

Abstract
Previous reports demonstrate that cell migration in the nervous system is associated with stereotypic changes in intracellular calcium concentration ([Ca2+]i), yet the target of these changes are essentially unknown. We examined chemotactic migration/invasion of human gliomas to study how [Ca2+]i regulates cellular movement and to identify downstream targets. Gliomas are primary brain cancers that spread exclusively within the brain, frequently migrating along blood vessels to which they are chemotactically attracted by bradykinin. Using simultaneous fura-2 Ca2+ imaging and amphotericin B perforated patch-clamp electrophysiology, we find that bradykinin raises [Ca2+]i and induces a biphasic voltage response. This voltage response is mediated by the coordinated activation of Ca2+-dependent, TRAM-34-sensitive KCa3.1 channels, and Ca2+-dependent, 4,4′-diisothiocyanato-stilbene-2,2′-disulfonic acid (DIDS)-sensitive and gluconate-sensitive Cl− channels. A significant portion of these Cl− currents can be attributed to Ca2+/calmodulin-dependent protein kinase II (CaMKII) activation of ClC-3, a voltage-gated Cl− channel/transporter, because pharmacological inhibition of CaMKII or shRNA-mediated knockdown of ClC-3 inhibited Ca2+-activated Cl− currents. Western blots show that KCa3.1 and ClC-3 are expressed in tissue samples obtained from patients diagnosed with grade IV gliomas. Both KCa3.1 and ClC-3 colocalize to the invading processes of glioma cells. Importantly, inhibition of either channel abrogates bradykinin-induced chemotaxis and reduces tumor expansion in mouse brain slices in situ. These channels should be further explored as future targets for anti-invasive drugs. Furthermore, these data elucidate a novel mechanism placing cation and anion channels downstream of ligand-mediated [Ca2+]i increases, which likely play similar roles in other migratory cells in the nervous system.
Introduction
Cell migration in the nervous system occurs during development, in response to disease/injury, and after malignant transformation of neural cells, and several studies suggest that an important regulator of this migration may be intracellular calcium concentration ([Ca2+]i). For example, in migrating cerebellar neurons and microglial cells, migration is Ca2+ dependent (Komuro and Rakic, 1996; Ifuku et al., 2007). Additionally, several stimuli that increase glioma cell migration also increase [Ca2+]i, including Ca2+-permeable AMPA receptors (Ishiuchi et al., 2002) and IP3-dependent pathways (Kang et al., 2010). Although these studies suggest that increases in [Ca2+]i enhance migration, a mechanistic linkage of Ca2+ changes to cell migration has been elusive. Here we ask how [Ca2+]i mechanistically regulates migration in the context of gliomas.
Malignant transformation of glial cells or their progenitors gives rise to gliomas, including grade IV glioblastomas, which are the most common and lethal form of primary brain cancer affecting adults (Wen and Kesari, 2008). The lethality of gliomas can be attributed to a variety of neoplastic features, including enhanced proliferation and angiogenesis, but a standout feature is their unusual invasiveness. The ability of glioma cells to migrate and invade into unaffected brain regions leads to a diffuse tumor mass lacking well-defined borders, making complete surgical resection challenging. Understanding the pathophysiological mechanisms underlying glioma cell migration may lead to the development of targeted inhibitors to reduce disease spreading and improve clinical prognosis, but may also inform us about mechanisms used by non-malignant cells as they migrate.
Recent evidence demonstrates that bradykinin increases human glioma cell motility (Kang et al., 2010; Montana and Sontheimer, 2011). Bradykinin activates the bradykinin receptor B2 (B2R), leading to increases in [Ca2+]i and enhanced migration of glioma cells along cerebral vasculature (Montana and Sontheimer, 2011). Interestingly, several ion channels expressed by glioma cells are Ca2+ sensitive and have been implicated as mediators of shape and volume changes that can facilitate cell movement. These include intermediate-conductance Ca2+-activated K+ channels (KCa3.1, IK channels) (Schwab et al., 2006) and ClC-3, a voltage-gated Cl− channel/transporter, (Mao et al., 2008) that is activated by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human glioma cells (Cuddapah and Sontheimer, 2010; Cuddapah et al., 2012). Because ions are osmolytes, permeation of K+ and Cl− through KCa3.1 and ClC-3 channels causes cytoplasmic water to move across the membrane, allowing for the robust shape and volume changes associated with migrating cells. Such hydrodynamic volume changes have been suggested to be necessary in migrating cells and, if inhibited, disrupt cell migration (Watkins and Sontheimer, 2011).
Here we hypothesize that bradykinin increases [Ca2+]i in human glioma cells, leading to the coordinated activation of Ca2+-sensitive K+ and Cl− channels. Using simultaneous [Ca2+]i measurements and electrophysiological recordings, we demonstrate that KCa3.1 and ClC-3 channels are activated as a result of bradykinin-dependent [Ca2+]i increases. Inhibition of KCa3.1 or ClC-3 channels decreases bradykinin-induced migration through murine cerebral parenchyma in situ. These results present a novel mechanism placing Ca2+-activated cation and anion channels under the control of receptor-mediated changes in [Ca2+]i, which may have broad applications to other migratory cells.
Materials and Methods
Cell culture.
D54, U87, U251, GBM 50, GBM 62, JX 22, and JX 39 human glioma cells were derived from human grade IV glioblastoma multiforme tumors. D54 cells were a gift from Dr. D. Bigner (Duke University, Durham, NC). Cells were maintained and passaged in DMEM–F-12 (Invitrogen). Medium was supplemented with 2 mm glutamine and 7% fetal bovine serum (Hyclone), and cells were incubated in 10% CO2 at 37°C. Unless otherwise stated, all reagents were purchased from Sigma-Aldrich.
Fura-2 calcium imaging.
D54 cells were washed with 0.1% serum-containing media. Cells were then loaded with Fura-2 Leakage Resistant (Teflabs) using 20% w/v pluronic acid in DMSO (Invitrogen) in 0.1% serum-containing media for 45 min at 37°C. Cells were then washed with 7% serum-containing media and allowed to recover for 45 min at 37°C. Cells were imaged with a CoolSNAP HQ2 CCD camera (Photometics) mounted on a Carl Zeiss Axiovert 200 inverted microscope. The excitation filters were controlled with a Lambda 10–2 Optical Filter Changer (Sutter Instruments), with light delivered from the X-Cite 120 microscope light source system (Lumen Dynamics). Fura-2 imaging was performed with filters exciting at 340 ± 12 and 387 ± 11 (Semrock). Imaging and analysis was done with Imaging Workbench (INDEC BioSystems) with 4 × 4 binning. Fura-2 was calibrated using the bath solution described below with 10 μm ionomycin to determine Rmax or 10 μm ionomycin and 100 nm thapsigargin to determine Rmin. For the formula [Ca2+]free = Kd × ((R − Rmin)/(Rmax − R)) × (F380, max/F380, min), we used the published Kd for fura-2, which is 145 nm, and measured the following values: Rmax = 2.9, Rmin = 0.76, F380, max = 3062, and F380, min = 454.
Electrophysiology.
Whole-cell patch-clamp recordings were performed as described previously (Cuddapah and Sontheimer, 2010). All experiments were performed in 2 μm paxilline (Santa Cruz Biotechnology) to block large-conductance Ca2+-activated K+ channels (BK channels). The pipette solution contained 140 mm KCl, 2 mm MgCl2,10 mm EGTA, and 10 mm HEPES sodium salt and was adjusted to pH 7.2 with Tris-base (302–304 mOsm). For a free [Ca2+]i = 0 nm, no CaCl2 was added. For a free [Ca2+]i = 65 nm, 2.7 mm CaCl2 was added. For a free [Ca2+]i = 180 nm, 4.9 mm, CaCl2 was added. These [Ca2+]i were calculated with the Ca–Mg–ATP–EGTA Calculator version 1.0 using constants from National Institute of Standards and Technology database #46 version 8 (maxchelator.stanford.edu). The bath solution contained the following (in mm): 130 NaCl, 5 KCl, 1 CaCl2, 10.55 glucose, and 32.5 HEPES acid, adjusted to pH 7.4 with NaOH (312–320 mOsm). For gluconate− substitution experiments, we exchanged the 130 mm NaCl with 130 mm Na-gluconate.
