessary for completion of myelination.
Figure 12.

Rescue of lethality but not CNS hypomyelination in Fig4−/−,Tg705 mice. A, B, Ultrastructural images of optic nerve cross-sections at P90. A, In the WT (Fig4+/+) optic nerve, 84 ± 2% axon myelination is observed. B, Transgenic Fig4−/−,Tg705 mice express low levels of Fig4I41T and survive beyond P90. Analysis of optic nerve cross-sections of Fig4−/−,Tg705 mice revealed that these mice fail to complete myelination by P90. Only 44 ± 2% of axons are myelinated in the Fig4−/−,Tg705 optic nerve at P90. C, Quantification of the percentage of myelinated axons in the optic nerve at P90 for Fig4+/+ (n = 3) and Fig4−/−,Tg705 (n = 3) mice. Results are presented as the mean ± SEM; **p < 0.01, Student's t test. Scale bar: A, B, 1 μm.
Discussion
We show that the PI(3,5)P2 phosphatase Fig4 is essential for normal CNS myelination. Biochemical, ultrastructural, and electrophysiological studies in Fig4−/− mice provide a consistent picture of a profound CNS myelinopathy. In the optic nerve, loss of Fig4 causes severe dysmyelination of small-caliber axons and frequent defects at nodes of Ranvier in larger-caliber axons that cause conduction delays. Although the number and tissue distribution of OPCs is normal in the Fig4−/− brain, the abundance of postmitotic OLs is significantly compromised. Cell death in the OL linage is not increased, suggesting that loss of Fig4−/− leads to an arrest in OL maturation. Rescue experiments with a neuron-specific Fig4 transgene revealed a non-cell-autonomous function of Fig4 in OPC maturation and proper CNS myelination, because neuron-specific expression is sufficient to rescue the defects. In a transgenic mouse model mimicking the genotype of human CMT4J patients, we found that ubiquitous expression of the human disease allele Fig4I41T can also rescue the CNS hypomyelination phenotype observed in Fig4−/− mice. The CNS of CMT4J patients may be similarly protected by the FIG4I41T allele. We conclude that proper regulation of neuronal PI(3,5)P2 levels by mammalian Fig4 is necessary for OL maturation and normal CNS myelination.
Impaired propagation of action potentials in the Fig4−/− optic nerve
Dysmyelination of the optic nerve in juvenile Fig4−/− mice results in faulty nerve conduction. CAP recordings revealed many more slow conducting axons in mutant mice. The loss of the contribution from the smallest group of myelinated axons and the large increase in the signal from unmyelinated axons are both explained by the structural studies at the light and TEM levels. The percentage of thin myelinated axons (≤0.5 μm) that contribute to fast conducting axons (peak 3 in Fig. 6A) is much decreased in Fig4−/− mutants, and many more unmyelinated fibers are seen (peak 4 in Fig. 6B). Furthermore, the CAP signal that is derived from larger myelinated axons is smaller and slower in Fig4−/− and may be a reflection of retracted paranodal myelin and segmental lack of myelin observed in a significant fraction of large-caliber axons. No signs of axon remyelination were observed in the optic nerve of Fig4−/− mice. Collectively, these studies show that the structural defects observed at nodes and deficiency in optic nerve myelination in Fig4−/− mice lead to greatly impaired nerve conduction.
Fig4 is not necessary for OPC development, migration, or viability
Before myelination, OL progenitors undergo cell division, migration, and differentiation into premyelinating OLs. In the healthy CNS, the number of newly formed premyelinating OLs exceeds that required for normal myelination, and only OLs that establish contact with axons survive (Barres et al., 1992; Trapp et al., 1997). Here we show that, in CNS white matter of Fig4−/− mice, there is no deficiency of OPCs. At P21, the number of NG2+ cells in optic nerve and the abundance of transcripts encoding PDGFRα and NG2 in whole brain are comparable, independent of Fig4 genotype. Cell death of postmitotic OLs appears not to be elevated. These observations suggest that “positive” signals are missing that normally promote OPC differentiation and axon myelination. Alternatively, “inhibitory” signals that block OPC differentiation and myelination may be elevated in the Fig4−/− CNS. As discussed below, the molecular mechanisms that initiate myelination in the CNS are complex and may depend on both positive signals (Park et al., 2008; Câmara et al., 2009; Emery et al., 2009) and release of inhibitory signals (Charles et al., 2000; Mi et al., 2005; Kremer et al., 2011; Mi et al., 2011).
