

Abstract
The immense molecular diversity of neurons challenges our ability to understand the genetic and cellular etiology of neuropsychiatric disorders. Leveraging knowledge from neurobiology may help parse the genetic complexity: identifying genes important for a circuit that mediates a particular symptom of a disease may help identify polymorphisms that contribute to risk for the disease as a whole. The serotonergic system has long been suspected in disorders that have symptoms of repetitive behaviors and resistance to change, including autism. We generated a bacTRAP mouse line to permit translational profiling of serotonergic neurons. From this, we identified several thousand serotonergic-cell expressed transcripts, of which 174 were highly enriched, including all known markers of these cells. Analysis of common variants near the corresponding genes in the AGRE collection implicated the RNA binding protein CELF6 in autism risk. Screening for rare variants in CELF6 identified an inherited premature stop codon in one of the probands. Subsequent disruption of Celf6 in mice resulted in animals exhibiting resistance to change and decreased ultrasonic vocalization as well as abnormal levels of serotonin in the brain. This work provides a reproducible and accurate method to profile serotonergic neurons under a variety of conditions and suggests a novel paradigm for gaining information on the etiology of psychiatric disorders.
Introduction
The CNS has remarkable cellular diversity with hundreds of distinct cell types based on morphology alone (Ramon y Cajal et al., 1899). Although nearly every one of these cell types has an identical genome, each cell only uses a subset of genes as required by its particular functional role. Understanding this molecular genetic diversity of cell types in the CNS may provide important insight, both for the particular roles of a given cell type as well as for the potential consequences of genetic polymorphisms to neural circuits implicated in human disease.
We have developed techniques that permit the global assessment of translation in genetically defined cell types in vivo (Doyle et al., 2008; Heiman et al., 2008). Here, we apply this methodology for the first time to the serotonergic system. The serotonergic system is thought to have important roles in regulation of basic physiological processes, such as breathing, thermoregulation, and sleep, as well as higher cognitive phenomena, such as mood and learning. Most importantly, its dysfunction is suspected in several neuropsychiatric diseases, including obsessive compulsive disorder and autism, among others (Veenstra-VanderWeele et al., 2000; Canli and Lesch, 2007; Deneris and Wyler, 2012).
Autism is a pervasive developmental disorder characterized by core symptoms, including impairment in social interactions and communication, as well as repetitive behaviors, restricted interests, and resistance to change (American Psychiatric Association, 2000; Fombonne, 2005). With a concordance rate reported from 60–90% for monozygotic twins, autism clearly has a remarkably strong genetic component, yet, as is the case for many psychiatric disorders, studies indicate that this genetic contribution is likely to be complex and polygenic. In autism, those genes that have been implicated thus far either explain only a small number of the cases or make relatively small contributions (Moldin and Rubenstein, 2006; Freitag, 2007; Wang et al., 2009; Weiss et al., 2009; Abrahams and Geschwind, 2010). One possible explanation for the difficulty in discovering genes contributing to complex psychiatric disorders, such as autism, would be distinct genetic causes in different groups of individuals. However, the commonality of the symptoms suggests that these distinct genes would still be impacting a common pathway or circuit in the brain. This suggests a candidate approach, focused on particular categories of genes, or genes expressed in particular cell types that are a priori suspected to contribute to a particular symptom, may increase statistical power by decreasing the number of tests. However, one risk with candidate gene studies is that choice of genes may be considered arbitrary or limited by our current knowledge of relevant genes for a specific biological process. Here, we propose that cell type specific translational profiling can be applied to provide guidance for genetic studies of the symptomatology of CNS disorders, and we test this approach in the case of serotonergic neurotransmission and autism.
To this end, we have applied the Translating Ribosome Affinity Purification (TRAP) methodology to identify the comprehensive in vivo suite of ribosome bound mRNA in serotonergic neurons in adult mice. Screening the TRAP-identified serotonergic genes using human data suggested polymorphisms in the CUGBP Elav-like family member 6 (CELF6) gene may contribute to autism risk. Consistent with these findings, Celf6 mutant mice exhibit some autism-related behaviors and abnormal brain levels of serotonin.
Materials and Methods
Generation and husbandry of Slc6a4 TRAP mice
All protocols involving animals were approved by the Institutional Animal Care and Use Committee of Rockefeller University and the Animal Studies Committee of Washington University. BAC transgenic mice were generated as described previously (Gong et al., 2002), using BAC RP24-335M24. Positive founders and subsequent eGFP-L10a-positive progeny were bred to C57BL/6J and pups were genotyped with tail-DNA PCR for eGFP at each generation. Lines from two independent founders were tested for accurate expression as described below. All serotonin-positive neurons labeled with GFP in both lines. A few nonserotonergic neurons showed trace labeling with GFP antibodies in the cingulate cortex, hypothalamus, inferior colliculi, lateral nucleus of the olfactory tract in one of the two lines (JD57), and in the dorsal part of the caudal spinal trigeminal nucleus in the other line (JD60, data not shown). We used line JD60 for all subsequent experiments.
Generation and characterization of Celf6 antibodies
Peptides (QPGSDTLYNNGVSPC and AASEGRGEDRKC) from Celf6, selected for cross-species conservation, relative uniqueness across the Celf family, and hydrophobicity, were synthesized, conjugated to KLH, and injected into New Zealand white rabbits following standard protocols (Green Mountain Antibodies). ELISA was used to confirm generation of antibodies specific to each peptide. High-titer rabbits were boosted, and blood sera were affinity purified following standard protocols. Antibodies were tested for effectiveness for Western blot, and immunofluorescence, using 3T3 cells transfected with GFP-tagged and untagged isoforms of Celf6, then fixed with 4% paraformaldehyde. Antibodies against both peptides exhibited immunoreactivity by immunoblot on protein from in Celf6 overexpressing 3T3 cells. Only antibodies against QPGSDTLYNNGVSPC were effective for immunofluorescence on fixed cells. Specificity was confirmed in Celf6 knock-out (Celf6−/−) mouse tissue.
Immunofluorescence and microscopy
Adult mice were killed and perfused transcardially with 15 ml PBS, then 25 ml 4% paraformaldehyde in PBS. Brains were extracted and then cryoprotected in 30% sucrose PBS overnight, frozen in Neg 50 mounting media (Richard Allen Scientific), and sectioned on a cryostat to 40 μm of thickness. Serial sections were collected in PBS with 0.1% sodium azide and stored at 4°C, protected from light, until use.
EGFP-L10a expression was examined both with and without chicken anti-GFP antibody (Abcam ab13970), and colocalization was confirmed with rabbit anti-serotonin antibody (Immunostar), mouse anti-Neun (Millipore Bioscience Research Reagents), detected with appropriate Alexa dye-conjugated secondary antibodies (Invitrogen). Images were captures on a LSM 510 Zeiss confocal microscope.
Immunohistochemistry
Mice were processed as above. For anti-GFP immunohistochemistry, brains were processed by Neuroscience Associates as described previously (Doyle et al., 2008), using custom goat anti-GFP antibodies. For Celf6 immunohistochemistry, brains were processed as above, incubated overnight with 1:1000 with purified rabbit anti-Celf6 antibody, then donkey anti-rabbit biotinylated secondary antibodies (Jackson ImmunoResearch Laboratories), and developed with the Vectashield Elite ABC kit (Vector Laboratories). Sections were digitized with a Zeiss Axioskop2 and customized macros.
Translating ribosome affinity purification
Three replicate pools of five adult mice of both sexes were killed, and brains were rapidly dissected in ice-cold dissection buffer, containing cycloheximide, to isolate the midbrain and brainstem. TRAP was conducted as described previously (Heiman et al., 2008). Briefly, each pool was homogenized for 12 strokes in a glass Teflon homogenizer on ice, in buffer (10 mm HEPES [pH 7.4], 150 mm KCl, 5 mm MgCl2, 0.5 mm dithiothreitol, 100 μg/ml cycloheximide, protease inhibitors, and recombinant RNase inhibitors). Nuclei and debris were removed with centrifugation at 2000 × g for 10 min at 4°C. DHPC (Avanti) and NP-40 (Ipgal-ca630, Sigma) were added to supernatant to final concentrations of 1% and 30 mm, respectively. After 5 min incubation of ice, supernatant was centrifuged for 15 min at 20,000 × g, and pellet was discarded. Supernatant was mixed with protein G-coated magnetic beads (Invitrogen), previously conjugated with a mix of two monoclonal anti-GFP antibodies (Doyle et al., 2008), and incubated with rotation for 30 min at 4°C. Beads were washed three times with high salt wash buffer (10 mm HEPES [pH 7.4], 350 mm KCl, 5 mm MgCl2, 1% NP-40, 0.5 mm dithiothreitol, and 100 μg/ml cycloheximide), and RNA was purified from ribosomes using Trizol (Invitrogen), following the manufacturer's protocols, followed by DNase treatment, further purification, and concentration with RNeasy min-elute columns, following the manufacturer's protocol (Qiagen). RNA was also harvested in parallel from each unbound fraction of affinity purification as a measure of total tissue RNA. RNA concentration of all samples was measured with Nanodrop spectrophotometer, and integrity was confirmed with PicoChips on the Agilent BioAnalyzer (RIN > 8).
Microarray experiments and statistical analysis
A total of 20 ng of each RNA sample was amplified with the Affymetrix Two-Cycle amplification kit following the manufacturer's instructions, and quality of labeled aRNA was assessed with Bioanalyzer. Labeled RNA from immunoprecipitated ribosomes and total tissue RNA was hybridized to separate Affymetrix Mouse Genome 430 2.0 arrays and scanned following the manufacturer's protocols. Data are available from the Gene Expression Omnibus (GSE36068).
Data were analyzed using the Bioconductor module within the statistical package R. Data quality was assessed by examining raw images of slides, boxplots, histograms, correlation coefficients, false-positive rates, and scatterplots comparing replicate experiments. Data were normalized as described previously (Dougherty et al., 2010) but using Affymetrix chip definition files. Briefly, GCRMA was used to normalize within replicates and to biotinylated spike in probes between conditions. Fold change, Specificity Index (SI), and Specificity Index statistic (pSI) were calculated for all genes with expression above background, as described (Dougherty et al., 2010). Probesets with pSI < 0.01 were selected as enriched in serotonergic neurons for the purposes of further analysis. This resulted in 196 probesets covering 174 distinct genes.
Heirarchical clustering was conducted using Bioconductor on any genes with pSI < 0.01 in any cell type from the current data and the initial survey (Doyle et al., 2008).
Mouse genes from above were mapped to human Unigenes genes using homologene, gene symbol, and BLAT of mouse mRNA against human genome sequence, with 147 genes having clear human homologs. Custom perl scripts were used to identify alignment coordinates of human Unigenes from UCSC genome browser, along with 10 kb of flanking sequence to cover potential regulatory regions, with 136 Unigenes having clear and unique genome coordinates.
Chromosomal distribution was analyzed by comparing number of regions expected to be found on each chromosome (Wilming et al., 2008), compared with observed number of regions.
Analysis of genome-wide association studies
The Rockefeller University Institutional Review Board and the Washington University Human Research Protection Office reviewed all human subjects related work in this manuscript.
Deidentified human genotype and phenotype data were downloaded with permission from the Autism Genetic Research Exchange (AGRE) consortium (Geschwind et al., 2001) for 3787 individuals in 943 multiplex families, genotyped for 550,000 SNPs with the Illumina Hap550 platform (Wang et al., 2009). SNP genotypes were called with BeadStudio 3.1.3.0, and genotypes and phenotypes were formatted for PLINK (Purcell et al., 2007) using Perl. Individuals with missing or poor data were excluded (MIND > 0.1, Geno > 0.1). Only those individuals with clearly diagnosed as autistic by AGRE were scored as affected. Those with ambiguous diagnosis (scored NQA or broad spectrum) were excluded. We selected all available SNPs within 10 kb of the transcription start or end site of the serotonergic neuron enriched genes, filtered to remove those with MAF < 0.05 and those that were out of Hardy-Weinburg equilibrium (p < 0.01). We further removed SNPs with an r2 > 0.5 as they would violate the assumption of independence for multiple testing corrections and would not provide any further information. This resulted in a total of 555 SNPs being tested with the transmission disequilibrium test. Tests were then corrected with the Bonferroni method for multiple testing. Tests were run for all probands (1142 trios); and because prior work had suggested that autism in males and females may have a separate genetic etiology (Stone et al., 2004), we further analyzed these SNPs with males (915 trios) and females (227 trios) independently.
To determine whether segregating probands by sex was meaningful, we performed an additional permutation analysis by randomly sampling 915 probands and repeating the analysis. Excluding SNPs in C1QTNF2 and CELF6, corrected p values <0.05 were observed in only 8 of 1000 iterations, suggesting that this sex-specific difference was not spurious.
Resequencing of candidate genes
Primers were designed for resequencing following standard methods (Montgomery et al., 2008), taking care to avoid SNPs and repetitive sequences in locations for the primers. Standard M13F and M13R sequences were incorporated into the primers. Deidentified DNAs from 384 male white probands from distinct families within the AGRE sample and 384 male white controls from the NIMH Human Genetic Initiative Controls (HGIC) set were amplified with PCR.
Amplicons were then purified and Sanger sequenced using M13F or M13R primers by Agencort Beckman Coulter Genomics. Sequences were trimmed to exclude low-quality reads and analyzed with Seqman software to identify known and novel SNPs. Any putative novel coding sequence mutations were confirmed by sequencing with the opposite primer.
Screening for stop mutation in additional cases and controls
An additional 864 probands from the Simons Simplex Collection (SSC) and 4992 controls from the HGIC set were screened as follows:
HGIC samples were first screened with Sequenom. The sequence variant was run in a 29-plex SNP Genotyping Assay using Sequenom iPLEX Gold Technology. The initial PCR, performed as per the manufacturer's instructions (Sequenom). The subsequent Shrimp Alkaline Phosphatase clean, the iPLEX reaction, and the resin clean were all performed as stated by Sequenom. Spectro chip arrays were spotted using a nanodispenser, according to the manufacturer's instructions. A Sequenom MALDI-TOF spectrophotometer was used to read the array. The SpectroAquire and MassARRAY Typer Software packages (Sequenom) were used for interpretation, and Typer analyzer Version 3.4.0.18 was used to review and analyze all data.
Any positive HGIC samples and all SSC samples were then screened with allele-specific PCR (Little, 2001). Primers that specifically amplify CELF6 only with the stop codon mutation were included in a multiplex reaction with internal control primers for β-actin for a final concentration of 500 nm each with Maxima SYBR Green quantitative RT-PCR Master Mix (Fermantas), and subjected to quantitative RT-PCR in an ABI Prism 7900, followed by a melt curve. β-Actin and CELF6 amplicons were distinguished on the basis of melting temperature (83°C and 88°C, respectively). Positive control DNA with stop codon was included on each plate of PCR.
