

Abstract
Background/Objective
Haploinsufficiency of PMP22 causes hereditary neuropathy with liability to pressure palsies (HNPP). However, the biological functions of PMP22 in humans are largely unexplored due to the absence of patients with PMP22 null mutations.
Design, Setting and Participants
We have evaluated a 7-year-old boy with PMP22 null. Findings were compared with those from nerves of Pmp22 null mice.
Results
Motor and sensory deficits in the proband were non-length dependent. Weakness was found in cranial muscles, but not in the limbs. Large fiber sensory modalities were profoundly abnormal, which started prior to the maturation of myelin. This is in line with the temporal pattern of PMP22 expression predominantly in cranial motor neurons and DRG during embryonic development, becoming undetectable in adulthood. Moreover, there were conspicuous maturation defects of myelinating Schwann cells that were more significant in motor nerve fibers than in sensory nerve fibers.
Conclusions
Taken together, these data suggest that PMP22 is important for the normal function of neurons that express PMP22 during early development, such as cranial motor neurons and spinal sensory neurons. Moreover, PMP22 deficiency differentially affects myelination between motor and sensory nerves, which may have contributed to the unique clinical phenotype in the patient with absence of PMP22.
INTRODUCTION
Peripheral myelin protein-22 (PMP22) is encoded by the PMP22 gene within the DNA segment of human chromosome 17p11.2. PMP22 is most abundantly expressed in the myelinating Schwann cells of peripheral nerves1, but its function is poorly understood.
During development, the transcription of PMP22 in motor neurons shows a rostral-caudal pattern with cranial motor nuclei first expressing the protein embryonically and spinal motor neurons expressing it after birth. In contrast, PMP22 transiently expresses in sensory neurons of cranial nuclei and DRG at embryonic stages, but the transcripts become undetectable in young adults2,3. The significance of this expression difference between sensory and motor neurons is unknown.
PMP22 is clinically important. Over-expression of PMP22 causes Charcot-Marie-Tooth disease type-1A (CMT1A), the most common heritable neuropathy afflicting 1:5000 people. Heterozygous deletion of the PMP22 causes a different disease, HNPP, which presents with transient, focal episodes of weakness or sensory loss. HNPP nerves show focal excessive myelin folds, the tomacula4. Pmp22 deficient (Pmp22+/−) mice recapitulate many abnormalities found in patients with HNPP, including the tomacula5. While those findings are important, the functional consequences of having no PMP22 have not been evaluated to date since no human patient with total absence of PMP22 has been described. We have evaluated such a patient, including his skin biopsy, and compared these results with those in pmp22−/− mice.
METHODS
Molecular analysis
Multiplex Ligation-dependent Probe Amplification (MLPA) assay was used to characterize the segment deleted in chromosome 17p. A MLPA kit (SALSA P033B) was used according to the manufacturer’s protocol (MRC-Holland, Netherlands)6. This kit contains eleven probes specific for sequences present in the CMT1A/HNPP region. There were 22 control probes interspersed throughout the genome, which were used for relative quantification purposes. More detailed information on probes, genes, and sequences can be found at www.mlpa.com.
Immunohistochemistry study on human skin biopsies and mouse tissues
This technique has been described before7. In brief, skin biopsies were fixed in 4% paraformaldehyde for 30 minutes – 12 hours, embedded in OCT medium, and cut in vertical with 10–60μm thickness. The sections were incubated with the primary antibodies (Table 1) overnight at 4°C, followed by incubation with the secondary antibodies overnight, and mounted onto the slides on the 3rd day. The slides were examined using a Nikon laser confocal microscope (Nikon D-Eclipse C1 confocal system).
Table 1.
Primary antibody list
| Antibody | Source | Species raised | Specific antigen | Type | Titers | Reference |
|---|---|---|---|---|---|---|
| MBP | Ultraclone | Mouse | purified MBP from human brain | Monoclonal IgG1 | 1:800 | Elfman et al., 1986 |
| PGP9.5 | AbD seroTec | Rabbit | Human PGP9.5 from brain | Polyclonal IgG | 1:1000 | Xue, L. et al. 2000 |
| LaminB | Santa Biotech. SC-6217 |
Goat | C-terminus of Lamin B1 of mouse origin | Polyclonal IgG | 1:100 | Ueda et al., 2004 |
| PERP | Abcam, ab3945 | Rabbit | amino acids 37–50 of mouse PERP | Polyclonal IgG | 1:50 | Attardi et al., 2000 |
Electron microscopy
Skin biopsies and mouse tissue were fixed in 2.5% glutaraldehyde overnight, osmicated for 2 hours in 1% osmium tetroxide, dehydrated, and then embedded in epoxy resin. Tissue blocks were sectioned, trimmed and examined by electron microscopy (Zeiss EM900)7.
Mouse genotyping
PMP22 deficient mice and genotyping
The pmp22−/− mouse was generated by using homologous recombination technique to inactivate the Pmp22 gene5. A breeding colony is maintained in Vanderbilt University animal facility. The Animal Investigational Committee in the institution has approved the use of animals for this study. Genotypes were determined as described in previous publications5.
RESULTS
Clinical evaluation
The proband is a 7-year and 10-month-old boy, who was first noticed to be floppy at the age of 4 months during a hospitalization for a non-related condition. He did not walk until three years of age. When he did walk he walked with an unsteady gait. Currently he requires a walker to take more than a few steps. His parents opine that he requires the walker because of unsteadiness rather than weakness. His parents first noted difficulty with him using his fingers when he began school at four years of age. His grip was weak and he began dropping objects. He currently cannot fasten buttons or close zippers but he can write. He states that he has normal sensation in his feet although he complains of occasional numbness in both hands.