Neuronal expression of Fig4 is sufficient for proper CNS myelination
Fig4 is broadly expressed in the CNS and not limited to myelinating glia. The importance of Fig4 in non-myelinating cells is underscored by the brain pathology observed before onset of myelin development (Ferguson et al., 2009). In the forebrain, for example, large numbers of hypertrophic astrocytes are observed at P7, a developmental stage when CNS myelination has not yet started. At the same age, p62 and LAMP2 are elevated in reactive astrocytes and neurons. To address the cell autonomy of Fig4−/− defects, we generated transgenic mice that selectively express WT Fig4 in neurons. Remarkably, neuron-specific expression of Fig4 on a Fig4−/− background is sufficient to rescue the optic nerve dysmyelination phenotype. Anatomical and biochemical studies revealed a virtually complete rescue of the myelination defects. We conclude that defects in OPCs maturation and CNS myelination in Fig4−/− mice are caused by indirect, non-OL-autonomous effects of Fig4.
How does neuronal Fig4 affect OPC differentiation and CNS myelination? One of the key functions of PI(3,5)P2 is in fragmentation/fission of endolysosomes into smaller compartments (Ho et al., 2011). Endosome–lysosome membrane trafficking and autophagy are the cellular processes most affected by mutations in Fig4 (Chow et al., 2007; Ferguson et al., 2009). Impaired vesicular trafficking along the endo–lysosomal axis or impaired lysosomal function is emerging as a common underlying mechanism for a growing number of hereditary disorders that cause CNS dysmyelination (Karten et al., 2009; Liao et al., 2009; Prolo et al., 2009; Schweitzer et al., 2009). We speculate that altered levels of PI(3,5)P2 in the Fig4−/− CNS either leads to a decrease in neuronal signals that promote OPC differentiation and axon ensheathment or an increase in signal(s) that suppress OPC differentiation. Several “direct” and “indirect” mechanisms that influence OPC differentiation and myelin development have been described (Kremer et al., 2011). One direct effect of neurons on OPC differentiation is mediated by axonal Jagged1. OPCs express the Notch receptor, and its activation by Jagged1 inhibits their differentiation (Wang et al., 1998). Neuronal electrical activity can influence myelination (Demerens et al., 1996). Mechanistic studies revealed that electrical activity induces the release of ATP from axons, which acts in a paracrine manner on astrocytes to release the cytokine LIF (leukemia inhibitory factor). LIF then stimulates premyelinating OLs to myelinate axons (Ishibashi et al., 2006). More recently, release of glutamate from synaptic vesicles along axons has been shown to control axo-glial signaling and induce myelination in OPC processes associated with axons (Wake et al., 2011). Mounting evidence suggests that astrocytes secrete numerous factors that promote OPC maturation and the rate of myelin ensheathment (Sorensen et al., 2008; Watkins et al., 2008; Gadea et al., 2009; Moore et al., 2011). Loss of neuronal Fig4 may indirectly affect astrocyte function, which then leads to impaired myelination. Additional studies are needed to identify the molecular mechanism(s) by which neuronal loss of FIG4 leads to impaired CNS myelinogenesis in vivo.
Abnormal CNS myelination in Fig4+/− heterozygous mice
ALS patients heterozygous for FIG4 null alleles (FIG4+/−) have been identified (Chow et al., 2009), but other heterozygous FIG4+/− individuals appear to be unaffected and lack neurological symptoms (Chow et al., 2007; Zhang et al., 2008; Nicholson et al., 2011). At the gross anatomical level, heterozygous Fig4+/− mice are indistinguishable from age-matched Fig4+/+ controls. However, we have identified a developmental delay of optic nerve myelination in Fig4+/− mice at P21 that is resolved by P90, indicating that one copy of WT Fig4 may be insufficient for proper timing of CNS myelination. This is the first functional deficit identified in Fig4+/− heterozygous mice. A similar insufficiency in human heterozygotes could act as a risk factor for development of late-onset neurological disease.
