

Abstract
Background and Purpose
Plasma thrombin concentration is increased following subarachnoid hemorrhage (SAH). However, the role of thrombin receptor (protease activated recptor-1, PAR-1) in endothelial barrier disruption has not been studied. The aims of this study were to investigate the role of PAR-1 in orchestrating vascular permeability and assess the potential therapeutics of a PAR-1 antagonist, SCH79797, through maintaining vascular integrity.
Methods
SCH79797 was injected intraperitoneally into male Sprauge-Dawley rats undergoing SAH by endovascular perforation. Assessment was conducted at 24 hours after SAH for brain water content, Evans blue content, and neurobehavioral testing. To explore the role of PAR-1 activation and the specific mechanism of SCH79797’s effect after SAH, Western blot, immunoprecipitation, and immunofluorescence of hippocampus tissue were performed. A p21-activated kinase1 (PAK1) inhibitor, IPA-3, was used to explore the underlying protective mechanism of SCH79797.
Results
At 24 hours after SAH, animals treated with SCH79797 demonstrated a reduction in brain water content, Evans blue content, and neurobehavioral deficits. SCH79797 also attenuated PAR-1 expression and maintained the level of VE-cadherin, an important component of adherens junctions. Downstream to PAR-1, c-Src dependent activation of PAK1 led to an increased serine/threonine phosphorylation of VE-cadherin; immunoprecipitation results revealed an enhanced binding of phosphorylated VE-cadherin with endocytosis orchestrator β-arrestin2. These pathological states were suppressed following SCH79797 treatment.
Conclusions
PAR-1 activation following SAH increases microvascular permeability, at least, partly through a PAR-1-c-Src-PAK1-VE-cadherin phosphorylation pathway. Through suppressing PAR-1 activity, SCH79797 plays a protective role in maintaining microvascular integrity after SAH.
Keywords: Subarachnoid hemorrhage, microvascular permeability, protease activated recptor-1, VE-cadherin, rat
Brain edema is one of the fatal pathologies following subarachnoid hemorrhage (SAH).1Previous research suggested increased microvascular permeability might be a primary contributor to brain edema. 2Normally, the vascular endothelial barrier is maintained by two key junctions between endothelial cells, tight junctions (TJs), and adherens junctions (AJs). 3In the last two decades, much attention has been paid to the involvement of TJs in the increased vascular permeability following SAH.4However, the integrity of AJs is also critical in maintaining microvascular permeability, and disruption of AJs can lead to interstitial edema.5AJs are comprised of transmembrane protein VE-cadherin and α, β, γ, and δ-catenins in the cytoplasm.3Inhibition of VE-cadherin by homophilic binding resulted in increased microvascular permeability.6
Protease activated receptor-1, -3, and -4(PAR-1, 3, and 4, but not PAR-2) are thrombin receptors, which are a subfamily of G protein-coupled receptors.7 PAR-1 is a major receptor on the endothelial cells.8 PAR-1 activation increases the activity of downstream protein kinases, such as c-Src and p21-activated kinase1 (PAK1), which leads to phosphorylation of key target proteins.9Before PAR-1 orchestrates its actions, it must be activated by thrombin, which increased in plasma following SAH.10-12 Although PAR-1 activation could lead to cerebral vasospasm,13 its role in increasing microvascular permeability following SAH is not clear yet.
In this study, we demonstrated that PAR-1 activation mediated an increase in microvascular permeability following SAH. Additionally, the protective effect of SCH79797, a specific PAR-1 antagonist, was also evaluated.
Materials and Methods
All procedures were approved by the Loma Linda University animal care committee.
SAH Model and Study Protocol
The endovascular perforation model of SAH was established in male Sprague-Dawley rats (300 to 320g; Harlan, Indianapolis, IN) as previously described.14With 2% to 3% isoflurane anesthesia, a sharpened 4-0 monofilament nylon suture was inserted rostrally into the right internal carotid artery from the external carotid artery stump and perforated the bifurcation of the anterior and middle cerebral arteries. Blood pressure and blood gas were recorded via the right femoral artery. Rectal temperature was maintained at 37°C during surgery. Sham-operated rats underwent the same procedures except the suture was withdrawn without puncture.
Firstly, 34 rats were randomly divided into four groups. Finally, 27 rats (described as 27/34, same below) were used after excluding the dead and unqualified animals according to the inclusion criteria (see below). The roles of a PAR-1 specific agonist, SFLLRN (Tocris, Ellisville, MO, 1mg/kg as reported by the others15,16), and a thrombin specific inhibitor, argatroban (Sigma-Aldrich, St. Louis, MO), were evaluated (Fig. 1A). Next, 53 rats were randomly divided into 6 groups (42 rats were used, 42/53). The dose-dependent effects of PAR-1 specific antagonist, SCH79797 (Tocris, Ellisville, MO), were evaluated (Fig. 1B). Subsequently, 91 rats were randomly divided into 3 groups (75 rats were used, 75/91) to assess SCH79797 on maintaining vascular integrity (Fig. 1C). Finally, 35 rats were randomly divided into 4 groups (28 rats were used, 28/35) to explore the mechanism of SCH79797’s effects (Fig. 1D). The time point, dose, and route of drug administration are shown in Fig. 1.