Analysis of 1000 genomes data
VCF files corresponding to exome sequence data from 1092 individuals (1000 Genomes Project Consortium, 2010) were downloaded into WASP (Dubin et al., 2010) and custom scripts used to extract all exonic variants, annotate these with regard to likelihood of being pathogenic (Sunyaev et al., 2001; Ng and Henikoff, 2002; Eddy, 2004; Adzhubei et al., 2010), and then determine minor allele frequencies. The DGV was queried in March 2012 in the UCSC genome browser using the UCSC annotated CELF6 transcriptional start and stop as a query.
Quantitative real-time RT-PCR
TRAP was conducted as above for an additional three male and three female slc6a4 bacTRAP mice. Each sample was used to synthesize cDNA from 50 ng of total RNA using Protoscript (NEB), primed with Oligo dT, following the manufacturer's instructions. Quantitative RT-PCR was conducted in a Bio-Rad IQ5 with Bio-Rad 2× Sybr Green Mastermix (Bio-Rad). Data were analyzed in the IQ5 software using the ddCT method, with β-actin as a control. Melt curves were conducted to assure specificity of product, and each product was sequenced to confirm accuracy of amplification.
Generation of Celf6 knock-out mice
Long and short arms with LoxP sites flanking exon 4 of Celf6 were cloned by PCR adjacent to an Frt flanked neomycin-resistance cassette gene. B6(Cg)-Tyrc-2j/j-derived ES cells were electroporated using standard methods, and neomycin-resistant colonies were screened by PCR and southern blot for proper integration. Positive colonies were injected into C57BL/6J mouse blastocysts, and chimeric mice were bred to germline Flpe expressing C57BL/6J mice to remove neomycin selection cassette, then actin-Cre C57BL/6J mice to create germline deletions of Celf6. Recombination was confirmed in progeny by PCR. Heterozygous progeny were crossed to generate knock-out (Celf6−/−) and wild-type (WT) littermate mice for behavioral and anatomical assays.
Behavioral tests
Animals and experimental design of behavioral studies.
A total of 46 C57BL/6J Celf6−/− (n = 23, 14 females and 9 males) and litter-matched WT (n = 23, 14 females and 9 males) mice were used for ultrasonic pup vocalization (USV) recording and analysis. The animal colony room lighting was a 12:12 h light/dark cycle; room temperature (∼20°C–22°C) and relative humidity (50%) were controlled automatically. Standard laboratory chow and water were available on a continuous basis.
Twenty experimentally naive male C57BL/6J Celf6−/− (n = 11) and litter-matched WT (n = 9) mice were used for adult behavioral testing. The sequence of behavioral testing was devised to minimize “carry-over” effects across behavioral tests as much as possible. Behavioral testing began when the mice were 3–5 months of age and included a 1 h locomotor activity/exploration test, followed 1 d later by an evaluation on a battery of sensorimotor measures, to assess possible disturbances in general activity and sensorimotor functions that might affect performance on subsequent tests. One day later, the mice were tested in the Morris water maze, which included cued (visible platform), place (hidden platform), and probe (platform removed) trials to assess potential nonassociative deficits as well as spatial learning and memory impairments. The following week, the mice were tested for their ability to learn another platform location (reversal and additional probe trials) to evaluate cognitive flexibility in terms of extinguishing one learned response and acquiring another. During the next week, sociability was assessed using the social approach test followed by an evaluation of sensorimotor reactivity and gating by quantifying acoustic startle and prepulse inhibition (PPI). Approximately 2–3 months later, anxiety-like behaviors were measured in the mice by testing them in the elevated plus maze (EPM). The following week, the mice were retested on the social approach test for further evaluation of sociability and preference for a novel conspecific (stimulus mouse). Nineteen days later, exploratory behaviors and olfactory preference were quantified in the holeboard exploration/olfactory preference test.
Ultrasonic vocalization recording and analysis.
WT and Celf6−/− pups were individually removed from their parents at postnatal day 8 and placed in a dark, enclosed chamber. Ultrasonic vocalizations were obtained using an Avisoft UltraSoundGate CM16 microphone, Avisoft UltraSoundGate 416H amplifier, and Avisoft Recorder software (gain = 6 dB, 16 bits, sampling rate = 250 kHz). Pups were recorded for 3 min, after which they were weighed and returned to their home cages. Frequency sonograms were prepared from recordings in MATLAB (frequency range = 40 kHz to 120 kHz, FFT size = 256, overlap = 50%), and individual syllables were identified and counted from the sonograms according to a previously published method (Holy and Guo, 2005).
One hour locomotor activity and sensorimotor battery.
Locomotor activity was evaluated in the mice using transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures and computerized photobeam instrumentation as previously described (Wozniak et al., 2004). General activity variables (total ambulations and vertical rearings) along with indices of emotionality, including time spent, distance traveled, and entries made in a 33 × 11 cm central zone and distance traveled in a 5.5 cm contiguous peripheral zone were analyzed. All mice were also evaluated on a battery of sensorimotor tests designed to assess balance (ledge and platform), strength (inverted screen), coordination (pole and inclined screens), and initiation of movement (walking initiation), as previously described (Wozniak et al., 2004; Grady et al., 2006). For the walking initiation test, the mouse was placed on a surface in the center of a 21 × 21 cm square marked with tape, and the time the mouse took to leave the square was recorded. During the balance tests, the time was recorded that the mouse remained on a Plexiglas ledge (0.75 cm wide) or a small circular wooden platform (3.0 cm in diameter) elevated 30 or 47 cm, respectively. The screen tests were conducted by placing a mouse head-oriented down in the middle of a mesh grid measuring 16 squares per 10 cm, elevated 47 cm, and angled at 60° or 90°. The time was recorded that the mouse required to turn 180° and climb to the top. For the inverted screen, the mouse was placed on the screen as described above; and when it appeared to have a secure grip, the screen was inverted 180°, and the time the mouse remained on to the screen was recorded. Each test had a maximum time of 60 s, except for the pole test, which had a maximum time of 120 s. The averaged time of two trials for each test was used for the analyses.
Morris water navigation.
Spatial learning and memory were evaluated in the Morris water maze using a computerized tracking system (ANY-maze, Stoelting) as previously described (Wozniak et al., 2004). Cued (visible platform, variable location) and place (submerged, hidden platform, constant location) trials were conducted, and escape path length, latency, and swimming speeds were computed. To assess nonassociative dysfunctions, the cued condition involved conducting 4 trials per day (60 s maximum per trial) for 2 consecutive days with the platform being moved to a different location for each trial using a 30 min intertrial interval, and with very few distal spatial cues being present to limit spatial learning. Performance was analyzed across four blocks of trials (2 trials/block). Three days later, place trials were conducted, which involved acquisition training to assess spatial learning where mice were required to learn the single location of a submerged platform in the presence of several salient distal spatial cues. During the 5 consecutive days of place trials, the mice received 2 blocks of 2 consecutive trials [60 s maximum for a trial; 30-s intertrial interval (spent on platform)] with each block being separated by ∼2 h and each mouse being released from a different quadrant for each trial. The place trials data were analyzed over five blocks of trials (4 trials/block) where each block represented the performance level for each of five consecutive days. A probe trial (60 s maximum) was administered ∼1 h after the last place trial on the fifth day of training with the platform being removed and the mouse being released from the quadrant opposite to where the platform had been located. Time spent in the various pool quadrants, including the target quadrant where the platform had been placed and crossings over the exact platform location, served as the dependent variables. Three days after completing the place and probe conditions, mice were tested on reversal trials, which involved the same procedures as the place trials, except that the hidden platform was moved to a new location in the pool for all trials. Another probe trial was conducted after completing the reversal trials.
Social approach.
Our protocol was adapted from methods previously described (Moy et al., 2004; Silverman et al., 2011) and involves quantifying sociability [tendency to initiate social contact with a novel conspecific (stimulus mouse) or cagemate] and preference for social novelty (tendency to initiate social contact with a novel vs a familiar stimulus mouse). The apparatus was a rectangular 3-chambered Plexiglas box (each chamber measuring 19.5 cm × 39 cm × 22 cm) containing Plexiglas dividing walls with rectangular openings (5 × 8 cm) covered by sliding Plexiglas doors. A small stainless-steel withholding cage (10 cm h × 10 cm diameter; Galaxy Pencil/Utility Cup, Spectrum Diversified Designs) was used to sequester a stimulus mouse. The withholding cage consisted of vertical bars, which allowed for minimal contact between mice but prevented fighting, and one was located in each outer chamber. A digital video camera connected to a PC loaded with a tracking software program (ANY-maze, Stoelting) recorded the movement of the mouse within the apparatus and quantified time spent in each chamber and investigation zone surrounding the withholding cages. The investigation zones were 12 cm in diameter, encompassing 2 cm around the withholding cages. An entry into the chambers was defined when a chamber contained 80% of the mouse's body while only the head was tracked in the zones surrounding the withholding cages to capture investigative social behaviors. Indirect lighting illuminated the test room, and the entire apparatus was cleaned with Nolvasan solution while the withholding cages were cleaned with 75% ethanol solution between tests.
The first test sequence consisted of 3 consecutive 10 min trials. For the first trial, each mouse was placed in the middle chamber with the doors to the outer chambers shut to become acclimated to the apparatus. During the second trial, the mouse was allowed to freely investigate and habituate to all three chambers, including the empty withholding cages (Page et al., 2009; Naert et al., 2011; Pobbe et al., 2012). Neither group showed prior preference for a particular withholding cage during habituation. The third (test) trial assessed sociability exhibited toward a novel stimulus mouse versus a familiar, empty withholding cage by placing an unfamiliar, gender-matched stimulus mouse in one withholding cage while the other was left empty, and the test mouse was allowed to freely explore the apparatus and investigate the novel mouse in the withholding cage. A second test sequence conducted ∼2 months later also consisted of 3 consecutive 10 min trials. For trial 1, each mouse was allowed to freely investigate and habituate to all three chambers, including the empty withholding cages. For the second trial, a gender-matched cagemate was placed in one withholding cage while the other remained empty, and the test mouse could freely explore the apparatus and investigate the cagemate in the withholding cage. During the third (test) trial, the familiar cagemate remained in the withholding cage, although a new, unfamiliar gender-matched stimulus mouse was now placed in the other withholding cage, and the test mouse was allowed to explore the apparatus and investigate the two mice contained in the withholding cages. The locations of the stimuli mice in the outer chambers for the first test sequence were counterbalanced within and across groups, and placement of the stimuli mice for the second test sequence was randomized. All stimuli mice were acclimated to the metal withholding cages for 10 min before beginning of testing.
Acoustic startle/prepulse inhibition.
Sensorimotor reactivity and gating were also evaluated in the mice by quantifying their acoustic startle response (ASR) and PPI because PPI has been reported to be abnormal in individuals with autism (Perry et al., 2007; Yuhas et al., 2011). The ASR and PPI were tested using computerized instrumentation designed specifically for mice (StartleMonitor, Kinder Scientific). The ASR to a 120 dBA auditory stimulus pulse (40 ms broadband burst) and PPI (response to a prepulse plus the startle pulse) were measured concurrently using our previously described methods (Hartman et al., 2001; Gallitano-Mendel et al., 2008). Beginning at stimulus onset, 65, 1 ms force readings were averaged to obtain an animal's startle amplitude for a given trial. A total of 20 startle trials were presented over a 20 min test period, during which the first 5 min served as an acclimation period when no stimuli above the 65 dB white noise background were presented. The session began and ended by presenting 5 consecutive startle (120 db pulse alone) trials unaccompanied by other trial types. The middle 10 startle trials were interspersed with PPI trials, consisting of an additional 30 presentations of 120 dB startle stimuli preceded by prepulse stimuli of 4, 12, or 20 dB above background (10 trials for each PPI trial type). A percent PPI score for each trial was calculated using the following equation: %PPI = 100 × (ASRstartle pulse alone − ASRprepulse + startle pulse)/ASRstartle pulse alone (see Hartman et al., 2001 for more details).
EPM.
Anxiety-like behaviors were evaluated in the EPM according to our previously described procedures (Schaefer et al., 2000; Boyle et al., 2006). Our apparatus consisted of two opposing open arms and two opposing enclosed arms (36 × 6.1 × 15 cm) that extended from a central platform (5.5 × 5.5 cm), which were constructed of black Plexiglas. The maze was equipped with pairs of photocells configured in a 16 (x-axis) × 16 (y-axis) matrix, the output of which was recorded by a computer and interface assembly (Kinder Scientific). A system software program (MotorMonitor, Kinder Scientific) enabled the beam-break data to be recorded and analyzed to quantify time spent, distance traveled, and entries made into the open and closed arms and center area. To adjust for differences in general activity, the percentage of distance traveled, time spent, and entries made into the open arms out of the totals (open arms + closed arms) for each variable were also computed. Test sessions were conducted in a dimly lit room where light was provided by two 13 W black-light bulbs (Feit EcoBulbs) and each session began by placing a mouse in the center of the maze and allowing it to freely explore the apparatus. Each test session lasted 5 min, and the mice were tested over 3 consecutive days.
Holeboard exploration/olfactory preference test.
Mice were evaluated for possible differences in exploratory behaviors and olfactory preferences using a slightly modified version of our previously published methods (Ghoshal et al., 2012) where hole poking served as the main behavioral response. Our modifications were based on procedures described by Moy et al. (2008) for Study 2 within their publication to assess shifts in olfactory preference in an autism mouse model. We used the same odorant, familiarization technique, and testing procedure, although our apparatus is slightly different from the one used by these authors. Our protocol involved the use of a computerized holeboard apparatus (41 × 41 × 38.5 cm high clear plastic chamber), containing 4 corner and 4 side holes in the floor, with a side hole being equidistant between the corner holes (Learning Holeboard; MotorMonitor, Kinder Scientific). Pairs of photocells were contained within each hole (27 mm in diameter) and were used to quantify the frequency and duration of pokes, whereby a poke that was at least 35 mm in depth was required to be registered as a hole-poke. Each mouse received one 30 min habituation session in the holeboard during which no holes contained any odorants. The following day (test session 1), a 20 min session was conducted during which hole-poke frequency and duration data were collected in the presence of empty and odorant-containing holes. All side holes were empty, while odorants were placed at the bottom of each of three corner holes, although access to the odorants was blocked, and one corner hole remained empty. The configuration of odorant-containing and empty corner holes (see Fig. 7C) was such that one hole contained a familiar odorant (fresh corn-cob bedding used in the home cages), which was placed in a corner hole opposite to a corner hole containing an unfamiliar odorant (woodchip bedding). Another novel odorant (chocolate chips) was placed in a corner hole opposite to the empty corner hole. This pattern was counterbalanced within and across groups. Immediately after completing test session 1, the mice were familiarized with the chocolate chips by placing 4–7 chocolate chips in each home cage. The same thing was done on the following day, and all but 1 chocolate chip were consumed before the start of the second test session. Test session 2 was conducted two days after test session 1 using the same procedures and pattern of odorant-containing and empty corner holes, except that the actual configuration used for test session 2 was opposite to the one used during test session 1 (see Fig. 8C). For example, the corner holes that contained bedding for test session 1 contained chocolate chips or were empty during test session 2, and vice versa. General exploratory behavior was evaluated by quantifying total hole-pokes as well as pokes into the side and corner holes for test sessions 1 and 2. Olfactory preferences were assessed by analyzing poke frequencies involving odorant-containing and empty corner holes within and between groups. Poke durations exhibited for the different types of holes were also analyzed to provide additional data on possible differences in the processing of olfactory stimuli.
