

Abstract
The role of the ubiquitin–proteasome system (UPS) in maintaining protein homeostasis has generated a demand for assays that quantify UPS function in the presence of chemical and protein UPS inhibitors. Here, we describe protocols that measure changes in UPS reporter levels in response to changes in the expression level, localization, or aggregation state of a second protein. We utilize cell lines stably expressing fluorescent UPS substrates that are transfected with a second protein tagged with a compatible fluorophore. We describe protocols to correlate levels of UPS substrates with changes in the levels or properties of the transfected protein.
Keywords: UPS function, Proteasome inhibition, Two-color flow cytometry, Fluorescent proteins, Degron, Live-cell imaging
1. Introduction
The ubiquitin–proteasome system (UPS) is the principal proteolytic system in the cytoplasm of eurkaryotic cells. The UPS regulates gene expression by limiting the lifetime and concentration of short-lived cell regulators such as transcription factors and cyclins. This proteolytic system also destroys folding-defective proteins that might otherwise adopt toxic non-native structures that characterize many degenerative disorders. Determining how the function and capacity of the UPS is influenced by genetic and environmental challenges is therefore critical to understanding the cellular basis of pathological conditions such as cancer, neurodegeneration, and immunoinflammatory disorders (1).
Short-lived fluorescent reporter proteins have been widely employed as noninvasive tools to monitor the status of the UPS. These synthetic “reporters” consist of fluorescent proteins (FP) fused to destabilizing signals called “degrons” that convert them from long-lived proteins to short-lived proteasome substrates (2, 3). Cells with a fully functional UPS maintain these reporters at low basal levels, and UPS impairment results in reporter accumulation. Consequently, microscopy or flow cytometry can be used to assess UPS function by measuring shifts in steady-state reporter levels. Turnover rates calculated by this method agree well with pulse-chase measurements (2).
Cells degrade different types of substrates by utilizing different degradation pathways. Since all UPS pathways converge at the 26S proteasome, impairment of the proteasome leads to accumulation of UPS substrates (2, 3). Reporters degraded by specific UPS pathways are useful tools for determining the effects of UPS inhibitors. Ubiquitin-dependent and -independent reporters (2–4), reporters that are degraded by endoplasmic reticulum-associated protein degradation (ERAD) (5, 6) and reporters targeted to nuclear or cytoplasmic compartments (7) or to synapses (8) have been developed to measure UPS function in specific pathways and compartments.
Use of fluorescent UPS reporters is subject to several technical considerations and potential artifacts. While clonal cell lines stably expressing UPS reporters are excellent tools for assessing the effects of pharmacological inhibition on proteasome function or upstream steps in the degradation pathway of UPS substrates (9, 10), using these lines to analyze the effects of transient protein-overexpression or knockdown can be confounded by cell-to-cell variation in expression level and transfection efficiency. This problem can be circumvented by use of fluorescent proteins co-expressed with the protein of interest as transfection markers (11). In some cases, the levels of the overexpressed proteins can be determined by directly fusing them to FPs with compatible excitation and emission spectra (12). This approach can be used to normalize different expression levels when comparing two or more transfected proteins.
Using steady-state fluorescence measurements to infer protein half-lives requires that the experimental manipulations under study do not influence the synthesis of the reporter protein. Indeed FP reporters have been used to measure transcription rates under conditions that are not thought to affect the UPS (4). It is therefore imperative to consider effects on reporter synthesis rates when using FP reporters to assess UPS function. In this regard, parallel measurement of stable, long-lived FPs can be used to assess and control for changes in transcription and translation rates.
Here, we describe protocols that utilize mCherry, a member of a class of improved fluorogenic proteins, displaying faster maturation, reduced aggregation, and better performance as FP fusion tags (13). Use of these proteins optimizes and simplifies existing protocols that use nonfluorogenic tags. Since fluorogenic proteins can be detected without cell permeabilization and fixation, it is also possible to perform some of the protocols described below in living cells. Tracking live cells allows assessment of UPS status over time and allows comparison between UPS reporter levels and levels of transiently expressed tagged proteins in individual cells.