For perforated-patch experiments, the pipette solution contained the following (in mm): 140 KCl, 2 MgCl2, and 10 HEPES sodium salt, adjusted to pH 7.2 with Tris-base (290–295 mOsm). We supplemented the pipette solution with 120 μg/ml Amphotericin B to permeabilize the cell and 2 μg/ml of Alexa Fluor 488 hydrazide sodium salt (Invitrogen) to visually ensure that the perforated patch was not ruptured. Myristoylated 10 μm autocamtide-2 related inhibitory peptide (AIP) (Enzo Life Sciences) was preincubated in the cell culture medium for 10 min at 37°C. Cells with membrane resistances up to 25 MΩ (with 80% compensation) were used for experiments. Bradykinin was applied at 10 μm to activate bradykinin receptors. TRAM-34 was applied at 10 μm to block KCa3.1 channels. BAPTA-AM was applied at 50 μm to chelate [Ca2+]i. DIDS was applied at 200 μm to block Cl− channels.
Glioma cell transfections.
D54 cells were transfected stably with EGFP as described previously (Habela et al., 2008). ClC-3 protein expression was constitutively knocked down using a pGIPZ-lentiviral small hairpin mir vector targeting CLCN3 (Open Biosystems) as described previously (Cuddapah et al., 2012). Multiple clones were created and screened to demonstrate consistent decreases in migration during ClC-3 shRNA transfection versus nontargeting (NT) shRNA transfection.
Western blotting.
Western blotting was performed as described previously (Cuddapah and Sontheimer, 2010). The following antibodies and concentrations were used to probe for ClC-3, KCa3.1, and B2R, respectively: rabbit anti-ClC-3 at 1:500 (ACL-001; Alomone Labs); rabbit anti-KCNN4 at 1:1000 (AV35098; Sigma-Aldrich); and rabbit anti-bradykinin B2R at 1:200 (sc-25671; Santa Cruz Biotechnology).
Immunocytochemistry.
D54 human glioma cells were cultured, fixed, and labeled as described previously (Cuddapah and Sontheimer, 2010). Cells were blocked with normal donkey serum and labeled with the following primary antibodies targeted against ClC-3, KCa3.1, and B2R, respectively: rabbit anti-ClC-3 at 1:100 (Alpha Diagnostic International; CLC31-A); mouse anti-KCa3.1 at 1:250 (Alomone); and goat anti-bradykinin B2R at 1:100 (sc-15050; Santa Cruz Biotechnology). The following secondary antibodies were purchased from Invitrogen and used at 1:500: donkey anti-rabbit Alexa Fluor 647, donkey anti-mouse Alexa Fluor 488, and donkey anti-goat Alexa Fluor 546. Confocal images were acquired as described previously (Haas et al., 2011) with a 60× water objective.
Transwell migration assay.
Migration assays were performed as described previously (Cuddapah and Sontheimer, 2010). We placed 0.3 μm bradykinin at the bottom of Transwell filters to induce migration. TRAM-34 at 1 μm and AIP at 1 μm were placed on the top and bottom of the filters to block KCa3.1 channels and CaMKII, respectively.
Glioma invasion in slice culture.
Slice cultures were prepared from brains of P13–P16 BALB/c SCID mice of either sex (The Jackson Laboratory). A vibratome was used to cut 300 μm coronal brain sections, and slices were then transferred into filter inserts on a polycarbonate membrane (pore size 0.45 μm; Falcon; BD Biosciences Discovery Labware). Filters were then placed into six-well plates containing 1 ml DMEM supplemented with 8% fetal bovine serum, 0.2 mm glutamine, 100 U/ml penicillin, and 100 mg/mL streptomycin. The following day, medium containing 25% heat-inactivated horse serum, 50 mm sodium bicarbonate, 2% glutamine, 25% HBSS, 1 mg/ml insulin (Invitrogen), 2.46 mg/ml glucose (Sigma-Aldrich), 0.8 mg/ml vitamin C (Sigma-Aldrich), 100 U/ml penicillin, 100 mg/ml streptomycin (Sigma-Aldrich), and 5 mm Tris in DMEM (Invitrogen) was added to slices. At day 3 of culturing, 3000 D54–EGFP cells in a volume of 1 μl were implanted per organotypic brain slice. Cells were injected into the right cortex over 30 s using a 1 μl Hamilton syringe mounted to a micromanipulator. Treatment of brain slices was started at day 3 of culturing with the following drugs: 1 μm bradykinin, 5 μm icatibant (Tocris Bioscience), and 10 μm TRAM-34. The media and drugs were exchanged every 2 d. Live brain slices were imaged at day 4 and day 11 with a Leica MZ 120 microscope. The fluorescence of the tumor area was determined using NIH ImageJ software. Tumor growth was calculated as percentage fluorescence increase at 488 nm excitation between day 4 and day 11 of culturing.
Data analyses.
Current responses to voltage changes were quantified with Clampfit (Molecular Devices). Raw data were compiled in Microsoft Excel and graphed in Origin 6.0 (MicroCal). Statistical analyses were completed with GraphPad Instat (GraphPad Software). We used one-way ANOVA or unpaired t test with Welch correction to determine the p value as appropriate. All data are reported as mean ± SE, and asterisks denote a significant difference with p < 0.05.
Results
Bradykinin induces [Ca2+]i increases in human glioma cells
Bradykinin has been shown to play a role in the chemotactic migration of human glioma cells (Montana and Sontheimer, 2011), yet the mechanism whereby it does so has been elusive. We hypothesized here that bradykinin causes increases in [Ca2+]i, which in turn modulate ion channels that regulate cell shape and volume to facilitate cell migration. To test this hypothesis, we first loaded human glioma cells with fura-2, a ratiometric Ca2+-indicator dye, and simultaneously performed electrophysiological recordings to monitor ion channel activity. Electrical access to glioma cells was obtained with an amphotericin B perforated-patch pipette, which allows for permeabilization of the cell membrane to monovalent cations and monovalent anions, without disturbing intracellular divalent cations, including Ca2+. This allowed simultaneous measurement of bradykinin-induced [Ca2+]i increases and ion channel activity. Figure 1 shows representative recordings obtained in two widely used human glioma cell lines derived from World Health Organization (WHO) grade IV glioblastoma tumors: D54 (Fig. 1A–C) and U87 (Fig. 1D–F). In Figure 1A, we simultaneously monitored [Ca2+]i and whole-cell current responses to a rapid voltage shift from −90 to +120 mV. Application of 10 μm bradykinin induced a rapid rise in [Ca2+]i in D54 glioma cells, peaking 20 s after application (Fig. 1B, top). This rapid rise in [Ca2+]i resulted in a concomitant leftward shift in the current reversal potential toward the equilibrium potential for K+ (EK+) (Fig. 1B, bottom). This shift suggests that the rise in [Ca2+]i caused an increased K+ conductance. By 202 s after application of bradykinin, [Ca2+]i dropped back to baseline levels (Fig. 1C, top). Current ramps taken at that time show a rightward shift in the reversal potential consistent with the delayed activation of a Cl− conductance (Fig. 1B, bottom). The equilibrium potential for Cl− was experimentally set at 0 mV using equivalent [Cl−] in the bath and pipette solutions; hence, a rightward shift in the reversal potential would be consistent with an increase in Cl− conductance. However, unlike the increase in K+ conductance, the increase in Cl− conductance appeared delayed and persisted after [Ca2+]i returned to basal levels. Qualitatively similar data was recorded in U87 human glioma cells (Fig. 1D–F). Application of 10 μm bradykinin resulted in a robust increase in [Ca2+]i and simultaneous leftward shift of the reversal potential toward EK+ (Fig. 1D,E). By 130 s, after [Ca2+]i returned closer to basal levels, there was a large rightward shift of the reversal potential toward the equilibrium potential of Cl− (ECl−) (Fig. 1F). Compared with D54 cells, the whole-cell conductance increased to a greater extent in U87 cells when the reversal potential was hyperpolarized. Additionally, the leftward shift in reversal potential preceded the maximal [Ca2+]i increase. In both cell types, bradykinin-induced [Ca2+]i increases temporally correlated with a biphasic leftward then rightward shift of the reversal potential, indicating the sequential activation of a K+ and Cl− conductance.
Figure 1.

Bradykinin induces [Ca2+]i elevations that correlate with electrophysiological changes in human glioma cells. A–C, D54 human glioma cells. D–F, U87 human glioma cells. A, D, At time = 0 s, fura-2 340/380 ratio is depicted in the top. Bottom trace depicts current–voltage curve for the same cell as measured with a perforated-patch pipette. B, E, After application of 10 μm bradykinin (BK), there is a large increase in [Ca2+]i (top) and a leftward shift of the reversal potential (bottom). C, F, After [Ca2+]i has returned to basal levels (top), there is a large rightward shift in the reversal potential (bottom).