Figure 7.
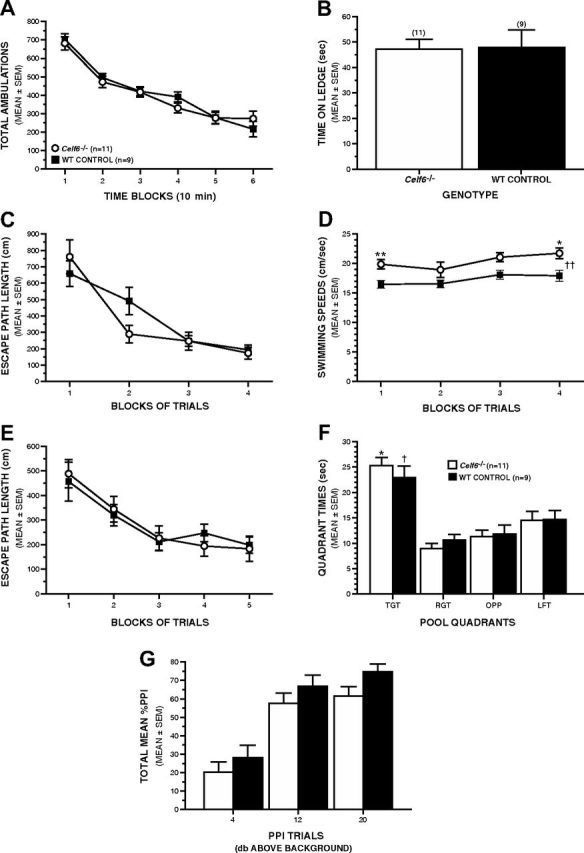
Adult male Celf6−/− mice demonstrate intact sensorimotor and spatial learning functions. A, B, Adult male Celf6−/− and WT mice performed similarly on several basic behavioral assays. A, Comparable levels of general ambulatory activity were observed in Celf6−/− and WT mice during the 1 h locomotor activity test. B, Celf6−/− mice did not exhibit any performance deficits on the ledge test or on any of the other 6 measures within the sensorimotor battery (data not shown). C–F, The Celf6−/− mice also generally performed at control-like levels on the Morris water maze. C, Celf6−/− and WT controls exhibited comparable path lengths to the escape platform during cued trials, indicating that the Celf6−/− mice did not display nonassociative dysfunctions that would interfere with subsequent learning and memory performance in the water maze. D, Celf6−/− mice swam significantly faster than the WT controls during the cued trials in the water maze (rmANOVA–genotype effect, ††p = 0.011, pairwise comparisons: **p = 0.004 and *p = 0.012). E, WT and Celf6−/− groups performed equally well during the place trials in the water maze, suggesting that spatial learning was not impaired. F, During the probe trial, both the Celf6−/− and WT mice spent more time in the target (TGT) quadrant where the platform had previously been (rmANOVA with planned comparisons, *p < 0.003 and †p < 0.025, respectively) compared with the right (RGT), opposite (OPP), and left (LFT) quadrants, suggesting that retention was not impaired. G, Equivalent levels of PPI were observed in the two groups of mice, indicating normal sensorimotor gating and startle responses. Data are mean ± SEM.
Figure 8.

Adult Celf6−/− mice exhibit control-like levels of sociability, social preference, and anxiety-like behaviors. A, During the first social approach test, adult Celf6−/− and WT mice both showed normal sociability preference for a novel stimulus mouse over an empty withholding cage during the test trial (rmANOVA with planned comparisons, *p < 0.00004). B, During the second social approach test, adult Celf6−/− mice spent more time investigating the cagemate compared with the empty withholding cage during test trial 1 (**p < 0.005), demonstrating no deficits in sociability toward a familiar cagemate. During test trial 2, adult Celf6−/− and WT mice both displayed a preference for social novelty by spending more time investigating the novel stimulus mouse compared with a familiar cagemate during test trial 2 (rmANOVA with planned comparisons, *p < 0.0003). C, D, Adult Celf6−/− and WT mice showed similar levels of anxiety-like behaviors as indexed by the amount of time spent (C) and distance traveled (D) in the open arms of the elevated plus maze. Data are mean ± SEM.
Statistical analyses for behavioral data.
ANOVA models were used to analyze the behavioral data. A two-way ANOVA model was used to analyze differences within and between genotype and sex for USV. Repeated measures (rm) ANOVA models containing one between-subjects variable (genotype) and one within-subjects (repeated measures) variable (e.g., blocks of trials) were typically used to analyze the learning and memory data. The Huynh-Feldt adjustment of α levels was used for all within-subjects effects containing more than two levels to protect against violations of sphericity/compound symmetry assumptions underlying rmANOVA models. Typically, one-way ANOVA models were used to analyze differences between groups for 1 h locomotor activity test and measures in the sensorimotor battery. Planned comparisons were conducted within ANOVA models for certain holeboard variables. In most other instances, pairwise comparisons were conducted following relevant, significant overall ANOVA effects, which were subjected to Bonferroni correction when appropriate.
Extraction for measurement of neurotransmitter levels
Ten Celf6−/− and seven WT whole adult mouse brains frozen at −80°C were dried on a lyophilizer for 72 h. The dried brains were crushed to a powder and stored at −80°C. A total of 10 mg of each was extracted as follows: vortexed 30 s; centrifuged 13,000 RPM, 4°C, 10 min; sonicated at 50°C in a bath sonicator; stored at −4°C; liquid nitrogen bath 1 min and then allowed to thaw. Acetone (600 μl at 4°C) was added to each tube. Tubes were vortexed, placed in liquid nitrogen and sonicated for 10 min; this was repeated three times. Samples were incubated for 30 min and then centrifuged. Supernatant was poured into new microcentrifuge tubes and stored. Methanol, water, and formic acid (400 μl at 86.5:12.5:1) were added to the remaining pellet. Tubes were vortexed, sonicated for 15 min, and incubated for 30 min. Samples were centrifuged, and the supernatant was combined with the previous 600 μl. The pooled supernatant was dried with a speedvac with no heat for 23 h. To each tube, 100 μl of water was added. Tubes were sonicated for 5 min and incubated for 1 h. Samples were centrifuged and the resulting supernatant was transferred to deactivated glass autosampler vials for MS analysis.
LC/MS analysis
Targeted LC/MS analyses were done using an HPLC system (Agilent 1260 Infinity) coupled to an Agilent 6460 Triple Quadrapole MS operated in positive ESI mode. Analytes were separated using a Zorbax 300 SB-C18 (5 μm, 150 × 0.5 mm) column. An isocratic elution was used with 0.1% v/v formic acid in water. The injection volume was 0.8 μl. A 30 min wash was performed between injections. Flow was set to 5 μl/min. Source conditions were gas temperature 300°C, gas flow 6 L/min, nebulizer 15 psi, capillary 4000 V. Acquisition settings were dwell time 200 ms, MS1 resolution unit, MS2 resolution unit, polarity positive, cell accelerator voltage 7. The 6460 Triple Quadrapole was operated in MRM mode monitoring two transitions for each analyte with the most intense transition being used for analysis. MRM transitions were integrated with Agilent Mass Hunter Qualitative Analysis B.04.00. Statistical analysis was conducted in MS Excel using 2-tailed t tests, with Bonferroni correction.
Results
Slc6a4 bacTRAP mice target eGFP-L10a specifically to serotonergic neurons
Classical studies using a variety of methodologies (Hillarp et al., 1966; Steinbusch, 1981, 1984; Ishimura et al., 1988), including in situ hybridization studies of the presynaptic serotonin transporter Slc6a4 (Bengel et al., 1997), have defined nine populations of serotonergic neurons, predominantly within the raphe nuclei. To enable translational profiling of these cells, we generated transgenic mouse lines using a Slc6a4 BAC to drive the expression of the TRAP construct (eGFP fused to the ribosomal protein L10a). Immunohistochemistry with anti-GFP antibodies demonstrated robust expression in a pattern consistent with expression in all serotonergic neurons (Fig. 1A, B). Neurotransmitter phenotype was confirmed using confocal immunofluorescence with antibodies to GFP and serotonin (Fig. 1C). In all cells, GFP signal was seen only in the cytoplasm and nucleolus, consistent with ribosomal incorporation of the fusion protein.
Figure 1.
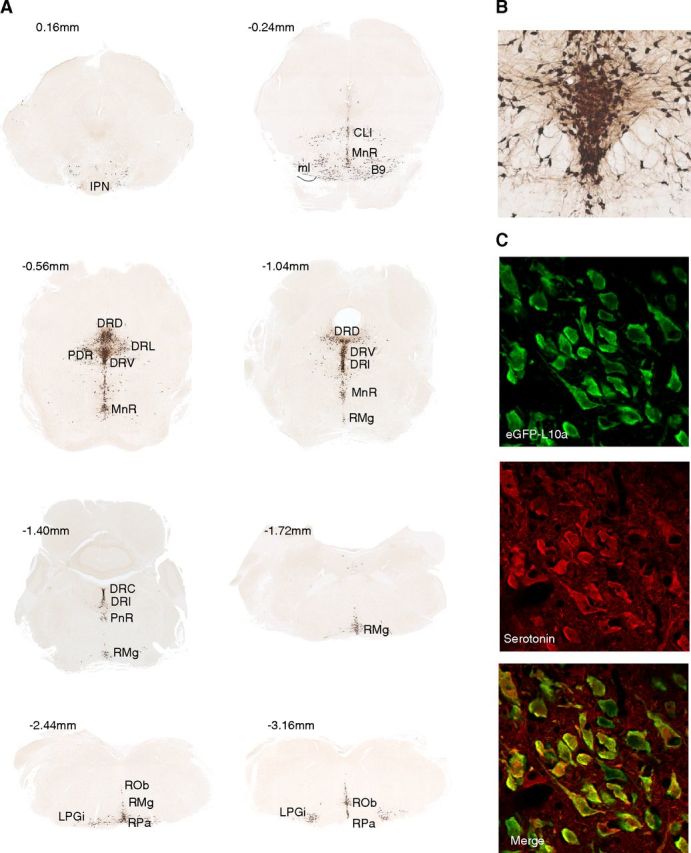
A bacTRAP line for serotonergic neurons. A, DAB immunohistochemistry with anti-GFP antibodies on Slc6a4 bacTRAP mice reveals eGFP-L10a transgene expression in the known serotonin cell-containing nuclei. B, Higher magnification of DRV shows neuronal morphology of eGFP-positive cells. C, Confocal immunofluorescence colocalization of eGFP-L10a transgene with neurotransmitter serotonin. Abbreviations following Paxinos (Franklin and Paxinos, 1997). Coordinates relative to interaural. IPN, Interpeduncular nucleus; ml, medial lemniscus; RMg, raphe magnus; ROb, raphe obscurus; RPa, raphe pallidus; LPGi, lateral paragigantocellular nucleus; DRD, dorsal raphe dorsal; DRI, dorsal raphe interfasicular; DRL, dorsal raphe lateral; DRV, dorsal raphe ventral; PDR, posterodorsal raphe; MnR, median raphe; CLi, caudal linear raphe.
Serotonergic neurons have a distinct translational profile
Using eGFP as an affinity tag, we purified the suite of ribosome bound mRNAs from serotonergic neurons (the TRAP methodology) (Doyle et al., 2008, Heiman et al., 2008) and interrogated them with microarrays. Independent biological replicates showed high reproducibility (Fig. 2A), with an average correlation >0.99. When these samples were compared with microarrays of the whole tissue (Fig. 2B), there was a clear, robust, and comprehensive enrichment of all mRNAs known to be specifically expressed in serotonergic neurons, including enzymes in the pathway for synthesizing serotonin in the CNS (Tph2, Ddc) (Goridis and Rohrer, 2002), transcription factors known to be important for specifying these neurons (Goridis and Rohrer, 2002; Pfaar et al., 2002) (Fev, Gata2, and Gata3), the serotonin autoreceptor (Htr1a; Korte et al., 1996), and the serotonin transporter itself (Slc6a4). There was also a clear depletion of mRNAs known to not be expressed by neurons, such as the glial genes Mobp, Aldh1L1, Mbp, and Plp. This suggests that we could indeed capture mRNA specifically from serotonergic neurons.
Figure 2.

Reproducible and accurate polysome purification from serotonergic neurons. A, Scatterplot of probeset signal intensity (log10) for independent biological replicate TRAP experiments reveals extremely high reproducibility. Black lines indicate fold change = 0.5 and 2. B, Scatterplot of mRNA TRAPed from serotonergic neurons (average of 3 replicates, x-axis), compared with mRNA from hindbrain (average of 3 replicates, y-axis), reveals a robust enrichment for genes expressed in serotonergic neurons (blue), but not for known glial genes (red). C, Blinded analysis of in situ hybridization patterns for probesets predicted to be enriched in serotonergic neurons (light gray, n = 197), compared with randomly selected probesets (dark gray, n = 197), reveals a robust overrepresentation of raphe-specific and enriched genes in serotonergic neuron gene list (p < 1E−66, χ2 test). D, Hierarchical clustering of TRAP data for serotonergic neurons with previously characterized cell types (Doyle et al., 2008).
To select for all mRNAs used in serotonergic neurons, we removed those probesets with median expression <50, or a fold change below a background threshold determined by the glial genes (Dougherty et al., 2010). These normalized data represent >5000 genes clearly expressed in serotonergic neurons.
To generate a comprehensive list of those mRNAs either highly enriched or specific for serotonergic neurons, we compared this translational profile to our previous survey of cell types in the CNS (Doyle et al., 2008) using the pSI statistic (Dougherty et al., 2010). We identified 196 probesets with pSI < 0.01 (Table 1), corresponding to 174 genes that included the known serotonergic cell genes listed above. To validate this list, we compared it to the online catalog of mouse in situ hybridizations of the Allen Brain Atlas (Lein et al., 2007) in a blinded and unbiased manner (Dougherty et al., 2010). Compared with a random list of genes from the array, the serotonergic cell list had nearly three times as many in situ hybridizations with a pattern of enrichment in the raphe nuclei. Seventeen of these genes scored as specific to serotonergic neurons, compared with none from the random list (Fig. 2C).
Table 1.