On neurological examination he could ambulate with an ataxic, narrow based gait, if his father held both of his hands. With someone holding his hands for balance he could stand up both on his heels and toes very easily. Cranial nerve testing revealed bilateral facial weakness with Bell signs confirming the lower motor neuron basis for this weakness. Mild bilateral ptosis was present. He had no visible muscle atrophy in upper or lower extremities; for example, he had good bulk of extensor digitorum brevis muscles. He had very mild hammer toes but no overt pes cavus. Strength was full to confrontation in proximal and distal muscles of both upper and lower extremities. He cooperated well with his sensory examination. There was bilateral, symmetric reduction of pinprick in the fingers and in the toes. Vibration was normal in the upper limbs but in the lower limbs it was reduced to the costal margins. Joint position sense was normal in the upper limbs but in the lower limbs was reduced up to his ankles bilaterally. He had mild pseudoathetosis in both hands. Deep tendon reflexes were unobtainable.
Because of the patient’s age only a limited electrodiagnostic study was performed which revealed very prolonged distal motor latencies and a slow peroneal motor conduction velocity. The only sensory nerve tested was the sural nerve and its response was unobtainable (Table 2). The proband’s parents are first cousins and both have symptoms and neurophysiologic studies consistent with HNPP (Table 2)8.
Table 2.
Neurophysiologic findings in the proband and his parents.
| Father | Mother | Proband | |
|---|---|---|---|
| Median DML (ms) | 6.0 | 5.7 | 11.2 |
| Ulnar DML (ms) | 3.8 | 4.6 | NP |
| Peroneal DML (ms) | 7.1 | 8.0 | 5.8 |
| Tibial DML (ms) | 7.0 | 6.7 | 10.7 |
| Ulnar CV BE (m/s) | 45.7 | 44.4 | NP |
| Ulnar CV AE (m/s) | 36.7 | 20.2 | NP |
| Peroneal bellow knee (m/s) | 39.0 | 37.0 | 12.6 |
| Peroneal above knee (m/s) | 40.0 | 40.6 | NP |
DML: Distal motor latency CV: Conduction velocity NP: Not performed
Molecular diagnosis