Bradykinin-induced rises in [Ca2+]i results in enhanced K+ and Cl− channel activity
After finding that bradykinin (1) increased [Ca2+]i, (2) led to an early-phase leftward shift in the reversal potential, and (3) led to a late-phase rightward shift of the reversal potential, we sought to determine whether bradykinin-induced increases in [Ca2+]i resulted in increased ion channel activity. Application of 10 μm bradykinin led to an increase in [Ca2+]i to 1.34 ± 0.04 (F/F0) in 80 ± 5% of D54 human glioma cells (n = 79 cells; Fig. 2A,B). As measured by simultaneous perforated-patch recordings, application of 10 μm bradykinin also resulted in a biphasic electrophysiological response. The early phase was characterized by a hyperpolarization of the reversal potential, peaking on average 14.57 ± 1.22 s (n = 21 cells) after bradykinin application (Fig. 2C). Thus, the early hyperpolarization response occurred while the [Ca2+]i was still elevated. After [Ca2+]i returned to basal levels, glioma cells maximally depolarized 148.19 ± 20.12 s (n = 21 cells) after bradykinin application (Fig. 2A,D). Figure 2E demonstrates this relationship between the bradykinin-induced [Ca2+]i increase and the biphasic voltage response in a representative cell. The top trace depicts the [Ca2+]i, and the bottom trace shows the voltage at which the current is equal to 0 pA, which is an indirect measure of the resting membrane potential (Fig. 2E). After bradykinin application, the [Ca2+]i rapidly increases, resulting in a concomitant rapid hyperpolarization of the membrane potential. After the [Ca2+]i began to return to basal levels, the membrane potential depolarized and was maximal after [Ca2+]i returned to baseline (Fig. 2E). On average, the early hyperpolarization was −6.38 ± 1.08 mV (n = 21 cells), and the late depolarization was 15.66 ± 2.46 mV (n = 21 cells; Fig. 2I,L). Thus, a single bradykinin-induced elevation in [Ca2+]i correlates with a biphasic voltage response.
Figure 2.
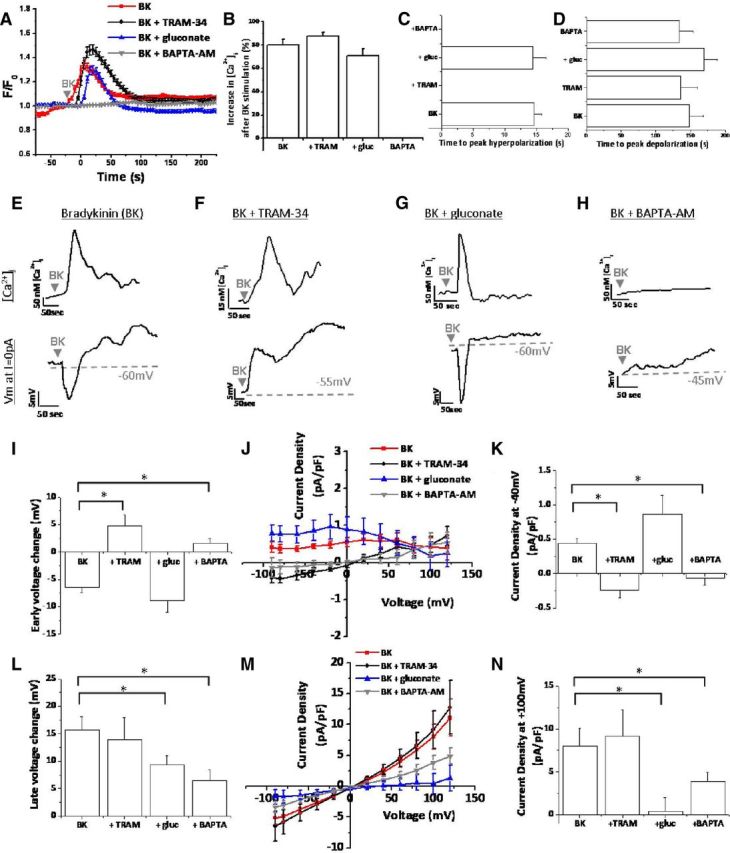
Bradykinin-induced [Ca2+]i elevations activate KCa3.1 channels and Cl− channels in human glioma cells. A, Fura-2 340/380 ratios after application of 10 μm bradykinin (BK). F/F0 indicates fluorescence normalized to fluorescence at t = 0 s. B, Percentage of cells responding to BK with a rise in [Ca2+]i. C, D, Time to BK-induced peak hyperpolarization and depolarization, respectively. E–H, Representative cells for BK, BK plus TRAM-34, BK plus gluconate (gluc), and BK plus BAPTA-AM conditions. Top traces depict [Ca2+]i versus time. For the same cell at the same time points, bottom traces depict membrane potential (Vm) at I = 0 pA versus time. I, Peak early-phase voltage change (millivolts). J, K, BK-induced current–voltage changes during early-phase response. L, Peak late-phase voltage change (millivolts). M, N, BK-induced current–voltage changes during late-phase response. n = 10–21 cells; *p < 0.05.
We next sought to determine which ion channels were responsible for the biphasic voltage response. Given that the early-phase hyperpolarization moved the reversal potential toward EK+ and correlated with increased [Ca2+]i, we hypothesized that Ca2+-activated K+ channels opened after bradykinin stimulation. Several recent reports demonstrate expression of intermediate-conductance Ca2+-activated K+ channels (KCa3.1 channels) in glioma cells (Fioretti et al., 2009; Sciaccaluga et al., 2010; Catacuzzeno et al., 2011). Therefore, we asked whether the bradykinin-induced early-phase hyperpolarization was attributable to KCa3.1 channel activity. Using 10 μm TRAM-34, a potent and selective inhibitor of KCa3.1 channels (Wulff et al., 2000), we found that the bradykinin-induced hyperpolarization was completely abolished by TRAM-34 and hence mediated by KCa3.1 channels (Fig. 2). In the presence of TRAM-34, the bradykinin-induced Ca2+ response was slightly larger (maximal F/F0 = 1.46 ± 0.04) compared with control in 88 ± 3% of cells (n = 78 cells; Fig. 2A,B). Although TRAM-34 application did not alter the timing of maximal depolarization (135.6 ± 24.85 s; n = 15 cells), it prevented bradykinin-induced membrane hyperpolarization (Fig. 2C,D). As seen in a representative cell (Fig. 2F), TRAM-34 did not prevent bradykinin-induced increases in [Ca2+]i but did eliminate membrane hyperpolarization. As in control conditions, the late-phase depolarization was maximal after [Ca2+]i returned to basal levels. Interestingly, instead of an early-phase hyperpolarization, bradykinin application in the presence of TRAM-34 produced an early-phase depolarization (Fig. 2F). This switch from an early-phase hyperpolarization to depolarization reveals that bradykinin activates K+ and Cl− channels simultaneously, and the depolarization caused by Cl− channels can only be fully appreciated after KCa3.1 channel blockade. Thus, the biphasic voltage response is explained by the different kinetics of KCa3.1 and Cl− channels. Although both channels are activated simultaneously, KCa3.1 dominates in the early-phase hyperpolarizing the cell, and Cl− channels predominate in the later phase depolarizing the cell. Additional quantification of these voltage responses demonstrated that the late-phase depolarization did not significantly change after TRAM-34 addition (13.95 ± 3.99 mV; p = 0.36; Fig. 2L), but the early-phase voltage significantly changed from −6.38 ± 1.08 mV (n = 21 cells) to +4.72 ± 2 mV (n = 15 cells; p < 0.0001; Fig. 2I). We also measured current responses normalized to cell capacitance (picoamperes per picofarads) during the early phase and late phase. Although 10 μm TRAM-34 had no effect on the late-phase current activation at +100 mV (p = 0.38; Fig. 2M,N), it did significantly reduce the early-phase currents at −40 mV (Fig. 2J,K). Again, inhibition of KCa3.1 channels appears to uncover an early-phase Cl− conductance. This is evident in Figure 2J in which blockage of KCa3.1 channels yields a current–voltage curve that reverses at 0 mV, the equilibrium potential for Cl− (ECl−).