Serotonin cell-specific and enriched genes
| Gene symbol | Probe set ID | pSI | Name |
|---|---|---|---|
| Acsl4 | 1451828_a_at | 6.54E-04 | Acyl-CoA synthetase long-chain family member 4 |
| Ankrd2 | 1419621_at | 1.72E-03 | Ankyrin repeat domain 2 (stretch responsive muscle) |
| Apoa2 | 1417950_a_at | 9.57E-03 | Apolipoprotein A-II |
| Arhgdig | 1448660_at | 4.87E-04 | Rho GDP dissociation inhibitor (GDI) gamma |
| Asb4 | 1433919_at | 1.91E-06 | Ankyrin repeat and SOCS box-containing protein 4 |
| Celf6 | 1429790_at | 8.40E-03 | Cugbp and Elav-like family member 6, RNA binding protein |
| C1qtnf2 | 1431079_at | 8.01E-03 | C1q and tumor necrosis factor related protein 2 |
| Calcr | 1418688_at | 1.91E-06 | Calcitonin receptor |
| Calr3 | 1453232_at | 2.20E-03 | Calreticulin 3 |
| Cckar | 1421195_at | 1.43E-03 | Cholecystokinin A receptor |
| Cd1d1 | 1449131_s_at, 1449130_at | 1.91E-06 | CD1d1 antigen |
| Ceacam10 | 1448573_a_at, 1417074_at | 1.91E-06 | CEA-related cell adhesion molecule 10 |
| Cep63 | 1436274_at | 1.91E-06 | Similar to protein C6orf117 |
| Chodl | 1451440_at | 3.57E-03 | Chondrolectin |
| Ciapin1 | 1438163 xat | 1.97E-03 | Rhomboid domain containing 2 |
| Cited1 | 1449031_at | 1.91E-06 | Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 1 |
| Col7a1 | 1419613_at | 1.91E-06 | Procollagen, type VII, alpha 1 |
| Cpa3 | 1448730_at | 1.91E-06 | Carboxypeptidase A3, mast cell |
| Cpne8 | 1430521_s_at, 1430520_at | 2.19 to 3.24E-03 | Copine VIII |
| Cryba2 | 1419011_at | 1.91E-06 | Crystallin, β A2 |
| Cthrc1 | 1452968_at | 5.54E-05 | Collagen triple helix repeat containing 1 |
| Ctla2b | 1416811_s_at, 1448471_a_at | 1.91E-06 | cytotoxic T lymphocyte-associated protein 2 β |
| Cxcl7 | 1418480_at | 1.91E-06 | Chemokine (C-X-C motif) ligand 7 |
| Ddc | 1430591_at, 1426215_at | 1.91E-06 | Dopa decarboxylase |
| Dlk1 | 1449939_s_at | 1.91E-06 | Delta-like 1 homolog (Drosophila) |
| Dnajc12 | 1417441_at | 3.44E-05 | DnaJ (Hsp40) homolog, subfamily C, member 12 |
| E2f5 | 1434493_at | 4.57E-04 | E2F transcription factor 5, p130-binding |
| En1 | 1418618_at | 1.34E-05 | Engrailed 1 |
| Esm1 | 1449280_at | 2.87E-05 | Endothelial cell-specific molecule 1 |
| Fbxw4 | 1442674_at | 7.26E-03 | F-box and WD-40 domain protein 4 |
| Fev | 1425886_at | 1.91E-06 | FEV (ETS oncogene family) |
| Fgd5 | 1460578_at | 1.91E-06 | FYVE, RhoGEF. and PH domain containing 5 |
| Foxa1 | 1418496_at | 7.42E-03 | Forkhead box A1 |
| Fxyd5 | 1418296_at | 1.91E-06 | FXYD domain-containing ion transport regulator 5 |
| Fxyd6 | 1417343_at | 9.95E-03 | FXYD domain-containing ion transport regulator 6 |
| Gas5 | 1449410_a_at | 2.42E-04 | Growth arrest-specific 5 (non-protein coding) |
| Gata2 | 1428816_a_at, 1450333_a_at | 1.91E-06 | GATA binding protein 2 |
| Gata3 | 1448886_at | 1.91E-06 | GATA binding protein 3 |
| Gch | 1429692_s_at, 1420499_at | 1.91E-06 | GTP cyclohydrolase 1 |
| Glra2 | 1434098_at | 2.58E-04 | Glycine receptor, α 2 subunit |
| Gm1574 | 1446591_at | 1.91E-06 | Gene model 1574, (NCBI) |
| Gnas | 1421740_at | 6.19E-04 | GNAS (guanine nucleotide binding protein, α stimulating) complex locus |
| Gng2 | 1418451_at | 6.11E-03 | Guanine nucleotide binding protein (G protein), γ 2 subunit |
| Gnpda2 | 1457230_at | 3.19E-04 | Glucosamine-6-phosphate deaminase 2 |
| Gpr1 | 1460123_at | 5.89E-04 | G protein-coupled receptor 1 |
| Gpr4 | 1457745_at | 1.91E-06 | G protein-coupled receptor 4 |
| Gpx3 | 1449106_at | 1.34E-05 | Glutathione peroxidase 3 |
| Gstm6 | 1422072_a_at | 1.83E-04 | Glutathione S-transferase, μ 1 |
| Guca1a | 1421061_at | 1.91E-06 | Guanylate cyclase activator 1a (retina) |
| Gus | 1430332_a_at | 3.05E-03 | Glucuronidase, β |
| H2-Q1 | 1418734_at | 1.91E-06 | histocompatibility 2, Q region locus 1 |
| Hcrtr1 | 1436295_at | 1.91E-06 | Hypocretin (orexin) receptor 1 |
| Hoxb3 | 1456229_at | 4.80E-03 | Homeo box B3 |
| Hs6st2 | 1420938_at, 1450047_at | 2.05E-04 to 1.91E-06 | Heparan sulfate 6-O-sulfotransferase 2 |
| Htr1a | 1438710_at | 1.91E-06 | 5-Hydroxytryptamine (serotonin) receptor 1A |
| Htr1d | 1440166_x_at, 1440741_at | 4.22E-04 to 1.91E-06 | 5-Hydroxytryptamine (serotonin) receptor 1D |
| Htr5b | 1422196_at | 1.91E-06 | 5-Hydroxytryptamine (serotonin) receptor 5B |
| Igh-6 | 1427351_s_at, 1427329_a_at | 5.75E-03 to 1.91E-06 | Immunoglobulin heavy chain 6 (heavy chain of IgM) |
| Igh-VJ558 | 1425763_x_at, 1421653_a_at | 3.82 to 1.91E-06 | Immunoglobulin heavy chain (J558 family) |
| Il1r1 | 1448950_at | 7.70E-04 | Interleukin 1 receptor, type I |
| Ing1 | 1456857_at | 2.10E-05 | Inhibitor of growth family, member 1 |
| Irs4 | 1441429_at | 1.91E-06 | Insulin receptor substrate 4 |
| Klk27 | 1421587_at | 1.91E-06 | Kallikrein 1-related peptidase b27 |
| Krt2–1 | 1422481_at | 6.65E-03 | Keratin complex 2, basic, gene 1 |
| Lgals8 | 1422661_at | 4.22E-03 | Lectin, galactose binding, soluble 8 |
| Lst1 | 1425548_a_at | 1.92E-03 | Leukocyte specific transcript 1 |
| Maob | 1434354_at | 1.91E-06 | Monoamine oxidase B |
| Mbd1 | 1430838_x_at, 1453678_at | 4.20E-03 to 7.45E-05 | Methyl-CpG binding domain protein 1 |
| Mcpt5 | 1449456_a_at | 7.07E-05 | Chymase 1, mast cell |
| Nanos1 | 1436648_at | 8.47E-03 | Nanos homolog 1 (Drosophila) |
| Nkx6-1 | 1425828_at | 1.10E-03 | NK6 transcription factor related, locus 1 (Drosophila) |
| Nos3 | 1434092_at | 8.57E-03 | Nitric oxide synthase 3 antisense |
| Nrl | 1450946_at | 1.91E-06 | Neural retina leucine zipper gene |
| Oas1e | 1416847_s_at | 1.91E-06 | 2′-5′ Oligoadenylate synthetase 1E |
| Pcbd | 1418713_at | 1.09E-04 | Pterin 4 α carbinolamine dehydratase |
| Pcdha4 | 1424341_s_at | 3.91E-03 | Protocadherin α 4 |
| Pcdha6 | 1451769_s_at | 7.28E-03 | Protocadherin α 6 |
| Pcdhac1 | 1425017_at | 1.91E-06 | Protocadherin α subfamily C, 1 |
| Pcsk1 | 1421396_at | 1.91E-06 | Proprotein convertase subtilisin/kexin type 1 |
| Pcsk5 | 1451406_a_at, 1437339_s_at | 2.89E-03 to 6.66E-03 | Proprotein convertase subtilisin/kexin type 5 |
| Pdzk8 | 1439088_at | 1.91E-06 | PDZ domain containing 8 |
| Prph1 | 1422530_at | 3.16E-03 | Peripherin 1 |
| Ptger3 | 1450344_a_at | 1.91E-06 | Prostaglandin E receptor 3 (subtype EP3) |
| Resp18 | 1417988_at | 2.03E-04 | Regulated endocrine-specific protein 18 |
| Rims3 | 1459042_at | 1.75E-03 | Regulating synaptic membrane exocytosis 3 |
| Rpgrip1 | 1454231_a_at | 1.91E-06 | Retinitis pigmentosa GTPase regulator interacting protein 1 |
| Rph3al | 1444409_at | 2.10E-05 | Rabphilin 3A-like (without C2 domains) |
| Rpl37a | 1416217_a_at | 9.57E-03 | Ribosomal protein L37a |
| Rps27 | 1460699_at | 2.10E-05 | Ribosomal protein S27 |
| Saa1 | 1419075_s_at | 1.51E-04 | Serum amyloid A1 |
| Scg2 | 1450708_at | 3.12E-04 | Secretogranin II |
| Scn9a | 1442333_a_at, 1442810_x_at, 1442809_at | 1.91E-06 | Sodium channel, voltage-gated, type IX, α |
| Siat8f | 1456147_at, 1456440_s_at | 9.94E-05 to 1.91E-06 | ST8 α-N-acetyl-neuraminide α-2,8-sialyltransferase 6 |
| Slc18a2 | 1437079_at | 1.91E-06 | Solute carrier family 18 (vesicular monoamine), member 2 |
| Slc22a3 | 1420444_at | 1.91E-06 | Solute carrier family 22 (organic cation transporter), member 3 |
| Slc6a4 | 1417150_at | 1.91E-06 | Solute carrier family 6 (neurotransmitter transporter, serotonin), member 4 |
| Sln | 1420884_at | 3.20E-03 | Sarcolipin |
| Sncg | 1417788_at | 1.91E-06 | Synuclein, γ |
| Stard3 nl | 1430274_a_at | 7.55E-03 | STARD3 N-terminal like |
| Stk32b | 1431236_at | 1.91E-06 | Serine/threonine kinase 32B |
| Swam2 | 1449191_at | 1.91E-06 | WAP four-disulfide core domain 12 |
| Tinag | 1419314_at | 1.91E-06 | Tubulointerstitial nephritis antigen |
| Tmie | 1441926_x_at, 1443964_at | 1.26E-03 to 5.76E-03 | Transmembrane inner ear |
| Tnfrsf11b | 1449033_at | 4.80E-03 | Tumor necrosis factor receptor superfamily, member 11b (osteoprotegerin) |
| Tph2 | 1435314_at | 1.91E-06 | Tryptophan hydroxylase 2 |
| Trh | 1418756_at | 1.91E-06 | Thyrotropin releasing hormone |
| Trpm4 | 1435549_at | 5.66E-03 | Transient receptor potential cation channel, subfamily M, member 4 |
| Twsg1 | 1441302_at | 8.37E-03 | Twisted gastrulation homolog 1 (Drosophila) |
| Zar1 | 1434494_at | 2.10E-05 | Zygote arrest 1 |
| Zfp622 | 1447775_x_at | 8.60E-04 | Zinc finger protein 622 |
| Zwint | 1444717_at | 7.90E-03 | ZW10 interactor |
Probesets corresponding to all named genes with pSI < 0.01 are listed.
We next compared our list of enriched and specific genes with the recently published data from flow-sorted embryonic serotonergic neurons from rostral and caudal nuclei (Wylie et al., 2010). There are numerous differences in methodology and design between the two studies, including age (embryonic vs adult), purification methodology (flow sorting vs TRAP), amplification methodology, normalization, and analytical strategy. Despite this, there was highly significant overlap between the two studies (χ2 test, p < 8.7E−227), suggesting good conservation of gene expression across development for these neurons. Thus, the lines generated here provide in vivo access to the translational profile of the adult serotonergic system and can be used in the future to study the response of these cells to whole animal manipulations in a reproducible and accurate manner.
Using hierarchical clustering, we compared the serotonergic translational profile to the cell types from our previous survey (Doyle et al., 2008; Fig. 2D). Serotonergic neurons are clearly distinct from other cells. However, remarkably, of the surveyed cell types, serotonergic neurons are most similar to cholinergic neurons of the basal forebrain. Although these two populations come from distinct regions and have distinct transmitter phenotypes, both have similar functional roles; that is, both cell types project widely through the CNS and function as neuromodulators, influencing the physiological properties of whole circuits. This suggests that the translational profile of a cell population is more strongly influenced by its particular roles within the CNS than the region in which it is found or the particular neurotransmitter it uses.
Next, we performed a standard Gene Ontologies analysis on the 174 serotonergic genes using DAVID (Dennis et al., 2003), which indicated, unsurprisingly, a role for a significant fraction of these genes in serotonin signaling (not shown). However, many other genes were unannotated, and we suspected that these 174 genes may provide a novel list of candidate genes for neuropsychiatric disorders thought to involve the serotonergic system.
Of the 174, 147 had clear human homologues based on homologene, BLAT, and probematchDB (Wang et al., 2002). These were distributed across 136 noncontiguous regions of the genome. Chromosomal analysis revealed a remarkably nonrandom distribution of these regions (χ2 test, p < 3.57E−7). In particular, 10.4% of these were found on the X chromosome, compared with the 4.5% that would be expected to be found there by chance given the size and gene density of the chromosomes. This is interesting considering the gender disparity in prevalence for autism (4:1 male to female; Fombonne, 2005) and depression (1: 1.7 male to female; Kessler et al., 1993), two of the disorders with suspected connections to the serotonergic system.
Transmission disequilibrium testing for polymorphisms in serotonergic genes in autism
We next sought to determine whether polymorphism in these 136 regions may be related to autism, using the Transmission Disequilibrium Test (Spielman and Ewens, 1996; Collins, 2007), applied to data generated by AGRE (Geschwind et al., 2001; Wang et al., 2009) from multiplex families with autism. We discovered two SNPs that met criteria for significance after multiple testing correction (Table 2): one in both males and females in the C1QTNF2 gene and one specifically in males in the gene encoding the CELF6 RNA-binding protein.
Table 2.