Figure 1.
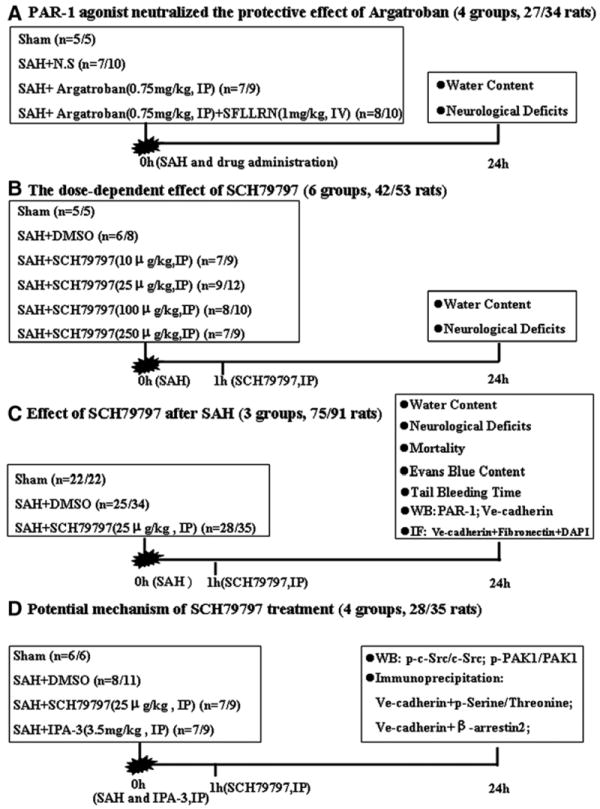
Experimental designs. (A) Experiment 1 was designed to investigate the effect of the PAR-1 agonist, SFLLRN. (B) Experiment 2 was designed to evaluate the dose-dependent effect of SCH79797. (C) Experiment 3 was designed to show the effects of SCH79797 treatment. (D) Experiment 4 was designed to explore the mechanism of SCH79797 treatment. IP, intraperitoneally injection; IV, intravenously injection; IF, immunofluorescence staining; WB, Western blot.
SAH Grade
The basal brains were divided into 6 segments with each segment allotted a grade from 0 to 3 depending on the amount of blood present. The animals received a total score ranging from 0 to 18 by summing the scores of each segment (sham group=0) 17. The similar scores amongst the groups indicated a similar injury induced by SAH. The animals with subdural hemorrhage, extradural hemorrhage, and mild hemorrhage were excluded. Only animals experiencing severe grade hemorrhage (scores >12) were included in this work.
Neurobehavioral Testing
Neurological outcomes were assessed by a blinded observer using the modified Garcia score.18This is an 18-point sensorimotor assessment system consisting of six tests with scores of 0-3 for each test (max score of 18). The tests included spontaneous activity, side stroking, vibrissa touch, limb symmetry, climbing, and forelimb walking. Additional testing was conducted blindly using the beam balance test, which assessed the animals’ ability to walk on a narrow wooden beam (22.5 mm in diameter) within 60 seconds; 4 points= walking ≥20 cm; 3 points=walking ≥10 cm but <20 cm; 2 points=walking ≥10 cm but falling; 1 point=walking <10 cm; and 0 points=falling with walking <10 cm. The mean score of three trials in a 5 minute interval was recorded.
Tail Bleeding Time
The tails of anesthetized rats were immersed in an isotonic (0.9%) saline solution at 37°C for five minutes and cut off at 4 mm from the distal end. The bleeding time was recorded blindly.19
Brain Water Content
After euthanasia, animals’ brains were divided into right and left hemispheres, brain stem, and cerebellum. These specimens were dried in an oven at 105°C for 72 hours. The following formula was used to calculate the percentage of water content: ([wet weight-dry weight]/wet weight) ×100%.
BBB Permeability
Under general anesthesia, Evans blue dye (2%; 5 mL/kg) was injected into the left femoral vein and allowed to circulate for 60 minutes. Rats were then euthanized by intracardiac perfusion with 0.01 mol/L phosphate-buffered saline (PBS), and brains were divided into the same regions as the water content study. The amount of extravasated Evans blue dye was measured by spectrofluorophotometry. 20
Western Blot Analyses
Protein samples (30μg) from the hippocampus were loaded on a Tris glycine gel, electrophoresed, and transferred to a nitrocellulose membrane. Membranes were blocked with a blocking solution, followed by incubation overnight at 4°C with the following primary antibodies(1:1000) (All antibodies were from Santa Cruz Biotechnology until specifically indicated): mouse anti-PAR-1, goat anti–VE-cadherin, goat anti–phosphorylated-c-Src and c-Src, rabbit anti–phosphorylated-PAK1 and PAK1, rabbit anti–β-arrestin2, mouse anti–phosphorylated-serine/threonine. Immunoblots were processed with appropriate secondary antibodies (1:2000) for 1 hour at 21°C. The bands were detected with a chemiluminescence reagent kit (Amersham Bioscience, Arlington Heights, IL) and quantified by densitometry with Image J software (National Institutes of Health, Bethesda, MD). ß-Actin was blotted on the same membrane as a loading control.