2. Materials
2.1. Flow Cytometry-Based Assays
FuGENE 6 Transfection Reagent (Roche/Promega).
Trypsin–EDTA: TrypLE Express for cell trypsinization (Invitrogen).
pmCherry-C1 vector (Clontech).
peGFP-C3 vector (Clontech).
PBS: 1× phosphate-buffered saline (PBS) (GIBCO).
FACS-buffer: PBS, 2% FBS, 0.1% sodium azide.
LSR II flow cytometer with 488 nm blue laser and 532 nm green laser (BD Biosciences).
FlowJo Flow Cytometry Analysis Software (Tree Star, Inc.).
2.2. Assays Based on Quantitative Microscopy
4%Paraformaldehyde(w/v)(methanol-free)(ThermoScientific) in PBS.
Fluormount-G mounting reagent (Southern Biotech).
Microslides.
Coverslips with poly-L-lysine (VWR).
3. Methods
3.1. Flow Cytometry-Based Assays
3.1.1. Assaying UPS-GFP Reporter Levels in Cells Transfected with an mCherry-Tagged Protein by Two-Color Flow Cytometry
The simplest way to quantify FP levels in medium-sized populations of 103–107 cells is to analyze total fluorescence intensity by flow cytometry. The advantage of this approach is that it is possible to interrogate multiple fluorophores with the UPS reporter. For these studies, it is optimal to select clonal stable cell lines that exhibit a narrow distribution of basal intensities yielding uniform histograms that are easy to quantify and permit the detection of small changes in mean fluorescence (see Note 1). Such cell lines have been successfully used in experiments that quantify proteasome inhibition by pharmacological agents (10, 14).
Transient transfection of a UPS inhibitor will lead to increased reporter fluorescence levels in the subpopulation of cells that have taken up and expressed the inhibitor (15). The extent of this effect will be proportional to the level at which cells express the inhibitory protein. Analysis of inhibitor proteins fused to a spectrally compatible FP by two-color flow cytometry can reveal the relationship between UPS reporter and inhibitor levels within the transfected subpopulation. The use of a second FP obviates the need for an epitope-tag that requires antibody detection, cell fixation and permeabilization, and several incubations followed by wash steps. This reduces experiment time and eliminates additional sources of error. Additionally, the transfection of untagged mCherry can control for effects of transfection and protein overexpression.
Maintain cells at subconfluent densities throughout the experiment. The number of cells that has to be analyzed depends on the percentage of cells in the gated subpopulations. Each gate has to contain enough events to generate statistically significant data.
Transfect UPS-GFP cells using FuGENE 6 or a lipid or calcium phosphate-based method with the mCherry-tagged gene of choice and an untagged mCherry control vector. Process samples 24–72 h after transfection.
Harvest adherent cells with 1 mL trypsin–EDTA (or PBS, 10 mM EDTA, if trypsin is undesirable). Pellet cells at 200 × g for 5 min in a 5-mL FACS tube in a swinging bucket rotor. Resuspend cells in 1 mL cold PBS or FACS-buffer.
Analyze the cells by flow cytometry. Use a 488-nm blue laser to excite GFP and a 532–598-nm yellow/green laser to excite mCherry (see Note 2). To determine whether levels of UPS-GFP are increased in cells expressing high levels of the investigated protein, it is necessary to gate for subpopulations of cells with low and high levels of mCherry (Fig. 1a) and then generate a GFP histogram for each of the subpopulations (Fig. 1b). Use FlowJo or some comparable software to compensate for bleed-through of mCherry emissions into the GFP channel. Perform the compensation by using mCherry single-color controls to calculate mean GFP fluorescence intensities for the same mCherry subpopulations not expressing UPS-GFP (see Note 3 for a list of controls). After performing compensation, compare the mean GFP fluorescence of each mCherry subpopulation.
Fig. 1.