Transmission disequilibrium in SNPs near two serotonergic neuron genes in human autistic probands
| Chromosome | SNP | A1 (minor allele) | A2 (major allele 2) | No. of observations of minor allele transmission to individual with autism | No. of observations of untransmitted minor allele | OR | p value | Bonferonni-corrected p value | Gene |
|---|---|---|---|---|---|---|---|---|---|
| Male probands | |||||||||
| 15 | rs2959930 | G | A | 273 | 378 | 0.72 | 3.87E-05 | 0.02521 | CELF6 |
| 5 | rs9313845 | A | G | 110 | 171 | 0.64 | 0.000274 | 0.1785 | C1QTNF2 |
| 14 | rs8003220 | A | G | 38 | 77 | 0.49 | 0.000276 | 0.18 | IGH |
| 5 | rs17409286 | A | G | 144 | 203 | 0.71 | 0.001539 | 1 | C1QTNF2 |
| 8 | rs11573856 | A | G | 113 | 165 | 0.68 | 0.001816 | 1 | TNFRSF11B |
| Female probands | |||||||||
| 17 | rs4470197 | G | A | 70 | 113 | 0.62 | 0.00148 | 0.9647 | CCDC55 |
| 16 | rs9929530 | C | A | 52 | 88 | 0.59 | 0.002346 | 1 | TMEM114 |
| 23 | rs4599945 | A | C | 6 | 22 | 0.27 | 0.002497 | 1 | DGKK |
| 5 | rs3762986 | A | G | 93 | 138 | 0.67 | 0.003069 | 1 | PCSK1 |
| 4 | rs11731545 | A | G | 90 | 56 | 1.61 | 0.004895 | 1 | TNFRSF11B |
| Male and female probands | |||||||||
| 5 | rs9313845 | A | G | 129 | 202 | 0.64 | 6.01E-05 | 0.03918 | C1QTNF2 |
| 14 | rs8003220 | A | G | 48 | 92 | 0.52 | 0.0002 | 0.1306 | IGH |
| 15 | rs2959930 | G | A | 352 | 451 | 0.78 | 0.000477 | 0.3107 | CELF6 |
| 8 | rs11573856 | A | G | 143 | 205 | 0.70 | 0.000889 | 0.5795 | TNFRSF11B |
| 10 | rs363341 | A | G | 400 | 498 | 0.80 | 0.001074 | 0.7005 | SLC18A2 |
We decided to first focus on the CELF6 gene because another RNA-binding protein, FMRP, is disrupted in fragile X syndrome, the most common syndromic cause of autism (Verkerk et al., 1991; Chonchaiya et al., 2009). CELF6 is a less well-studied member of an RNA binding protein family known as the CELF family (Cugbp and Etr-3 like factors) (Barreau et al., 2006). CELF proteins are known to regulate RNA in a variety of manners. In Drosophila, this family has been studied in the context of regulation of translation (Good et al., 2000); whereas in Xenopus, homologous proteins are implicated in mRNA localization and stability (Barreau et al., 2006). In mammals, family members are known to both positively and negatively regulate splicing of a variety of transcripts (Ladd et al., 2004), and members of the CELF family seem to be in a dynamic competition with other splicing factors to regulate cell type-specific alternative splicing (Charlet et al., 2002).
SNP rs2959930 is found near an alternative transcriptional start site of CELF6
The CELF6 gene has 16 exons (Fig. 3A) and is highly homologous to other CELF genes, particularly CELF3 (TNRC4), CELF4, and CELF5. Their long isoforms each contain three RNA recognition motifs (RRMs) that are nearly identical across family members, and a divergent domain, required in other family members for both splicing activity and repression (Han and Cooper, 2005). In other family members, the first two RRMs are required for RNA binding (Good et al., 2000), whereas the third may be involved in nuclear localization (Chapple et al., 2007).
Figure 3.

CELF6 gene, exons, domains, and regions of interest. A, UCSC genome browser view of the CELF6 gene in human, showing 16 exons (blue numbers), 5 putative isoforms (a–e), and alignment of the 3 RRMs. B, First candidate region, covered by two amplicons (B1 and B2), encompassing short poly-glutamine sequence (polyQ) and putative human-specific exon (red arrow). Human CELF6 exons are shown condensed in blue, and aligned mouse mRNAs are shown below in black. C, Second candidate region, covered by two amplicons (A3 and A4), encompassing the alternative first exon, and highly conserved putative promotor region (purple, brown) and the associated SNP RS2959930. D, Illustration of CELF6 protein isoforms. Isoform “a” of CELF6, which includes the alternative first exon (1b), does not contain the first RRM.
The SNP associated with autism in the AGRE male probands, rs2959930, is found in the 5′ UTR of an alternative first exon (Fig. 3C). Inclusion of this exon precludes the incorporation of the first RRM (Fig. 3D). In family member CELF2, truncations removing the first RRM reduce the inclusion of GRIN1 exon 21 in a minigene splicing assay (Han and Cooper, 2005). This would suggest that polymorphisms that impact the transcription, translation, or stability of the different isoforms of CELF6 may have an impact on the splicing, localization, and translation of mRNA within the cell. However, it is unclear whether SNP rs2959930 is a relevant functional polymorphism or whether it is in linkage disequilibrium with a more profound variation. First, we sought to determine whether both isoforms are expressed in serotonergic neurons and to confirm that CELF6 is indeed being translated in these cells. Then, we initiated a screen for more profound polymorphisms that may be influencing this gene in autism.
Celf6 is found in serotonergic neurons
To determine which isoforms might be used in serotonergic neurons, we designed primers specific to the long and short isoforms. Quantitative RT-PCR clearly confirmed an enrichment of Celf6 mRNA in serotonergic neurons (data not shown), with both isoforms detectable. To investigate whether Celf6 protein is also found in serotonergic neurons, we generated antibodies to Celf6. Quantitative RT-PCR of 3T3 cells suggested no endogenous expression of Celf6. Experiments with these cells transiently expressing long or short isoforms, or a GFP fusion, demonstrate that these antibodies can detect Celf6 by SDS-PAGE (Fig. 4A) or in fixed cells (Fig. 4B).
Figure 4.

Celf6-like immunoreactivity in serotonergic nuclei. A, Celf6 immunoblot of whole-cell lysates from 3T3 fibroblasts transfected with a long isoform of Celf6, a short isoform of Celf6, or a short isoform of Celf6 fused to GFP demonstrates specificity of antibody to target. B, GFP and Celf6 immunofluorescence in 3T3 fibroblasts transfected with Celf6-GFP (as in A). C, Celf6 immunofluorescence in Slc6a4 bacTRAP mice demonstrates colocalization of Celf6 with eGFP-L10a in serotonergic neurons of dorsal raphe.
Immunofluorescence in Slc6a4 bacTRAP mice reveals colocalization of eGFP with Celf6 immunoreactivity in raphe neurons (Fig. 4C). Celf6 immunoreactivity is also seen in a few other neuromodulatory populations, including the noradrenergic locus ceruleus, populations of cells in the hypothalamus, including presumptive dopaminergic cells of the arcuate nucleus, and a few, scattered, dimly labeled cells in cortex (data not shown). Unlike other presumptive neuronal splicing factors, which are predominantly nuclear (Underwood et al., 2005), labeling is largely cytoplasmic, suggesting that in the brain Celf6 may have roles beyond splicing. Having confirmed Celf6 protein is found in serotonergic neurons, we next conducted a screen to identify additional variants that may be associated with human autism.
Resequencing of candidate exons in CELF6 identifies rare premature stop codon
Toward an understanding of the extent to which CELF6 tolerates genetic variation in humans, we used publicly available exome data from 1092 individuals, to compare SNV rates here with those in all other RefSeq protein coding genes (n = 19,032). Although tolerance for nonsynonymous variation was seen to vary, the large majority of annotated genes (n = 14,022) were observed to harbor one or more SNVs predicted to be deleterious (2+ of BLOSUM62, Polyphen2, and SIFT) (Sunyaev et al., 2001; Ng and Henikoff, 2002; Eddy, 2004; Adzhubei et al., 2010) and present at an appreciable frequency in this population (>1%). Consistent with intolerance to nonsynonymous variation, no such events were observed within CELF6. Similarly, no structural variants overlapping with CELF6 were observed in the DGV (Iafrate et al., 2004). Although power issues currently limit a precise ranking of how tolerant different genes are to genetic variation, consideration of all coding variation put CELF6 at the 97th percentile of all annotated genes, a result consistent with biological intolerance. Based on this result, we reasoned that a focused resequencing effort, enabling deep characterization of key functional regions, should be used.
Toward this end, we selected three regions for our first screen of CELF6. This included the area around the alternative first exon 1b, where the common variant was identified, as well as the exon 12, which contains a short CAG repeat (Fig. 3B). Finally, we also targeted the exon 11 because available EST data suggest that it is used exclusively by humans: no other available species show any mRNA or ESTs aligning to it (Fig. 3B). Of course, as human, mouse, and rat sequences dominate the EST databases, these data could represent a loss of the exon in rodents, rather than a gain in humans.
Thus, we sequenced the promoter and alternative first exon 1b, as well as exons 11–13 in 384 male white probands (randomly selected from previous AGRE probands) and 384 normal male white controls (new samples). In this semi-independent sample, we found no common alleles showing stronger association than rs2959930 (Fisher's exact test, p < 0.051), although one novel low-frequency intronic variant was seen only in controls (Table 3, p < 0.0012). We did not detect any instances of triplet repeat expansion in exon 12, although extraordinarily large repeats would not be assayable with PCR-based methods.
Table 3.
Results of Celf6 resequencing study
| DBsnp | Amplicon | Position within amplicon | SNP | AGRE, % (no.) of samples with SNP detected, of interpretable reads | Controls, % (no.) of samples with SNP detected, of interpretable reads | p value (Fisher's test), deviating from expected (Controls) | Type | Confirmed |
|---|---|---|---|---|---|---|---|---|
| Novel | B1 | 129 | C → CG | 0.27% (1/370) | 0% (0/355) | 0.510 | Stop | Yesa |
| Novel | B1 | 150 | C → CG | 0.27% (1/370) | 0% (0/355) | 0.510 | Intron | None |
| Novel | B1 | 247 | T → CT | 0.81% (3/370) | 0.57% (2/353) | 0.522 | Intron | None |
| Novel | B1 | 263 | T → CT | 0.27% (1/370) | 0.57% (2/353) | 0.469 | Intron | None |
| Novel | B2 | 88 | C → CG | 0.53% (2/375) | 0.29% (1/341) | 0.536 | Intron | None |
| Novel | B2 | 104 | T → TC | 0% (0/375) | 0.29% (1/341) | 0.476 | Intron | None |
| Novel | B2 | 200 | G → AG | 0.28% (1/357) | 0% (0/341) | 0.511 | Intron | None |
| Novel | B2 | 362 | G → GA | 0% (0/375) | 0.29% (1/341) | 0.476 | Intron | None |
| Novel | B2 | 391 | T → CT | 0% (0/375) | 0.29% (1/341) | 0.476 | Intron | None |
| Novel | B2 | 459 | G → AG | 0% (0/375) | 2.64% (9/341) | 0.001 | Intron | None |
| Novel | B2 | 513 | C → CT | 0% (0/375) | 0.29% (1/341) | 0.476 | Intron | None |
| rs4625684 | A3 | 60 | T → C | 100% (282/282) | 100% (357/357) | 1.000 | Promoter/intron | In dbSNP |
| rs2959930 | A3 | 147 | C → CT | 29.43% (83/282) | 35.85% (128/357) | 0.051 | 5′ UTR | In dbSNP |
| rs2959930 | A3 | 147 | C → T | 6.74% (19/282) | 3.64% (13/357) | 0.055 | 5′ UTR | In dbSNP |
| Novel | A3 | 151 | C → CT | 0.71% (2/282) | 1.12% (4/357) | 0.459 | 5′ UTR | None |
| rs74026061 | A3 | 165 | G → CG | 0.35% (1/282) | 0% (0/357) | 0.441 | 5′ UTR | In dbSNP |
| Novel | A3 | 166 | C → CT | 0% (0/282) | 0.28% (1/357) | 0.559 | 5′ UTR | None |
| Novel | A3 | 234 | C → CT | 0.35% (1/282) | 0% (0/357) | 0.441 | 5′ UTR | None |
| rs2959928 | A4 | 91 | C → CT | 1.06% (4/377) | 0% (0/363) | 0.067 | Promoter/intron | In dbSNP |
| rs2959929 | A4 | 159 | G → AG | 4.26% (16/376) | 2.48% (9/363) | 0.129 | Promoter/intron | In dbSNP |
| Novel | A4 | 233 | C → CT | 0.27% (1/376) | 0% (0/363) | 0.509 | Promoter/intron | None |
| Novel | A4 | 50 | G → AG | 0% (0/376) | 0.28% (1/363) | 0.491 | Promoter/intron | None |
| Novel | A4 | 169 | C → AC | 0% (0/376) | 0.28% (1/363) | 0.491 | Promoter/intron | None |
a Confirmed in proband and father with bidirectional Sanger sequencing, as well as sequenome and allele-specific PCR assays.
We did, however, discover a premature stop codon in one autistic patient (Fig. 5A) and no controls. This SNP is found in the putative human-specific exon 11 and would be predicted to lead nonsense-mediated decay, and thus is likely a loss of function allele. No coding sequence variations were identified in the 384 controls. The presence of this rare variant in an autistic patient provides further evidence implicating this gene in autism, although it clearly cannot account for the original transmission disequilibrium findings. Sequencing of the entire pedigree (AU1397) reveals that the variant was inherited from the father, suggesting incomplete penetrance if the allele is involved in autism. There is also additional strong matrilineal contribution for autism in this family, as the proband has affected twin siblings and matrilineal cousins not harboring this allele (Fig. 5B). Although not included in the AGRE consortium, the father's extended family also included additional individuals with intellectual disability and reportedly odd behavior.
Figure 5.

Premature stop codon in CELF6 in a family with autism. A, Novel coding SNP identified in human-specific exon. Top row, Proband 303 shows C->C/G. Bottom row, Normal DNA. Translation shown beneath each. Red arrow indicates heterozygote Y->stop SNP. B, Pedigree of Family AU1397. **Not sequenced. *Has stop Y-> Stop. C, Example of quantitative RT-PCR melt curves from 96 allele specific PCRs. 95 amplified only β-actin (genomic control). Positive control DNA (proband 303) amplified both β-actin and mutant Celf6 allele. D, Summary of all assays for mutant Celf6 allele.
To determine whether this rare variant is found in any additional cases or controls, we designed both a primer-extension (Sequenom) assay as well as an allele-specific PCR (Fig. 5C, D). Analysis of 4992 normal individuals, as well as 864 autistic individuals from the Simons Simplex Collection, identified no additional cases or controls harboring this allele, highlighting the rarity of the variant. Likewise, this variant was not seen in the 1092 individuals above, or the ∼5000 individuals currently available in the Exome Variant Server (http://evs.gs.washington.edu/EVS/)[3/2012].
Like other cases of extremely rare variants detected in autism, such as many of the exonic deletions (Bucan et al., 2009) and other copy number variants (Pinto et al., 2010), it is difficult to amass sufficient power to draw statistical conclusions regarding the relevance of such an extremely rare variant. Also, similar to what is seen in deletions of NRXN1 and 16p11, this polymorphism, if contributing to causation, is not showing complete penetrance in the afflicted family (Abrahams and Geschwind, 2010).
Further statistical validation of this allele now awaits more complete resequencing studies of CELF6, a byproduct of ongoing large-scale autism exome sequencing efforts. However, even with these studies, the human genetics alone will still likely be underpowered. In addition, reanalysis of published microarray datafrom human lymphoblasts reveals that CELF6 is not detectable in these cells (Luo et al., 2012), precluding any straightforward patient-based allelic expression imbalance analyses. Therefore, we decided to investigate this allele functionally by creating a mutation in mouse and testing for autism-like behaviors.