Immunoprecipitation
The samples were mixed and rotated for 2h at 4°C with corresponding primary antibody, and incubated overnight with protein G-agarose. The immunoprecipitated proteins were collected by centrifugation, the pellets were washed and boiled in loading buffer and analyzed as described above for immunoblotting.
Immunofluorescence Staining
The coronal sections (10 μm thickness) containing the bilateral hippocampus were cut on a cryostat (Leica Microsystems, Bannockburn, IL) and mounted on poly-L-lysine–coated slides. Sections were incubated overnight at 4°C with goat anti-VE-cadherin, rabbit anti-Fibronectin primary antibodies and DAPI. Appropriate fluorescence dye–conjugated secondary antibodies (Jackson Immunoresearch, West Grove, PA) were applied in the dark for 1 hour at 21°C. For negative controls, the primary antibodies were omitted and the same staining procedures were performed. The sections were visualized with a fluorescence microscope, and the photomicrographs were saved and merged with Image Pro Plus software (Olympus, Melville, NY).
Statistical Analyses
The data of neurological scores and tail bleeding time were expressed as median±25th to 75th percentiles and analyzed using the Kruskal-Wallis test followed by the Steel-Dwass multiple comparisons. The mortality was analyzed by the Chi square test. Other values were expressed as mean±standard deviation, and one way analysis of variance (ANOVA) with the Tukey-Kramer post-hoc tests was used. A P-value of less than 0.05 was considered significant.
Results
PAR-1 Agonist Neutralized the Protective Effects of Argatroban
Following SAH, PAR-1 expression in the hippocampus was significantly increased and peaked at 2-6 hours and 24 hours (Fig. 2A). Argatroban, a thrombin specific inhibitor, significantly decreased brain water content (left hemisphere: SAH+argatroban 79.23%±0.25% vs. SAH+N.S 79.63%±0.24%, P<0.05; right hemisphere: SAH+argatroban 79.21%±0.09% vs. SAH+N.S 79.79%±0.14%, P<0.01) and reduced neurological deficits (P<0.05) at 24 hours following SAH. These protective effects were neutralized by simultaneously applying a PAR-1 agonist, SFLLRN (brain water content: left hemisphere, SAH+argatroban+SFLLRN 79.93%±0.69% vs. SAH+argatroban 79.23%±0.25%,P<0.05; right hemisphere: SAH+argatroban+SFLLRN 80.29%±0.75% vs. SAH+argatroban 79.21%±0.09%, P<0.01)(Fig. 2B, C and D). These results indicated that, as a major thrombin receptor on endothelial cells, PAR-1 activation could lead to increased microvascular permeability and neurological impairments following SAH.
Figure 2.
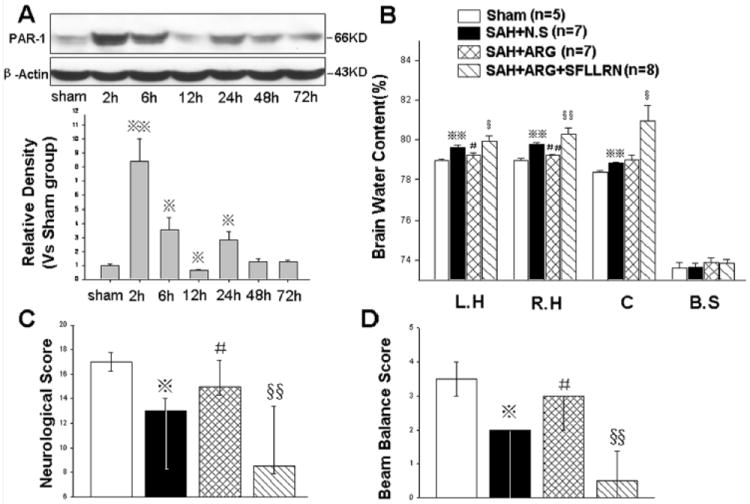
The effects of the PAR-1 agonist, SFLLRN. There were two peak levels of PAR-1 expression at 2-6 hours and 24 hours following SAH (Fig. A, n=6 each group). SFLLRN neutralized the protective effects of the thrombin inhibitor, argatroban, on attenuating brain edema (Fig. B) and neurological deficits (Fig. C and D) at 24 hours following SAH. In Figures B, C, and D, n=5,7,7,8 in sham, SAH+N.S, SAH+argatroban, and SAH+argatroban+SFLLRN groups. *P<0.05, **P<0.01 compared with the sham group; #P<0.05, ##P<0.01 compared with the SAH+N.S group; §P<0.05, §§P<0.01 compared with the SAH+argatroban group. The Western blot results were representative of three independent experiments. N.S, normal saline; ARG, argatroban; L.H, left hemisphere; R.H, right hemisphere; C, cerebellum; B.S, brain stem.