After finding that bradykinin activates TRAM-34-sensitive KCa3.1 channels leading to hyperpolarization of the membrane potential, we sought to determine the channels responsible for the bradykinin-induced late-phase membrane depolarization. Because the late-phase current–voltage curve reversed at 0 mV (Fig. 2M), the equilibrium potential of Cl−, we hypothesized that the late-phase depolarization was attributable to increased Cl− channel activity. We attempted to isolate the contribution of Cl− channels by using several pharmacological inhibitors, including 200 μm 5-nitro-2–3-phenylpropylaminobenzoic acid (NPPB), 200 μm DIDS, 10 μm tamoxifen, and 100 μm niflumic acid. However, as reported by others (Fioretti et al., 2004), NPPB also blocks KCa3.1 channels, precluding its use to specifically block Cl− channels. DIDS could not be used because it is fluorescent at overlapping excitations with fura-2, preventing visualization of bradykinin-induced [Ca2+]i increases. Both 10 μm tamoxifen and 100 μm niflumic acid blocked bradykinin-induced [Ca2+]i increases in addition to the related biophysical changes. Because none of these inhibitors could be used reliably, we reverted to an ion replacement strategy, whereby we swapped the extracellular 137 mm Cl− with 130 mm gluconate− plus 7 mm Cl− to eliminate the majority of the outward current through Cl− channels. (The 7 mm Cl− was retained as charge carrier.) Gluconate− has reduced or no permeability through Cl− channels (Olsen et al., 2003; Lee et al., 2010), allowing us to determine whether Cl− channels were indeed responsible for the bradykinin-induced depolarization of the cell membrane. Importantly, gluconate− replacement did not significantly change bradykinin-induced [Ca2+]i increases, elevating the maximal F/F0 to 1.30 ± 0.03 in 71 ± 6% of cells (n = 100 cells; Fig. 2A,B). Additionally, gluconate− replacement did not significantly change the time to early-phase hyperpolarization (14.38 ± 2.13 s) and late-phase depolarization (169 ± 20 s; n = 16 cells; Fig. 2C,D). Figure 2G depicts a representative response to 10 μm bradykinin with extracellular gluconate− replacement. While the Ca2+ response and early-phase hyperpolarization were similar to control conditions, the late-phase depolarization was significantly inhibited. Quantification in a number of cells (n = 16 cells) demonstrated that gluconate− replacement did not significantly inhibit the early-phase KCa3.1 channel activation (Fig. 2I–K) but did significantly decrease the late voltage change to 9.37 ± 1.59 mV (p < 0.02; Fig. 2L). Because we replaced extracellular Cl− with gluconate−, we quantified changes in the outward current density compared with control. Gluconate− replacement significantly reduced the late-phase current density at +100 mV (p < 0.003; Fig. 2M,N). These data indicate that the membrane depolarization after bradykinin stimulation was mediated by Cl− channels.
Bradykinin has been shown to activate B2R expressed by human glioma cells (Montana and Sontheimer, 2011). B2R is a G-protein-coupled receptor (GPCR) that signals through a variety of mechanisms, including Gq mediators, to increase phospholipase C activity, leading to IP3-dependent Ca2+ release from intracellular stores (Kang et al., 2010). We hypothesized that activation of KCa3.1 channels and Cl− channels primarily occurred via bradykinin-induced elevations in [Ca2+]i. To test this hypothesis, we preincubated human glioma cells in 50 μm BAPTA-AM for 45 min to chelate Ca2+ inside of glioma cells and prevent bradykinin-induced elevations in [Ca2+]i (Fig. 2A,B). Although BAPTA-AM inhibited bradykinin-induced hyperpolarization, it did not delay the average time-to-peak depolarization (133.6 ± 20.39 s; n = 10; Fig. 2D). While BAPTA-AM-loaded cells depolarized, the average amplitude of such depolarizations were significantly reduced (15.66 ± 2.46 mV in control vs 6.39 ± 2.08 mV in BAPTA-AM; p < 0.004; Fig. 2L). Interestingly, inhibition of the early-phase hyperpolarization again revealed an early-phase depolarization, albeit reduced in amplitude (Fig. 2H–K). BAPTA-AM also significantly reduced the late-phase Cl− current density at +100 mV from 8.03 ± 2.04 pA/pF to 3.87 ± 1.1 pA/pF (p < 0.04). Cumulatively, these data suggest that bradykinin-induced increases in [Ca2+]i lead to biphasic activation of KCa3.1 channels and Cl− channels.
Changes in [Ca2+]i modulate KCa3.1 channel and Cl− channel activity
Given the above findings demonstrating that bradykinin-induced elevation in [Ca2+]i enhanced ion channel activity, we next questioned whether small experimental increases in intracellular Ca2+ via the patch pipette would be sufficient to activate KCa3.1 channels and Cl− channels. To test this hypothesis, we performed whole-cell patch-clamp experiments with 0, 65, or 180 nm free [Ca2+] in the pipette solution. We held the cells at −40 mV and stepped from −100 to +120 mV in 20 mV increments. To isolate KCa3.1 channel activity, we performed the experiments in 2 μm paxilline to block large-conductance Ca2+-activated K+ channels (BK channels) and replaced the extracellular 137 mm Cl− with 130 mm gluconate− plus 7 mm Cl−. We then washed on 10 μm TRAM-34 to selectively inhibit KCa3.1 channels and subtracted out TRAM-34-sensitive currents. At 0 and 65 nm [Ca2+]i, there was little to no TRAM-34-sensitive current (Fig. 3A–C), indicating that TRAM-34 is not strongly activated at resting [Ca2+]i in human glioma cells. However, at elevated [Ca2+]i, there was a large activation of TRAM-34-sensitive KCa3.1 currents (Fig. 3A–C). [Ca2+]i = 180 nm potentiated KCa3.1 currents that inactivated at depolarized potentials (Fig. 3A). These currents reversed at −89 mV, close to the predicted EK+ of −84 mV (Fig. 3B). At −40 mV, the resting membrane potential of glioma cells (Olsen and Sontheimer, 2004), the TRAM-34 current density significantly increased from 0.42 ± 0.2 pA/pF at 65 nm [Ca2+] to 2.21 ± 0.51 pA/pF at 180 nm [Ca2+] (n = 5–8 cells; p < 0.006; Fig. 3B,C). These data indicate that experimentally increasing the [Ca2+]i in glioma cells enhances KCa3.1 channel activity.
Figure 3.

Increases in [Ca2+]i activate TRAM-34-sensitive KCa3.1 channels and DIDS- and gluconate−-sensitive Cl− channels. A, Representative TRAM-34-sensitive KCa3.1 currents elicited after a step protocol from −100 to 120 mV at each of the [Ca2+]i listed. B, TRAM-34-sensitive current–voltage relationship at 0, 65, and 180 nm [Ca2+]i. C, TRAM-34-sensitive KCa3.1 current density at −40 mV. n = 5–8 cells. D, Representative DIDS-sensitive Cl− currents elicited after a step protocol from −100 mV to 120 mV at each of the [Ca2+]i listed, with or without extracellular gluconate− replacement. E, DIDS-sensitive current–voltage relationship at 0, 65, and 180 nm [Ca2+]i, with or without extracellular gluconate− replacement. F, DIDS-sensitive Cl− current density at 40 mV. n = 5–10 cells; *p < 0.05. gluc, Gluconate.
We then asked whether elevating [Ca2+]i also increased Cl− channel activity. We again patched onto human glioma cells with 0, 65, or 180 nm free [Ca2+] in the pipette solution. Cl− currents were isolated by washing on 200 μm of the Cl− channel blocker DIDS. Remarkably, at [Ca2+]i = 0 nm, we observed little to no Cl− current (Fig. 3D–F). This is the first indication that Cl− channel activity in human glioma cells is Ca2+ dependent. There was an increase in the amplitude of DIDS-sensitive Cl− currents from 0.29 ± 0.14 pA/pF at 0 nm [Ca2+]i to 6 ± 1.89 pA/pF at 65 nm [Ca2+]i at +40 mV (p < 0.01; n = 8–10; Fig. 3D–F), indicating that Cl− channels are active at resting [Ca2+]i. Stepping the [Ca2+]i to 180 nm further increased DIDS-sensitive currents at +40 mV (p < 0.03; n = 8–10; Fig. 3D–F). These Ca2+-activated Cl− currents reversed at ECl−, inactivated at depolarized potentials, and were slightly outwardly rectifying (Fig. 3D–F). Currents meeting these characteristics have been ascribed previously to ClC-3, a voltage-gated Cl− channel expressed in glioma cells (Shimada et al., 2000; Olsen et al., 2003; Cuddapah and Sontheimer, 2010). Because gluconate− replacement was used to inhibit Cl− channel activity in Figure 2, we asked whether gluconate−-sensitive Cl− currents were mediated by the same channels as DIDS-sensitive Cl− currents. We again replaced the extracellular 137 mm Cl− with 130 mm gluconate− plus 7 mm Cl− and found that DIDS-sensitive Cl− currents were greatly reduced (Fig. 3D–F). At [Ca2+]i = 180 nm, gluconate− replacement decreased DIDS-sensitive Cl− currents from 14.14 ± 3.19 pA/pF to 1.63 ± 0.8 pA/pF at +40 mV (p < 0.002; n = 5–10 cells; Fig. 3D–F). Thus, gluconate− replacement significantly ablates DIDS-sensitive Cl− currents in human glioma cells.