MLPA of the proband’s DNA (Figure 1) revealed a homozygous deletion of a segment of at least 1.1Mb in chromosome 17p. This segment included all five exons of the PMP22, and the TEKT3 and FLJ genes flanking the PMP22. Both copies of the COX10 gene were spared. MLPA analysis of the proband parents’ DNA revealed a heterozygous deletion of the same segment in both parents, confirming the diagnosis of HNPP.
Figure 1. PMP22, but not COX10, is absent in both alleles of the proband.

MLPA revealed absent peaks (red arrows) in the proband (A), corresponding to all five exons of PMP22 as determined in the control sample (B). Peaks corresponding to TEKT3, BX089850 and FLJ were also absent on the proband, but COX10 was retained in both alleles (green arrows). (C) Table showing the ratios of the analyzed peaks (relative peak areas) compared to the normal controls. (D) Schematic of the segment of chromosome 17p deleted in the proband.
Human skin biopsy analysis
Glabrous skin biopsies were taken from the index finger of the proband, as we have previously described7. Immunohistochemistry of the biopsy with antibodies against MBP and PGP9.5 revealed a profound reduction in myelinated fiber density. In the few fibers positive for MBP staining, Schwann cells were surrounding PGP9.5 positive axons but did not form well-organized internodes or compact myelin (Figure 2A and 2B). In contrast, Schwann cells from control biopsy formed regular internodes, ensheathed by compact myelin (Figure 2C) as previously reported9. We next performed electron microscopy. We were unable to identify any compact myelin; rather there were only Schwann cell processes loosely wrapped around a space where the axon presumably has degenerated. In many cases these Schwann cells form “onion bulb”-like structures but axons usually were absent (Figure 3). Taken together, these changes were consistent with axonal loss. Schwann cells around degenerated axons were proliferative and unable to form myelinating internodes along regenerating axons.