The Dose-Dependent Effect of SCH79797
At 24 hours post injury, the dose of 25μg/kg SCH79797 (PAR-1 antagonist) was most effective at decreasing brain water content (left hemisphere: SAH+25μg/kg SCH 79.34%±0.15% vs. SAH+DMSO 79.65%±0.23%, P<0.05; right hemisphere: SAH+25μg/kg SCH 79.25%±0.19% vs. SAH+DMSO 79.62%±0.36%, P<0.05, Fig. 3A) and neurological deficits (P<0.05, Fig. 3B and C). In addition, SAH grades were not significantly different amongst the groups (Supplemental Figure 1, please see http://stroke.ahajournals.org).
Figure 3.
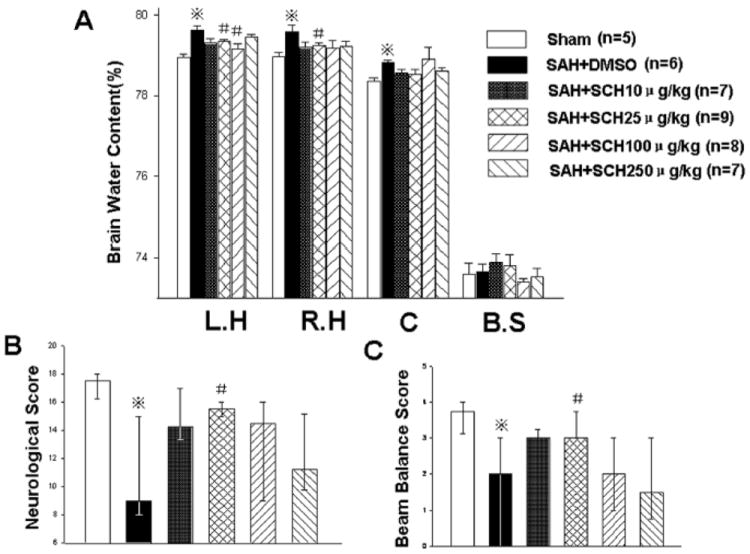
The dose-dependent effects of SCH79797. Compared with other doses, the dose of 25μg/kg SCH79797 was more effective on decreasing brain water content (Fig. A) and neurological deficits (Fig. B and C) at 24 hours following SAH. n=5,6,7,9,8,7 in sham, SAH+DMSO, SAH+ 10,25,100,250 μg/kg SCH79797 groups. *P<0.05 compared with the sham group; #P<0.05 compared with the SAH+DMSO group.
SCH79797 Provided Significant Neuroprotection following SAH
At 24 hours after SAH, in addition to reducing brain edema and neurological impairments (Fig. 3), SCH79797 also decreased Evans blue extravasations in the brain(μg/g) (left hemisphere: SAH+25μg/kg SCH 1.21±0.24 vs. SAH+DMSO 2.08±0.68,P<0.05; right hemisphere: SAH+25μg/kg SCH 1.87±0.08 vs. SAH+DMSO 3.25±0.96,P<0.05, Fig. 4A).
Figure 4.
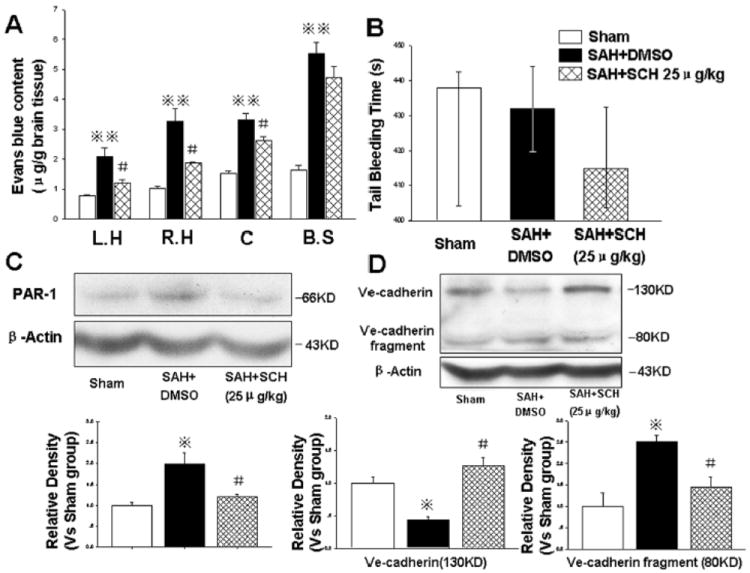
SCH79797 maintained microvessel integrity. At 24 hours following SAH, 25μg/kg SCH79797 markedly decreased Evans blue content (Fig. A, n=5,7,7 in sham, SAH+DMSO, SAH+ 25μg/kg SCH79797 groups) without prolonging the tail bleeding time (Fig. B, n=5,6,9 in sham, SAH+DMSO, SAH+ 25μg/kg SCH79797 groups). Western blot results revealed that SCH79797 suppressed PAR-1 expression (Fig. C) and maintained VE-cadherin level by reducing the cleavage of VE-cadherin (about 80KD fragment, Fig. D) (in Fig. C and D, n=6 each group). *P<0.05,**P<0.01 compared with sham group; #P<0.05 compared with SAH+DMSO group.