B2R, ClC-3, and KCa3.1 are expressed on the leading edges of glioma cells
Figure 3 demonstrates that increases in [Ca2+]i activate KCa3.1 channels and Cl− channels in human glioma cells. Because the electrophysiological characteristics of the Ca2+-activated Cl− currents matched ClC-3 (Fig. 3D), we assessed where ClC-3 and KCa3.1 channels were expressed with respect to B2R in human glioma cells. We cultured D54 human glioma cells, labeled ClC-3, KCa3.1, and B2R with antibodies, and then took confocal images of labeled cells. As seen in a representative field of view, ClC-3 (blue channel), KCa3.1 (green channel), and B2R (red channel) all colocalized on the leading edges of glioma cells (Fig. 4A). Digital zooms of individual cells demonstrate the typical punctuate labeling of ClC-3, characteristic of proteins expressed on intracellular vesicles (Fig. 4B,C). Both KCa3.1 and B2R labeling was more diffuse throughout the cell bodies (Fig. 4B,C). Remarkably, ClC-3, KCa3.1, and B2R all strongly labeled the ruffled lamellipodiums of polarized migrating cells (Fig. 4B,C, arrows). This elevated labeling could be explained by increased trafficking of the proteins to the lamellipodium, nonspecific accumulation of the plasma membrane attributable to membrane ruffling, or even decreased light scattering secondary to the lack of organelles in the lamellipodium (Dewitt et al., 2009). Although we did not investigate these possibilities in further detail, the data nevertheless demonstrate that B2R are located in the same subcellular domains as ClC-3 and KCa3.1 and could regulate channel activity.
Figure 4.

ClC-3, KCa3.1, and B2R are expressed on the leading edges of human glioma cells. A, Field of view of cultured D54 human glioma cells labeled with antibodies targeted against ClC-3 (blue), KCa3.1 (green), and B2R (red). Zooms of boxed cells depicted in B and C. Scale bar, 20 μm. B, C, Zooms of individual cells depicted in A. Scale bar, 10 μm. n = 3. D, Several human glioma lines express ClC-3, KCa3.1, and B2R protein. n = 3.
We then assessed potential differences in protein expression between human glioma cell lines by probing Western blots for ClC-3, KCa3.1, and B2R. Along with D54, we probed U87 and U251 cells, which are commercially available WHO grade IV glioblastoma cell lines. We also probed GBM 50 and GBM 62 cells, which we derived from WHO grade IV glioblastomas. Finally, we probed JX 22 and JX 39 cells, which were derived from grade IV tumors that were propagated in the flanks of mice, thereby avoiding in vitro culturing and exposure to serum. All the aforementioned human glioma cell lines expressed ClC-3 (Fig. 4D). In U87 and JX 22 cells, the ClC-3 band produced a smear, suggesting posttranslational modifications leading to differences in molecular mass. Six of seven human glioma lines expressed KCa3.1 channels (Fig. 4D). All glioma lines also expressed B2R (Fig. 4D). Therefore, ClC-3, KCa3.1, and B2R protein expression is shared among human glioma cells, and these proteins localize to the leading edges of migrating cells.
A subset of Ca2+-activated Cl− currents are mediated by CaMKII-dependent ClC-3 channels in glioma cells
Given that glioma cells express ClC-3 and have Ca2+-activated Cl− currents that match the electrophysiological characteristics of ClC-3 (Fig. 3), we asked whether bradykinin-induced [Ca2+]i increases activate ClC-3. We and others have reported previously that ClC-3 activity is enhanced by CaMKII phosphorylation (Huang et al., 2001; Cuddapah and Sontheimer, 2010); therefore, Ca2+ elevations in glioma cells may be enhancing Cl− channel activity via a Ca2+-sensitive kinase, such as CaMKII. To answer this question, we again performed whole-cell patch-clamp experiments with 0, 65, or 180 nm [Ca2+] in the pipette solution. We found that there were little to no DIDS-sensitive Cl− currents at 0 nm [Ca2+]i (Fig. 5A–C). Elevation of [Ca2+]i to 65 nm, which is close to basal [Ca2+]i, significantly increased DIDS-sensitive Cl− currents at +40 mV (p < 0.01; n = 10 cells; Fig. 5A–C). As described previously for ClC-3, these currents inactivated at depolarized potentials, were slightly outwardly rectifying (Fig. 5A), and reversed at ECl− (Fig. 5B). Importantly, Cl− currents at 65 nm [Ca2+]i were completely inhibited by 10 μm AIP, a potent and specific inhibitor of CaMKII (Ishida et al., 1995) (Fig. 5). At +40 mV and [Ca2+]i = 65 nm, current density was reduced in 10 μm AIP (p < 0.04; n = 10 cells; Fig. 5A–C), indicating that all DIDS-sensitive Cl− currents in basal [Ca2+]i are CaMKII dependent. However, at elevated [Ca2+]i, most of the Cl− currents are not CaMKII dependent. AIP did not significantly decrease Cl− current density when [Ca2+]i = 180 nm (p > 0.2; n = 10 cells; Fig. 5A–C). These data indicate that Cl− currents in human glioma cells are Ca2+ dependent, and a subset is also CaMKII dependent.
Figure 5.

A subset of Ca2+-activated Cl− currents are mediated by CaMKII-dependent ClC-3 channels. A, Representative DIDS-sensitive Cl− currents elicited after a step protocol from −100 to 120 mV at each of the [Ca2+]i listed, with or without AIP preincubation to inhibit CaMKII. B, DIDS-sensitive current–voltage relationship at 0, 65, and 180 nm [Ca2+]i, with or without AIP. C, DIDS-sensitive Cl− current density at 40 mV. n = 10 cells. D, Representative Western blot demonstrating knockdown of ClC-3 expression. GAPDH used as a loading control. E, Representative DIDS-sensitive Cl− currents elicited after a step protocol from −100 to 120 mV at each of the [Ca2+]i listed, with NT or ClC-3 shRNA transfected cells. F, DIDS-sensitive current–voltage relationship at 0, 65, and 180 nm [Ca2+]i, with or without ClC-3 expression. G, DIDS-sensitive current–voltage relationship at 65 and 180 nm [Ca2+]i, with simultaneous ClC-3 knockdown and CaMKII inhibition with AIP. H, DIDS-sensitive Cl− current density at 40 mV. n = 5–13 cells; *p < 0.05.
To directly asses the contribution of ClC-3 to Ca2+-activated Cl− currents, we transfected cells with constitutively expressed NT shRNA or ClC-3 shRNA. Endogenous ClC-3 expression was knocked down to 67% of control levels as normalized to GAPDH expression (Fig. 5D). As observed in previous experiments, 0 nm [Ca2+] in the pipette solution resulted in little to no DIDS-sensitive Cl− currents (Fig. 5E,F,H). However, 65 nm [Ca2+] in the pipette solution only increased DIDS-sensitive Cl− currents when ClC-3 was expressed. ClC-3 knockdown reduced current density from 3.35 ± 1.16 pA/pF at 40 mV to 1.07 ± 0.38 pA/pF (p < 0.05; n = 13 cells; Fig. 5E,F,H). Therefore, basal levels of [Ca2+]i (i.e., 65 nm) activate CaMKII and ClC-3 to increase Cl− currents. Unexpectedly, ClC-3 knockdown alone did not significantly decrease Cl− current density at 180 nm [Ca2+]i (Fig. 5E,F,H). Simultaneous inhibition of CaMKII with AIP and knockdown of ClC-3 expression also resulted in smaller DIDS-sensitive Cl− currents when [Ca2+]i = 65 nm (p < 0.05; n = 12–13 cells; Fig. 5G,H). Interestingly, simultaneous inactivation of CaMKII and ClC-3 decreased DIDS-sensitive Cl− currents when [Ca2+]i = 180 nm. Current density was reduced from 8.24 ± 1.9 pA/pF in control conditions to 3.61 ± 1.49 pA/pF after CaMKII and ClC-3 inhibition (p < 0.05; n = 10–12 cells; Fig. 5G,H). Although inhibition of ClC-3 or CaMKII is sufficient to decrease Ca2+-activated Cl− currents at basal [Ca2+]i levels, both CaMKII and ClC-3 must be inhibited to decrease Ca2+-activated Cl− currents at elevated [Ca2+]i levels. This may be secondary to incomplete CaMKII inhibition or incomplete ClC-3 knockdown. Nevertheless, these data indicate that CaMKII-dependent ClC-3 channels partially mediate Ca2+-activated Cl− currents.