Figure 2. Abnormal internodes in skin biopsies from the patient with PMP22 null.
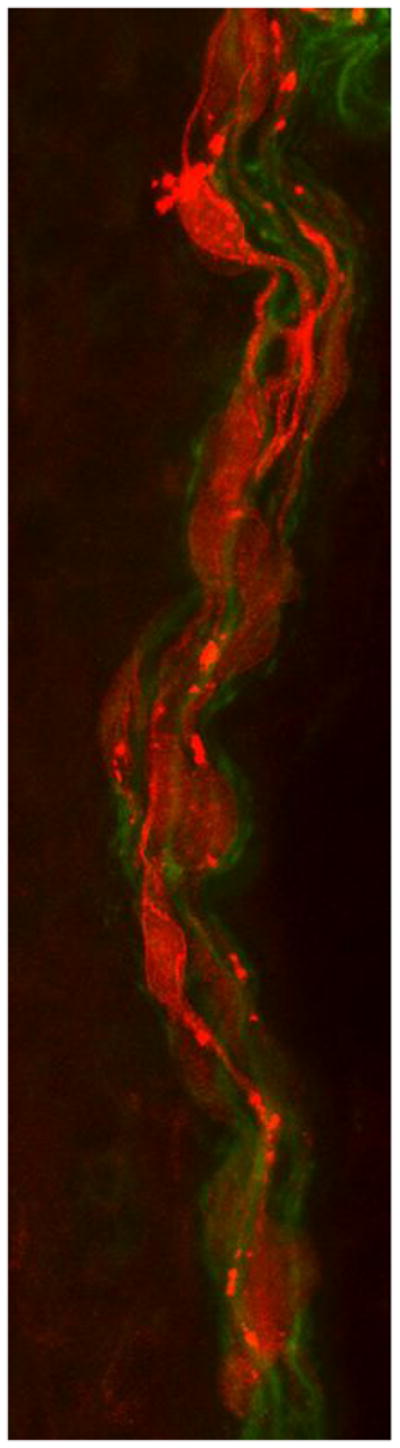
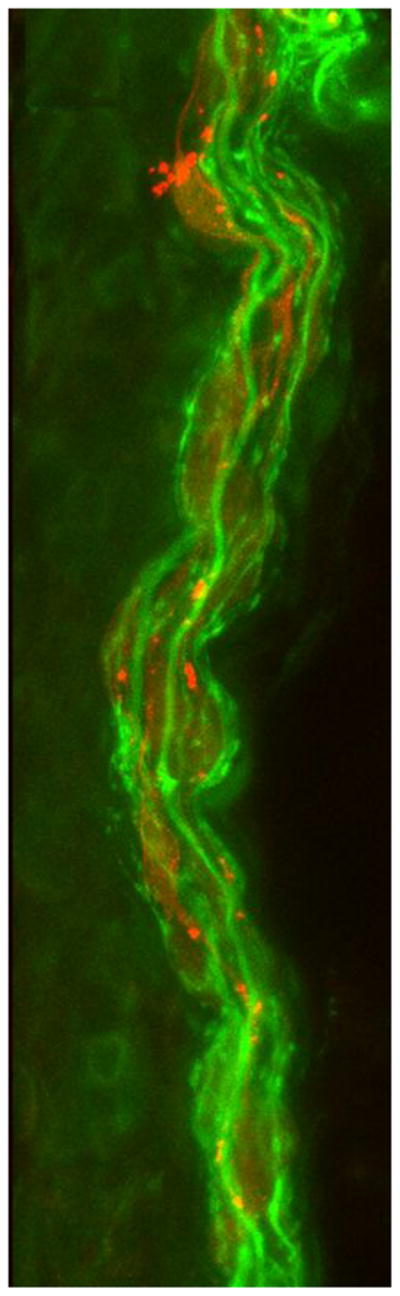
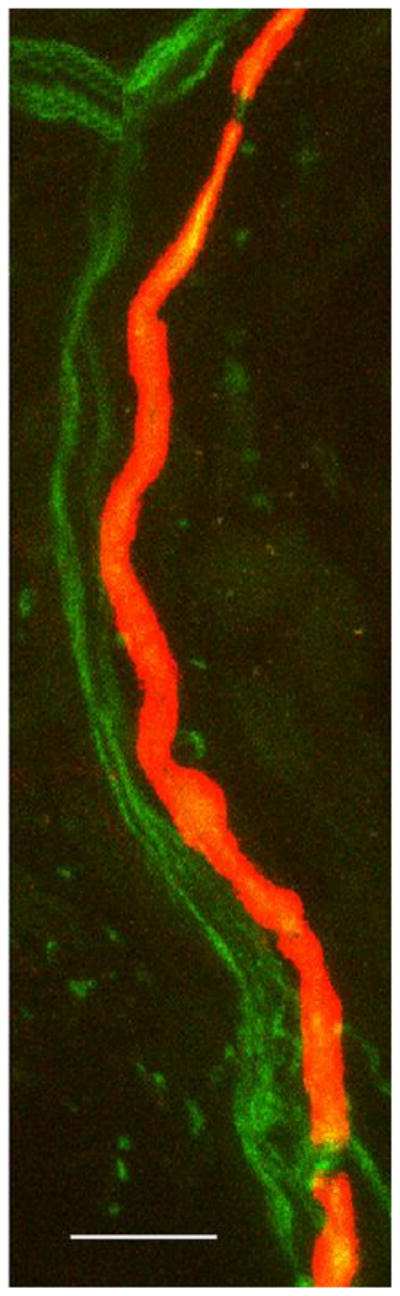
(A&B) Immunohistochemistry using MBP (red) and PGP9.5 (green) antibodies reveals multiple MBP-positive Schwann cells (red) surrounding axons (green) without forming internodes in dermal myelinated fibers in the patient’s skin sample. In A, red channel gain was artificially increased to highlight MBP positive Schwann cells. In B, the same image as A with the green channel gain increased to highlight PGP9.5 positive axons. (C) A control skin biopsy discloses normal internode of dermal myelinated fibers flanked by MBP negative regions corresponding to nodes of Ranvier. Scale bar = 10 um.
Figure 3. Axonal loss and basal lamina redundancy in skin biopsies from the patient with PMP22 null.
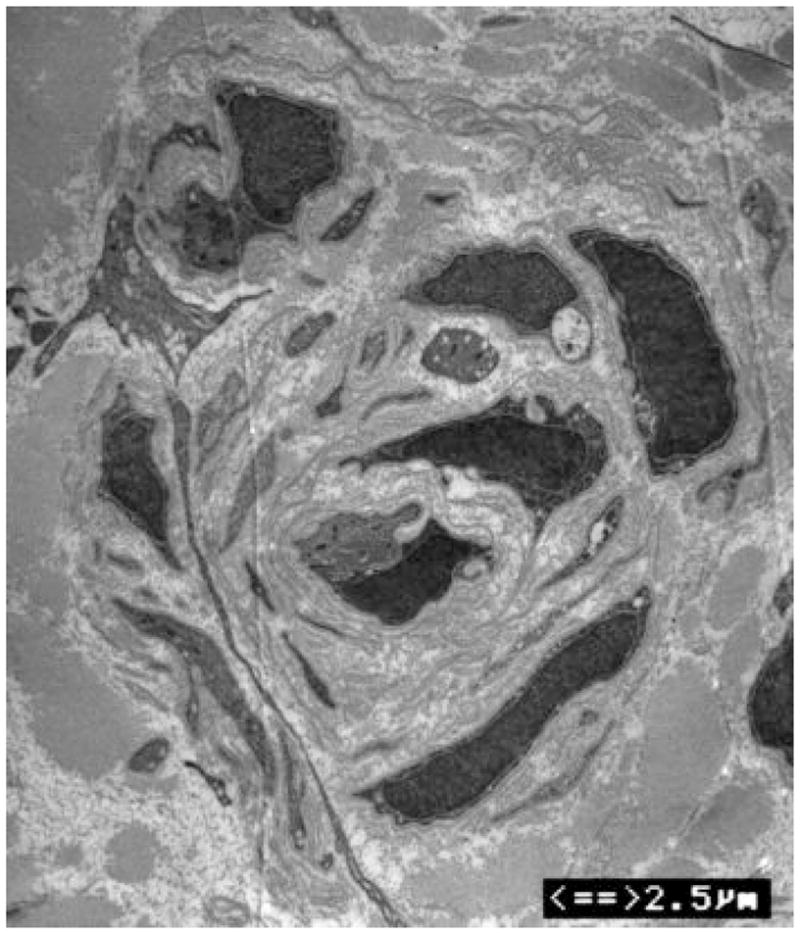

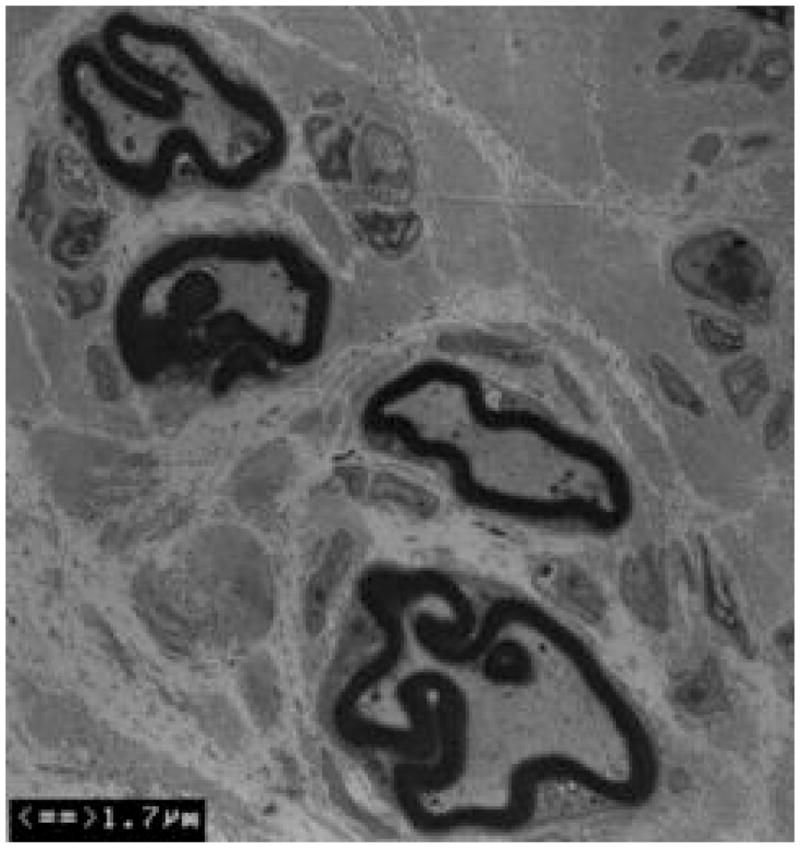
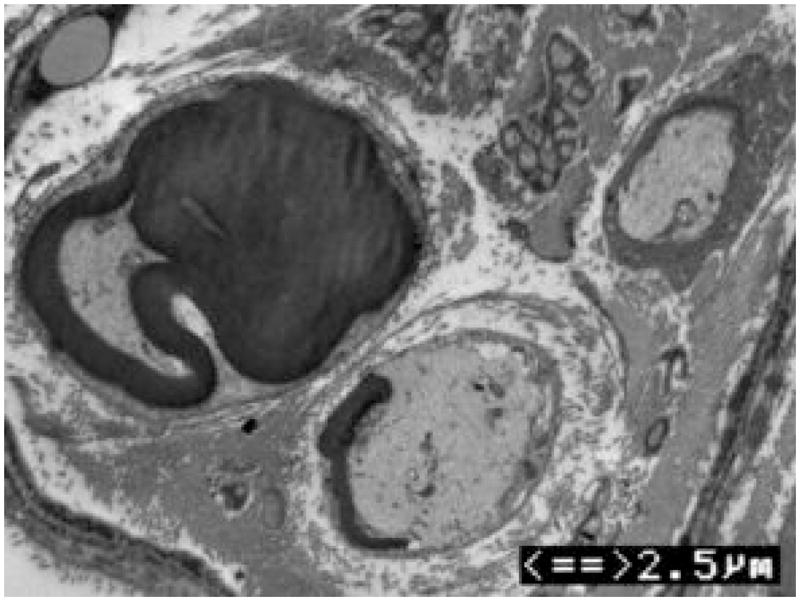
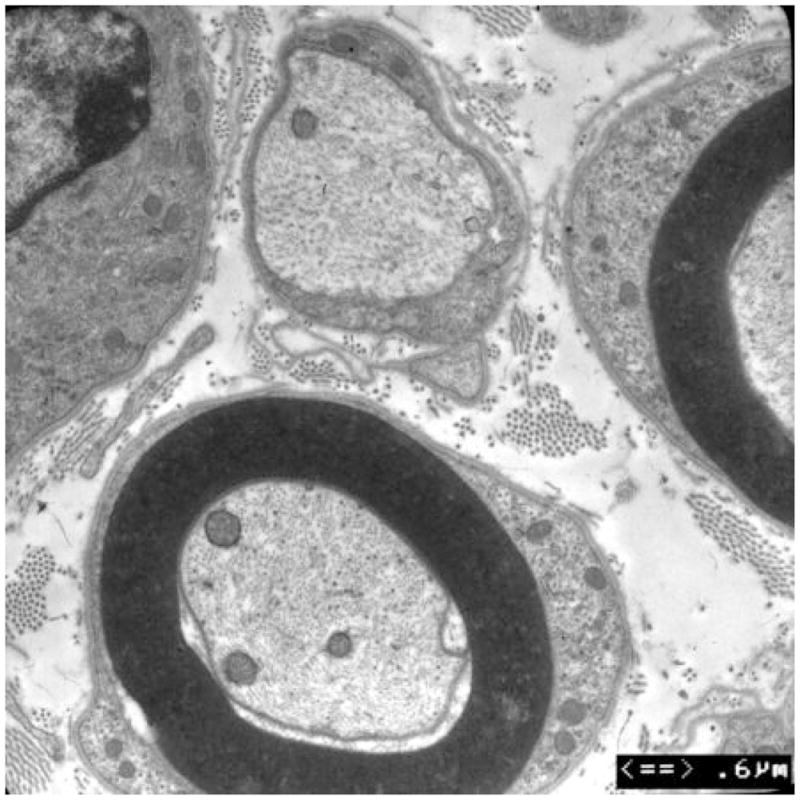
(A) Electron microscopy of the patient’s skin biopsy reveals Schwann cell processes loosely wrapping around an area where the axon appears to have degenerated. The overall appearance resembles an onion bulb. (B) The area circled by asterisks in A is enlarged to reveal redundant basal lamina of the Schwann cells (arrows). (C) A representative skin biopsy from a healthy control demonstrates the compacted myelin with normal thickness. (D) Skin biopsy of a pmp22−/− mouse reveals tomaculous formation (asterisk). Tomacula could not be identified in the patient’s dermal nerves since myelinated nerve fibers are degenerated. Two axons on the right side fail to form compact myelin. E. EM of pmp22−/− mouse sciatic nerve reveals a number of relatively large axons devoid of compact myelin, as exemplified by the axon on the top. In contrast, an axon in the bottom has a comparable diameter to the one on the top but its myelin is well formed. The excessive basal membrane redundancy is readily identifiable (arrow).