Mortality occurred within 6 hours after surgery, and the mortality of the SAH+DMSO group (26.47%, 9 of 34 rats) was not significantly different from the SAH+25μg/kg SCH79797 group (20.00%, 7 of 35 rats; P=0.52, chi square tests). In addition, the tail bleeding time was not significantly altered following SCH79797 treatment (P>0.05, Fig. 4B).
SCH79797 significantly decreased PAR-1 expression (Fig. 4C and Supplemental Figure 2) and maintained the level of VE-cadherin by reducing the cleavage of VE-cadherin (about 80KD fragment) (P<0.05, Fig. 4D).
At 24 hours after SAH, immunofluorescence staining of microvessels in the hippocampus was performed. In the sham group, VE-cadherin was abundantly distributed in the borders of endothelial cells (Fig. 5A1 and a). Fibronectin, a well established indicator for evaluating microvascular integrity, 21 is a high-molecular weight glycoprotein normally distributed in the vessel walls and blood. In the sham group, fibronectin was exclusively distributed in the vessel walls (Fig. A2). After SAH, the level of VE-cadherin was significantly decreased with numerous fibronectin extravasations scattered around the microvessels (Fig. B1, b and B2). Following SCH79797 treatment, these pathological findings were markedly attenuated (Fig.C1, c and C2).
Figure 5.
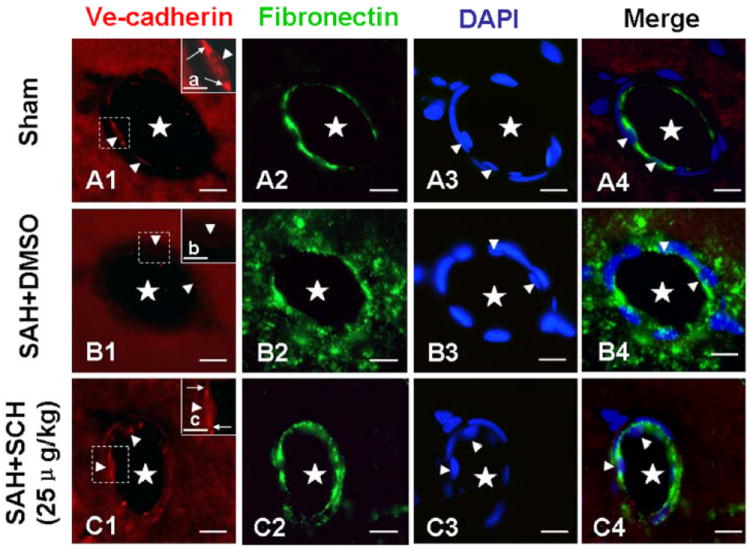
Immunofluorescence staining of mircovessels in the hippocampus. In the sham group, VE-cadherin was abundantly distributed in the borders of endothelial cells (Fig. A1 and a), while fibronectin was distributed exclusively in the vessel walls (Fig. A2). At 24 hours after SAH, the level of VE-cadherin was significantly decreased (Fig. B1 and b), and numerous fibronectin positive extravasations were scattered around the microvessels (Fig. B2). These pathologies were attenuated by SCH79797 treatment (Fig. C1, c and C2). DAPI staining showed the nuclei of endothelial cells. The “star” means microvessel lumen; the “arrowhead” indicates endothelial cell; the “arrow” (in Fig. a and c) indicates VE-cadherin. The figures a, b, and c were the magnification of the dashed area in the Figures A1, B1, and C1, respectively. In Fig. A, B, and C, scale bar=20μm; in Fig. a, b, and c, scale bar=5μm. n=6 each group.
SCH79797 Maintained VE-cadherin Level through Blocking PAR-1-c-Src-PAK1 Pathway
At 24 hours following SAH, the levels of phosphorylated c-Src and PAK1 were significantly increased, which were reversed by SCH79797 treatment (P<0.05, Fig. 6A and B). A PAK1 inhibitor, IPA-3, decreased the level of phosphorylated PAK1 (Fig. 6B) but not phosphorylated c-Src (Fig. 6A), however, a c-Src inhibitor, PP2 (1mg/kg, IP), significantly decreased the level of phosphorylated PAK1 (P<0.05, Supplemental Figure 3), which indicated c-Src is an upstream regulator of PAK1 in the PAR-1 signaling pathway.
Figure 6.