After determining that experimental increases in [Ca2+]i were sufficient to activate CaMKII-dependent ClC-3 channels, we asked whether bradykinin-induced [Ca2+]i elevations could do the same. Again using an amphotericin B perforated-patch configuration, we washed on 10 μm bradykinin and found that glioma cells transfected with NT shRNA depolarized by 19.51 ± 3.49 mV after 165.33 ± 19.12 s (Fig. 6A,B). ClC-3 knockdown or CaMKII inhibition with 10 μm AIP did not significantly alter the amplitude of peak depolarization or time-to-peak depolarization (Fig. 6A,B). However, both ClC-3 knockdown and CaMKII inhibition significantly decreased bradykinin-induced Cl− currents at 100 mV (Fig. 6C,D). Current density decreased from 14.62 ± 3.01 to 7.62 ± 2.74 pA/pF after CaMKII inhibition, to 7.66 ± 2.35 pA/pF after ClC-3 knockdown and to 6.78 ± 1.72 pA/pF after both CaMKII inhibition and ClC-3 knockdown (p < 0.05; n = 15 cells; Fig. 6C,D). Thus, bradykinin-induced [Ca2+]i elevations activate CaMKII-dependent ClC-3 channels in human glioma cells.
Figure 6.

Bradykinin-induced Cl− currents are partially mediated by CaMKII activation of ClC-3. A, Time to BK-induced peak depolarization after inhibition of CaMKII with AIP and knockdown of ClC-3. B, Peak late-phase voltage change (millivolts) after inhibition of CaMKII with AIP and knockdown of ClC-3. C, D, BK-induced current–voltage changes during late-phase response. CaMKII or ClC-3 inhibition decreases bradykinin-induced Cl− currents. n = 15 cells; *p < 0.05.
Bradykinin activation of ion channels enhances glioma cell invasion into cerebral parenchyma
Ion channels have been implicated previously in facilitating chemotactic migration by promoting the shape and volume changes associated with motile cells (Cuddapah and Sontheimer, 2011). Given that bradykinin activates KCa3.1 channels and Cl− channels, we hypothesized that bradykinin-induced increases in glioma cell migration are partially mediated by enhanced activity of these two classes of ion channels acting in concert. To test this hypothesis, we plated D54 human glioma cells onto a Transwell filter containing 8 μm pores (Fig. 7A). We then placed bradykinin on the opposite side of the filter and counted the number of cells that migrated toward bradykinin. Bradykinin induced a 1.5 ± 0.14-fold increase in migration compared with control (p < 0.05; n = 4; Fig. 7B). When bradykinin was placed on both sides of the filter, there was no significant increase in migration (data not shown), indicating that the observed effects were attributable to chemotaxis and not solely chemokinesis. Importantly, inhibition of either KCa3.1 channels, CaMKII, or ClC-3 channels significantly decreased bradykinin-induced migration (p < 0.05; n = 4; Fig. 7B). KCa3.1 channel inhibition with TRAM-34, CaMKII inhibition with AIP, or ClC-3 knockdown decreased migration to 0.98 ± 0.07, 0.75 ± 0.12, and 0.58 ± 0.07, respectively (Fig. 7B). Additionally, CaMKII inhibition and ClC-3 knockdown did not have additive effects (p > 0.05), suggesting that CaMKII and ClC-3 act along the same pathway in bradykinin-induced migration.
Figure 7.

Bradykinin-induced glioma cell migration requires KCa3.1 channel and CaMKII-dependent ClC-3 channel activity. A, Diagram depicting glioma cells plated onto a Transwell barrier containing 8 μm pores. Bradykinin (BK) loaded on opposite side of filter to assess chemotactic migration. B, Normalized number of glioma cells migrated under various conditions. Ion channel inhibition eliminates bradykinin-induced migration. n = 4; *p < 0.05.
To extend these findings to glioma expansion in brain tissue, we cultured organotypic brain slices from P13–P16 SCID mice and engrafted EGFP-expressing D54 human glioma cells into the cortex, leading to the formation of a large tumor (Fig. 8A). Eight days after transplantation, we measured the area of bradykinin-induced tumor expansion. Incubating tumor-containing brain slices in bradykinin increased tumor spreading from 80.45 ± 15.94 to 212.08 ± 46.26% of starting tumor volume (p < 0.05; n = 13–14; Fig. 8A,B). We verified that glioma cells were inside the slices by imaging with higher magnification and finding that glioma cells migrated along the vasculature, as has been reported previously (Montana and Sontheimer, 2011). In line with the previous study, bradykinin increases chemotaxis by promoting the association of glioma cells with the vasculature, which releases bradykinin and serves as a substratum for migration (Montana and Sontheimer, 2011). Bradykinin-induced tumor spreading was significantly inhibited to 54.33 ± 8.05% of starting tumor volume by application of TRAM-34 to inhibit KCa3.1 channels (p < 0.05; n = 6–14; Fig. 8A,B). Additionally, knockdown of ClC-3 decreased tumor volume from 173.84 ± 12.04 to 28.62 ± 30.97% of starting tumor volume (p < 0.05; n = 3–4; Fig. 8A,C). These data indicate that bradykinin-induced tumor growth requires the downstream activation of ion channels and that tumor growth in situ can be contained by blockade of these channels.
Figure 8.
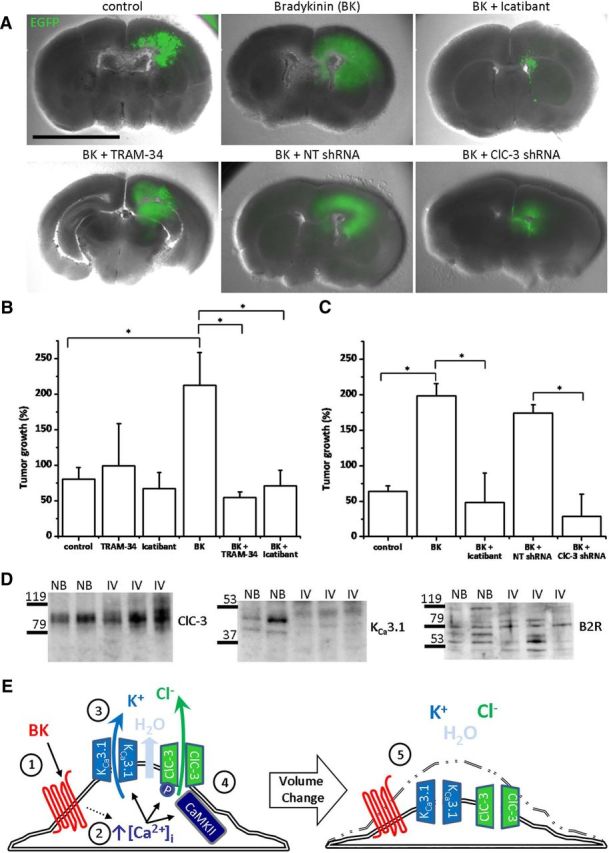
Bradykinin-induced glioma cell invasion through cerebral parenchyma requires ion channel activity. A, Representative mouse brain slices containing large bradykinin (BK)-induced EGFP-labeled human glioma tumor, which is inhibited by TRAM-34, icatibant, and ClC-3 knockdown. B, C, Quantification demonstrates that BK-induced tumor spreading and growth is suppressed by ion channel or B2R inhibition. n = 3–14; *p < 0.05. D, Human tissue lysates probed for ClC-3, KCa3.1, and B2R. NB, Normal brain; IV, grade IV glioblastoma. n = 3. E, BK binds to GPCR (1) and leads to increases in [Ca2+]i (2). These increases in [Ca2+]i activate KCa3.1 channels and K+ efflux (3). Increases in [Ca2+]i also activate Cl− channels, including CaMKII activation of ClC-3, leading to Cl− efflux (4). K+ and Cl− efflux lead to osmotic loss of cytosolic water. A loss in cytosolic ions and water enables cellular volume and shape changes, facilitating glioma cell migration through narrow extracellular spaces (5).
We also applied 5 μm icatibant (HOE-140; Firazyr) to tumor-containing slices. Icatibant is a specific B2R antagonist that received Food and Drug Administration approval as an orphan drug in 2011 for the treatment of hereditary angioedema. Importantly, icatibant significantly reversed bradykinin-induced increases in tumor volume from 212.08 ± 46.26 to 70.89 ± 22.06% of starting tumor volume (p < 0.05; n = 9–14; Fig. 8A–C). These results appear to be clinically relevant, because we probed three human WHO grade IV glioblastoma samples for ClC-3, KCa3.1, and B2R protein expression and found expression of all three proteins in all samples (Fig. 8D). Cumulatively, these data suggest that bradykinin-induced activation of ClC-3 and KCa3.1 channels increases glioma cell migration and tumor expansion.