An additional finding was a striking redundancy of the basal lamina with several loose layers or empty pockets of the lamina around or nearby Schwann cells (Figure 3). Similar findings were also described in a recent model of Pmp22 null mice10. As with our patient, mouse skin biopsies showed a few myelinated nerve fibers (Figure 3). The redundancy of basal lamina was prominent in the biopsies from the mice (Figure 3).
Differential effect of myelination between motor and sensory nerve fibers in pmp22 null mice
Because of the predominance of proprioception loss in our patient, we wondered whether PMP22 might be particularly important in sensory rather than motor myelinated nerve fibers. Our previous study has demonstrated that axonal loss affects both dorsal and ventral roots equally in pmp22−/− mice at the ages of 10–13 months11. However, differential effect between sensory and motor nerves could still be present in younger mice. We analyzed both dorsal and ventral roots of three pmp22−/− and three wild-type animals. There were a large number of immature Schwann cells that have established 1:1 ratio with axons, but did not form myelin in pmp22−/− mice (Table 3 and Figure 4). The immature Schwann cells failed to produce tomacula since tomacula only can form in myelinated internodes. This defect appeared particularly prominent in ventral roots. All three mice showed abundant immature Schwann cells in ventral roots. In dorsal roots, Schwann cells were either fully differentiated or differentiated in a majority of them. These findings are consistent with our previous observation11. In addition, we noticed a small group of swollen axons in the roots, consistent with axonal loss shown in skin biopsies. Overall, as documented in our previous publication,11 myelinated nerve fibers appeared significantly reduced. Taken together, the data suggest PMP22 deficiency delays maturation of Schwann cells predominantly in motor nerve fibers.
Table 3.
Pathological changes in pmp22−/− spinal roots.
| Mouse No | Age | Sex | Slide No | Root | Axonal Swelling | Amyelinated axons |
|---|---|---|---|---|---|---|
| 1 | 4m | F | 18 | VR | 2+ | 2+ |
| 19 | VR | 2+ | 2+ | |||
| 16 | VR | 2+ | 2+ | |||
| 15 | VR | 0 | 3+ | |||
| 17 | VR | 1+ | 3+ | |||
|
| ||||||
| 11 | DR | 2+ | 2+ | |||
| 8 | DR | 2+ | 1+ | |||
| 7 | DR | 2+ | 1+ | |||
| 9 | DR | 2+ | 1+ | |||
| 10 | DR | 2+ | 0 | |||
|
| ||||||
| 2 | 4m | F | D5 | VR | 2+ | 3+ |
| C4 | VR | 1+ | 3+ | |||
| C5 | VR | 1+ | 3+ | |||
| D2 | VR | 3+ | 2+ | |||
| C3 | VR | 2+ | 3+ | |||
|
| ||||||
| B2 | DR | 1+ | 0 | |||
| B3 | DR | 2+ | 1+ | |||
| B6 | DR | 2+ | 1+ | |||
|
| ||||||
| 3 | 2.5m | F | C6 | VR | 2+ | 3+ |
| D2 | VR | 1+ | 1+ | |||
| D5 | VR | 1+ | 1+ | |||
|
| ||||||
| 33 | DR | 1+ | 0 | |||
| B5 | DR | 1+ | 0 | |||
| B2 | DR | 2+ | 0 | |||
| B1 | DR | 2+ | 0 | |||
| A6 | DR | 2+ | 0 | |||
m=months VR = Ventral roots DR = Dorsal roots
0 = none of the pathological change; 1+ = The pathological change was in <5 myelinated fibers; 2+ = The pathological change was in >5 fibers and readily detectable in all examined fields; 3+ = The pathological change were numerous in any examined field.
Figure 4. Delayed maturation of myelinating Schwann cells in spinal roots of pmp22−/− mice.