Assaying UPS-GFP reporter levels in cells transfected with an mCherry-tagged protein by two-color flow cytometry. (a) Histogram displaying frequency distribution as a function of mCherry-fluorescence of cells transiently transfected with mCherry fluorescent protein (dashed line) and the mCherry-tagged protein under investigation (solid line). Populations of cells with low and high mCherry levels are indicated. (b ) Histograms displaying the UPS-GFP fluorescence intensities of cells in the low and high mCherry populations. The two mCherry populations can be compared by their mean GFP fluorescence intensities.
3.1.2. Assaying UPS-GFP Reporter Levels in Cells Transfected with an mCherry-Tagged Protein According to Levels of mCherry Expression
This is a variation of step 4 of the protocol provided in Subheading 3.1. Instead of simply comparing two populations that have different levels of mCherry-tagged proteins, it is informative to further subdivide the cells by mCherry expression. This makes it possible to distinguish between effects caused by different levels of the overexpressed proteins. One possible effect of overexpression of an inhibitor is that UPS impairment is linearly correlated with levels of the mCherry-tagged protein. Another possibility is that UPS impairment occurs only in cells that express the protein beyond some threshold level. This type of analysis also identifies conditions that change the linear relationship between UPS-GFP and mCherry levels in the former case or shift the threshold in the latter.
Follow steps 1–3 of Subheading 3.1. Note that the resolution of this assay is inversely correlated to the width of the mCherry gates. It is therefore desirable to minimize the size of each gate. To still be able to collect enough events in each gate, it is necessary to analyze a higher number of cells than in Subheading 3.1. Therefore, analyze at least 2 × 105 cells per condition to ensure that enough data points are collected at the highest expression levels.
To plot levels of the reporter-protein versus levels of the transfected protein, subdivide the logarithmic axis for the transfected protein into as many equally weighted gates as possible (Fig. 2a).
Calculate the mean fluorescence of the GFP-UPS reporter for the cells that fall into each gate (Fig. 2b). Compensate for bleed through from mCherry into the GFP channel for each gate using the corresponding gates in the single color control without GFP-UPS. Do not include gates with less than 100 events to avoid statistically nonsignificant results.
Plot the gate number (arbitrary units corresponding to the log of fluorescence intensity of the transfected protein) on the abscissa and the compensated reporter fluorescence on the ordinate (Fig. 2c).
Fig. 2.

Assaying UPS-GFP reporter levels in cells transfected with an mCherry-tagged protein according to levels of mCherry expression. (a ) Histogram displaying frequency distribution as a function of mCherry fluorescence. The mCherry axis has been split into a series of gates of equal width. (b) Histograms displaying frequency distribution (Y-axis) of UPS-GFP fluorescence intensities (X-axis) of cells in each gated mCherry population (Z-axis). (c) X–Y scatter plot displaying the relationship between the level of the overexpressed protein and UPS impairment. The compensated mean GFP fluorescence intensity for each mCherry gate is plotted on the Y-axis versus the gate number on the X-axis.
3.2. Assays Based on Quantitative Microscopy
Two-color flow cytometry measures total GFP intensities versus total mCherry intensities for large numbers of cells on an individual cell basis. This allows analysis of enough events to compare large populations of cells with matched levels of the investigated protein. However, this assay does not detect changes in UPS function that are due to other parameters of the protein under investigation such as its localization or aggregation state. These questions can be addressed by quantitative microscopy (7, 16). Microscopy-based assays that do not rely on antibody staining and fixation can also measure changes in UPS reporter levels over time in individual cells (16).