Discussion
Using human malignant glioma, a clinically relevant model system in which to study cell migration and invasion, we present a novel mechanism demonstrating how changes in [Ca2+]i secondary to ligands, such as bradykinin, translate into altered cell movement in a three-dimensional and spatially restricted environment. Specifically, we show that the synchronous activation of a Ca2+-activated K+ channel, KCa3.1, and a CaMKII-activated Cl− channel, ClC-3, regulate the flux of osmotically active K+ and Cl− ions, which cause dynamic changes in cytoplasmic volume required for cellular movement (Fig. 8E). This ligand-activated modulation of ion channels leading to the flux of osmotically active ions may be a mechanism conserved among migratory cell types in the context of both chemotaxis and chemokinesis.
[Ca2+]i is a regulator of ion channels, cell volume, and migration
The regulation of cellular migration by [Ca2+]i has been documented previously for both malignant and non-malignant neural cells. For example, Komuro and Rakic (1996) published a series of studies demonstrating that immature granule cells show oscillatory changes in [Ca2+]i, and these fluctuations dictate the rate of migration. The movement of neuronal growth cone extensions toward netrin-1 requires the influx of Ca2+ through transient receptor potential TRPC channels (Wang and Poo, 2005). In human glioma cells, glutamate activates Ca2+-permeable AMPA receptors, thereby inducing [Ca2+]i oscillations, and their disruption impairs cell migration and tumor spread in vivo (Lyons et al., 2007). The central role of Ca2+ for motility in this context is illustrated by the fact that, if the AMPA receptors are mutated and rendered Ca2+ impermeable, glioma cell invasion is inhibited in vivo (Ishiuchi et al., 2002).
These examples illustrate that Ca2+ signaling appears to be a conserved feature among migratory cells. However, the molecular targets of such [Ca2+]i changes and how they affect cell movement are by and large not well understood. Clearly changes in [Ca2+]i can influence actin–myosin molecular motors (Martini and Valdeolmillos, 2010), regulate the dynamics of the tubulin cytoskeleton, and alter the adhesion of cells to substrates via focal adhesion kinases (Giannone et al., 2004). In addition, our data suggest that [Ca2+]i changes regulate the flux of ions and thereby the cytoplasmic water content of a cell. In support of this, a previous study found that invading glioma cells must move through narrow extracellular spaces and maximize their chances to do so by releasing essentially all free cytoplasmic water at once, thereby reducing the total cellular volume by 33% (Watkins and Sontheimer, 2011). This volume condensation is independent of barrier size, because glioma cells decreased in volume by 33% while migrating through 3, 5, or 8 μm pores (Watkins and Sontheimer, 2011). Additionally, migrating glioma cells oscillate in total cellular volume as leading edges are extended and lagging edges are retracted (Watkins and Sontheimer, 2011). Importantly, these volume oscillations were inhibited by Cl− channel blockers, which also impaired cell migration (Watkins and Sontheimer, 2011). Therefore, the pro-migratory effects of bradykinin may partially be mediated by simultaneous Ca2+-dependent activation of K+ and Cl− channels, which facilities shape and volume changes characteristic of migrating cells (Fig. 8E).
Our present findings suggest that Ca2+-mediated activation of both K+ and Cl− channels are required for cellular volume change. We propose that the K+ movement is mediated by KCa3.1 channels, which are Ca2+ sensitive and facilitate volume and shape changes (Schwab et al., 2007). A portion of the K+ movement may also be mediated by the KCa1.1 channel (BK channel), which is expressed by human glioma cells, promotes migration, and is activated by bradykinin (Ransom and Sontheimer, 2001; Weaver et al., 2006). The anion movement is at least in part mediated by ClC-3, which is highly expressed in human glioma cells (Olsen et al., 2003; Cuddapah and Sontheimer, 2010). ClC-3 has been shown to mediate volume regulation and has been implicated in the migration of other malignant and non-malignant cell types, including neutrophils and nasopharyngeal carcinoma cells (Mao et al., 2008; Volk et al., 2008). Temporally, CaMKII phosphorylation of ClC-3 in glioma cells may be responsible for potentiating Cl− currents several minutes after [Ca2+]i has returned to resting levels (Fig. 2D), analogous to neuronal CaMKII activation of NMDA receptors minutes after [Ca2+]i elevations during long-term potentiation (Lengyel et al., 2004). Our findings here elucidate a novel mechanism for bradykinin-induced migration: bradykinin increases [Ca2+]i to simultaneously activate KCa3.1 and ClC-3 channels, which facilitate the volume and shape changes associated with migrating cells (Fig. 8E). In accordance, we observed bradykinin-induced changes in cell volume as measured by changes in fura-2 fluorescence after excitation with 360 nm light, which is the isosbestic point of fura-2 producing a Ca2+-insensitive emission (data not shown).
Bradykinin modulates ion channel activity in several cell types
In this study, we were particularly interested in bradykinin, which has been shown to be an essential signal for the chemotactic migration of glioma cells toward blood vessels (Montana and Sontheimer, 2011). Bradykinin predominantly increases [Ca2+]i through GPCR signaling, subsequently leading to downstream IP3 receptor isoform 3 (IP3R3)-mediated Ca2+ release from intracellular Ca2+ stores (Kang et al., 2010). Additionally, inhibition of the B2R receptor (Montana and Sontheimer, 2011) or IP3R3-mediated Ca2+ release by caffeine significantly reduces in vivo tumor size and improves animal survival (Kang et al., 2010). Thus, bradykinin-induced [Ca2+]i increases play a critical role in glioma cell migration and in vivo tumor spreading. Our data suggest that this effect is at least in part attributable to the activation of KCa3.1 and ClC-3, enhancing cell motility via changes in cell volume dynamics.
Interestingly, inhibition of both KCa3.1 and ClC-3 reduced Transwell migration to below control levels, when bradykinin was not present (Fig. 7). This indicates that KCa3.1 and ClC-3 are critical for the mechanics of glioma cell migration, even independent of bradykinin-induced migration. In accordance, previous studies have demonstrated that ClC-3 and CaMKII activity is required for glioma cell migration in the absence of ligands (Cuddapah and Sontheimer, 2010). KCa3.1 and ClC-3 flux osmotically active ions, promoting the shape and volume changes necessary in migrating cells (Schwab et al., 2007; Volk et al., 2008). Our findings demonstrate that bradykinin enhances glioma cell migration by increasing the activation of these known modulators of cellular volume, allowing cells to migrate through narrow and tortuous spaces in the brain.
Bradykinin also activates Cl− channels in several non-malignant cells, including fibroblast-like satellite cells (England et al., 2001), murine epithelial cells (Tiwari et al., 2007), and neurons (Lee et al., 2005). Additionally, unidentified factors in fetal calf serum activate Cl− currents in a Ca2+-independent manner in human glioma cells. This serum also simultaneously activates Ca2+-dependent KCa3.1 channels (Catacuzzeno et al., 2011), as we see here in response to bradykinin, suggesting that bradykinin in serum may be responsible for the previously observed effects.
Most of the Ca2+-induced Cl− currents we observed does not appear to be mediated by the bestrophin 1 anion channel, which has a gluconate−/Cl− permeability of 0.4 (Lee et al., 2010). Here we found that gluconate− replacement at [Ca2+]i = 180 nm ablated Cl− currents by 89% (Fig. 3D–F). Although CaMKII-dependent Cl− currents can be attributed to ClC-3 (Fig. 5) (Cuddapah and Sontheimer, 2010), future studies will attempt to identify which channels are responsible for the large Ca2+-dependent and CaMKII-independent Cl− currents (Fig. 5) by using genetic knockdown strategies. Although CaMKII or ClC-3 inhibition decreased bradykinin-induced Cl− currents (Fig. 6C,D), the peak depolarization was unchanged (Fig. 6B), indicating that other Cl− channels were able to compensate and flux enough Cl− to depolarize the resting membrane potential. One potential candidate may be anoctamin (TMEM16) channels, which are activated by physiological [Ca2+]i and overexpressed in several cancer types (Hartzell et al., 2009).