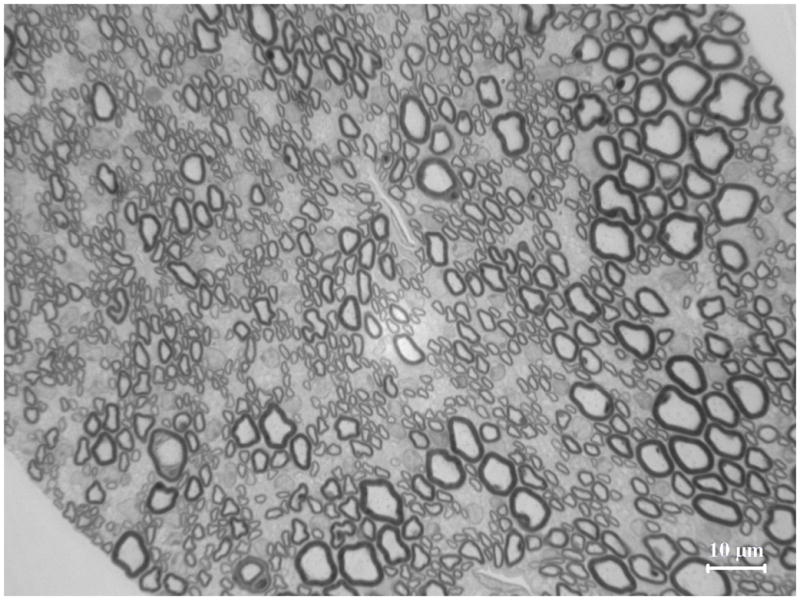
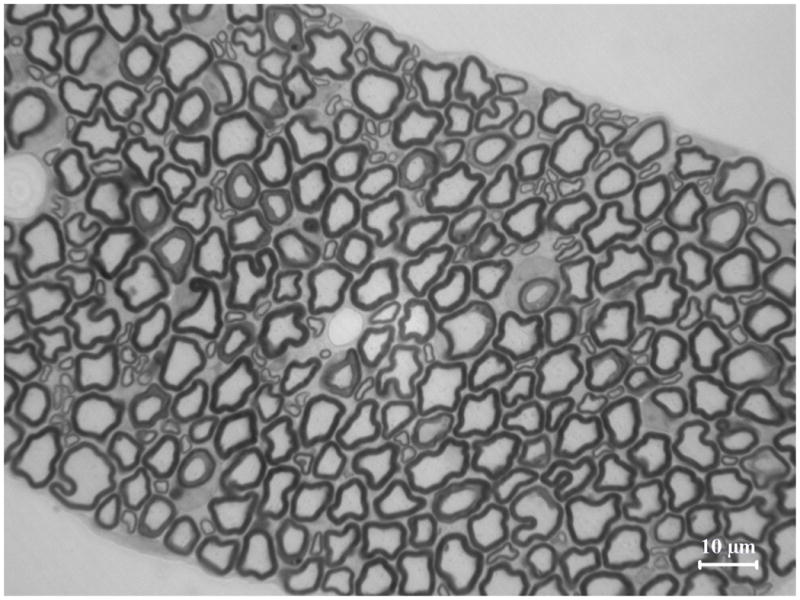
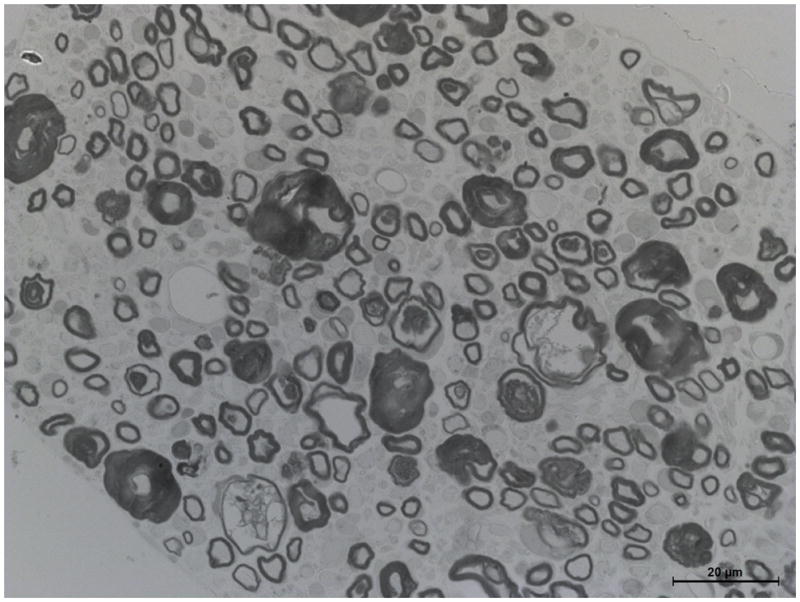
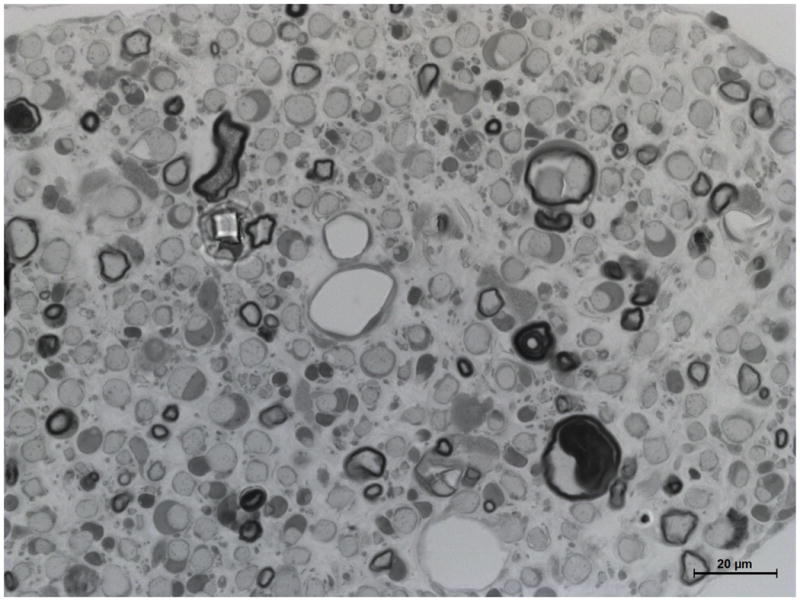
A. Spinal roots were dissected from a 4-week-old mouse and processed for semithin section studies. A dorsal root was visualized under oil lens and showed numerous myelinated nerve fibers with variable diameters. B. In contrast, diameters of myelinated nerve fibers are relatively uniform in the ventral roots. Thus, Schwann cells are well differentiated to myelinate the axons by this age. However, when pmp22−/− ventral roots were examined at 4-month-old age, numerous myelinating Schwann cells still remained at an immature stage after establishing 1:1 relationship with axons (arrows in D). Since most myelinating Schwann cells failed to form myelin, tomacula were few in the ventral root (arrowhead in D). Overall axonal density appeared comparable to the wild-type ventral root. C. In contrast, most myelinating Schwann cells have formed myelin in the dorsal roots by the age of 4 months, and tomacula were common (arrowheads). Axonal density of myelinated nerve fibers appeared reduced. Some of the axons were swollen (arrows), suggesting axonal degeneration.