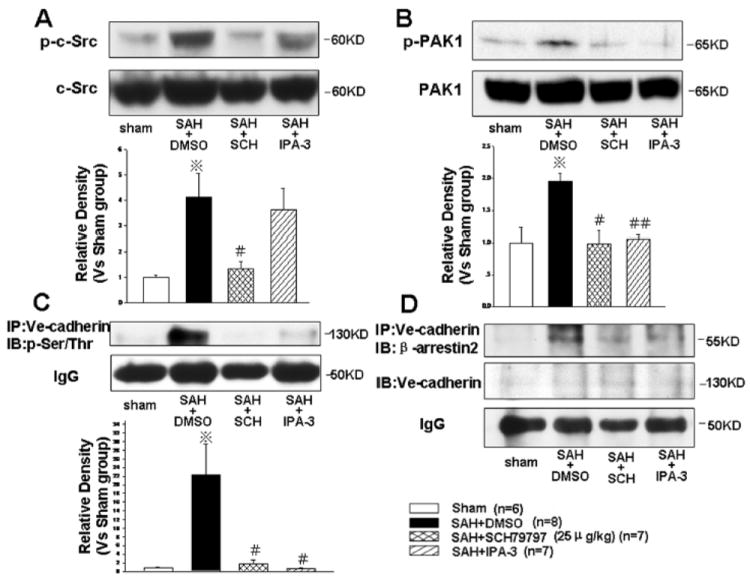
Endocytosis of VE-cadherin induced by c-Src dependent PAK1 activation. At 24 hours following SAH, SCH79797, but not PAK1 inhibitor, IPA-3, decreased the phosphorylated c-Src level (Fig. A). Both SCH79797 and IPA-3 decreased the levels of phosphorylated-PAK1 (Fig. B), phosphorylated-VE-cadherin on serine/threonine residues (Fig. C), and VE-cadherin binding with β-arrestin2 (Fig. D). *P<0.05 compared with the sham group; #P<0.05, ##P<0.01 compared with the SAH+DMSO group. n=6,8,7,7 in sham, SAH+DMSO, SAH+25μg/kg SCH79797, and SAH+IPA-3 groups. “IP” indicates “immunoprecipitation”; “IB” indicates “immunoblot”.
The immunoprecipitation results revealed that the phosphorylation of VE-cadherin on its serine/threonine residues facilitates its binding with endocytosis orchestrator β-arrestin2, a cytoplasmic adaptor protein that orchestrates the endocytosis of membrane proteins. This binding was attenuated by SCH79797 and IPA-3 (P<0.05, Fig. 6C and D). These results indicated activated PAK1 modulates phosphorylation and consequent endocytosis (degradation) of VE-cadherin after SAH, and SCH79797 decreases VE-cadherin endocytosis by suppressing c-Src dependent PAK1 activation in the PAR-1 signaling pathway.
Discussion
In this study, we investigated the role of PAR-1 in orchestrating increased microvascular permeability following SAH. We also found the PAR-1 antagonist, SCH79797, preserved microvascular integrity and provided neurobehavioral protection, which was partly mediated via suppression of VE-cadherin endocytosis induced by c-Src dependent PAK1 activation.
Thrombin plays roles in both coagulation processes and receptor-dependent inflammation, some of which are mediated via PAR activation. Thrombin leads to brain edema after forebrain ischemia22 and intracerebral hemorrhage.23 The increased plasma thrombin following SAH is partly attributed to the disruption of arteries, endogenous thrombin production, and transfer from CSF to veins.10-12 As a major thrombin receptor on endothelial cells,8 the role of PAR-1 in inflammation after SAH was not clear. In this study, we found the expression of PAR-1 was significantly increased after SAH (Fig. 2A), which indicated increased thrombin in plasma and PAR-1 on endothelial cells leads to some pronounced pathologies in the vessels, such as vasospasm as reported previously.13
In addition, we explored the role of PAR-1 in the formation of brain edema. We found the protective role of the thrombin inhibitor, argatroban, was reversed by the PAR-1 agonist, SFLLRN (Fig. 2B, C and D). In addition, the PAR-1 antagonist, SCH79797, significantly reduced brain water content (Fig. 3A). These results indicated PAR-1 activation did play a vital role in increasing microvessel permeability, regardless of the roles of other thrombin receptors (for example, PAR-4).
After determining the role of PAR-1 in increasing vessel permeability, we examined the effects of PAR-1 inhibition following SAH. In fact, the protective role of PAR-1 antagonists in other cardiovascular disease models have been reported.24We found 25μg/kg SCH79797 [a potent, specifically selective PAR-1 receptor antagonist25] was the most effective in attenuating brain edema, Evans blue extravasations, and neurobehavioral deficits. Additionally, PAR-1 expression was decreased after SCH79797 treatment, although the mechanism was not clear. The unactivated PAR-1 may be delivered to the cell surface and cycles constitutively between the plasma membrane and an early endosomal recycling compartment26, forming a cytoplasmic pool. This PAR-1 internalization is dependent on clathrin and dynamin27.SCH79797 inhibited PAR-1 activation after SAH, which may indirectly promote the internalization and degradation of PAR-1 in lysosomes. Therefore, the level of PAR-1 was reduced after SCH79797 treatment. Furthermore, tail bleeding time, a well-established parameter for evaluating coagulation function,28was not significantly prolonged after SCH79797 treatment, which was similar to the results reported by the others.29 These results suggested SCH79797 plays a protective role in maintaining microvascular integrity, clinically, without interfering with coagulation function and increasing the risk of further hemorrhage in SAH patients.