The disease allele FIG4I41T can rescue CNS myelination in a dose-dependent manner
Unlike Fig4 null mice, CMT4J patients contain residual FIG4 activity from their I41T allele and are therefore less severely affected than the null mice (Chow et al., 2007; Lenk et al., 2011). In a recent study of patients with CMT4J, four patients were examined by brain MRI with no evidence of myelination defects, and one patient (A6) had MRI evidence of abnormal white matter (Nicholson et al., 2011, their supplement). Clinical examination of CMT4J patients revealed no cognitive dysfunction or upper motoneuron signs (Zhang et al., 2008; Nicholson et al., 2011). FIG4I41T is catalytically active (Ikonomov et al., 2010) but fails to interact with the scaffold protein Vac14 and, as a result, is rapidly degraded via the proteasome (Lenk et al., 2011). Additional mechanistic studies will be needed to elucidate why the mutant protein Fig4I41T is sufficient to protect the CNS in CMT4J patients but fails to protect the PNS. Other mutations in the FIG4 gene have been identified, and it is possible that some of them contribute to hypomyelination in other human disorders.
In summary, we have demonstrated for the first time that Fig4 is required for normal CNS myelination and that neuron-specific expression can provide this essential function. With regards to the pathophysiology of CMT4J patients, we demonstrate that global transgenic expression of the Fig4I41T transcript at five times the WT level rescues the CNS hypomyelination phenotype of Fig4−/− mice. These studies reveal that loss of the PI(3,5)P2 phosphatase Fig4 leads to an arrest of OPC differentiation and failure of the OL lineage to produce sufficient numbers of postmitotic OLs. This causes profound CNS dysmyelination, tremor, and impaired propagation of action potentials in Fig4 null mice. Patients with CMT4J disease, and transgenic mice expressing the pathogenic allele FIG4I41T, are protected from CNS dysmyelination by a low level of Fig4 function from the mutant allele. Fig4 mouse models developed here may provide new opportunities to study molecular mechanisms of OL maturation and CNS myelination in vivo.
Footnotes
This work was supported by fellowships from the Rackham School of Graduate Studies (J.J.W.), Medical Scientists Training Program Grant T32 GM07863 and Training Program in Systems and Integrative Biology Grant T32 GM008322 (C.J.F.), the Hartwell Foundation (G.M.L.), National Institute of Neurological Disorders and Stroke Grant R56 NS047333 (R.J.G., P.S.), the Dr. Miriam and Sheldon G. Adelson Medical Foundation on Neural Repair and Rehabilitation (R.J.G.), National Institute of General Medical Sciences Grant R01 GM24872 (M.H.M), the Schmitt Program on Integrative Brain Research (P.S.), and Bridging Funds from the University of Rochester (P.S.). Margaret Youngman provided excellent technical assistance.
The authors declare no competing financial interests.