Characterization of Celf6 mutant mice
Autism is characterized by abnormal social interactions and language delay, as well as restricted interests and resistance to change. To determine whether mutations in Celf6 could contribute to analogous abnormalities in mouse behavior, we deleted the constitutive fourth exon of Celf6. This deletion introduces premature stop codons in all known isoforms of Celf6 and, like the human mutation, would be predicted to result in nonsense-mediated decay of mRNA. PCR confirmed the deletion of the locus in the genome. Quantitative RT-PCR from mouse brain confirmed decrease of Celf6 mRNA (data not shown). Examination of the brain using nuclear stains (DAPI) and neuronal staining (NeuN) did not reveal any gross morphological differences. However, immunofluorescence using Celf6 antibodies confirms a loss of Celf6 signal from cells in all regions of the brain, including raphe neurons (Fig. 6A). Behavioral studies were then conducted to characterize Celf6−/− and littermate WT control mice during both development and later in adulthood. It was our goal not only to study behaviors that may be related to autism but also to broadly characterize the phenotype of Celf6−/− mice also, as this is the first report of deletion in mice.
Figure 6.

Celf6 mutant mice show loss of Celf6 signal in brain, and Celf6−/− mouse pups show deficits in early communicative behaviors. A, Immunofluorescent confocal microscopy of dorsal raphe neurons, labeled with GFP antibody (green), reveals loss of Celf6 antibody staining (red) in Celf6−/− mice. B, Celf6−/− mouse pups produce significantly fewer ultrasonic syllables per 3 min recording session compared with WT mice (ANOVA, *p = 0.001). Data are mean ± SEM.
Celf6−/− pups have decreased ultrasonic vocalizations
To determine whether Celf6−/− mice exhibited any alterations in communication-related behaviors during early development, we conducted the standard maternal isolation test, which is frequently used to evaluate mouse models of autism (Hofer et al., 2002; Chadman et al., 2008; Scattoni et al., 2008; Nakatani et al., 2009). Eight-day-old mouse pups exhibit robust stereotyped vocalizations in the ultrasonic range when separated from their dam. These calls (“syllables”) (Holy and Guo, 2005) can be considered a form of social communication as they elicit a retrieval response from the dam. Celf6−/− pups produced 60% fewer syllables per 3 min recording session compared with WT littermates (Fig. 6B, ANOVA; F(1,42) = 12.794, p = 0.001). The significant reduction in vocalization in Celf6−/− pups was further observed between genotypes within each sex separately (F(1,42) = 5.792, p = 0.02 for females, F(1,42) = 7.049, p = 0.01 for males; not shown) with no significant difference between sexes. There was no difference in body weights between groups, suggesting that the vocalization effect was not the result of gross developmental delay (data not shown). Results from this assay can be interpreted as reflecting alterations in anxiety levels or communicative function (Hofer et al., 2002). However, we think that the decreased USV in Celf6−/− mouse pups probably reflects communication deficits because we found no evidence of abnormally high levels of anxiety-like behaviors in Celf6−/− mice when they were tested as adults in the elevated plus maze (EPM), at ages when this test has been well validated (see results described below for the EPM).
Adult male Celf6−/− mice exhibit several normal behavioral/cognitive functions
Adult male Celf6−/− and WT mice performed similarly on several basic behavioral assays, including a 1 h locomotor activity test and a battery of sensorimotor measures (e.g., Fig. 7A, B). For example, no differences were observed between groups in terms of general ambulatory activity (Fig. 7A) or vertical rearing (not shown) during a 1 h locomotor activity test. In addition, no performance deficits were observed in the Celf6−/− mice on the ledge test (Fig. 7B) or on any of the other 6 sensorimotor measures that were conducted (not shown). The Celf6−/− mice also generally performed at control-like levels on the Morris water maze. Specifically, there were no effects involving genotype for either escape path length (Fig. 7C) or latency (not shown) during the cued trials, although the Celf6−/− mice swam significantly (25%) faster than the WT control group (Fig. 7D; rmANOVA–genotype effect: F(1,18) = 8.13, p = 0.011). Importantly, no differences were observed between groups with regard to spatial learning and memory; no effects involving genotype were found for path length (Fig. 7E) or latency (not shown) during the place (hidden platform) trials, and no retention performance deficits were evident during the probe trial in terms of spatial bias (Fig. 7F), platform crossings, or time in the target quadrant (data not shown). The increased swimming speeds in the Celf6−/− mice during the cued trials were a transitory effect because no differences were observed between groups on this variable during the place (spatial learning) trials. Sensorimotor reactivity and gating were also evaluated in the mice, but no significant effects involving genotype were found for either the acoustic startle response (data not shown) or PPI (Fig. 7G). In summary, the above results suggest that Celf6−/− mice did not have demonstrable deficits in nonassociative (visual, sensorimotor, or motivational) functions and that their spatial learning and memory appeared to be intact, at least for reference memory-based capabilities.
The mice were also evaluated on standard social interaction assays involving the social approach test (Moy et al., 2004; Silverman et al., 2011), the results of which showed that the Celf6−/− mice performed like WT controls. In the first social approach test sequence, neither Celf6−/− nor WT control mice demonstrated investigation zone biases during the habituation trial (empty vs target zone, Fig. 8A), presenting no confounding issues for the interpretation of the test trial results. We assessed sociability exhibited toward a novel stimulus mouse versus an empty withholding cage during the test trial, and rmANOVAs conducted on the relevant variables did not reveal significant effects involving genotype. More importantly, planned comparisons conducted within each group showed that both groups spent significantly more time investigating a novel stimulus mouse compared with an empty withholding cage, whether measured by time in the investigation zone surrounding the cage (Fig. 8A; F(1,18) = 37.30, p = 0.000009 for Celf6−/−, F(1,18) = 43.45, p = 0.000003 for WT), or by time spent in the chamber (F(1,18) = 17.30, p = 0.0006 for Celf6−/−, F(1,18) = 25.75, p = 0.0008 for WT not shown). A second social approach test conducted at a slightly later age involved quantifying social preference for a same-sex cagemate versus a novel stimulus mouse. Again, neither Celf6−/− nor WT control mice demonstrated investigation zone biases during the habituation trial (empty vs target zone, Fig. 8B). The Celf6−/− mice showed no deficits in sociability toward a cagemate during test trial 1. No differences were found between Celf6−/− and WT control mice during test trial 2 in that both groups of mice spent significantly more time in the investigation zone (Fig. 8B; F(1,16) = 30.46, p = 0.00005 for Celf6−/−, F(1,16) = 23.51, p = 0.0002 for WT) and chamber (F(1,16) = 12.59, p = 0.003 for Celf6−/−, F(1,16) = 9.28, p = 0.008 for WT) containing a novel stimulus mouse compared with the times spent in the same areas where a cagemate was contained.
Given the robust decreases in USV observed in the Celf6−/− mouse pups, we were interested to determine whether they showed high levels of anxiety-like behaviors in the EPM because it might have relevance to interpreting the vocalization results. Specifically, the Celf6−/− mice exhibited control-like levels in terms of time spent (Fig. 8C), distance traveled (Fig. 8D), and entries made into the open arms, as well as the percentages of these variables computed with reference to totals observed in both sets of arms (not shown). Thus, analyzing the classic variables thought to represent anxiety-like behaviors in the EPM did not reveal any differences between the Celf6−/− and WT control mice. These results suggest that the USV deficit was not likely the result of any consistent lifelong differences in anxiety but instead may reflect an impairment in early social communication.
Celf6−/− mice show evidence of resistance to change
Another diagnostic criterion of autism involves resistance to change. Although Celf6−/− mice exhibited intact spatial learning and retention during in the Morris water maze, they swam faster than the WT control group when introduced into the pool during the cued trials, an effect that disappeared with extended exposure during the place trials. This initial increase in swimming speeds in the Celf6−/− mice suggests an altered response in these mice to a significant change in environmental conditions. To evaluate the mice more formally for resistance to change, we evaluated their performance during reversal trials in the water maze. In the first block of trials, the Celf6−/− mice exhibited a tendency toward considerably longer escape latencies and path lengths (Fig. 9A, B), although planned comparisons showed that these differences were not significantly different (F(1,18) = 3.88, p = 0.065 and F(1,18) = 3.45, p = 0.080, respectively). Consistent with the data from the place trials, the groups performed in a similar manner in the later blocks of trials, with regard to swimming speeds, and during the probe trial (data not shown).
Figure 9.
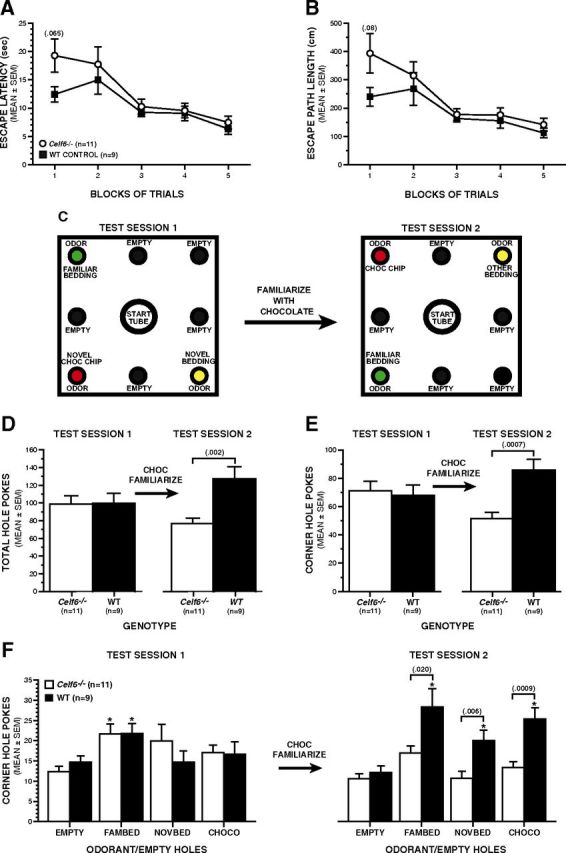
Adult Celf6−/− mice showed evidence of resistance to change. A, B, There were no significant performance differences between groups during the reversal trials in the water maze, although there were nonsignificant trends for the Celf6−/− mice to have longer escape latencies (A, rmANOVA with pairwise comparisons, p = 0.065) and path lengths (B, rmANOVA with pairwise comparisons, p = 0.080) compared with the WT group. C–F, Holeboard assay for resistance to change. C, A schematic of the holeboard showing the location of odorants and empty holes for test session 1 and test session 2 (after familiarization with chocolate). D, Celf6−/− and WT mice showed equivalent exploratory behavior (hole-poke frequencies) during test session 1. Celf6−/− mice failed to show the potentiation of exploratory behavior after familiarization with chocolate that was exhibited by the WT controls in terms of total hole-pokes (ANOVA, p = 0.002). E, The increase in exploratory behavior by WT mice occurred largely in the corner holes (ANOVA, p = 0.0007). F, Before familiarization, Celf6−/− and WT mice showed equivalent exploratory behavior as both groups showed preference for holes containing familiar bedding compared with the empty corner holes (rmANOVA with planned comparisons, *p < 0.010), suggesting normal olfactory capacities for both groups (test session 1). After familiarization, WT mice poked significantly more often than the Celf6−/− mice into the holes containing novel bedding (rmANOVA with planned comparisons, p = 0.006) and, particularly, chocolate chips (p = 0.0009), with large differences into the hole containing familiar bedding (p = .020). Overall, the WT group also showed significant preferences for the holes containing odorants over the empty corner hole (*p < 0.007), whereas the Celf6−/− mice failed to show any change in exploratory hole poking or evidence of olfactory preferences following familiarization to the chocolate. Data are mean ± SEM.
Based on the differences observed during the first block of reversal trials, we decided that a less stressful behavioral task, which involved exploratory behavior using the primary sensory system of mice (olfaction), might provide a more sensitive test of the resistance-to-change phenotype. A type of holeboard task has been used to study behaviors in mice considered to be analogous to the resistance to change and restricted interests found in people with autism-spectrum disorder (Moy et al., 2008; Silverman et al., 2011). Our modified version of this holeboard test (Ghoshal et al., 2012) was used to evaluate both exploratory and olfactory preference behaviors and whether familiarization with the putative reward value of an odorant could alter behavior. Figure 9C shows a schematic of the holeboard apparatus as well as the placement of odorants into the corner holes for test sessions 1 and 2. The results from ANOVAs showed that, initially, Celf6−/− and WT mice displayed similar general hole-poke frequencies with regard to total and corner hole-pokes (Fig. 9D, E, left), as well as side (empty) hole-pokes (not shown). Within-subjects comparisons revealed that both groups also poked significantly more often (F(1,18) = 17.83, p = 0.0005 for Celf6−/−, F(1,18) = 8.58, p = 0.009 for WT) into the hole containing a familiar odorant (fresh homecage bedding) than the empty corner hole (Fig. 9F, left), suggesting that both Celf6−/− and WT mice have intact olfactory abilities, and a baseline preference for familiar bedding, as do other strains of mice (Moy et al., 2008). Poke frequencies did not differ between groups for any odorant or the empty corner hole at baseline.
After test session 1, the mice were familiarized with the chocolate chips by permitting consumption over a two-day period before they were tested on the holeboard again, with the odorants being placed in the corner holes as depicted in Figure 9C (right, test session 2). Familiarization with the chocolate chips had a profound effect on exploratory hole-poking and olfactory preference behaviors in the WT control mice but had no effect on the Celf6−/− group. For example, the results of ANOVAs showed that, after familiarization, WT mice made significantly greater numbers of total hole-pokes (Fig. 9D, F(1,18) = 12.46, p = 0.002), corner hole-pokes (Fig. 9E, F(1,18) = 16.71, p = 0.0007), and side hole-pokes (F(1,18) = 6.185, p = 0.023; data not shown) compared with the Celf6−/− group. Most importantly, familiarization with the chocolate also had a major impact on differences in poke frequencies into the odorant-containing and empty corner holes (Fig. 9F, right). For example, a significant genotype by hole interaction (F(3,54) = 2.814, p = 0.048) for test session 2, followed by subsequent comparisons showed that the WT mice poked significantly more often into the holes containing the novel bedding (F(1,18) = 9.90, p = 0.006), and particularly the chocolate chips (F(1,18) = 15.91, p = 0.0009), compared with the Celf6−/− group, with large differences also observed between groups in hole-pokes into the hole containing familiar bedding (F(1,18) = 6.51, p = 0.020, Bonferroni-corrected critical p = 0.013). Moreover, within-subjects comparisons revealed that the Celf6−/− mice showed no preference for any odorant over the empty corner hole during test session 2, whereas the WT group showed significant preferences (F(3,16) = 14.13, p = 0.00009), for the familiar bedding (p = 0.0002), novel bedding (p = 0.006), and chocolate (p = 0.00005) over the empty corner hole (Bonferroni corrected critical p = 0.017). Thus, familiarization with the chocolate had a robust effect on WT control mice in terms of increasing their general exploratory hole poking and changing their olfactory preferences, whereas the Celf6−/− group showed a resistance to change on either of these two behavioral dimensions after exposure to the chocolate.