A protocol published earlier (17) can easily be adapted to incorporate a second fluorescent protein marker. The use of a second marker in this protocol eliminates the need for cell permeabilization, several wash steps, and incubations with antibodies. The published protocol measures the total integrated fluorescence intensity of individual cells and relies on the generation of spherical cells by trypsinization to generate equatorial, circular cross-sections of nearly uniform area (Fig. 3a). A variation of this protocol measures the mean fluorescence intensity of one area of the cell (Fig. 3b). While the differences in the mean fluorescence intensity are smaller than the differences in the total fluorescence intensity, this approach has several advantages. Measuring the mean fluorescence of a small region enables analysis of UPS-GFP reporter levels in specific cellular regions or compartments and enables the detection of effects specific to those compartments. This approach can also be used to exclude regions where high levels of mCherry-tagged protein can colocalize with UPS-GFP, causing bleed through of mCherry into the GFP channel. Since it is not necessary to generate spherical cells in this protocol, it is possible to monitor changes in UPS reporter levels in live adherent cells over time. In this case, cells should not be fixed and mounted.
Fig. 3.
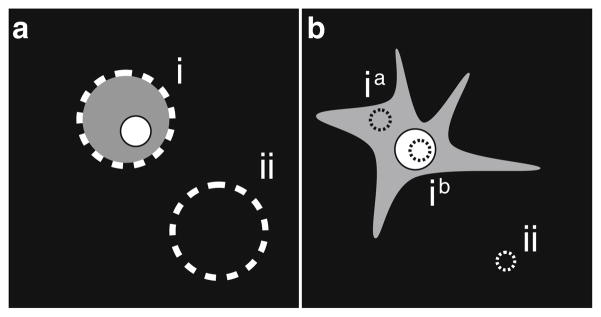
Assaying UPS-GFP reporter levels by quantitative microscopy. (a) Equatorial image of a trypsinized, spherical cell transfected with a protein of interest. To determine the fluorescence intensity of the UPS reporter, subtract the total integrated pixel intensity of region (ii) from the total integrated pixel intensity of region (i). (b) Adherent cell transfected with protein of interest. To determine the mean fluorescence of the UPS reporter in the cellular compartment of interest (e.g., cytosol ia or nucleus ib), subtract the mean intensity of the background region (ii) from the mean intensity of the compartment investigated.
Maintain cells at subconfluent densities throughout the experiment.
Transfect cells stably expressing the UPS-GFP reporter (see Note 1) using FuGENE 6 or a lipid or calcium phosphate-based method with the mCherry-tagged gene of choice and an mCherry control vector.
Transfer cells to lysine-coated coverslips 24–48 h before analysis.
At the desired time point wash the coverslips three times with 1 mL PBS.
Incubate coverslips in 4% paraformaldehyde for 15 min at room temperature (see Note 4).
Wash coverslips three times with 1 mL PBS.
Mount coverslips with mounting media with an antiphoto-bleaching reagent onto microscopy slides.
Use the red channel on the microscope to scan for mCherry-positive cells with the desired phenotype to minimize GFP photobleaching. Then image cells in the GFP channel to detect UPS-GFP. Use an exposure time that captures image intensities within the linear range of the CCD camera with the highest grayscale bit rate.
Use a suitable software program to collect and analyze images. Draw a circular region inside the cellular compartment of interest (e.g., ia or ib), and a second, identical region in a blank part of the field (ii). Calculate the final mean pixel intensity by subtracting the mean pixel intensity in region (i) from the mean pixel intensity in region (ii).
Footnotes
Reporter cell lines expressing destabilized fluorescent proteins can be created using standard tissue culture techniques (18). A HEK293 cell line stably expressing GFP destabilized by the CL-1 degron is available through American Type Culture Collection (ATCC, #CRL-2794). A HEK293 cell line expressing the green fluorescent protein ZsGreen, destabilized by a proteasome targeting sequence is available through Clontech (#631 535).
This setup minimizes emission overlap between the fluorophores (12).
Measuring levels of fluorescent reporters, destabilized by degrons, is a well-established technique used to determine the status of the UPS. However, levels of UPS reporters are also influenced by many other factors such as the rate of protein synthesis (4) and different forms of stress (11). In addition to actual changes in UPS reporter levels, other effects can lead to observed changes in total reporter fluorescence. Cross-talk/bleed-through between a pair of different fluorescent proteins can artificially elevate the observed fluorescence intensity or quench the fluorescence of one of the proteins. For instance, GFP and mCherry are a FRET-pair, and emissions from GFP can be quenched by excitation of mCherry when the two proteins are in close proximity. The observed fluorescence can also be altered by the excitation/emission spectra of any chemical substances that are used in the experiment. Therefore, it is necessary to control for these effects when performing fluorescence measurements.