References
- Barres BA, Raff MC. Control of oligodendrocyte number in the developing rat optic nerve. Neuron. 1994;12:935–942. doi: 10.1016/0896-6273(94)90305-0. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Botelho RJ. Changing phosphoinositides “on the fly”: how trafficking vesicles avoid an identity crisis. Bioessays. 2009;31:1127–1136. doi: 10.1002/bies.200900060. [DOI] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Câmara J, Wang Z, Nunes-Fonseca C, Friedman HC, Grove M, Sherman DL, Komiyama NH, Grant SG, Brophy PJ, Peterson A, ffrench-Constant C. Integrin-mediated axoglial interactions initiate myelination in the central nervous system. J Cell Biol. 2009;185:699–712. doi: 10.1083/jcb.200807010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A. 2000;97:7585–7590. doi: 10.1073/pnas.100076197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL. Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci. 2004;24:4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, Everett L, Lenk GM, McKenna-Yasek DM, Weisman LS, Figlewicz D, Brown RH, Meisler MH. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, Zalc B, Lubetzki C. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci U S A. 1996;93:9887–9892. doi: 10.1073/pnas.93.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P(2) Controls membrane traffic by direct activation of mucolipin Ca release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J. 2009;419:1–13. doi: 10.1042/BJ20081950. [DOI] [PubMed] [Google Scholar]
- Dubourg O, Azzedine H, Verny C, Durosier G, Birouk N, Gouider R, Salih M, Bouhouche A, Thiam A, Grid D, Mayer M, Ruberg M, Tazir M, Brice A, LeGuern E. Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8:75–86. doi: 10.1385/nmm:8:1-2:75. [DOI] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009;18:4868–4878. doi: 10.1093/hmg/ddp460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- Gadea A, Aguirre A, Haydar TF, Gallo V. Endothelin-1 regulates oligodendrocyte development. J Neurosci. 2009;29:10047–10062. doi: 10.1523/JNEUROSCI.0822-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger RJ, Wolfer DP, De Wit GM, Verhaagen J. Anatomy of rat semaphorin III/collapsin-1 mRNA expression and relationship to developing nerve tracts during neuroembryogenesis. J Comp Neurol. 1996;375:378–392. doi: 10.1002/(SICI)1096-9861(19961118)375:3<378::AID-CNE3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000;25:29–41. doi: 10.1016/s0896-6273(00)80869-7. [DOI] [PubMed] [Google Scholar]
- Gold R, Hartung HP, Toyka KV. Animal models for autoimmune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- Hines M. A program for simulation of nerve equations with branching geometries. Int J Biomed Comput. 1989;24:55–68. doi: 10.1016/0020-7101(89)90007-x. [DOI] [PubMed] [Google Scholar]
- Hines M, Shrager P. A computational test of the requirements for conduction in demyelinated axons. Restor Neurol Neurosci. 1991;3:81–93. doi: 10.3233/RNN-1991-3205. [DOI] [PubMed] [Google Scholar]
- Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3,5-Bisphosphate: no longer the poor PIP(2) Traffic. 2011 doi: 10.1111/j.1600-0854.2011.01246.x. [DOI] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Fligger J, Delvecchio K, Shisheva A. ArPIKfyve regulates Sac3 Protein abundance and turnover: disruption of the mechanism by Sac3141T mutation causing Charcot-Marie-Tooth 4J disorder. J Biol Chem. 2010;285:26760–26764. doi: 10.1074/jbc.C110.154658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Chow CY, Liu L, Zolov SN, Bronson R, Davisson M, Petersen JL, Zhang Y, Park S, Duex JE, Goldowitz D, Meisler MH, Weisman LS. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B, Peake KB, Vance JE. Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta. 2009;1791:659–670. doi: 10.1016/j.bbalip.2009.01.025. [DOI] [PubMed] [Google Scholar]
- Kaye EM. Update on genetic disorders affecting white matter. Pediatr Neurol. 2001;24:11–24. doi: 10.1016/s0887-8994(00)00232-0. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- Kremer D, Aktas O, Hartung HP, Küry P. The complex world of oligodendroglial differentiation inhibitors. Ann Neurol. 2011;69:602–618. doi: 10.1002/ana.22415. [DOI] [PubMed] [Google Scholar]