We also measured duration of hole-pokes to determine whether it was likely that the Celf6−/− and WT groups were processing the olfactory stimuli in a different manner. Our analyses showed that there were no differences between the groups with regard to the durations of total hole-pokes or for pokes made into odorant-containing or empty holes for either test session (data not shown). These findings along with the similar hole-poking performances of the groups before familiarization with the chocolate provide evidence that the groups did not differ in their abilities to discriminate odors. Rather, our holeboard results suggest that mutations in CELF6 may contribute autism risk by influencing the propensity to modify behavior in response to a rewarding experience. More generally, our results provide evidence that Celf6 contributes to a subset of the autism-like behaviors that were assayed in the present study.
Celf6−/− mice have abnormal levels of CNS serotonin
Finally, our initial bacTRAP screen was to identify genes that may be important for the regulation of the serotonergic system in the brain (Figs. 1, 2), and we determined that Celf6 protein in mice was indeed found fairly specifically in monoamine-containing neurons within the brainstem (Fig. 4). To determine whether the changes in behavior seen might indeed be the result of perturbations of monoaminergic systems, we used mass spectrometry to measure total levels of three monoamines in the brains of Celf6−/− and WT mice. We observed a 30 ± 4% (mean ± SEM) decrease in serotonin levels (p < 0.002, Bonferroni corrected), and similar trends in norepinephrine and dopamine (p < 0.08, < 0.04, respectively, before Bonferroni corrections, not significant after) in brains of Celf6−/− mice (Fig. 10). There was no effect on levels of the GABA, glycine, or glutamate (not shown) neurotransmitters (p > 0.88, p > 0.23, and p > 0.28, respectively, no Bonferroni correction), demonstrating the specificity of the effect to neurons previously seen to be expressing Celf6.
Figure 10.
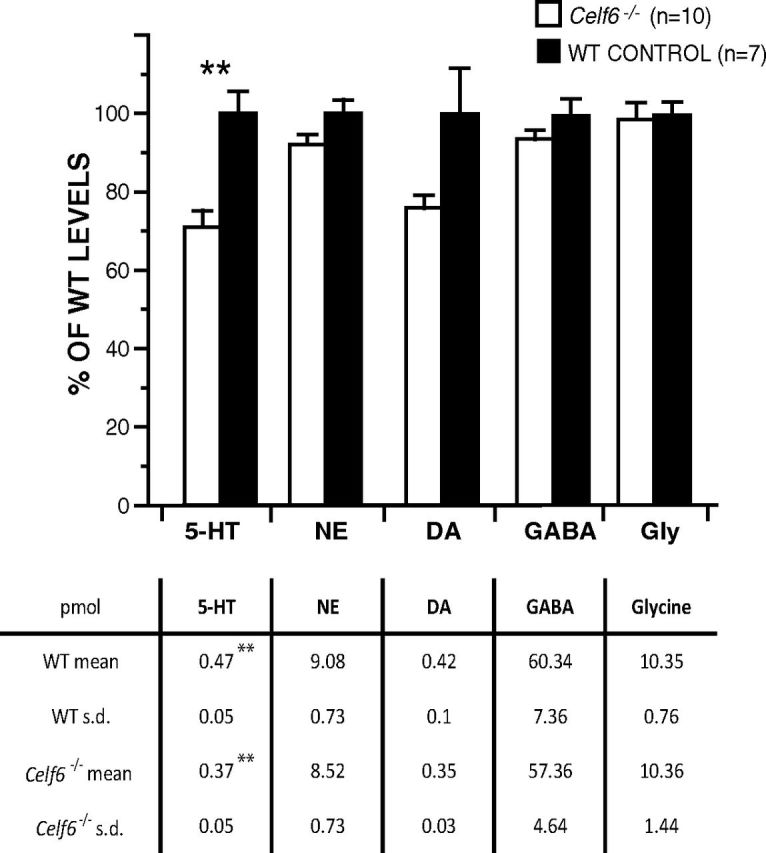
Celf6−/− mice show decreased brain serotonin levels. LC-MS/MS measures of levels of neuromodulatory neurotransmitters serotonin (5HT), norepinephrine (NE), and dopamine (DA) reveal a significant decrease (**p < 0.002) in 5HT levels, and nonsignificant trend for NE and DA in whole brains from n > 7 WT and Celf6−/− mice. There is no change in the negative control transmitters GABA and glycine. Data are plotted as a percentage of WT ion count levels; error bars represent SEM. Tables provide absolute levels in pMol.
Discussion
The paradigms of the previous era of human genetics, typified by observations of a suspicious looking variant in a few families and nothing similar in a few hundred families, are insufficient to cope with the overwhelming catalogs of variants being discovered now (Neale et al., 2012; O'Roak et al., 2012; Sanders et al., 2012). Results like those reported here are becoming standard: an inherited variant within a compelling candidate gene is observed only once in thousands of families. This highlights the need not only to increase our sample sizes, but also to find new analytical approaches to this complexity to overcome issues of power. We propose here that a broader adoption of blended strategies, drawing simultaneously on statistical genetics and empirical biology, are required.
There is a growing consensus that many psychiatric disorders are strongly influenced by a diverse set of rare, nearly private mutations (McClellan and King, 2010; Pinto et al., 2010). Thus, it is likely that there are multiple genetic routes to developing a particular disorder, or even individual symptoms within a disorder. Yet these diverse genetic routes must converge at some smaller number of common pathways in their molecular or cellular neurobiology to engender a common behavioral phenotype. Therefore, we have combined a unique and unbiased method for generating key biological priors in mice, with analysis of genetic variation in humans, and functional validation in murine models, to identify a gene contributing to some of the essential diagnostic features of autism. We propose that this kind of paradigm, leveraging information at multiple analytical levels, may be required to dissect complex genetic disorders. By working from the hypotheses that serotonergic abnormalities may contribute to a reasonable fraction of the cases of autism, we were able to reduce the penalties taken by looking at common variation genome-wide, as well as appropriately direct our functional assays in mice. More broadly, although the evidence linking serotonergic neurons to some cases of autism is alluring, the true extent of the contribution of this cell type remains to be established. Several lines of evidence suggest that a subset of cases of autism may involve a perturbation of the serotonergic system. Multiple studies have detected abnormally high levels of serotonin in whole blood of a subset (>25%) of autistic patients (Schain and Freedman, 1961; Takahashi et al., 1976; Anderson et al., 1987; Cook, 1990). A combination of genetic investigation, neural imaging, and drug treatment have all implicated corresponding dysregulation of serotonin-related genes and pathways in affected individuals (McDougle et al., 1996b, 1997; McDougle et al., 1998; Chugani et al., 1999; Chandana et al., 2005; Coon et al., 2005; Hollander et al., 2005). For example, selective serotonin reuptake inhibitors have been shown to alleviate rigid/compulsive behaviors in some cases (Gordon et al., 1993; McDougle et al., 1996b; Hollander et al., 2011) and depletion of serotonin in autistic individuals exacerbates these symptoms (McDougle et al., 1996a). Likewise, genetic or pharmacological perturbation of the serotonergic system in rodents alters behavioral responses which characterize animal models of autism, such as altered USV, repetitive behavior, social interaction, and resistance to change (Olivier et al., 1998; Fish et al., 2000; Lopez-Rubalcava et al., 2000; Whitaker-Azmitia, 2001; McNamara et al., 2008; Nakatani et al., 2009). Most recently, Veenstra-VanderWeele et al. (2012) have shown that introduction of an autism-associated human coding variant in the serotonin transporter gene into mice resulted in a number of autism-related behaviors, such as impaired USV, decreased social dominance, and increased repetitive behaviors.
Our behavioral results suggest that, although Celf6−/− and WT mice performed similarly on several different tests, the Celf6−/− mice exhibited certain selective functional deficits that may be analogous to the behavioral impairments that exist in a subset of people with autism. Specifically, Celf6−/− and WT mice performed similarly on several behavioral assays, including: a 1-h locomotor activity test; a battery of sensorimotor measures; spatial learning and memory performance in the Morris water maze; various tests of social interaction as indexed by the social approach test; and anxiety-like behaviors quantified in the elevated plus maze. However, reduction of isolation-induced USV suggests that Celf6−/− pups show early communication deficits. The measurement of USV in response to maternal isolation is an established assay that has been used in several other mouse models of autism, including the BTBR, Shank1 null, and neuroligin models (Chadman et al., 2008; Scattoni et al., 2008; Nakatani et al., 2009). Although genotypic differences in pup USV may be the result of either communication deficits or changes to anxiety, we feel the latter is unlikely as adult Celf6−/− mice did not exhibit differences compared with littermate WT control mice in anxiety-like behaviors measured during elevated plus maze testing. However, we acknowledge that it will be important to evaluate vocalization in Celf6−/− mice at later stages, such as during juvenile play and adult mating, and this is one of our future research aims. Our behavioral data also suggest that Celf6−/− mice react abnormally to certain environmental alterations and may be resistant to changing their behavior under conditions that robustly alter WT mouse behavior. For example, Celf6−/− swam faster than controls when first introduced into the water maze during cued trials, but this effect habituated and was not present during the place condition. Also, familiarizing the mice with chocolate chips during the holeboard exploration/olfactory preference test led to robust changes in the behavioral responses of the WT control group, whereas Celf6−/− mice showed a resistance to such changes. Specifically, familiarization with the chocolate in WT mice led to greatly increased exploratory hole poking and distinct olfactory preferences for several odorants, whereas Celf6−/− showed no such changes. These results are similar to those reported by Moy et al., 2008 who used a slightly different procedure to show that BTBR mice exhibit a similar resistance to change after familiarization with chocolate. Our holeboard results provide evidence that mutations in CELF6 may promote autism risk by modifying behavioral responses to rewarding experiences. However, it will be necessary to further characterize the resistance to change in olfactory preference in Celf6−/− mice after exposure to various rewarding odorant stimuli, and we are conducting additional studies to this end. Our initial behavioral data suggest that Celf6−/− mice exhibit selective deficits and may serve as a model for understanding a subset of autism-like behaviors.
Still, additional mechanistic studies are needed to inform both the Celf6 and the serotonergic contribution to autism in general. Although Celf6 is clearly enriched in these cells, and serotonin is decreased in the brains of Celf6−/− mice, conditional deletion and rescue studies are needed to confirm the behavioral abnormalities are cell-autonomous to the serotonergic system. Likewise, although we have provided evidence for this gene at multiple levels (a common 5′UTR variant and a rare heterozygous stop codon in humans, and homozygous deletions in mouse), each of these levels warrants additional investigation to generate a comprehensive view of the contribution of this gene. Although the common variant is linked with autism in the AGRE sample, this finding does not replicate in the Simons Simplex Collection (J. K. Lowe, personal communication), although that sample was smaller and optimized for finding idiopathic and especially de novo events rather than inherited, familial causes. The most parsimonious interpretation of our CELF6 findings thus far is that CELF6 levels may influence some behaviors relevant to autism; thus, there are alleles in this gene that contribute to risk of developing autism in a subset of patients. This would be consistent with autism being a highly heterogeneous and polygenic disorder; there are likely to be many genes with this type and magnitude of effect. This may also explain the incomplete penetrance seen in the family with the CELF6 mutation: resistance to change, in the absence of social deficits, would be insufficient to generate a diagnosis for autism. This would be consistent with recent studies, which suggest that the restricted interests and sociability components are factorially separable and partially genetically independent (Happe and Ronald, 2008; Frazier et al., 2012; Robinson et al., 2012). Thus, other genes and cell types likely contribute more to the social symptoms. An unbiased screen using this same approach systematically across many cell types may permit inferences regarding circuitry important to the neurobiology of autism.
Footnotes
This work was supported by the Howard Hughes Medical Institute (N.H.), the Simons Foundation (N.H.), the Mallinkrodt Foundation (J.D.D.), and National Institutes of Health Grant 4R00NS067239-03 to J.D.D. and Grant P30 HD062171 to D.F.W. We thank Wenxiang Zhang, Connie Zhao, Rada Norinsky, Chingwen Yang, Sujata Bupp, Wendy Yang, Rajiv Shah, Eric Klein, Nilambari Pisat, A. Gulhan Ercan-Sencicek, and Afua Akuffo for their assistance. We thank Shiaoching Gong, Jennifer Stone, John Constantino, Tim Holy, Terra Barnes, and Don Conrad for advice. We gratefully acknowledge the resources provided by the Autism Genetic Resource Exchange (AGRE) Consortium and the participating AGRE families. AGRE is a program of Autism Speaks and National Institute of Mental Health Grant 1U24MH081810 [Clara M. Lajonchere, Principal Investigator (PI)]. We are equally grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, and PIs (A. Beaudet, R. Bernier, J. Constantino, E. Cook, E. Fombonne, D. Geschwind, R. Goin-Kochel, E. Hanson, D. Grice, A. Klin, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, E. Wijsman).
The authors declare no competing financial interests.