Bradykinin activation of ion channels as a therapeutic target for glioma treatment
We found that bradykinin-activation of KCa3.1 and Cl− channels facilitates glioma cell migration through cerebral parenchyma and that inhibition of bradykinin signaling or KCa3.1 activity decreases migration. This has important implications for the management and treatment of glioblastoma multiforme, the most common and deadly primary brain cancer affecting adults. Icatibant is a promising drug candidate for the treatment of gliomas and is already being used clinically in the United States and Europe for the treatment of hereditary angioedema. Icatibant is a specific antagonist of B2R, the primary receptor for bradykinin in gliomas (Montana and Sontheimer, 2011), and our data suggest that it has clinical promise. Alternatively, inhibition of ion channels downstream of bradykinin, including KCa3.1 channels and Cl− channels, could be used to decrease glioma cell migration and disease burden. Chlorotoxin induces internalization of glioma ClC-3 channels (Sontheimer, 2008), significantly hampers glioma cell migration (Soroceanu et al., 1999), and was well tolerated in a phase I study (Mamelak et al., 2006). KCa3.1 channels could also be targeted, because our data are similar to other findings (Sciaccaluga et al., 2010) demonstrating that TRAM-34 inhibits glioma cell migration. To inhibit activation of both KCa3.1 and Cl− channels, caffeine may be efficacious because it decreases bradykinin-induced [Ca2+]i elevations and glioma invasiveness (Kang et al., 2010). Thus, this novel mechanism of bradykinin-induced activation of ion channels presents several druggable targets to improve clinical management of gliomas.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants RO1 NS036692 and RO1 NS031234 (H.S.) and F31 NS073181 (V.A.C.).
References
- Catacuzzeno L, Aiello F, Fioretti B, Sforna L, Castigli E, Ruggieri P, Tata AM, Calogero A, Franciolini F. Serum-activated K and Cl currents underlay U87-MG glioblastoma cell migration. J Cell Physiol. 2011;226:1926–1933. doi: 10.1002/jcp.22523. [DOI] [PubMed] [Google Scholar]
- Cuddapah VA, Sontheimer H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J Biol Chem. 2010;15:11196. doi: 10.1074/jbc.M109.097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Sontheimer H. Ion channels and the control of cancer cell migration. Am J Physiol Cell Physiol. 2011;301:C541–C549. doi: 10.1152/ajpcell.00102.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah VA, Habela CW, Watkins S, Moore LS, Barclay TT, Sontheimer H. Kinase activation of ClC-3 accelerates cytoplasmic condensation during mitotic cell rounding. Am J Physiol Cell Physiol. 2012;302:C527–C538. doi: 10.1152/ajpcell.00248.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitt S, Darley RL, Hallett MB. Translocation or just location? Pseudopodia affect fluorescent signals. J Cell Biol. 2009;184:197–203. doi: 10.1083/jcb.200806047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Heblich F, James IF, Robbins J, Docherty RJ. Bradykinin evokes a Ca2+-activated chloride current in non-neuronal cells isolated from neonatal rat dorsal root ganglia. J Physiol. 2001;530:395–403. doi: 10.1111/j.1469-7793.2001.0395k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioretti B, Castigli E, Calzuola I, Harper AA, Franciolini F, Catacuzzeno L. NPPB block of the intermediate-conductance Ca2+-activated K+ channel. Eur J Pharmacol. 2004;497:1–6. doi: 10.1016/j.ejphar.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Fioretti B, Catacuzzeno L, Sforna L, Aiello F, Pagani F, Ragozzino D, Castigli E, Franciolini F. Histamine hyperpolarizes human glioblastoma cells by activating the intermediate-conductance Ca2+-activated K+ channel. Am J Physiol Cell Physiol. 2009;297:C102–C110. doi: 10.1152/ajpcell.00354.2008. [DOI] [PubMed] [Google Scholar]
- Giannone G, Rond é P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, Takeda K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem. 2004;279:28715–28723. doi: 10.1074/jbc.M404054200. [DOI] [PubMed] [Google Scholar]
- Haas BR, Cuddapah VA, Watkins S, Rohn KJ, Dy TE, Sontheimer H. With-No-Lysine Kinase 3 (WNK3) stimulates glioma invasion by regulating cell volume. Am J Physiol Cell Physiol. 2011;301:C1150–C1160. doi: 10.1152/ajpcell.00203.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habela CW, Olsen ML, Sontheimer H. ClC3 is a critical regulator of the cell cycle in normal and malignant glial cells. J Neurosci. 2008;28:9205–9217. doi: 10.1523/JNEUROSCI.1897-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl− channels. J Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 2001;276:20093–20100. doi: 10.1074/jbc.M009376200. [DOI] [PubMed] [Google Scholar]
- Ifuku M, Färber K, Okuno Y, Yamakawa Y, Miyamoto T, Nolte C, Merrino VF, Kita S, Iwamoto T, Komuro I, Wang B, Cheung G, Ishikawa E, Ooboshi H, Bader M, Wada K, Kettenmann H, Noda M. Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J Neurosci. 2007;27:13065–13073. doi: 10.1523/JNEUROSCI.3467-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, Sasaki T, Ozawa S. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8:971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, Yoo SH, Chung CK, Park SH, Paek SH, Roh EJ, Lee SJ, Park JY, Traynelis SF, Lee CJ. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010;70:1173–1183. doi: 10.1158/0008-5472.CAN-09-2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Lee MG, Macglashan DW, Jr, Undem BJ. Role of chloride channels in bradykinin-induced guinea pig airway vagal C-fibre activation. J Physiol. 2005;566:205–212. doi: 10.1113/jphysiol.2005.087577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Lengyel I, Voss K, Cammarota M, Bradshaw K, Brent V, Murphy KP, Giese KP, Rostas JA, Bliss TV. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur J Neurosci. 2004;20:3063–3072. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamelak AN, Rosenfeld S, Bucholz R, Raubitschek A, Nabors LB, Fiveash JB, Shen S, Khazaeli MB, Colcher D, Liu A, Osman M, Guthrie B, Schade-Bijur S, Hablitz DM, Alvarez VL, Gonda MA. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24:3644–3650. doi: 10.1200/JCO.2005.05.4569. [DOI] [PubMed] [Google Scholar]
- Mao J, Chen L, Xu B, Wang L, Li H, Guo J, Li W, Nie S, Jacob TJ, Wang L. Suppression of ClC-3 channel expression reduces migration of nasopharyngeal carcinoma cells. Biochem Pharmacol. 2008;75:1706–1716. doi: 10.1016/j.bcp.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Martini FJ, Valdeolmillos M. Actomyosin contraction at the cell rear drives nuclear translocation in migrating cortical interneurons. J Neurosci. 2010;30:8660–8670. doi: 10.1523/JNEUROSCI.1962-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Mislocalization of Kir channels in malignant glia. Glia. 2004;46:63–73. doi: 10.1002/glia.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expresssion of voltage-gated chloride channels in human glioma cells. J Neurosci. 2003;23:5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Schwab A, Wulf A, Schulz C, Kessler W, Nechyporuk-Zloy V, Römer M, Reinhardt J, Weinhold D, Dieterich P, Stock C, Hebert SC. Subcellular distribution of calcium-sensitive potassium channels (IK1) in migrating cells. J Cell Physiol. 2006;206:86–94. doi: 10.1002/jcp.20434. [DOI] [PubMed] [Google Scholar]
- Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Arch. 2007;453:421–432. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- Sciaccaluga M, Fioretti B, Catacuzzeno L, Pagani F, Bertollini C, Rosito M, Catalano M, D'Alessandro G, Santoro A, Cantore G, Ragozzino D, Castigli E, Franciolini F, Limatola C. CXCL12-induced glioblastoma cell migration requires intermediate conductance Ca2+-activated K+ channel activity. Am J Physiol Cell Physiol. 2010;299:C175–C184. doi: 10.1152/ajpcell.00344.2009. [DOI] [PubMed] [Google Scholar]
- Shimada K, Li X, Xu G, Nowak DE, Showalter LA, Weinman SA. Expression and canalicular localization of two isoforms of the clc-3 chloride channel from rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2000;279:G268–G276. doi: 10.1152/ajpgi.2000.279.2.G268. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. An unexpected role for ion channels in brain tumor metastasis. Exp Biol Med (Maywood) 2008;233:779–791. doi: 10.3181/0711-MR-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl− and K+ ion channel blockers. J Neurosci. 1999;19:5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari MM, Stimers JR, Mayeux PR. Bradykinin-induced chloride conductance in murine proximal tubule epithelial cells. Mol Cell Biochem. 2007;297:1–8. doi: 10.1007/s11010-006-9315-3. [DOI] [PubMed] [Google Scholar]
- Volk AP, Heise CK, Hougen JL, Artman CM, Volk KA, Wessels D, Soll DR, Nauseef WM, Lamb FS, Moreland JG. ClC-3 and IClswell are required for normal neutrophil chemotaxis and shape change. J Biol Chem. 2008;283:34315–34326. doi: 10.1074/jbc.M803141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- Watkins S, Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31:17250–17259. doi: 10.1523/JNEUROSCI.3938-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AK, Bomben VC, Sontheimer H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia. 2006;54:223–233. doi: 10.1002/glia.20364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]