The permeability between endothelial cells is mainly determined by TJs andAJs.3 In fact, TJs and AJs are sometimes intermingled together.30 AJs form earlier than TJs during embryogenesis and are also established prior to TJs.31The integrity of AJs is critical in regulating microvascular permeability, and the disruption of AJs leads to interstitial edema.5 AJs in microvessels are mainly composed of VE-cadherin, β-catenin, α-catenin, and p120.3 The extracellular domain of VE-cadherin mediates homophilic interactions,32 and the inhibition of VE-cadherin homophilic binding results in the increased microvascular permeability.6 In this study, at 24 hours following SAH, with increased expression of PAR-1, the level of VE-cadherin was significantly decreased due to cleavage (Fig. 4D). In addition, VE-cadherin was absent from the borders of endothelial cells, while microvascular permeability was enhanced (Fig. 5). These pathological changes were alleviated by SCH79797 treatment. These results demonstrated SCH79797 preserved microvascular integrity by maintaining the level of VE-cadherin via attenuating its cleavage induced by PAR-1 activation.
Finally, we explored the mechanism of VE-cadherin cleavage and the neuroprotective roles of SCH79797. It had been reported that the phosphorylation of VE-cadherin, induced by c-Src and PAK1, resulted in VE-cadherin undergoing endocytosis and degradation by binding with β-arrestin2 in vitro.9 In this study, both c-Src and PAK1 were activated after SAH (Fig. 6A and B). Meanwhile, the serine/threonine phosphorylation and endocytosis of VE-cadherin, orchestrated by β-arrestin2, were also markedly increased (Fig. 6C and D), which was decreased by the PAK1 inhibitor, IPA-3. SCH79797 alleviated the phosphorylation and endocytosis of VE-cadherin by decreasing the levels of phosphorylated c-Src and PAK1. These results indicated PAR-1 induced phosphorylation and endocytosis of VE-cadherin via c-Src dependent PAK1 activation, which partly mediated VE-cadherin loss following SAH. Through blocking the PAR-1-c-Src-PAK1 pathway, SCH79797 maintained VE-cadherin levels by suppressing the endocytosis of VE-cadherin after SAH.
In conclusion, we showed that the increased endocytosis of VE-cadherin, induced by the PAR-1-c-Src-PAK1 pathway, might be partly responsible for increased microvascular permeability after SAH. The PAR-1 antagonist, SCH79797, played a neuroprotective role by maintaining microvascular integrity via blocking the PAR-1-c-Src-PAK1 signaling pathway.
Supplementary Material
Acknowledgments
Sources of Funding
This study was partially supported by a grant from the NIH/National Institute of Neurological Disorders and Stroke (NS053407 to J.H.Z.).
Footnotes
Disclosures
None.
References
- 1.Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–1232. doi: 10.1161/01.str.0000015624.29071.1f. [DOI] [PubMed] [Google Scholar]
- 2.Yan J, Li L, Khatibi NH, Yang L, Wang K, Zhang W, et al. Blood-brain barrier disruption following subarachnoid hemorrhage may be facilitated through PUMA induction of endothelial cell apoptosis from the endoplasmic reticulum. Exp Neurol. 2011;230:240–247. doi: 10.1016/j.expneurol.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 3.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 4.Yan J, Chen C, Hu Q, Yang X, Lei J, Yang L, et al. The role of p53 in brain edema after 24 h of experimental subarachnoid hemorrhage in a rat model. Exp Neurol. 2008;214:37–46. doi: 10.1016/j.expneurol.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Corada M, Mariotti M, Thurston G, Smith K, Kunkel R, Brockhaus M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito T, Bunnett NW. Protease-activated receptors: regulation of neuronal function. Neuromolecular Med. 2005;7:79–99. doi: 10.1385/NMM:7:1-2:079. [DOI] [PubMed] [Google Scholar]
- 8.Lang NN, Guethmundsdottir IJ, Newby DE. Vascular PAR-1: Activity and Antagonism. Cardiovasc Ther. 2011;29:349–361. doi: 10.1111/j.1755-5922.2010.00140.x. [DOI] [PubMed] [Google Scholar]
- 9.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 10.Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Koike T, Tanaka R. Serial changes of hemostasis in aneurysmal subarachnoid hemorrhage with special reference to delayed ischemic neurological deficits. J Neurosurg. 1997;86:594–602. doi: 10.3171/jns.1997.86.4.0594. [DOI] [PubMed] [Google Scholar]