References
- Abrahams BS, Geschwind DH. Genetics of autism. In: Speicher MR, Antonarakis SE, Motulsky AG, editors. Vogel and Motulsky's human genetics: problems and approaches. Ed 4. Berlin: Springer; 2010. pp. 699–714. [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. Ed 4. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Anderson GM, Freedman DX, Cohen DJ, Volkmar FR, Hoder EL, McPhedran P, Minderaa RB, Hansen CR, Young JG. Whole blood serotonin in autistic and normal subjects. J Child Psychol Psychiatry. 1987;28:885–900. doi: 10.1111/j.1469-7610.1987.tb00677.x. [DOI] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Méreau A, Osborne HB. Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie. 2006;88:515–525. doi: 10.1016/j.biochi.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Bengel D, Jöhren O, Andrews AM, Heils A, Mössner R, Sanvitto GL, Saavedra JM, Lesch KP, Murphy DL. Cellular localization and expression of the serotonin transporter in mouse brain. Brain Res. 1997;778:338–345. doi: 10.1016/s0006-8993(97)01080-9. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci. 2006;26:1971–1978. doi: 10.1523/JNEUROSCI.2173-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, Kim C, Gidaya NB, Lindquist I, Hutman T, Sigman M, Kustanovich V, Lajonchere CM, Singleton A, Kim J, Wassink TH, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canli T, Lesch KP. Long story short: the serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10:1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- Chadman KK, Gong S, Scattoni ML, Boltuck SE, Gandhy SU, Heintz N, Crawley JN. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 2008;1:147–158. doi: 10.1002/aur.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandana SR, Behen ME, Juhász C, Muzik O, Rothermel RD, Mangner TJ, Chakraborty PK, Chugani HT, Chugani DC. Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int J Dev Neurosci. 2005;23:171–182. doi: 10.1016/j.ijdevneu.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Chapple JP, Anthony K, Martin TR, Dev A, Cooper TA, Gallo JM. Expression, localization and tau exon 10 splicing activity of the brain RNA-binding protein TNRC4. Hum Mol Genet. 2007;16:2760–2769. doi: 10.1093/hmg/ddm233. [DOI] [PubMed] [Google Scholar]
- Charlet-B N, Logan P, Singh G, Cooper TA. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell. 2002;9:649–658. doi: 10.1016/s1097-2765(02)00479-3. [DOI] [PubMed] [Google Scholar]
- Chonchaiya W, Schneider A, Hagerman RJ. Fragile X: a family of disorders. Adv Pediatr. 2009;56:165–186. doi: 10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J, Chugani HT. Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol. 1999;45:287–295. doi: 10.1002/1531-8249(199903)45:3<287::aid-ana3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Collins A. Linkage disequilibrium and association mapping: analysis and applications. Totowa, NJ: Humana; 2007. [Google Scholar]
- Cook EH. Autism: review of neurochemical investigation. Synapse. 1990;6:292–308. doi: 10.1002/syn.890060309. [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, Haas R, Courchesne E, Leventhal BL. Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry. 1997;2:247–250. doi: 10.1038/sj.mp.4000266. [DOI] [PubMed] [Google Scholar]
- Coon H, Dunn D, Lainhart J, Miller J, Hamil C, Battaglia A, Tancredi R, Leppert MF, Weiss R, McMahon W. Possible association between autism and variants in the brain-expressed tryptophan hydroxylase gene (TPH2) Am J Med Genet B Neuropsychiatr Genet. 2005;135B:42–46. doi: 10.1002/ajmg.b.30168. [DOI] [PubMed] [Google Scholar]
- Deneris ES, Wyler SC. Serotonergic transcriptional networks and potential importance to mental health. Nat Neurosci. 2012;15:519–527. doi: 10.1038/nn.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, Heintz N. Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 2010;38:4218–4230. doi: 10.1093/nar/gkq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin R, Jing Q, O'Broin P, Calder B, McLellan A, Moskowitz D, Suzuki M, Greally JM. WASP: Wiki-based Automated Sequence Processor for Epigenomics and Genomics Applications. J Biomol Techniques. 2010;21:S11. [Google Scholar]
- Eddy SR. Where did the BLOSUM62 alignment score matrix come from? Nat Biotechnol. 2004;22:1035–1036. doi: 10.1038/nbt0804-1035. [DOI] [PubMed] [Google Scholar]
- Fish EW, Sekinda M, Ferrari PF, Dirks A, Miczek KA. Distress vocalizations in maternally separated mouse pups: modulation via 5-HT(1A), 5-HT(1B) and GABA(A) receptors. Psychopharmacology (Berl) 2000;149:277–285. doi: 10.1007/s002130000370. [DOI] [PubMed] [Google Scholar]
- Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66(suppl 10):3–8. [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Frazier TW, Youngstrom EA, Speer L, Embacher R, Law P, Constantino J, Findling RL, Hardan AY, Eng C. Validation of proposed DSM-5 criteria for autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2012;51:28–40.e3. doi: 10.1016/j.jaac.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12:2–22. doi: 10.1038/sj.mp.4001896. [DOI] [PubMed] [Google Scholar]
- Gallitano-Mendel A, Wozniak DF, Pehek EA, Milbrandt J. Mice lacking the immediate early gene Egr3 respond to the anti-aggressive effects of clozapine yet are relatively resistant to its sedating effects. Neuropsychopharmacology. 2008;33:1266–1275. doi: 10.1038/sj.npp.1301505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ. The autism genetic resource exchange: a resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69:463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45:395–408. doi: 10.1016/j.nbd.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good PJ, Chen Q, Warner SJ, Herring DC. A family of human RNA-binding proteins related to the Drosophila Bruno translational regulator. J Biol Chem. 2000;275:28583–28592. doi: 10.1074/jbc.M003083200. [DOI] [PubMed] [Google Scholar]
- Gordon CT, State RC, Nelson JE, Hamburger SD, Rapoport JL. A double-blind comparison of clomipramine, desipramine, and placebo in the treatment of autistic disorder. Arch Gen Psychiatry. 1993;50:441–447. doi: 10.1001/archpsyc.1993.01820180039004. [DOI] [PubMed] [Google Scholar]
- Goridis C, Rohrer H. Specification of catecholaminergic and serotonergic neurons. Nat Rev Neurosci. 2002;3:531–541. doi: 10.1038/nrn871. [DOI] [PubMed] [Google Scholar]
- Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR. Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J Neurosci. 2006;26:2841–2851. doi: 10.1523/JNEUROSCI.4823-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Cooper TA. Identification of CELF splicing activation and repression domains in vivo. Nucleic Acids Res. 2005;33:2769–2780. doi: 10.1093/nar/gki561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happé F, Ronald A. The ‘fractionable autism triad’: a review of evidence from behavioural, genetic, cognitive and neural research. Neuropsychol Rev. 2008;18:287–304. doi: 10.1007/s11065-008-9076-8. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L, Holtzman DM. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillarp NA, Fuxe K, Dahlström A. Demonstration and mapping of central neurons containing dopamine, noradrenaline, and 5-hydroxytryptamine and their reactions to psychopharmaca. Pharmacol Rev. 1966;18:727–741. [PubMed] [Google Scholar]
- Hofer MA, Shair HN, Brunelli SA. Ultrasonic vocalizations in rat and mouse pups. Curr Protoc Neurosci Chapter. 2002;8 doi: 10.1002/0471142301.ns0814s17. Unit 8.14. [DOI] [PubMed] [Google Scholar]
- Hollander E, Phillips A, Chaplin W, Zagursky K, Novotny S, Wasserman S, Iyengar R. A placebo controlled crossover trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology. 2005;30:582–589. doi: 10.1038/sj.npp.1300627. [DOI] [PubMed] [Google Scholar]
- Hollander E, Soorya L, Chaplin W, Anagnostou E, Taylor BP, Ferretti CJ, Wasserman S, Swanson E, Settipani C. A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. Am J Psychiatry. 2012;169:292–299. doi: 10.1176/appi.ajp.2011.10050764. [DOI] [PubMed] [Google Scholar]
- Holy TE, Guo Z. Ultrasonic songs of male mice. PLoS Biol. 2005;3:e386. doi: 10.1371/journal.pbio.0030386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Ishimura K, Takeuchi Y, Fujiwara K, Tominaga M, Yoshioka H, Sawada T. Quantitative analysis of the distribution of serotonin-immunoreactive cell bodies in the mouse brain. Neurosci Lett. 1988;91:265–270. doi: 10.1016/0304-3940(88)90691-x. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey: I. Lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29:85–96. doi: 10.1016/0165-0327(93)90026-g. [DOI] [PubMed] [Google Scholar]
- Korte SM, Meijer OC, de Kloet ER, Buwalda B, Keijser J, Sluyter F, van Oortmerssen G, Bohus B. Enhanced 5-HT1A receptor expression in forebrain regions of aggressive house mice. Brain Res. 1996;736:338–343. doi: 10.1016/0006-8993(96)00723-8. [DOI] [PubMed] [Google Scholar]
- Ladd AN, Nguyen NH, Malhotra K, Cooper TA. CELF6, a member of the CELF family of RNA-binding proteins, regulates muscle-specific splicing enhancer-dependent alternative splicing. J Biol Chem. 2004;279:17756–17764. doi: 10.1074/jbc.M310687200. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Little S. Current Protocols in Human Genetics. New York: Wiley; 2001. Amplification-refractory mutation system (ARMS) analysis of point mutations. [DOI] [PubMed] [Google Scholar]
- López-Rubalcava C, Hen R, Cruz SL. Anxiolytic-like actions of toluene in the burying behavior and plus-maze tests: differences in sensitivity between 5-HT(1B) knockout and wild-type mice. Behav Brain Res. 2000;115:85–94. doi: 10.1016/s0166-4328(00)00241-2. [DOI] [PubMed] [Google Scholar]
- Luo R, Sanders SJ, Tian Y, Voineagu I, Huang N, Chu SH, Klei L, Cai C, Ou J, Lowe JK, Hurles ME, Devlin B, State MW, Geschwind DH. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am J Hum Genet. 2012;91:38–55. doi: 10.1016/j.ajhg.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- McDougle CJ, Naylor ST, Cohen DJ, Aghajanian GK, Heninger GR, Price LH. Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch Gen Psychiatry. 1996a;53:993–1000. doi: 10.1001/archpsyc.1996.01830110029004. [DOI] [PubMed] [Google Scholar]
- McDougle CJ, Naylor ST, Cohen DJ, Volkmar FR, Heninger GR, Price LH. A double-blind, placebo-controlled study of fluvoxamine in adults with autistic disorder. Arch Gen Psychiatry. 1996b;53:1001–1008. doi: 10.1001/archpsyc.1996.01830110037005. [DOI] [PubMed] [Google Scholar]
- McDougle CJ, Epperson CN, Price LH, Gelernter J. Evidence for linkage disequilibrium between serotonin transporter protein gene (SLC6A4) and obsessive compulsive disorder. Mol Psychiatry. 1998;3:270–273. doi: 10.1038/sj.mp.4000391. [DOI] [PubMed] [Google Scholar]
- McNamara IM, Borella AW, Bialowas LA, Whitaker-Azmitia PM. Further studies in the developmental hyperserotonemia model (DHS) of autism: social, behavioral and peptide changes. Brain Res. 2008;1189:203–214. doi: 10.1016/j.brainres.2007.10.063. [DOI] [PubMed] [Google Scholar]
- Moldin SO, Rubenstein JLR. Understanding autism: from basic neuroscience to treatment. Boca Raton, FL: CRC/Taylor and Frances; 2006. [Google Scholar]
- Montgomery KT, Lartchouck O, Li L, Perera A, Yassin Y, Tamburino A, Loomis S, Kucherlapati R. Current protocols in human genetics. New York: Wiley; 2008. Mutation detection using automated fluorescence-based sequencing. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Poe MD, Nonneman RJ, Young NB, Koller BH, Crawley JN, Duncan GE, Bodfish JW. Development of a mouse test for repetitive, restricted behaviors: relevance to autism. Behav Brain Res. 2008;188:178–194. doi: 10.1016/j.bbr.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naert A, Callaerts-Vegh Z, D'Hooge R. Nocturnal hyperactivity, increased social novelty preference and delayed extinction of fear responses in post-weaning socially isolated mice. Brain Res Bull. 2011;85:354–362. doi: 10.1016/j.brainresbull.2011.03.027. [DOI] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, Iwamoto K, Kato T, Okazawa M, Yamauchi K, Tanda K, Takao K, Miyakawa T, Bradley A, Takumi T. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–13 duplication seen in autism. Cell. 2009;137:1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier B, Molewijk HE, van der Heyden JA, van Oorschot R, Ronken E, Mos J, Miczek KA. Ultrasonic vocalizations in rat pups: effects of serotonergic ligands. Neurosci Biobehav Rev. 1998;23:215–227. doi: 10.1016/s0149-7634(98)00022-0. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DT, Kuti OJ, Sur M. Computerized assessment of social approach behavior in mouse. Front Behav Neurosci. 2009;3:48. doi: 10.3389/neuro.08.048.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Pfaar H, von Holst A, Vogt Weisenhorn DM, Brodski C, Guimera J, Wurst W. mPet-1, a mouse ETS-domain transcription factor, is expressed in central serotonergic neurons. Dev Genes Evol. 2002;212:43–46. doi: 10.1007/s00427-001-0208-x. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bölte S, Bolton PF, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobbe RL, Pearson BL, Defensor EB, Bolivar VJ, Young WS, 3rd, Lee HJ, Blanchard DC, Blanchard RJ. Oxytocin receptor knockout mice display deficits in the expression of autism-related behaviors. Horm Behav. 2012;61:436–444. doi: 10.1016/j.yhbeh.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon y Cajal S, Pasik P, Pasik T. Texture of the nervous system of man and the vertebrates. New York: Springer; 1899. [Google Scholar]
- Robinson EB, Koenen KC, McCormick MC, Munir K, Hallett V, Happé F, Plomin R, Ronald A. A multivariate twin study of autistic traits in 12-year-olds: testing the fractionable autism triad hypothesis. Behav Genet. 2012;42:245–255. doi: 10.1007/s10519-011-9500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Gandhy SU, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PloS One. 2008;3:e3067. doi: 10.1371/journal.pone.0003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, Nardi A, Hartman RE, Vogt SK, Luedke CE, Storm DR, Muglia LJ. Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J Neurosci. 2000;20:4809–4820. doi: 10.1523/JNEUROSCI.20-13-04809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schain RJ, Freedman DX. Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatr. 1961;58:315–320. doi: 10.1016/s0022-3476(61)80261-8. [DOI] [PubMed] [Google Scholar]
- Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY, Sheng M, Crawley JN. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011;1380:120–137. doi: 10.1016/j.brainres.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- Steinbusch HW. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience. 1981;6:557–618. doi: 10.1016/0306-4522(81)90146-9. [DOI] [PubMed] [Google Scholar]
- Steinbusch HW. Serotonin-immunoreactive neurons and their projections in the CNS. In: Björklund A, Hökfelt T, Kuhar MJ, editors. Handbook of chemical neuroanatomy, Vol 3: Classical transmitters and transmitter receptors in the CNS, Part II. Amsterdam: Elsevier; 1984. pp. 1pp. 4pp. 6–16.–18.–20. [Google Scholar]
- Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, Nelson SF. Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet. 2004;75:1117–1123. doi: 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kanai H, Miyamoto Y. Reassessment of elevated serotonin levels in blood platelets in early infantile autism. J Autism Child Schizophr. 1976;6:317–326. doi: 10.1007/BF01537909. [DOI] [PubMed] [Google Scholar]
- Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol. 2005;25:10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Anderson GM, Cook EH., Jr Pharmacogenetics and the serotonin system: initial studies and future directions. Eur J Pharmacol. 2000;410:165–181. doi: 10.1016/s0014-2999(00)00814-1. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, Cohen J, Mannangatti P, Jessen T, Thompson BJ, Ye R, Kerr TM, Carneiro AM, Crawley JN, Sanders-Bush E, McMahon DG, Ramamoorthy S, Daws LC, Sutcliffe JS, Blakely RD. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci U S A. 2012;109:5469–5474. doi: 10.1073/pnas.1112345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Ding F, Chiang H, Thompson RC, Watson SJ, Meng F. ProbeMatchDB: a web database for finding equivalent probes across microarray platforms and species. Bioinformatics. 2002;18:488–489. doi: 10.1093/bioinformatics/18.3.488. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res Bull. 2001;56:479–485. doi: 10.1016/s0361-9230(01)00615-3. [DOI] [PubMed] [Google Scholar]
- Wilming LG, Gilbert JG, Howe K, Trevanion S, Hubbard T, Harrow JL. The vertebrate genome annotation (Vega) database. Nucleic Acids Res. 2008;36:D753–D760. doi: 10.1093/nar/gkm987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wylie CJ, Hendricks TJ, Zhang B, Wang L, Lu P, Leahy P, Fox S, Maeno H, Deneris ES. Distinct transcriptomes define rostral and caudal serotonin neurons. J Neurosci. 2010;30:670–684. doi: 10.1523/JNEUROSCI.4656-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhas J, Cordeiro L, Tassone F, Ballinger E, Schneider A, Long JM, Ornitz EM, Hessl D. Brief report: sensorimotor gating in idiopathic autism and autism associated with fragile X syndrome. J Autism Dev Disord. 2011;41:248–253. doi: 10.1007/s10803-010-1040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]