- Lenk GM, Ferguson CJ, Chow CY, Jin N, Jones JM, Grant AE, Zolov SN, Winters JJ, Giger RJ, Dowling JJ, Weisman LS, Meisler MH. Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet. 2011;7:e1002104. doi: 10.1371/journal.pgen.1002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Cheung S, Galeano J, Ji AX, Qin Q, Bi X. Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1−/− mouse brain. Brain Res. 2009;1270:140–151. doi: 10.1016/j.brainres.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegoz M, Gaspar P, Le Bert M, Galvez T, Burgaya F, Palfrey C, Ezan P, Arnos F, Girault JA. Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron. 1997;19:319–331. doi: 10.1016/s0896-6273(00)80942-3. [DOI] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Hu Y, Ji B, Shao Z, Yang W, Huang G, Walus L, Rhodes K, Gong BJ, Miller RH, Pepinsky RB. Death receptor 6 negatively regulates oligodendrocyte survival, maturation and myelination. Nat Med. 2011;17:816–821. doi: 10.1038/nm.2373. [DOI] [PubMed] [Google Scholar]
- Moore CS, Milner R, Nishiyama A, Frausto RF, Serwanski DR, Pagarigan RR, Whitton JL, Miller RH, Crocker SJ. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J Neurosci. 2011;31:6247–6254. doi: 10.1523/JNEUROSCI.5474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JA, Blakemore WF. The relationship between internodal length and fibre diameter in the spinal cord of the cat. J Neurol Sci. 1980;45:29–41. doi: 10.1016/s0022-510x(80)80004-9. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Lenk GM, Reddel SW, Grant AE, Towne CF, Ferguson CJ, Simpson E, Scheuerle A, Yasick M, Hoffman S, Blouin R, Brandt C, Coppola G, Biesecker LG, Batish SD, Meisler MH. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P2 phosphatase FIG4. Brain. 2011;134:1959–1971. doi: 10.1093/brain/awr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Liu B, Chen T, Li H, Hu X, Gao J, Zhu Y, Zhu Q, Qiang B, Yuan J, Peng X, Qiu M. Disruption of Nectin-like 1 cell adhesion molecule leads to delayed axonal myelination in the CNS. J Neurosci. 2008;28:12815–12819. doi: 10.1523/JNEUROSCI.2665-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, Schlessinger J. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J. 1997;16:978–988. doi: 10.1093/emboj/16.5.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poccia D, Larijani B. Phosphatidylinositol metabolism and membrane fusion. Biochem J. 2009;418:233–246. doi: 10.1042/BJ20082105. [DOI] [PubMed] [Google Scholar]
- Prolo LM, Vogel H, Reimer RJ. The lysosomal sialic acid transporter sialin is required for normal CNS myelination. J Neurosci. 2009;29:15355–15365. doi: 10.1523/JNEUROSCI.3005-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, Oldstone MB. Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron. 1995;15:1183–1191. doi: 10.1016/0896-6273(95)90105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robak LA, Venkatesh K, Lee H, Raiker SJ, Duan Y, Lee-Osbourne J, Hofer T, Mage RG, Rader C, Giger RJ. Molecular basis of the interactions of the Nogo-66 receptor and its homolog NgR2 with myelin-associated glycoprotein: development of NgROMNI-Fc, a novel antagonist of CNS myelin inhibition. J Neurosci. 2009;29:5768–5783. doi: 10.1523/JNEUROSCI.4935-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudge SA, Anderson DM, Emr SD. Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15:24–36. doi: 10.1091/mbc.E03-05-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer JK, Krivda JP, D'Souza-Schorey C. Neurodegeneration in Niemann-Pick Type C disease and Huntington's disease: impact of defects in membrane trafficking. Curr Drug Targets. 2009;10:653–665. doi: 10.2174/138945009788680437. [DOI] [PubMed] [Google Scholar]
- Shrager P. Axonal coding of action potentials in demyelinated nerve fibers. Brain Research. 1993;619:278–290. doi: 10.1016/0006-8993(93)91622-y. [DOI] [PubMed] [Google Scholar]
- Shrager P, Rubinstein CT. Optical measurement of conduction in single demyelinated axons. J Gen Physiol. 1990;95:867–889. doi: 10.1085/jgp.95.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaugenhaupt SA. The molecular basis of mucolipidosis type IV. Curr Mol Med. 2002;2:445–450. doi: 10.2174/1566524023362276. [DOI] [PubMed] [Google Scholar]
- Sorensen A, Moffat K, Thomson C, Barnett SC. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia. 2008;56:750–763. doi: 10.1002/glia.20650. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/s0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- Watkins TA, Emery B, Mulinyawe S, Barres BA. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron. 2008;60:555–569. doi: 10.1016/j.neuron.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chow CY, Sahenk Z, Shy ME, Meisler MH, Li J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain. 2008;131:1990–2001. doi: 10.1093/brain/awn114. [DOI] [PMC free article] [PubMed] [Google Scholar]