The following are controls for an experiment that measures the effects of Protein A fused to mCherry (A-mCherry) expressed in a cell line stably expressing destabilized GFP (UPS-GFP) in the presence or absence of drug X:
UPS-GFP, baseline GFP control; UPS-GFP plus proteasome inhibitor, single-color GFP control; UPS-GFP plus drug X, control for how drug X affects UPS-GFP baseline levels; UPS-GFP plus mCherry, control for mCherry overexpression effects on UPS-GFP levels; UPS-GFP plus mCherry plus drug X, control for combinatorial effects between mCherry and drug X; UPS-GFP plus A-mCherry, effects of the tagged protein on UPS-GFP levels; UPS-GFP plus A-mCherry plus X, combinatorial effects of the tagged protein and drug X on UPS-GFP levels.
These conditions should also be analyzed in the parental cell line to generate single-color controls to compensate for cross-talk/bleed-through between the GFP and mCherry channels. These controls also correct for optical effects caused by drug X, including quenching and autofluorescence. Additionally, a line expressing a stable variant of GFP can be used to indicate whether changes in UPS-GFP levels are due to altered transcription/translation rates. The effects of FRET or quenching can be controlled by switching the fluorophores (UPS-mCherry and A-GFP) or by using a non-FRET pair of fluorophores.
Perform the incubation in the dark to avoid bleaching the chromophores.
References
- 1.Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 2.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 3.Dantuma NP, Lindsten K, Glas R, et al. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Zhao X, Fang Y, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- 5.Fiebiger E, Story C, Ploegh HL, et al. Visualization of the ER-to-cytosol dislocation reaction of a type I membrane protein. EMBO J. 2002;21:1041–1053. doi: 10.1093/emboj/21.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLaBarre B, Christianson JC, Kopito RR, et al. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22:451–462. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 7.Bennett EJ, Bence NF, Jayakumar R, et al. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Wang CE, Orr A, et al. Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol. 2008;180:1177–1189. doi: 10.1083/jcb.200709080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiebiger E, Hirsch C, Vyas JM, et al. Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell. 2004;15:1635–1646. doi: 10.1091/mbc.E03-07-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keyomarsi K, Efuet ET, Bui TN. Semi-high throughput method of measuring proteasome inhibition in vitro and in cultured cells. Cell Biol Toxicol. 2010 doi: 10.1007/s10565-010-9175-1. [DOI] [PubMed] [Google Scholar]
- 11.Salomons FA, Menendez-Benito V, Bottcher C, et al. Selective accumulation of aggregation-prone proteasome substrates in response to proteotoxic stress. Mol Cell Biol. 2009;29:1774–1785. doi: 10.1128/MCB.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riley BE, Kaiser SE, Shaler TA, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010;191:537–552. doi: 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaner NC, Campbell RE, Steinbach PA, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 14.Kessler BM, Tortorella D, Altun M, et al. Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic beta-subunits. Chem Biol. 2001;8:913–929. doi: 10.1016/s1074-5521(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 15.Lindsten K, de Vrij FM, Verhoef LG, et al. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol. 2002;157:417–427. doi: 10.1083/jcb.200111034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitra S, Tsvetkov AS, Finkbeiner S. Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J Biol Chem. 2009;284:4398–4403. doi: 10.1074/jbc.M806269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bence NF, Bennett EJ, Kopito RR. Application and analysis of the GFPu family of ubiquitin-proteasome system reporters. Methods Enzymol. 2005;399:481–490. doi: 10.1016/S0076-6879(05)99033-2. [DOI] [PubMed] [Google Scholar]
- 18.Davis JM, editor. Basic Cell Culture. 2. Oxford University Press; 2002. [Google Scholar]