Because PMP22 expresses in spinal sensory and motor neurons during early development, we questioned whether PMP22 deficiency affects the survival of these neurons. TUNEL staining was performed in mouse spinal cords and DRG. There was no increase of apoptosis (For spinal anterior horns: 0.33±0.58 positive cells in 3 wt mice vs 0.33±0.58 positive cells in 3 pmp22−/− mice; for DRG: 2.33±2.52 vs 2.67±2.08; Supplementary Figure 1). In addition, we have examined the spinal cord and DRG with semithin sections, H&E staining and immunohistochemistry with antibodies against nuclear envelope protein laminin-B. No abnormality was detected in pmp22−/− mice (Supplementary Figure 2).
DISCUSSION
This 7-year-old boy is the first reported patient unable to express any PMP22. This null mutation has revealed several features that have crucial implication for PMP22 functions. First, this patient presented with a predominantly large fiber sensory loss resulting in decreased proprioception and sensory ataxia. This differs from many length-dependent inherited neuropathies that present with symmetric distal sensory loss, but with minimal sensory ataxia. Moreover, his motor deficits are also non-length dependent. While his muscles in upper and lower limbs have normal strength, weakness was found in cranial muscles. These features suggest a proximal pathology. This notion is supported by pathological changes in the spinal roots of pmp22−/− mice. Our previous study has also quantitatively demonstrated that axonal loss in pmp22 null mice is more severe in roots than in distal nerves11.
The pattern of sensory/motor deficits in the patient mirrors the expression pattern of PMP22 during development. Sensory loss and ataxia were severe, and cranial motor nerves were significantly afflicted in the patient. In contrast, functions related to spinal motor neurons were largely spared. Interestingly, PMP22 is highly expressed in rodent DRG sensory and cranial motor neurons during embryonic development and declines after birth2,3. However, expression of PMP22 in spinal motor neurons initiates only after P10 and persists until adulthood12,13. Taken together, these data suggest that transient expression of PMP22 in developing DRG and cranial motor neurons may be important for the survival of these neurons during early development. Dysfunction of these neurons would manifest early when PMP22 is deficient. We speculate that the late-expression of PMP22 in the spinal motor neurons makes these neurons less dependent on the protein during development. This notion is consistent with no spinal motor neuron loss observed in young pmp22−/− mice14. However, PMP22-null spinal motor neurons may degenerate later in life. Alternatively, predominant sensory neuropathy could just be an individual variation of phenotype. These changes in the PMP22 null patient are not seen in patients with heterozygous deletion of PMP22, HNPP4, suggesting that PMP22 null produces phenotypes distinct from those in PMP22 haploinsufficiency.
PMP22 function appears shifted after the early phase of development. Robust expression of PMP22 occurs in the PNS along with the maturation of myelin during the first 3 weeks of development1. Deficiency of PMP22 causes numerous tomacula in the myelinated nerve fibers of peripheral nerves. Axons encased by tomacula become constricted or deformed which may impair action potential propagation or axonal transport, leading to axonal degeneration5,15. We observed a profound delay of myelination that appears more severe in pmp22−/− motor nerves than in pmp22−/− sensory nerves. The second striking feature of this patient is that the motor nerve conduction was extremely slow in the range of 10 m/s. Schwann cells in spinal roots of pmp22−/− mice are often immature and fail to form normal compact myelin. These amyelinated nerve fibers should alter the normal saltatory conduction of action potential to continuous propagation, which would substantially reduce the conduction velocity. Finally, PMP22 deficient Schwann cells form shorter internodes, which may also slow conduction16,17.
A previous report identified another 7-year-old boy who had a compound heterozygous mutation of PMP22 in which there was a 1.4Mb HNPP deletion in one allele and a smaller deletion of PMP22 exons 2 and 3 in the other18. The motor deficits of this child were more severe than that in our patient. It is possible that this patient is not expressing any PMP22 and that the differences between his and our patient’s phenotypes simply represent phenotypic variability. Alternatively, truncated PMP22 from one of the alleles may cause “toxic” gain of function that leads to a more severe phenotype than if there was no PMP22 at all. Consistent with this hypothesis, a patient with the relatively benign PMP22 T118M mutation19 was much more severely affected when this was combined with the HNPP deletion on the other allele20.
We observed a striking redundancy of the basal lamina in our patient’s skin biopsy. This redundancy has been identified in Pmp22−/− mice. The authors demonstrated a direct interaction between Pmp22 and α6β4 integrin, a laminin receptor localized adjacent to the basal lamina. Beta-4 integrin levels are reduced in sciatic nerves of Pmp22 deficient mice, leading the authors to hypothesize that the abnormalities in the basal lamina are the result of decreased interactions between PMP22 and α6β4 integrin10. One function of PMP22, therefore, may be to stabilize the linkage between the extracellular matrix and the abaxonal surface of the myelin sheath.
Recently, we have demonstrated a predisposition for Pmp22+/− mice to develop conduction block and focal constrictions of axons underneath tomacula that may increase axial resistance to action potential propagation15. Tomacula and axonal constrictions were predominantly localized at paranodes, important areas of interactions between the axon and myelin15. Therefore, PMP22 deficiency also appears to disrupt interactions between the adaxonal surface of myelin and the underlying axon. Taken together, these data suggest that an important role for PMP22 is to regulate myelin functions at both the abaxonal surface, where it interacts with the extracellular matrix, and at the adaxonal surface, where it interacts with the axon.
Supplementary Material
Supplementary Figure 1. Absence of apoptosis in sensory and motor neurons. Pmp22+/− Mice were crossbred with Yfp transgenic mice to label the neurons as described in our previous publication16. Sections of DRG or spinal cord were studied using TUNEL staining to detect apoptosis. Cells with positive TUNEL signals were rare in both pmp22+/+ and pmp22−/− mice (arrow in A).