- 11.Nina P, Schisano G, Chiappetta F, Luisa Papa M, Maddaloni E, Brunori A, et al. A study of blood coagulation and fibrinolytic system in spontaneous subarachnoid hemorrhage. Correlation with hunt-hess grade and outcome. Surg Neurol. 2001;55:197–203. doi: 10.1016/s0090-3019(01)00402-5. [DOI] [PubMed] [Google Scholar]
- 12.Fujii Y, Takeuchi S, Sasaki O, Minakawa T, Koike T, Tanaka R. Hemostasis in spontaneous subarachnoid hemorrhage. Neurosurgery. 1995;37:226–234. doi: 10.1227/00006123-199508000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Kai Y, Hirano K, Maeda Y, Nishimura J, Sasaki T, Kanaide H. Prevention of the hypercontractile response to thrombin by proteinase-activated receptor-1 antagonist in subarachnoid hemorrhage. Stroke. 2007;38:3259–3265. doi: 10.1161/STROKEAHA.107.487769. [DOI] [PubMed] [Google Scholar]
- 14.Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26:1086–1091. doi: 10.1161/01.str.26.6.1086. [DOI] [PubMed] [Google Scholar]
- 15.Chintala MS, Chiu PJ, Bernadino V, Tetzloff GG, Tedesco R, Sabin C, et al. Disparate effects of thrombin receptor activating peptide on platelets and peripheral vasculature in rats. Eur J Pharmacol. 1998;349:237–243. doi: 10.1016/s0014-2999(98)00200-3. [DOI] [PubMed] [Google Scholar]
- 16.Hwa JJ, Ghibaudi L, Williams P, Chintala M, Zhang R, Chatterjee M, et al. Evidence for the presence of a proteinase-activated receptor distinct from the thrombin receptor in vascular endothelial cells. Circ Res. 1996;78:581–588. doi: 10.1161/01.res.78.4.581. [DOI] [PubMed] [Google Scholar]
- 17.Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 19.Arruzazabala ML, Molina V, Carbajal D, Mas R. D-003 and warfarin interaction on the bleeding time and venous thrombosis experimentally induced in rats. J Med Food. 2004;7:260–263. doi: 10.1089/1096620041224003. [DOI] [PubMed] [Google Scholar]
- Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- 21.Yeung D, Manias JL, Stewart DJ, Nag S. Decreased junctional adhesion molecule-A expression during blood-brain barrier breakdown. Acta Neuropathol. 2008;115:635–642. doi: 10.1007/s00401-008-0364-4. [DOI] [PubMed] [Google Scholar]
- 22.Ohyama H, Hosomi N, Takahashi T, Mizushige K, Kohno M. Thrombin inhibition attenuates neurodegeneration and cerebral edema formation following transient forebrain ischemia. Brain Res. 2001;902:264–271. doi: 10.1016/s0006-8993(01)02354-x. [DOI] [PubMed] [Google Scholar]
- 23.Nagatsuna T, Nomura S, Suehiro E, Fujisawa H, Koizumi H, Suzuki M. Systemic administration of argatroban reduces secondary brain damage in a rat model of intracerebral hemorrhage: histopathological assessment. Cerebrovasc Dis. 2005;19:192–200. doi: 10.1159/000083466. [DOI] [PubMed] [Google Scholar]
- 24.Létienne R, Leparq-Panissié A, Bocquet A, Calmettes Y, Culié C, Le Grand B. PAR1 antagonist mediated antithrombotic activity in extracorporeal arterio-venous shunt in the rat. Thromb Res. 2010;125:257–261. doi: 10.1016/j.thromres.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Perez M, Lamothe M, Maraval C, Mirabel E, Loubat C, Planty B, et al. Discovery of novel protease activated receptors 1 antagonists with potent antithrombotic activity in vivo. J Med Chem. 2009;52:5826–5836. doi: 10.1021/jm900553j. [DOI] [PubMed] [Google Scholar]
- 26.Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. 2006;26:3231–3242. doi: 10.1128/MCB.26.8.3231-3242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trejo J, Altschuler Y, Fu HW, Mostov KE, Coughlin SR. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem. 2000;275:31255–31265. doi: 10.1074/jbc.M003770200. [DOI] [PubMed] [Google Scholar]
- 28.Wollny T, Iacoviello L, Buczko W, de Gaetano G, Donati MB. Prolongation of bleeding time by acute hemolysis in rats: a role for nitric oxide. Am J Physiol. 1997;272:H2875–H2884. doi: 10.1152/ajpheart.1997.272.6.H2875. [DOI] [PubMed] [Google Scholar]
- 29.Nadal-Wollbold F, Bocquet A, Bourbon T, Létienne R, Le Grand B. Protease-activated receptor 1 antagonists prevent platelet aggregation and adhesion without affecting thrombin time. Eur J Pharmacol. 2010;644:188–194. doi: 10.1016/j.ejphar.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009;335:17–25. doi: 10.1007/s00441-008-0694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 32.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.