Supplementary Figure 2. Pathological changes were not detected in neuronal cell bodies of pmp22−/− mice. A–D. Paraffin sections of mouse DRG and spinal cord were stained with H&E. No abnormality was identified in pmp22−/− neuronal cell bodies. E–H. These sections were also stained with antibodies against Laminin-B, a marker of nuclear envelope. No abnormality was found in pmp22−/− neurons (arrows in E–H). Perp is a protein that belongs to PMP22 family and has been found to play a role in cell death. Perp was stained in these sections and showed similar expression between pmp22+/+ and pmp22−/− neurons.
{kind=link}
Acknowledgments
This work is, in part, supported by grants from Veterans Affairs (B6243R) and NIH (R01NS066927). Work in the lab of US is also supported by the Swiss National Science Foundation and the National Center for Competence in Research (NCCR) Neural Plasticity and Repair. MMR is grateful to the Medical Research Council (MRC), the Muscular Dystrophy Campaign and the NIH for their support.
References
- 1.Jetten AM, Suter U. The peripheral myelin protein 22 and epithelial membrane protein family. Prog Nucleic Acid Res Mol Biol. 2000;64:97–129. doi: 10.1016/s0079-6603(00)64003-5. [DOI] [PubMed] [Google Scholar]
- 2.Parmantier E, Cabon F, Braun C, et al. Peripheral myelin protein-22 is expressed in rat and mouse brain and spinal cord motoneurons. Eur J Neurosci. 1995;7:1080–1088. doi: 10.1111/j.1460-9568.1995.tb01095.x. [DOI] [PubMed] [Google Scholar]
- 3.Parmantier E, Braun C, Thomas JL, et al. PMP-22 expression in the central nervous system of the embryonic mouse defines potential transverse segments and longitudinal columns. J Comp Neurol. 1997;378:159–172. [PubMed] [Google Scholar]
- 4.Li J, Krajewski K, Lewis RA, et al. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29:205–210. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- 5.Adlkofer K, Martini R, Aguzzi A, et al. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 6.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Bai Y, Ghandour K, et al. Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain. 2005;128:1168–1177. doi: 10.1093/brain/awh483. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Krajewski K, Shy ME, et al. Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology. 2002;58:1769–1773. doi: 10.1212/wnl.58.12.1769. [DOI] [PubMed] [Google Scholar]
- 9.Saporta MA, Katona I, Lewis RA, et al. Shortened internodal length of dermal myelinated nerve fibres in Charcot-Marie-Tooth disease type 1A. Brain. 2009 doi: 10.1093/brain/awp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amici SA, Dunn WA, Jr, Murphy AJ, et al. Peripheral myelin protein 22 is in complex with alpha6beta4 integrin, and its absence alters the Schwann cell basal lamina. J Neurosci. 2006;26:1179–1189. doi: 10.1523/JNEUROSCI.2618-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancho S, Magyar JP, Aguzzi A, et al. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain. 1999;122 (Pt 8):1563–1577. doi: 10.1093/brain/122.8.1563. [DOI] [PubMed] [Google Scholar]
- 12.Maier M, Berger P, Nave KA, et al. Identification of the regulatory region of the peripheral myelin protein 22 (PMP22) gene that directs temporal and spatial expression in development and regeneration of peripheral nerves. Mol Cell Neurosci. 2002;20:93–109. doi: 10.1006/mcne.2002.1116. [DOI] [PubMed] [Google Scholar]
- 13.Ohsawa Y, Murakami T, Miyazaki Y, et al. Peripheral myelin protein 22 is expressed in human central nervous system. J Neurol Sci. 2006;247:11–15. doi: 10.1016/j.jns.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Nattkamper H, Halfter H, Khazaei MR, et al. Varying survival of motoneurons and activation of distinct molecular mechanism in response to altered peripheral myelin protein 22 gene dosage. J Neurochem. 2009;110:935–946. doi: 10.1111/j.1471-4159.2009.06200.x. [DOI] [PubMed] [Google Scholar]
- 15.Bai YH, Zhang XB, Katona I, et al. Conduction block in PMP22 deficiency. J Neurosci. 2009 doi: 10.1523/JNEUROSCI.4264-09.2010. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amici SA, Dunn WA, Jr, Notterpek L. Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J Neurosci Res. 2007;85:238–249. doi: 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- 17.Court FA, Sherman DL, Pratt T, et al. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature. 2004;431:191–195. doi: 10.1038/nature02841. [DOI] [PubMed] [Google Scholar]
- 18.Al-Thihli K, Rudkin T, Carson N, et al. Compound heterozygous deletions of PMP22 causing severe Charcot-Marie-Tooth disease of the Dejerine-Sottas disease phenotype. Am J Med Genet A. 2008;146A:2412–2416. doi: 10.1002/ajmg.a.32456. [DOI] [PubMed] [Google Scholar]
- 19.Shy ME, Scavina MT, Clark A, et al. T118M PMP22 mutation causes partial loss of function and HNPP-like neuropathy. Ann Neurol. 2006;59:358–364. doi: 10.1002/ana.20777. [DOI] [PubMed] [Google Scholar]
- 20.Roa BB, Garcia CA, Pentao L, et al. Evidence for a recessive PMP22 point mutation in Charcot- Marie-Tooth disease type 1A. Nat Genet. 1993;5:189–194. doi: 10.1038/ng1093-189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Absence of apoptosis in sensory and motor neurons. Pmp22+/− Mice were crossbred with Yfp transgenic mice to label the neurons as described in our previous publication16. Sections of DRG or spinal cord were studied using TUNEL staining to detect apoptosis. Cells with positive TUNEL signals were rare in both pmp22+/+ and pmp22−/− mice (arrow in A).
Supplementary Figure 2. Pathological changes were not detected in neuronal cell bodies of pmp22−/− mice. A–D. Paraffin sections of mouse DRG and spinal cord were stained with H&E. No abnormality was identified in pmp22−/− neuronal cell bodies. E–H. These sections were also stained with antibodies against Laminin-B, a marker of nuclear envelope. No abnormality was found in pmp22−/− neurons (arrows in E–H). Perp is a protein that belongs to PMP22 family and has been found to play a role in cell death. Perp was stained in these sections and showed similar expression between pmp22+/+ and pmp22−/− neurons.